

Abstract
The vasoconstrictor peptide endothelin (ET-1) exerts its physiological and pathological effects via activation of ETA and ETB receptor (ET-R) subtypes. In this study, we demonstrate that both ET-R subtypes are highly expressed in rat astrocytes in vivo, indicating that these cells are potential targets of the biological effects of ET-1 in the brain. In cultured cortical astrocytes, both ET-R subtypes are expressed, and selective stimulation of ETB-R with ET-1 induces phosphorylation of cAMP response element-binding protein (CREB). The signal transduction pathway activated by ET-1 includes the Rap1/B-Raf and the Ras/Raf-1 complexes, protein kinase C (PKC) together with extracellular signal-regulated kinases (ERK), and the ribosomal S6 kinase (RSK) isoforms RSK2 and RSK3, two kinases that lie immediately downstream of ERK and are able to phosphorylate CREB. Moreover, ET-1 activates the p38 mitogen-activated protein kinase (MAPK)-dependent, but not the c-jun N-terminal kinase (JNK)-dependent pathway. By using selective protein kinase inhibitors and expression of dominant-negative Rap1 protein, we also found that the Rap1/PKC/ERK-dependent pathway induces the phosphorylation of activating transcription factor-1, CREB, and Elk-1, whereas the p38MAPK-dependent pathway only causes CREB phosphorylation. ET-1-induced transcription of the immediate early genec-fos requires the concomitant activation of both the PKC/ERK- and p38MAPK-dependent pathways, because inhibitors of either pathway block the ET-1-induced increase of c-fos mRNA. Our findings indicate that changes in the expression of cAMP response element-dependent immediate and delayed response genes could play a pivotal role in the physiological effects elicited by ET-1 in astrocytes.
Keywords: glia, Rap1, Elk-1, protein kinase C, Raf kinase, ribosomal S6 kinase
The three endothelin peptides (ET-1, ET-2, and ET-3) are the most powerful vasoconstrictive agents and are involved in several physiological activities, either in vascular or nonvascular tissue (Masaki et al., 1992). In the mammalian CNS, ETs regulate cerebral blood vessel function, modulate neuronal activity, and are involved in distinct physiological and pathological responses (Kuwaki et al., 1997).
The ET system plays a crucial role in the physiology and pathology of glial cells, because ET synthesis is increased in astrocytes during proliferation, differentiation, and after brain lesions such as infarct, transient forebrain ischemia, and neurodegeneration (Nie and Olsson, 1996). Moreover, the role of ETs seems to be particularly intriguing in brain tumors, because peritumoral reactive astrocytes display increased ET immunoreactivity (Zhang and Olsson, 1997). These findings indicate that ETs may act as transmitters or growth factors in the brain during development, injury, and regeneration.
The effects of ETs are mediated by two distinct receptors (ETA-R and ETB-R) coupled to heterotrimeric G-proteins. Stimulation of ET-Rs causes inositol phosphate turnover, intracellular calcium mobilization, activation of protein kinase C (PKC), inhibition of intracellular cAMP accumulation, and activation of different mitogen-activated protein kinase (MAPK)-dependent signaling pathways (Rubanyi and Polokoff, 1994).
Previous studies have shown that ET-1 activates the extracellular signal-regulated kinase (ERK) in astrocytes (Cazaubon et al., 1997) and causes phosphorylation of the transcription factor cAMP response element-binding protein (CREB) in Schwann cells (Tabernero et al., 1998) and in human melanocytes (Böhm et al., 1995). Furthermore, ETs increase the rate of transcription of the immediate early gene (IEG) c-fos in both astrocytes (Ladenheim et al., 1993;Leach et al., 1999) and the glioma C6 cell line (Yin et al., 1992;Ladenheim et al., 1993).
Activation of the ERK signaling pathway induces c-fostranscription by primarily regulating two cis elements in the c-fos promoter, the cAMP response element (CRE) and the serum response element (SRE) (Karin et al., 1997). The two transcription factors that bind to CRE, CREB, and activating transcription factor-1 (ATF-1) can be indirectly phosphorylated by MAPKs via the ribosomal protein kinases S6 RSK1, RSK2, and RSK3 (Frodin and Gammeltoft, 1999). In contrast, activation and subsequent nuclear translocation of MAPKs, such as the c-jun N-terminal protein kinase (JNK), p38MAPK, and ERKs, may directly phosphorylate the SRE-bound ternary complex factor Elk-1 (Mielke and Herdegen, 2000).
The physiological and pathological responses elicited by ETs in astrocytes ultimately require changes in the expression of delayed response genes, whose transcription is modulated by the IEGc-fos (Karin et al., 1997). Therefore, understanding the signal transduction pathways associated with the activation of ET-Rs and responsible for c-fos induction is of pivotal importance to delineate the effects of ETs on astrocyte function. These signal transduction pathways have been investigated predominantly in cell lines, and it is unknown whether they are also active in glia under more physiological conditions.
The purpose of this study was first to define the expression of ET-Rs in astrocytes in vivo and in vitro. Second, we wanted to elucidate the intracellular signaling cascade triggered by ET-R activation in astrocytes, in particular the MAPK-dependent pathways and the transcription factors involved in ET-inducedc-fos transcription.
MATERIALS AND METHODS
Materials. Anti-ET-R antibodies were from Alexis Corporation (San Diego, CA). Antibodies against S100-β were from Sigma (St. Louis, MO). The TSA Fluorescence System kits (fluorescein green or tetramethylrhodamine red) were from NEN (Boston, MA). The antibodies directed against phosphorylated Ser 133 CREB (P-CREB), phosphorylation state-independent CREB, and B-Raf, together with glutathione S-transferase (GST)-Raf-1-Rac-binding domain (RBD) agarose beads and GST- MAP kinase kinase (MEK), were purchased from Upstate Biotechnology (Lake Placid, NY). The following antibodies were all from Cell Signaling Technology (Beverly, MA): dually phosphorylated (Thr 202/Tyr 204) ERK, phosphorylation state-independent ERK, dually phosphorylated (Ser 217/Ser 221) MEK1/2, phosphorylation state-independent MEK1/2, phosphorylated (Ser 381) p90RSK, dually phosphorylated (Thr 180/Tyr 182) p38MAPK, phosphorylation state-independent p38MAPK, dually phosphorylated (Thr 183/Tyr 185) JNK, phosphorylation state-independent JNK, phosphorylated (Ser 383) Elk-1, and phosphorylation state-independent Elk-1. Polyclonal anti-Rap1 (sc-65), anti-B-Raf (sc-166), anti-Raf-1 (sc-133), anti-hemoagglutinin (HA) (sc-805), anti-RSK1 (sc-231), anti-RSK2 (sc-1430), anti-RSK3 (sc-1431), anti-ATF-1 (sc-243), and anti-Elk-1 (sc-355) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal antibodies directed against Rap1 and Ras were from Transduction Laboratories (Lexington, KY). Trizol, fetal bovine serum, horse serum, and all reagents for cell cultures were from Life Technologies (Gaithersburg, MD). The protein kinase inhibitors calmidazolium chloride and KN-62 were purchased fromBiomol (Plymouth Meeting, PA). Gö6369, U-73122, and PD98019 were from Alexis Corporation. All of the other reagents were purchased from Sigma. Protein A agarose and GST-Sepharose beads were from Amersham Pharmacia Biotech (Uppsala, Sweden). The bacterial lysate containing the GST fusion protein of the minimal Rap1-binding domain of ralGDS (GST-ral-RBD) was a generous gift from Dr. M. Freissmuth (Institute of Pharmacology, University of Vienna, Vienna, Austria).
Cell cultures. Purified astrocyte cultures were prepared from 20-d-old Sprague Dawley rat embryos. The animals were killed following the NIH Animal Welfare guidelines. Cortices were dissected and then mechanically dissociated by means of a fire-polished Pasteur pipette. Cells were then plated in 60 mm tissue culture dishes in DMEM high-glucose medium containing 2 mm glutamine, 5% fetal bovine serum, and 5% horse serum. Approximately 24 hr after plating, the medium was completely replaced, and the cells were grown for 10 d in vitro (DIV) with a complete medium change every 48 hr. The cultures comprised >95% glial fibrillary acidic protein (GFAP)-positive cells (Gallo and Armstrong, 1995).
Immunocytochemistry in tissue sections and cultured cells.Rats were perfused with 4% paraformaldehyde, and brains were dissected out. For double immunostaining, vibratome tissue sections (sagittal; 35 μm) were prepared from postnatal day 8 (P8) and P30 rats. Sections were incubated with 50% methanol–6% H2O2 in PBS for 20 min, followed by 0.2% Triton X-100 in PBS for 30 min. Sections were then incubated with TNB blocking buffer (0.1 mTris-HCl, pH 7.5, 0.15 m NaCl, and 0.5% blocking reagent from the TSA Fluorescence System kit) for 60 min at room temperature, followed by an incubation with anti ET-R antibodies (1:50 dilution) in combination with anti-S100-β antibodies for 12 hr at 4°C. Tissue sections were then washed with TNT buffer (3m Tris-HCl, pH 7.5, 0.15 mNaCl, and 0.05% Tween 20) and incubated with rabbit and sheep biotin and FITC-goat anti-mouse IgG for 2 hr at room temperature. After extensive washing, TNT buffer sections were incubated with streptavidin-HRP (1:100) for 60 min at room temperature, washed, and incubated with tetramethylrhodamine tyramide reagent (red) or fluorescein tyramide reagent (green). After extensive washing, sections were finally mounted in Vectashield (Vector Laboratories, Burlingame, CA).
For staining of cell cultures, anti-S100-β antibodies and anti-ETA-R and ETB-R antibodies were used. Double-indirect immunofluorescence experiments were performed as described previously (Gallo and Armstrong, 1995). All secondary fluorochrome-conjugated antibodies were from Organon Teknika-Cappel (Durham, NC). Cells were then fixed in 4% paraformaldehyde and 0.2% glutaraldehyde (pH 7.4, in PBS) for 15 min, permeabilized in 95% ethanol–5% acetic acid for 10 min at −20°C, and incubated with anti-S100-β antibodies (1: 200) for 1 hr at room temperature. Cells were then incubated with anti-ETA-R and ETB-R antibodies (1: 50) overnight at 4°C. After incubation with anti-sheep fluorescein-conjugated IgG for 60 min, cells were mounted in Vectashield. Controls for antibody specificity were performed by sequentially omitting each of the primary antibodies in the immunostaining protocols. The immunofluorescence micrographs presented are representative of at least two experiments.
Immunoprecipitation and Western blots. For immunoprecipitation, cells were first rinsed twice with ice-cold PBS and then lysed for 45 min on ice in 500 μl of ice-cold lysis buffer (50 mm Tris-HCl, pH 7.5, 140 mm NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mm sodium-orthovanadate, 1 mm NaF, 1 mmphenylmethylsulfonylfluoride (PMSF), 2 μg/ml aprotinin, 2 μg/ml pepstatin, 2 μg/ml leupeptin, and 1 μmmicrocystin-LR). The lysates were centrifuged at 16,000 ×g for 5 min at 4°C, and aliquots were taken for protein determination using the Pierce (Rockford, IL) BCA protein assay kit. The supernatants containing equal protein amounts (500 μg) were then incubated for 15 hr at 4°C with primary antibodies and then with 15 μl of protein A-agarose for 1 hr. Immunoprecipitates were washed four times with lysis buffer and resuspended in 50 μl of SDS sample buffer for Western blot experiments. For Western blotting with antibodies against ET-Rs, 25 μg of the cell and tissue extracts were first dissolved in cell lysis buffer and then resolved on a 4–20% mini-SDS polyacrylamide gel, followed by transfer to Immobilon polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA). Equal protein loading was verified by Ponceau S solution (Sigma) reversible staining of the blots. Blots were processed as described previously (Ghiani et al., 1999). Anti-ETA-R and ETB-R antibodies were used at a 1:100 dilution.
Western blot analysis of ET-stimulated astrocytes was performed either on 60 μg of total cell lysate (for P-CREB, CREB, P-ERK1/2, ERK1/2, P-MEK1/2, MEK1/2, P-p38MAPK, p38MAPK, P-JNK, JNK, B-Raf, and ATF-1) or on immunoprecipitates for Raf-1, RSK2, RSK3, and Elk-1. Proteins were resolved by 15% (Ras and Rap1), 10% (P-CREB, CREB, P-ERK1/2, ERK1/2, P-MEK 1/2, MEK 1/2, P-JNK, JNK, and GST-MEK), 8% (B-Raf and Raf-1), or 7% (RSKs) SDS-PAGE and transferred to nitrocellulose or Immobilon PVDF membranes by tank blotting (0.8 A constant current) in transfer buffer (25 mm Tris, 192 mm glycine, and 20% v/v methanol, pH 8.3) for 16 hr at 4°C. The membranes were rinsed twice in Tris-buffered saline (TBS) (25 mm Tris-HCl, pH 7.5, 140 mm NaCl, and 0.05% Tween 20), incubated for 1 hr in TBS containing 4% nonfat dry milk (TBSM), and then incubated for 16 hr at 4°C with primary antibodies in TBSM. After washing three times for 15 min each with TBS, the membranes were incubated in TBSM for 1 hr at room temperature with either horseradish peroxidase-conjugated goat polyclonal anti-rabbit IgG for polyclonal primary antibodies or horseradish peroxidase-conjugated goat polyclonal anti-mouse for mouse monoclonal primary antibodies. The chemiluminescent signals were detected using the Super signal kit (Pierce) or ECL plus kit (Amersham Pharmacia Biotech, Piscataway, NJ). x-Ray films were then scanned using an Agfa T1200 scanner with Photolook software.
Immunocomplex protein kinase assay. Raf-1- and B-Raf-associated kinase activity was determined as reported previously (Ghiani and Gallo, 2001) with minor modifications. Briefly, the Raf-1 and B-Raf kinases were immunoprecipitated from total cell lysates by using goat anti-B-Raf and anti-Raf-1 antibodies (see Immunoprecipitation and Western blots above). Immunoprecipitates were washed three times with 500 μl of cell lysis buffer and two additional times with kinase assay buffer (50 mmHEPES, pH 7.4, 50 mm MgCl2, and 1 mm DTT). The immunoprecipitated proteins were then dissolved in 20 μl of kinase assay buffer. Kinase assays were performed for 20 min at 30°C in 20 μl of kinase reaction mixture containing 10 μl of immunoprecipitated proteins, 0.5 μg of inactive GST-MEK1 (Upstate Biotechnology) as substrate, 100 μm ATP for B-Raf kinase assay, or the same reagents plus 2 μCi [γ-32P]ATP (NEN) for Raf-1 assay. Reactions were stopped by adding 20 μl of 2× SDS loading buffer and heating at 95°C for 3 min. Proteins were then resolved on 4–20% mini-SDS polyacrylamide gels. In the B-Raf kinase assay, phosphorylated GST-MEK1 bands were detected with rabbit phospho-MEK1/2 antibodies, and, in Raf-1 kinase assay, phosphorylated GST-MEK1 bands were visualized and quantified by Phosphorimager (Molecular Dynamics, Sunnyvale, CA). The amount of immunoprecipitated B-Raf and Raf-1 was detected by using the same antibodies used for immunoprecipitation.
Reverse transcription-PCR of B-Raf isoforms. Astrocyte cultures were rinsed twice with PBS and treated with the RNA extraction reagent Trizol (Life Technologies, Grand Island, NY). After extraction with 1:10 vol of chloroform, the upper aqueous phase was precipitated overnight at −20°C with 1:1 vol of isopropanol. Total RNA was precipitated by centrifugation at 20,000 × gfor 15 min at 4°C, washed twice with ethanol, and quantified by UV spectrophotometry at 260 nm. Total RNA was treated with 10 U RNase-free DNase I for 15 min at 37°C to eliminate contaminating genomic DNA. The DNase was then digested with 15 mg of proteinase K at 70°C for 20 min, and the RNA was phenol–chloroform extracted and ethanol precipitated. An aliquot of RNA (1 μg) was retrotranscribed into cDNA in a final volume of 20 μl of reverse transcription (RT) buffer containing 0.5 mm each dNTP, 15 U of ribonucleasin, 1 μm random hexamers, and 15 U of avian myeloblastoma virus reverse transcriptase. The reaction was performed for 10 min at room temperature, 60 min at 42°C, and was blocked by heating at 95°C for 5 min before chilling on ice. The RT mixture was diluted to 100 μl with water, and 5 μl was used for PCR amplification. PCR reagents (in a total volume of 50 μl) were as follows: 200 μm dNTP, 0.2 μm each primer, 1.5 μmMgCl2, and 2.5 U of Taq polymerase. Primer composition, designed as reported previously (Barnier et al., 1995) and based on mouse partial mRNA B-Raf (GenBank accession number AJ 276307) were as follows: forward primer, 5′-CCAATTCCACAGCCCTTCCG-A-3′ (position 822–842); and reverse primer, 5′-CATCCGACTTCTGT-CCTCCGA-3′ (position 1101–1121). PCR conditions were 94°C for 2 min, followed by 40 cycles, each one performed at 94°C for 45 sec, 60°C for 45 sec, and 72°C for 45 sec. The reaction was terminated by a final extension step of 10 min at 72°C. Aliquots of the amplification products were separated on 2% agarose gels and visualized by ethidium bromide staining. The PCR experiments were repeated twice with two distinct cell cultures and RNA preparations.
Pull-down assay for the determination of Rap1 and Ras activation. The pull-down assays for the determination of Rap1 and Ras activation were performed as reported previously (Seidel et al., 1999). Briefly, the GST fusion protein of the minimal Rap1-binding domain of ralGDS (GST-ral-RBD; a generous gift from Dr. M. Freissmuth) was induced in Escherichia coli (strain BL21DE3) by isopropyl-1-thio-β-d-galactopyranoside, and bacterial lysates were prepared as described previously (Seidel et al., 1999). The GST fusion protein was immobilized by incubating the bacterial lysate for 1 hr at 4°C with glutathione-Sepharose preequilibrated in pull-down buffer (50 mm Tris, 200 mm NaCl, 2 mmMgCl2, 1 mm PMSF, 10% glycerol, 1% Nonidet P-40, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 2 μg/ml pepstatin). Sepharose beads were washed three times to remove excess GST fusion protein.
After stimulation with agonists for the indicated time, cells were rinsed twice with stimulation buffer and then chilled immediately in pull-down buffer; cell lysates were cleared by centrifugation at 10,000 × g for 10 min at 4°C, and 5% of the supernatants were analyzed to normalize for the total amount of Rap1 present in the cell lysates. The remaining supernatants (500 μg of proteins each) were incubated with the GST-Sepharose beads (50 μl of a 1:1 slurry containing ∼10 μg of immobilized GST fusion protein) for 2 hr, to allow for binding of activated Rap1 with the effector-GST fusion protein. Samples were washed twice with modified lysis buffer, resuspended in Laemli's sample buffer and applied to SDS polyacrylamide gels. The same procedure was used for Ras activation, except that GST-Raf-1-RBD agarose (Upstate Biotechnology) was used instead of GST-ral-RBD. Rap1 and Ras bands were then visualized using specific antibodies in a 1:1000 dilution. The chemiluminescent signals were detected as described for Western blot, and x-ray films were scanned as reported for Western blot.
Plasmids and transfection. Rat cortical astrocytes grown for ∼10 DIV were trypsinized and seeded on 60 mm tissue culture dishes at a density of 50,000 cells/cm2. After 24 hr, subconfluent astrocytes (70–80% confluency) were rinsed twice with DMEM and transfected with lipofectamine reagent (Life Technologies). Cortical astrocytes were transfected with pMT2HA mammalian expression vector expressing hemoagglutinin (HA)-tagged dominant-negative Rap1 (RapN17). This vector has been described previously (Reedquist et al., 2000) and was a generous gift from Dr. J. L. Bos (Laboratory for Physiological Chemistry and Center for Biomedical Genetics, Utrecht University, Utrecht, The Netherlands). To rule out nonspecific or toxic effects of vector–liposome complexes, control cells were transfected with pMT2HA empty vector. For each dish, 8 μg of vector were added to 92 μl of DMEM, while, in a separate tube, 25 μl of lipofectamine were added to 75 μl of DMEM; the two suspensions were mixed and incubated for 30 min at room temperature to allow the formation of vector–liposome complexes. The transfection mixture (200 μl) was added to cells in tissue culture dishes containing 800 μl of DMEM. After 8 hr, 1 ml of fresh DMEM medium containing 10% FBS and 10% horse serum was added to the cells, without removing the transfection mixture, for an additional 12 hr. The medium was then completely replaced with fresh DMEM containing 5% FBS and 5% horse serum, and cells were allowed to recover for 48 hr. Cells were then starved overnight (∼16 hr) in serum-free DMEM before treatment with the indicated agents and lysis for Western blot analysis.
Northern blot analysis. Astrocyte cultures, previously treated with appropriate stimuli for the time indicated, were rinsed twice with PBS and treated with 3 ml/dish the RNA extraction reagent Trizol (Life Technologies). After an extraction with a 1:10 vol of chloroform, the upper aqueous phase was precipitated overnight at −20°C with a 1:1 vol of isopropanol. Total RNA was pelleted spinning the tubes at 20,000 × g for 15 min at 4°C, washed twice with ethanol, and quantified by UV spectrophotometry at 260 nm. Total RNA (20 μg) was then extracted as reported above (in Reverse transcription-PCR of B-Raf isoforms) and was size resolved on a 1.2% denaturing agarose gel containing 1 mparaformaldehyde in 20 mm3-(N-morpholino) propanesulfonic acid buffer, pH 7.0, 5 mm sodium acetate, and 1 mmEDTA and electroblotted overnight in 10× SSC onto a GeneScreen nylon membrane (NEN). The membranes were baked under vacuum at 80°C for 2 hr and prehybridized for 30 min at 65°C in 50% formamide, 0.6m NaCl, 1% SDS, and 0.1 mg/ml denatured herring sperm DNA. Blots were then hybridized overnight at 65°C in prehybridization solution containing ∼1 × 106 cpm/ml full-length c-fos or 18S ribosomal gene (internal loading control) cDNA random-primed [32P]-labeled probe. Blots were washed to a final stringency of 2× 0.15 m sodium chloride–0.015 m SCC at 65°C, and the radioactivity associated with the membranes was visualized and quantified with a Storm phosphorimager (Molecular Dynamics).
RESULTS
Both endothelin receptor subtypes ETA-R and ETB-R are expressed by glial cells in vivoand in culture
We first wanted to determine which ET-R subtype was expressed by the major glial cell types in vivo and in culture. Immunocytochemical and biochemical analysis was performed using anti-ETA- and ETB-R antibodies in brain tissue and in purified astrocyte cultures.
The staining pattern for ETA- and ETB-Rs in tissue and in purified cells was very similar, and no preferential expression of either receptor subtype could be detected. Double immunocytochemistry with astroglial markers revealed expression of both ETA- and ETB-Rs in astrocytes in situ (Fig.1). In brain sections from P8 rats, most of the ET-R immunoreactivity was localized in white matter regions. Figure 1A–F shows strong ETA-R (Fig. 1A–C) and ETB-R (Fig.1D–F) staining of S100-β+ astrocytes in subcortical white matter. In cerebellum, S100-β+astrocytes and Bergmann glial cells were the major cell types expressing both ETA- and ETB-Rs (data not shown). ET-R expression was also detected in rat white matter regions at P30, as shown in Figure1G–L. Similar to the staining pattern at P8, the vast majority of S100-β+ cells were also immunostained with both anti-ETA- and ETB-R antibodies. No apparent differences were detected between ETA- and ETB-Rs in either the proportion of astrocytes stained or the intensity of the staining. Also in P30 cerebellum, S100-β+ astrocytes and Bergmann glial cells were the major cell types expressing both ETA- and ETB-Rs (data not shown).
Fig. 1.
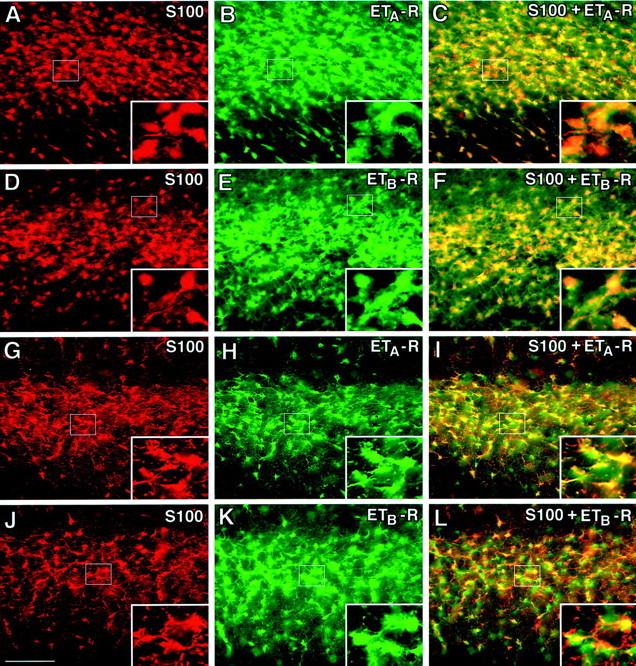
Rat astrocytes express ETA- and ETB-Rs in vivo. In the immature (P8) subcortical white matter (A–F), S100-β+ astrocytes express both ETA- and ETB-Rs. A similar staining pattern was also found in the adult (P30) white matter (G–L). Double immunostaining with antibodies against ETA-R (B, H; green) and ETB-R (E, K;green) and the astrocyte marker S100-β+(A, D, G,J; red) reveals coexpression in the white matter (yellow and orangestaining). C and I are overlays of S100-β+ and ETA-R, whereas Fand L are overlays of S100-β+ and ETB-R. Note that the staining pattern of ETA- and ETB-R in S100-β+ astrocytes of the developing and adult cerebellar white matter was very similar to that shown here for the subcortical white matter (data not shown). Scale bar: A–L, 100 μm; insets, 33 μm.
ETA- and ETB-R expression was analyzed in purified astrocyte cultures. ETA-R and ETB-R immunoreactivity was detected in all S100-β+ (Fig.2A–F) and GFAP+ (Fig. 2G–L) cells. Western blot analysis showed identical molecular weights for both ETA- and ETB-R proteins in cultured astrocytes and in vivo (Fig.3). Interestingly, in brain tissue, both ETA- and ETB-R relative levels increased between P8 and P30 (Fig. 3), i.e., during gliogenesis and glial cell maturation. Together, these results indicate that ET-Rs are expressed in astrocytes at different developmental stages, and that receptor expression is not limited to subpopulations of cells but rather uniformly detected in all S100-β+/GFAP+astrocytes.
Fig. 2.
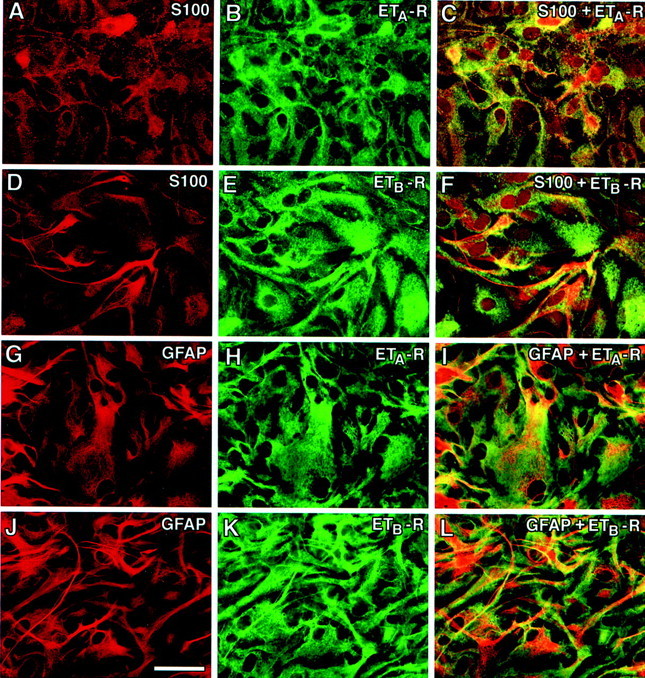
Astrocytes express ETA- and ETB-Rs in culture. S100-β+ astrocytes express both ETA- and ETB-Rs (A–F). A similar staining pattern was observed when astrocytes were stained with anti-GFAP antibodies (G–L). Double labeling of cultured astrocytes with antibodies against ETA-R (B,H; green) and ETB-R (E, K; green) and astrocyte markers (red). C andF are overlays of S100-β and ETA-R, and S100-β and ETB-R, respectively. I andL are overlays of GFAP and ETA-R, and GFAP and ETB-R, respectively. All astrocytes coexpress both ETA- and ETB-Rs. Scale bar, 50 μm.
Fig. 3.

ETA- and ETB-R proteins are expressed in astrocytes in culture and in cerebral cortex in vivo. Western blot analysis demonstrates that ETA- and ETB-R proteins with the same molecular weight are detected in cultured cells and in vivo. Lane 1, Cultured astrocytes (ASTR). Lane 2, Tissue extract from postnatal day 8 rat cerebral cortex (CX-P8). Lane 3, Brain tissue extracts from postnatal day 30 rat cerebral cortex (CX-P30). Eachlane contained 25 μg of total protein.Arrows indicate the bands corresponding to ETA-R (46 kDa) and ETB-R (39 kDa).
ET-1 induces CREB phosphorylation in astrocytes via ETB receptors
Previous studies have demonstrated that, in cultured astrocytes, ET-1 activates the MAPK pathways (Lazarini et al., 1996, Cazaubon et al., 1997). Because in several experimental models MAPK activation leads to CREB phosphorylation, we examined, using an antibody that recognizes CREB phosphorylated at Ser 133, whether a similar signaling transduction pathway was activated by ET-1 in cortical astrocytes. Time course analysis of CREB phosphorylation induced by ET-1 revealed that P-CREB was detectable after 3 min of stimulation with the peptide. The P-CREB signal reached a plateau between 10 and 30 min and then decreased between 30 and 60 min after stimulation (Fig.4A, top panel). Analysis of the same protein extracts using another antibody that recognizes CREB independently of its phosphorylation state demonstrated no difference in the total amount of CREB in cells stimulated with ET-1 (Fig. 4A, bottom panel).
Fig. 4.

ET-1 induces CREB phosphorylation via stimulation of ETB-R in astrocytes. A, Time course of ET-1-induced CREB phosphorylation. Cortical astrocytes were stimulated with 50 nm ET-1 for the indicated times, and total cell lysates were analyzed by Western blot using an anti-P-CREB antibody (top panel) or an anti-CREB antibody (bottom panel) to normalize for the total (phosphorylated plus nonphosphorylated) CREB. B, Effects of specific ETA- and ETB-R antagonists on ET-1-induced CREB phosphorylation. Astrocytes were preincubated for 15 min with the ETA-R antagonist BQ123 (A; 500 nm) or with the ETB-R antagonist BQ788 (B; 50 nm) and then stimulated for 10 min with 50 nm ET-1. The samples were then processed for CREB phosphorylation analysis as in A. Data inA and B are representative of three independent experiments.
Our immunocytochemical and immunoblot data demonstrated expression of both ETA-R and ETB-R subtypes in astrocytes in vitro and in vivo. We therefore investigated which receptor subtype mediates ET-1-induced CREB phosphorylation in astrocytes. Preincubation of cells for 15 min with the ETA-R antagonist BQ123 did not significantly modify CREB phosphorylation induced by ET-1 (Fig.4B). In contrast, under the same experimental conditions, the ETB-R antagonist BQ788 strongly inhibited the effect of ET-1 on CREB phosphorylation (Fig.4B), indicating that, in cortical astrocytes, CREB phosphorylation induced by ET-1 is mainly mediated by activation of ETB-R.
ET-1 induces CREB phosphorylation via a PKC/MAPK-dependent pathway
Stimulation of both ET receptor subtypes leads to protein kinase C activation via a G-protein/phospholipase C (PLC)-mediated mechanism involving the formation of the second-messengers inositol phosphates, diacylglycerol, and calcium (Rubanyi and Polokoff, 1994). Therefore, we investigated the possible involvement of PKC- and ERK-dependent pathways in ET-1-induced CREB phosphorylation by using specific protein kinase inhibitors. Preincubation of cortical astrocytes for 30 min with the specific PKC inhibitor Gö6976 or the ERK kinase inhibitor PD98059 completely prevented the stimulatory effect of ET-1 on CREB phosphorylation (Fig. 5A).
Fig. 5.

ET-1 induces CREB phosphorylation via a PKC/ERK-dependent pathway. A, PKC and MEK inhibitors prevented ET-1-induced CREB phosphorylation. Astrocytes were preincubated for 30 min with the PKC inhibitor Gö6976 (Gö; 5 μm) or with the MEK inhibitor PD98059 (PD; 50 μm) and then stimulated for 10 min with 50 nm ET-1. Total cell lysates were then analyzed by Western blot using an anti-P-CREB antibody.B, PKC inhibition blocks ET-1-induced ERK activation. Astrocytes were preincubated for 30 min with the PKC inhibitor Gö6976 (Gö; 5 μm) and then stimulated for 10 min with 50 nm ET-1. Total cell lysates were then analyzed by Western blot using an anti-P-ERK antibody (top panel) or an anti-ERK antibody (bottom panel) to normalize for the total (phosphorylated plus nonphosphorylated) ERK. C, ET-1 activates the ERK kinase MEK. Astrocytes were stimulated as inA, and total cell lysates were analyzed by Western blot using an anti-P-MEK antibody (top panel) or an anti-MEK antibody (bottom panel) to normalize for the total (phosphorylated plus nonphosphorylated) MEK.D, ET-1 activates the CREB kinases RSK2 and RSK3. Astrocytes were stimulated as in A, and RSK2 and RSK3 were immunoprecipitated from total cell lysates using specific anti-RSK2 and anti-RSK3 antibodies. The amount of P-RSK2 and P-RSK3 in the immunoprecipitates was then analyzed by Western blot using an anti-P-RSK antibody (top panel). The amount of total (phosphorylated plus nonphosphorylated) RSK2 and RSK3 in the immunoprecipitates (bottom panel) was analyzed with the same specific anti-RSK2 or anti-RSK3 antibodies used for the immunoprecipitation. Data in A–D were representative of three independent experiments.
To establish whether PKC and ERK belong to the same signal transduction pathway or are involved in independent pathways converging on CREB phosphorylation, we investigated the effect of ET-1 on ERK phosphorylation using an antibody that recognizes the dually phosphorylated (Thr 202 and Tyr 204) ERK isoforms. Preincubation of astrocytes with Gö6976, under the same experimental conditions adopted for CREB phosphorylation, completely abolished ERK2 phosphorylation induced by ET-1 (Fig. 5B). ET-1-induced ERK phosphorylation is mediated by the ERK kinases MEK1 and MEK2. In fact, incubation of astrocytes with ET-1 resulted in phosphorylation of both MEK1/2 isoforms, as revealed by Western blot with an antibody that recognizes MEK1/2 isoforms dually phosphorylated at Ser 217 and 221 (Fig. 5C).
To gain additional insights into the mechanism by which ET-1 induces CREB phosphorylation in astrocytes, we examined the activation of kinases downstream of ERKs. Activated ERK kinases may translocate to the nucleus and phosphorylate directly target transcription factors (such as Elk-1) or, alternatively, ERK may phosphorylate members of the 90 kDa ribosomal S6 kinases family (RSK1–RSK3). Because in some experimental models RSKs phosphorylate CREB (Shaywitz and Greenberg, 1999), RSKs were immunoprecipitated from ET-1-treated astrocytes by specific anti-RSK isoform antibodies, and the amount of immunoprecipitated phosphorylated RSKs was assessed by a different antibody recognizing all RSK isoforms phosphorylated in the Ser 381 residue. Although cortical astrocytes express all three RSK isoforms (data not shown), ET-1 induced phosphorylation only of RSK2 and RSK3 (Fig. 5D). This was assessed by either a phospho-RSK antibody (Fig. 5D, top panel) or the presence of an upper-shifted band using anti-RSK isoform antibodies (Fig. 5D, bottom panel).
In several cellular models, calcium/calmodulin-dependent protein kinases (CaMKs) mediate, directly or indirectly, calcium-dependent ERK activation, CREB phosphorylation, and CRE-dependent gene expression (Vanhoutte et al., 1999). The recent finding that CaMKs exert a positive control on glutamate-induced CREB phosphorylation in striatal slices (Vanhoutte et al., 1999), together with the fact that ET-1 raises intracellular calcium levels (Rubanyi and Polokoff, 1994), prompted us to investigate a possible role of CaMKs in cortical astrocytes. Preincubation of the cells with the two CaMK inhibitors KN62 and calmidazolium chloride did not interfere with CREB phosphorylation induced by ET-1 (data not shown). This result indicates that CaMK does not participate in ET-1-mediated regulation of CREB phosphorylation in astrocytes.
ET-1 induces ERK phosphorylation via a Rap1/B-Raf-dependent signaling pathway
Several studies have clearly demonstrated a role for the small G-protein Ras in coupling stimulation of ET-Rs to ERK activation (Chiloeches et al., 1999). However, several reports have emphasized recently the role of another small G-protein, Rap1 and its effector B-Raf, in the propagation of receptor-mediated extracellular stimuli to ERK activation (York et al., 1998; Pizon and Baldacci, 2000; Schmitt and Stork, 2000).
We first sought to determine whether the Rap1/B-Raf coupling system is activated by ET-1 in cortical astrocytes. Using a pull-down assay to separate the activated GTP-bound form of Rap1, we found that treatment of cortical astrocytes with ET-1 induced the formation of the GTP-bound active form of Rap1 (Fig.6A, top panel). This mechanism requires activation of PLC, because Rap1 activation was blocked by the PLC inhibitor U-73122 (Fig.6A, top panel). In a variety of cellular models, Rap1 interacts with its downstream effector B-Raf, and several B-Raf isoforms have been described previously (Hagemann and Rapp, 1999). Therefore, we investigated the expression of B-Raf isoforms in cortical astrocytes. RT-PCR of RNA extracted from cortical astrocytes, using primers flanking exon 8 and 10 of the mouse B-Raf gene, detected the presence of at least four different transcripts (Fig. 6B), likely corresponding to alternative spliced isoforms of the rat B-Raf gene (Barnier et al., 1995). The expression of B-Raf isoforms was then analyzed by Western blot, using a sheep polyclonal antibody raised against a synthetic peptide corresponding to residues 10–22 of human B-Raf and a rabbit polyclonal antibody raised against a recombinant protein corresponding to amino acid 12–156 mapping at the N terminus of B-Raf of human origin. Both antibodies detected an identical pattern of expression of B-Raf isoforms: a large band of ∼95–97 kDa (probably including more than a single protein), another band of ∼65–68 kDa, and a faint band with a lower molecular mass (Fig. 6C).
Fig. 6.

ET-1 activates the Rap1/B-Raf complex in astrocytes. A , ET-1 activates Rap1. Astrocytes were preincubated for 30 min with the PLC inhibitor U-73122 (10 μm) and then stimulated for 10 min with 50 nmET-1. Total cell lysates were processed for the pull-down assay of Rap1-GTP, as described in Materials and Methods. The amount of GTP-Rap1 (top panel) and total Rap1 (bottom panel) in the cell lysates was analyzed by Western blot with an anti-Rap1 antibody. Note that only 5% of the lysates that was used to determine GTP-Rap1 was used to determine the total amount of Rap1 (bottom panel). B, Expression of B-Raf isoforms in cortical astrocytes. RT-PCR of B-Raf transcripts. Total RNA extracted from cortical astrocytes was retrotranscribed to cDNA (RT+ lane). Untreated RNA was used as a negative control (RT− lane). cDNAs were then amplified by PCR, as reported in Materials and Methods. PCR products were analyzed on a 3% agarose gel. Size markers (M lane) are indicated in base pairs. C, Expression of B-Raf proteins. Total cell lysates were analyzed by Western blot using an antibody raised against a synthetic peptide corresponding to residues 10–22 of human B-Raf (lane 1) or an antibody raised against a recombinant protein corresponding to amino acid 12–156, mapping at the N terminus of human B-Raf (lane 2). Data in A–C are representative of at least three independent experiments.
We then investigated the effect of ET-1 treatment on B-Raf activation and the functional consequences of mutated Rap1 on ERK phosphorylation. Immunocomplex kinase assay of B-Raf, using inactive MEK as substrate, demonstrated that activation of Rap1 induced by ET-1 also leads to B-Raf activation (Fig. 7A). Also, cortical astrocytes transfected with a vector coding for HA-tagged dominant-negative Rap1 (Rap1N17) expressed the exogenous Rap1 protein, as assessed by Western blot using anti-HA antibodies (Fig.7B, top panel) or anti-Rap1 antibodies (Fig. 7B, second panel). Compared with mock-transfected cells, expression of Rap1N17 blunted the increase in P-ERK levels elicited by ET-1 (Fig. 7B, third panel), indicating that Rap1 is directly involved in ET-1-induced P-ERK activation.
Fig. 7.

ET-1 activates the B-Raf and Raf-1 kinases.A, ET-1 activates the B-Raf kinase. Astrocytes were stimulated for 10 min with 50 nm ET-1. B-Raf activity was measured by an immunocomplex kinase assay, as described in Materials and Methods. The amount of the phosphorylated substrate GST-MEK was then analyzed by Western blot using an anti-P-MEK antibody (top panel). The amount of immunoprecipitated B-Raf (bottom panel) was analyzed with the same B-Raf antibody used for the immunoprecipitation. B, Rap1 is involved in ET-1-induced ERK activation. Astrocytes were transfected with a vector coding for HA-tagged dominant-negative Rap1 (Rap1N17), as described in Materials and Methods. Control cells (dash) were transfected with pMT2HA empty vector. Cells were then stimulated for 10 min with 50 nmET-1, and total cell lysates were analyzed by Western blot using an anti-HA antibody (top panel), an anti-Rap1 antibody (second panel), an anti P-ERK antibody (third panel), and an anti-ERK antibody (bottom panel) to normalize for the total (phosphorylated plus nonphosphorylated) ERK. C, ET-1 induces Ras activation. Astrocytes were stimulated for 10 min with 50 nm ET-1. Total cell lysates were processed for the pull-down assay of GTP-Ras, as described in Materials and Methods. The amount of GTP-Ras (top panel) and total Ras (bottom panel) in the cell lysates was analyzed by Western blot with an anti-Ras antibody. Note that only 5% of the lysates that was used to determine GTP-Ras was used to determine the total amount of Ras (bottom panel).D, ET-1 activates Raf-1 kinase. Astrocytes were stimulated for 10 min with 50 nm ET-1, and Raf-1 activity was measured by an immunocomplex kinase assay, as described in Materials and Methods. The substrate GST-MEK was separated by SDS-PAGE, and the radioactive phosphorylated band was measured by Phosphorimager (Molecular Dynamics) analysis (top panel). The amount of immunoprecipitated Raf-1 (bottom panel) was analyzed with the same Raf-1 antibody used for the immunoprecipitation (IP).
In several cellular models, Rap1 might either interfere with Ras-dependent pathways or be involved in signaling pathways distinct from Ras, while sharing similar or identical downstream effector kinases (Bos, 1998). Because ET-1 may activate the Ras/Raf-1 coupling system in different cell types (Foschi et al., 1997; Chiloeches et al., 1999), we investigated whether ET-1 could also trigger this mechanism in cortical astrocytes. A pull-down assay of Ras (Fig.7C, top panel) and an immunocomplex kinase assay for Raf-1 (Fig. 7D, top panel), together with a retarded mobility of Raf-1 band by immunoblot analysis (data not shown), indicate that ET-1 activates also the Ras/Raf-1 coupling system in cortical astrocytes. Together, these data show that both the Rap1/B-Raf and the Ras/Raf-1 effector systems are activated by ET-1 in cortical astrocytes. Furthermore, we demonstrate that Rap1 is directly involved in the propagation of ET-1 signaling to downstream kinases.
ET-1 induces CREB phosphorylation also via a p38MAPK-dependent but not a JNK-dependent pathway
The activation of a wide array of G-protein-coupled receptors may stimulate, via cross-talk mechanisms, not only the ERK pathway but also the p38MAPK- and JNK-dependent pathways (Gutkind, 1998). Because these pathways could be activated in a PKC-dependent manner, we investigated whether ET-1 was able to induce p38MAPK and JNK phosphorylation in cortical astrocytes. Using antibodies recognizing dually phosphorylated p38MAPK at Thr 180/Tyr 182 residues and dually phosphorylated JNK at Thr 183/Tyr 185, we found that ET-1 activated p38MAPK (Fig.8A, top panel) but not JNK (Fig. 8B, top panel). The P-JNK antibody detected a 54 kDa band in whole-cell lysates of anisomycin-treated NIH/3T3 cells used as a positive control (Fig. 8B, pc). The absence of phosphorylated JNK was not attributable to a lack of expression of JNKs in cortical astrocytes, because a different antibody directed against phosphorylation-state independent JNK recognized the two 54 and 46 kDa JNK isoforms (Fig. 8B, bottom panel).
Fig. 8.
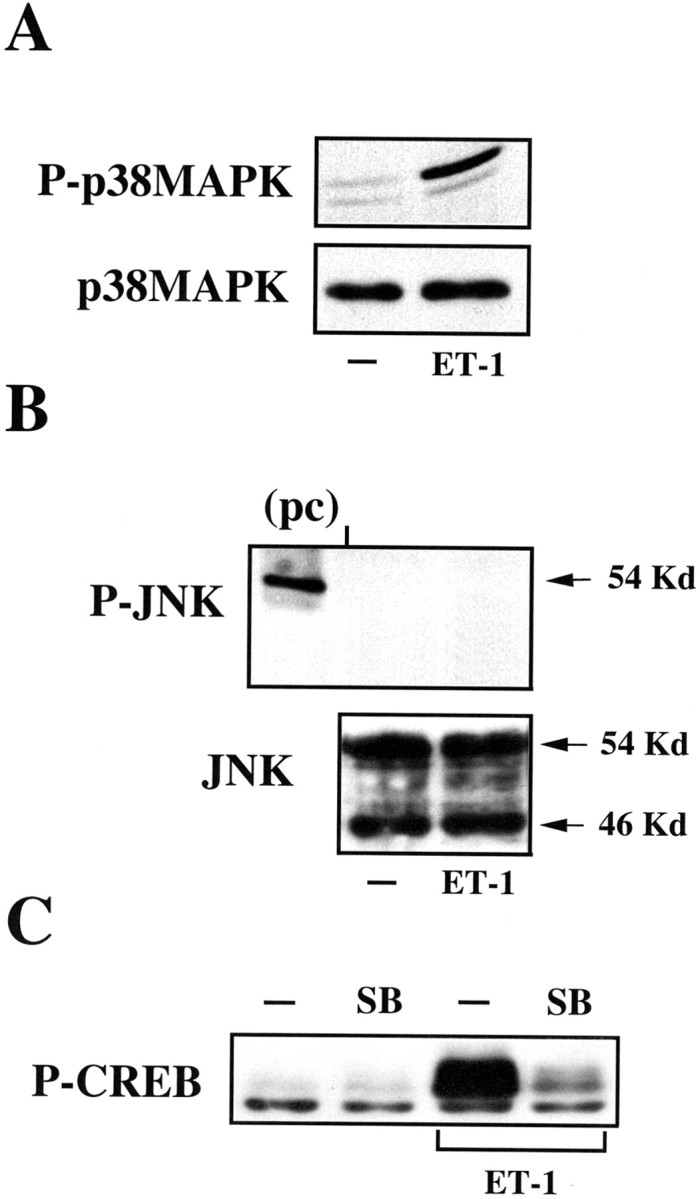
ET-1 induces CREB phosphorylation via the p38MAPK-dependent pathway. A, ET-1 activates p38MAPK. Astrocytes were stimulated for 10 min with 50 nm ET-1, and total cell lysates were analyzed by Western blot using an anti-P-p38MAPK antibody (top panel) or an anti-p38MAPK antibody (bottom panel) to normalize for the total (phosphorylated plus nonphosphorylated) p38MAPK.B, ET-1 does not activate the JNK-dependent pathway. Astrocytes were stimulated as in A, and total cell lysates were analyzed by Western blot using an anti-P-JNK antibody (top panel) or an anti-JNK antibody (bottom panel) to normalize for the total (phosphorylated plus nonphosphorylated) JNK. A whole-cell lysate of anisomycin-treated NIH/3T3 cells was analyzed as a positive control (pc) for the 54 kDa JNK isoform.C, The p38MAPK inhibitor SB203580 (SB) blocks ET-1-induced CREB phosphorylation. Astrocytes were pretreated for 30 min with the p38MAPK inhibitor SB203580 (10 μm) and stimulated as in A. Total cell lysates were analyzed by Western blot using an anti-P-CREB antibody. Data inA–C were representative of at least three independent experiments.
The finding that p38MAPK activation mediates CREB phosphorylation induced by stimulation of tyrosine kinase receptors (Tan et al., 1996) led us to investigate whether this signaling cascade participates in ET-1-induced CREB phosphorylation in astrocytes. Preincubation of cells with the specific p38MAPK inhibitor SB203580 prevented CREB phosphorylation induced by ET-1 (Fig. 8C), indicating not only that more than one MAPK pathway is activated by ET-1 but also that these pathways may converge to regulate the phosphorylation and functional state of a transcription factor.
ET-1 induces ATF-1 and Elk-1 phosphorylation
In addition to converging on common nuclear targets, single or multiple MAPK-dependent pathways may also diverge to phosphorylate distinct transcription factors. For example, ERK activation directly phosphorylates Elk-1 (Schaeffer and Weber, 1999), a member of the ternary factor complex that binds the SRE (Whitmarsh and Davis, 1996).
The transcription factor ATF-1 shares some interesting features with CREB; ATF-1 may dimerize with CREB to interact with the CRE element and contains a Ser 63 residue in its sequence, which is surrounded by the same phosphorylation consensus site found in CREB. Indeed, in SK-N-MC cells, fibroblast growth factor causes not only CREB phosphorylation but also ATF-1 phosphorylation through p38MAPK (Tan et al., 1996).
We therefore investigated whether ET-1 also phosphorylated ATF-1 and Elk-1 and the signaling cascades responsible for this phosphorylation. ET-1 induced ATF-1 phosphorylation in astrocytes, and this effect was inhibited by the PKC inhibitor Gö6369 or by the MEK inhibitor PD58059 but not by the p38MAPK inhibitor SB203508 (Fig.9A, top panel). The identity of the P-ATF-1 band was confirmed by using an antibody recognizing phosphorylation state-independent ATF-1 that does not cross-react with CREB (Fig. 9A, bottom panel).
Fig. 9.

ET-1 induces ATF-1 and Elk-1 phosphorylation in astrocytes. A, ET-1 induces ATF-1 phosphorylation via a PKC/ERK-dependent but not a p38MAPK-dependent pathway. Astrocytes were pretreated for 30 min with the p38MAPK inhibitor SB203580 (SB; 10 μm), the PKC inhibitor Gö6976 (Gö; 5 μm), or the MEK inhibitor PD98059 (PD; 50 μm) and then stimulated for 10 min with 50 nm ET-1. Total cell lysates were analyzed by Western blot using an anti-P-CREB antibody that recognizes also the phosphorylation consensus sequence of ATF-1 (top panel). P-ATF-1 was identified based on its molecular weight. Total ATF-1 (phosphorylated plus nonphosphorylated) was determined with an anti-ATF-1 antibody. B, ET-1 induces Elk-1 phosphorylation via an ERK-dependent pathway. Astrocytes were pretreated for 30 min with the MEK inhibitor PD98059 (PD; 50 μm) and then stimulated for 10 min with 50 nm ET-1. Elk-1 was immunoprecipitated from total cell lysate and then analyzed by Western blot using an anti-P-Elk-1 antibody (top panel) or the same anti-Elk-1 (phosphorylated plus nonphosphorylated) antibody (bottom panel) used for immunoprecipitation. Data inA and B are typical of at least three independent experiments.
Because of its very low levels of expression in astrocytes, Elk-1 was immunoprecipitated from total cell lysate, and its phosphorylated form was detected by immunoblot using an antibody recognizing phosphorylated Elk-1 at Ser 383. Treatment of cortical astrocytes with ET-1 induced Elk-1 phosphorylation that was blocked by the MEK inhibitor PD58059 (Fig. 9B, top panel). These data indicate that ET-1-induces Elk-1 phosphorylation via an ERK-dependent pathway.
ET-1 induces c-fos transcription via multiple MAPK-dependent pathways
As a result of phosphorylation, activated transcription factors can bridge between the RNA polymerase complex and their cognatecis elements in the gene promoter to trigger transcription. The c-fos promoter contains several cis elements that bind CREB, ATF-1, and Elk-1 transcription factors (Karin et al., 1997). In the attempt to characterize the relative contribution of different signaling pathways and cis elements activated by ET-1, we assessed the effect of different kinase inhibitors on ET-1-induced c-fos transcription by measuring the levels ofc-fos mRNA.
Treatment of cortical astrocytes with ET-1 induced a large increase inc-fos mRNA (Fig. 10). This increase was strongly suppressed by the PKC inhibitor Gö6976, the MEK inhibitor PD58059, or the p38MAPK inhibitor SB203508 (Fig. 10). These data indicate that maximal ET-1-induced transcription ofc-fos requires the concomitant phosphorylation of the transcription factors CREB, ATF-1, and Elk-1 via PKC/ERK- and p38MAPK-dependent pathways. These results are consistent with the finding that maximal expression of the c-fos gene, which contains in its promoter both an SRE and a CRE element, requires the concomitant activation of several transcription factors and their cognate cis elements present in the promoter region (Robertson et al., 1995).
Fig. 10.

ET-1 induces c-fos transcription via multiple MAPK-dependent pathways. Astrocytes were pretreated for 30 min with the p38MAPK inhibitor SB203580 (SB; 10 μm), the PKC inhibitor Gö6976 (Gö; 5 μm), or the MEK inhibitor PD98059 (PD; 50 μm) and then stimulated for 30 min with 50 nm ET-1. Total RNA was analyzed by Northern blot using a radiolabeled probe for c-fos(top panel) or a radiolabeled probe for 18S ribosomal RNA (bottom panel), and the signals were detected by autoradiography. Data are representative of three independent experiments.
DISCUSSION
This study demonstrates that astrocytes express both subtypes of ET-Rs, ETA and ETB,in vivo and in cell culture. Importantly, ET-Rs were detected in this glial cell type both at early developmental stages and in the mature brain, suggesting that ET could play a role not only after a CNS lesion (Nie and Olsson, 1996) but also as a developmental neuron–glial signal. In this study, we identified a potentially important physiological signaling pathway for ET-Rs in the brain.
Our finding that ETA- and ETB-Rs are highly expressed in astrocytes indicates that these are cellular targets of the physiological effects of ET-1 in the brain. The finding that ET-R expression is maintained in cultured cells allowed us to investigate the intracellular signal transduction pathways activated by ET-1 in astrocytes, with a focus on transcription factor phosphorylation and gene expression. Our study shows that ET-1 induces CREB phosphorylation in cortical astrocytes through two different MAPK cascades, a PKC/ERK- and a p38MAPK-mediated signaling pathway (Fig. 11).
Fig. 11.
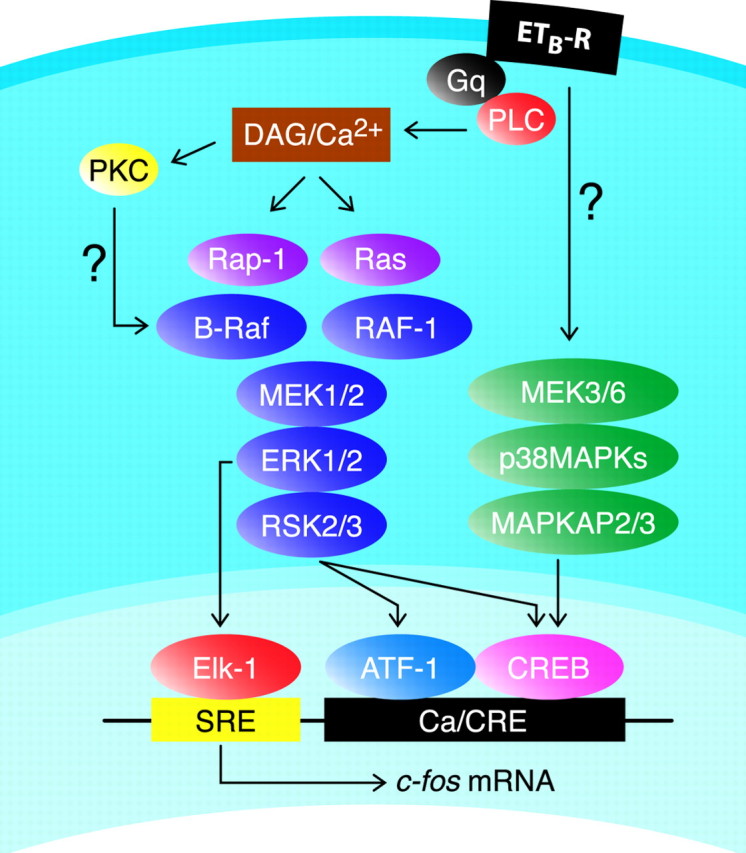
Representative scheme of signaling pathways involved in ET-1-induced/ETB-R-mediated CREB, ATF-1, and Elk-1 phosphorylation and c-fos expression in astrocytes. Question marks indicate unknown links between elements of signaling pathways. DAG, Diacylglycerol; MAPKAP2/3, MAP kinase-activated protein kinase 2/3.
Another novel finding reported here is that ET-1 induces activation of the Ras-like small G-protein Rap1, with consequent activation of the downstream kinase B-Raf (Figs. 7A, 11). In a previous report, B-Raf expression was not detected in astrocytes (Dugan et al., 1999), however our study shows that (1) several B-Raf isoforms are expressed in cultured cortical astrocytes at both the RNA and protein levels (Fig. 6B,C), and (2) B-Raf is functionally activated, as determined by immunocomplex kinase assay (Fig. 7A).
In early reports, only cAMP was thought to activate Rap1, but recent studies have challenged this view, demonstrating that also other second-messenger systems can be involved. For example, ET-1 induces Rap1 activation in fibroblasts via a PLC-dependent mechanism (Zwartkruis et al., 1998), and both calcium and diacylglycerol can activate Rap1 in platelets (Franke et al., 1997) and in B-cells (McLeod et al., 1998). In our cellular model, we also show that Rap1 activation occurs through a PLC-dependent mechanism and that this activation is required to propagate ET-1-induced signaling to ERK, as demonstrated by the experiments with mutated Rap1 (Fig. 7B). The PLC-dependent mechanism indicates that Rap1 activation in astrocytes might be regulated by the recently identified Ca2+- and diacylglycerol-dependent Rap1-specific guanine exchange factor (Bos, 1998).
Our results showing that ET-1 induces both Rap1/B-Raf and Ras/Raf-1 in astrocytes are in agreement with previous findings that both of these pathways might need to be recruited for maximal ERK activation (York et al., 1998; Garcia et al., 2001). Additional experiments will clarify whether Rap1/B-Raf and Ras/Raf-1 activation occurs simultaneously or with a different time course (York et al., 1998).
An important finding of our study is that PKC is a key intermediate in the signaling pathway responsible for ET-1-induced CREB phosphorylation and CRE-dependent gene expression (Figs. 5A, 11). Several studies have demonstrated that the interaction between small G-proteins and their target Raf kinases is insufficient to stimulate kinase activity. Additional events are required, and, among these, the phosphorylation of Raf kinases by other kinases appears to be critical. In particular, because PKC can phosphorylate and activate directly Raf-1 (Campbell et al., 1998), it is conceivable to assume that, in astrocytes, ETB-R-induced PKC activation could trigger B-Raf and Raf-1 phosphorylation either directly or indirectly (Fig. 11).
Previous reports demonstrated that ET-R stimulation in astrocytes activates the ERK pathway (Lazarini et al., 1996; Cazaubon et al., 1997), but the kinases downstream of ERK and responsible for CREB phosphorylation and of other transcription factors involved inc-fos expression were not investigated in detail. Our study shows that, in astrocytes, ET-1-induced CREB phosphorylation is likely to be mediated by the two RSK isoforms, RSK2 and RSK3, because the contribution of RSK1 seems to be negligible. The importance of RSK2-mediated CREB phosphorylation was also demonstrated in a different cellular model (Wang and Prywes, 2000).
In addition to the ERK pathway, other MAPK-dependent pathways are thought to be involved in stimulus-mediated CREB phosphorylation (Shaywitz and Greenberg, 1999). For example, in PC12, cells nerve growth factor induces CREB phosphorylation (Xing et al., 1998), and fibroblast growth factor treatment of SK-N-MC cells causes CREB and ATF-1 phosphorylation (Tan et al., 1996). These two stimuli induce transcription factor phosphorylation via MAP kinase-activated protein kinase 2, an enzyme that lies immediately downstream of p38MAPK.
The mechanism by which ET-1 triggers activation of the p38MAPK-dependent pathway is unclear. It is possible that this occurs through members of the Rho family of guanine nucleotide binding proteins (Tibbles and Woodgett, 1999), because (1) Rho activation is required for ET-1-induced transcription of c-fos through the SRE (Kim et al., 1997), and (2) Rho is activated by ET-1 in astrocytes (Cazaubon et al., 1997). Alternatively, p38MAPK could be activated by PKC, because this pathway has been identified during stimulation of another class of G-protein-coupled receptors (Naor et al., 2000).
Our results obtained with the inhibitor SB203580 indicate that, in astrocytes, a p38MAPK-dependent pathway mediates ET-1-induced CREB phosphorylation but is not involved in ATF-1 phosphorylation (Fig.9A). These differences in the relative contribution of the p38MAPK-dependent pathway in transcription factor phosphorylation might be attributable to a cell type-specific expression or mechanism of activation of different kinase isoforms lying downstream of p38MAPK and directly responsible for CREB and ATF-1 phosphorylation.
In contrast to CREB and ATF-1, the transcription factor Elk-1 could be directly phosphorylated by nuclear translocation of ERKs, p38MAPKs, and JNKs (Schaeffer and Weber, 1999). In astrocytes, ET-1-induced Elk-1 phosphorylation appears to be predominantly mediated by an ERK-dependent pathway (Fig. 9B), because the JNK-dependent pathway does not seem to be activated (Figs. 8B, 11). The ET-1-induced regulation of the JNK pathway appears to be cell-type specific, because ET-1 can activate the JNK pathway in smooth muscle cells (Fei et al., 2000), in glomerular mesangial cells (Isono et al., 1998), and in cardiomyocytes (Choukroun et al., 1998). Future studies in astrocytes are required to define the relative contribution of JNK- and p38MAPK-dependent pathways in ET-1-stimulated CREB phosphorylation.
The effect of ET-1 on c-fos transcription requires the participation of the PKC/ERK-dependent or the p38MAPK-dependent pathway, because pharmacological blockade of either of these pathways prevents the increase in c-fos mRNA triggered by ET-1. A possible explanation of these results is that, although the ERK pathway plays a major role in the phosphorylation of target transcription factors in the c-fos promoter, maximal c-fostranscription is achieved only when concomitant activation of two MAPK pathways occurs (Figs. 10, 11).
In conclusion, we demonstrated that ET-Rs are expressed in astrocytes both in vivo and in vitro and that the ETB-R subtype is selectively coupled to CREB phosphorylation. Distinct and parallel signal transduction pathways are induced by ET-1 and converge on regulation of CRE-dependent genes (Fig.11). Given the wide array of physiological and pathological responses elicited by ETs in neural cells and the central role played by ETB-Rs in the brain (Kuwaki et al., 1997), the use of selective ETB-R agonists and antagonists, or agents that interfere with the ETB-R-associated pathways, will further clarify the molecular mechanisms associated with the physiological action of ETs in the brain.
Recent findings indicated that endothelial cells, which actively synthesize and release ET-1 (Vanhoutte, 2000), stimulate astrocyte differentiation (Mi et al., 2001). Our study might shed new light on the possible role of ET-1 on the development of different classes of neural cells and might contribute to our understanding of the intracellular molecular mechanism mediating endothelial cell-induced astrocyte differentiation.
Footnotes
This work was supported by the National Institute of Child Health and Human Development. We are particularly grateful to Dr. J. L. Bos (Laboratory for Physiological Chemistry and Center for Biomedical Genetics, Utrecht University, Utrecht, The Netherlands) for providing the Rap1N17 plasmid and Dr. M. Freissmuth (Institute of Pharmacology, University of Vienna, Vienna, Austria) for providing the bacterial lysate containing the GST fusion protein of the minimal Rap1-binding domain of ralGDS (GST-ral-RBD). We thank Cristina Ghiani for help with cell culture and for discussion. We thank Ye Chen for assistance with Western blot and Xiaoqing Yuan for help with immunostaining. We thank Enrico V. Avvedimento for c-fosNorthern blots. We thank Shibeshih Belachew, Douglas Fields, Chris McBain, and Beth Stevens for critically reading this manuscript.
Correspondence should be addressed to Dr. Vittorio Gallo, Laboratory of Cellular and Synaptic Neurophysiology, National Institute of Child Health and Human Development, National Institutes of Health, Building 49, Room 5A-78, Bethesda, MD 20892-4495. E-mail: vgallo@helix.nih.gov.
REFERENCES
- 1.Barnier JV, Papin C, Eychene A, Lecoq O, Calothy G. The mouse B-raf gene encodes multiple protein isoforms with tissue-specific expression. J Biol Chem. 1995;270:23381–23389. doi: 10.1074/jbc.270.40.23381. [DOI] [PubMed] [Google Scholar]
- 2.Böhm M, Moellmann G, Cheng E, Alvarez-Franco M, Wagner S, Sassone-Corsi P, Halaban R. Identification of p90RSK as the probable CREB-Ser133 kinase in human melanocytes. Cell Growth Differ. 1995;6:291–302. [PubMed] [Google Scholar]
- 3.Bos JL. All in the family? New insight and questions regarding interconnectivity of Ras, Rap 1 and Ral. EMBO J. 1998;17:6776–6782. doi: 10.1093/emboj/17.23.6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395–1413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 5.Cazaubon S, Chaverot N, Romero IA, Girault JA, Adamson P, Strosberg AD, Couraud PO. Growth factor activity of endothelin-1 in primary astrocytes mediated by adhesion-dependent and -independent pathway. J Neurosci. 1997;15:6203–6212. doi: 10.1523/JNEUROSCI.17-16-06203.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiloeches A, Paterson HF, Marais R, Clerk A, Marshall CJ, Sugden PH. Regulation of ras-GTP loading and ras-raf association in neonatal rat ventricular myocites by G protein-coupled receptor agonists and phorbol ester. J Biol Chem. 1999;274:19762–19770. doi: 10.1074/jbc.274.28.19762. [DOI] [PubMed] [Google Scholar]
- 7.Choukroun G, Hajjar R, Kyriakis JM, Bonventre JV, Rosenzweig A, Force T. Role of the stress-activated protein kinases in endothelin-induced cardiomyocyte hypertrophy. J Clin Invest. 1998;102:1311–1320. doi: 10.1172/JCI3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dugan LL, Kim JS, Zhang Y, Bart RD, Sun Y, Holtzman DM, Gutmann DH. Differential effects of cAMP in neurons and astrocytes. J Biol Chem. 1999;274:25842–25848. doi: 10.1074/jbc.274.36.25842. [DOI] [PubMed] [Google Scholar]
- 9.Fei J, Viedt C, Soto U, Elsing C, Jahn L, Kreuzer J. Endothelin-1 and smooth muscle cells: induction of jun amino-terminal kinase through an oxygen radical-sensitive mechanism. Arterioscler Thromb Vasc Biol. 2000;20:1244–1249. doi: 10.1161/01.atv.20.5.1244. [DOI] [PubMed] [Google Scholar]
- 10.Foschi M, Chari S, Dunn MJ, Sorokin A. Biphasic activation of p21ras by endothelin-1 sequentially activates the ERK cascade and phosphatidylinositol 3-kinase. EMBO J. 1997;16:6439–6451. doi: 10.1093/emboj/16.21.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franke B, Akkerman JW, Bos JL. Rapid Ca2+-mediated activation of Rap1 in human platelets. EMBO J. 1997;16:252–259. doi: 10.1093/emboj/16.2.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol. 1999;25:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 13.Gallo V, Armstrong R. Developmental and growth factor induced regulation of nestin in oligodendrocyte lineage cells. J Neurosci. 1995;15:394–406. doi: 10.1523/JNEUROSCI.15-01-00394.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia J, de Gunzburg J, Eychene A, Gisselbrecht S, Porteau F. Thrombopoietin-mediated sustained activation of extracellular signal-regulated kinase in UT7-Mp1 cells requires both Ras-Raf-1- and Rap1-B-Raf-dependent pathways. Mol Cell Biol. 2001;21:2659–2670. doi: 10.1128/MCB.21.8.2659-2670.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghiani CA, Gallo V. Inhibition of cyclin E-cyclin-dependent kinase 2 complex formation and activity is associated with cell cycle arrest and withdrawal in oligodendrocyte progenitor cells. J Neurosci. 2001;21:1274–1282. doi: 10.1523/JNEUROSCI.21-04-01274.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghiani CA, Eisen AM, Yuan X, DePinho RA, McBain CJ, Gallo V. Neurotransmitter receptor activation triggers p27Kip1 and p21CIP1 accumulation and G1 cell cycle arrest in oligodendrocyte progenitors. Development. 1999;126:1077–1090. doi: 10.1242/dev.126.5.1077. [DOI] [PubMed] [Google Scholar]
- 17.Gutkind JS. The pathways connecting G protein-coupled receptors to the nucleus through divergent mitogen-activated protein kinase cascades. J Biol Chem. 1998;273:1839–1842. doi: 10.1074/jbc.273.4.1839. [DOI] [PubMed] [Google Scholar]
- 18.Hagemann C, Rapp UR. Isotype specific functions of Raf kinases. Exp Cell Res. 1999;253:34–46. doi: 10.1006/excr.1999.4689. [DOI] [PubMed] [Google Scholar]
- 19.Isono M, Haneda M, Maeda S, Omatsu-Kanbe M, Kikkawa R. Atrial natriuretic peptide inhibits endothelin-1-induced activation of JNK in glomerular mesangial cells. Kidney Int. 1998;53:1133–1142. doi: 10.1046/j.1523-1755.1998.00869.x. [DOI] [PubMed] [Google Scholar]
- 20.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 21.Kim JH, Cho YS, Kim BC, Kim YS, Lee GS. Role of Rho GTPase in the endothelin-1-induced nuclear signaling. Biochem Biophys Res Commun. 1997;232:223–226. doi: 10.1006/bbrc.1997.6261. [DOI] [PubMed] [Google Scholar]
- 22.Kuwaki T, Kurihara H, Cao WH, Kurihara Y, Unekawa M, Yazaki Y, Kumada M. Physiological role of brain endothelin in the central autonomic control: from neuron to knockout mouse. Prog Neurobiol. 1997;51:545–579. doi: 10.1016/s0301-0082(96)00063-9. [DOI] [PubMed] [Google Scholar]
- 23.Ladenheim RG, Lacroix I, Foignant-Chaverot N, Strosberg AD, Couraud PO. Endothelins stimulate c-fos and nerve growth factor expression in astrocytes and astrocytoma. J Neurochem. 1993;60:260–266. doi: 10.1111/j.1471-4159.1993.tb05846.x. [DOI] [PubMed] [Google Scholar]
- 24.Lazarini F, Strosberg AD, Courad PO, Cazaubon SM. Coupling of ETB endothelin receptor to mitogen-activated protein kinase stimulation and DNA synthesis in primary cultures of rat astrocytes. J Neurochem. 1996;66:459–465. doi: 10.1046/j.1471-4159.1996.66020459.x. [DOI] [PubMed] [Google Scholar]
- 25.Leach K, Turner D, Zhang W, Mulholland MW. Endothelin-1 stimulates c-fos mRNA expression in C6 glioma cells via MAPKinase pathway. Peptides. 1999;20:907–914. doi: 10.1016/s0196-9781(99)00080-7. [DOI] [PubMed] [Google Scholar]
- 26.Masaki T, Yanagisawa M, Goto K. Physiology and pharmacology of endothelins. Med Res Rev. 1992;12:391–421. doi: 10.1002/med.2610120405. [DOI] [PubMed] [Google Scholar]
- 27.McLeod SJ, Ingham RJ, Bos JL, Kurosaki T, Gold MR. Activation of the Rap1GTPase by the B cell antigen receptor. J Biol Chem. 1998;273:29218–29223. doi: 10.1074/jbc.273.44.29218. [DOI] [PubMed] [Google Scholar]
- 28.Mi H, Haeberle H, Barres BA. Induction of astrocyte differentiation by endothelial cells. J Neurosci. 2001;21:1538–1547. doi: 10.1523/JNEUROSCI.21-05-01538.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mielke K, Herdegen T. JNK and p38 stress kinase—degenerative effectors of signal-transduction-cascades in the nervous system. Prog Neurobiol. 2000;61:45–60. doi: 10.1016/s0301-0082(99)00042-8. [DOI] [PubMed] [Google Scholar]
- 30.Naor Z, Benard O, Seger R. Activation of MAPK cascades by G-protein-coupled receptors: the case of gonadotropin-releasing hormone receptor. Trends Endocrinol Metab. 2000;11:91–99. doi: 10.1016/s1043-2760(99)00232-5. [DOI] [PubMed] [Google Scholar]
- 31.Nie XJ, Olsson Y. Endothelin peptides in brain diseases. Rev Neurosci. 1996;7:177–186. doi: 10.1515/revneuro.1996.7.3.177. [DOI] [PubMed] [Google Scholar]
- 32.Pizon V, Baldacci G. Rap1A protein interferes with various MAP kinase activating pathways in skeletal myogenic cells. Oncogene. 2000;19:6074–6081. doi: 10.1038/sj.onc.1203984. [DOI] [PubMed] [Google Scholar]
- 33.Reedquist KA, Ross E, Koop EA, Wolthuis RMF, Zwartkruis FJT, van Kooyk Y, Salmon M, Buckley CD, Bos JL. The small GTPase, Rap1, mediates CD31-induced integrin adhesion. J Cell Biol. 2000;148:1151–1158. doi: 10.1083/jcb.148.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robertson LM, Kerppola TK, Vendrell M, Luk D, Smeyne RJ, Bocchiaro C, Morgan JI, Curran Regulation of c-fos expression in transgenic mice requires multiple interdependent transcription control elements. Neuron. 1995;14:241–252. doi: 10.1016/0896-6273(95)90282-1. [DOI] [PubMed] [Google Scholar]
- 35.Rubanyi GM, Polokoff MA. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev. 1994;46:325–415. [PubMed] [Google Scholar]
- 36.Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmitt JM, Stork PJS. β2-Adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein Rap1 and the serine/threonine kinase B-Raf. J Biol Chem. 2000;275:25342–25350. doi: 10.1074/jbc.M003213200. [DOI] [PubMed] [Google Scholar]
- 38.Seidel MG, Klinger M, Freissmuth M, Holler Activation of mitogen-activated protein kinase by the A(2A)-adenosine receptor via a Rap1-dependent and via a p21(ras)-dependent pathway. J Biol Chem. 1999;274:25833–25841. doi: 10.1074/jbc.274.36.25833. [DOI] [PubMed] [Google Scholar]
- 39.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 40.Tabernero A, Stewart HJS, Jessen KR, Mirsky R. The neuron-glia signal beta neuregulin induces sustained CREB phosphorylation on Ser-133 in cultured rat Schwann cells. Mol Cell Neurosci. 1998;10:309–322. [PubMed] [Google Scholar]
- 41.Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ. FGF and stress regulate CREB and ATF-1 via pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996;15:4629–4642. [PMC free article] [PubMed] [Google Scholar]
- 42.Tibbles LA, Woodgett JR. The stress-activated protein kinase pathways. Cell Mol Life Sci. 1999;55:1230–1254. doi: 10.1007/s000180050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vanhoutte P. Say NO to ET. J Auton Nerv Syst. 2000;81:271–277. doi: 10.1016/s0165-1838(00)00126-0. [DOI] [PubMed] [Google Scholar]
- 44.Vanhoutte P, Barnier JV, Guibert B, Pages C, Besson MJ, Hipskind RA, Caboche J. Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol. 1999;19:136–146. doi: 10.1128/mcb.19.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Prywes R. Activation of the c-fos enhancer by the erk MAP kinase pathway through two sequence elements: the c-fos AP-1 and p62TCF sites. Oncogene. 2000;19:1379–1385. doi: 10.1038/sj.onc.1203443. [DOI] [PubMed] [Google Scholar]
- 46.Whitmarsh AJ, Davis RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med. 1996;74:589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- 47.Xing J, Kornhauser JM, Xia Z, Thiele EA, Greenberg ME. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol. 1998;18:1946–1955. doi: 10.1128/mcb.18.4.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin J, Lee JA, Howells RD. Stimulation of c-fos and c-jun gene expression and down-regulation of proenkephalin gene expression in C6 glioma cells by endothelin-1. Brain Res Mol Brain Res. 1992;14:213–220. doi: 10.1016/0169-328x(92)90176-c. [DOI] [PubMed] [Google Scholar]
- 49.York RD, Yao H, Dillon T, Ellig CL, Eckert SP, McCleskey EW, Stork PJS. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature. 1998;392:622–625. doi: 10.1038/33451. [DOI] [PubMed] [Google Scholar]
- 50.Zhang M, Olsson Y. Reactions of astrocytes and microglial cells around hematogenous metastases of the human brain. Expression of endothelin-like immunoreactivity in reactive astrocytes and activation of microglial cells. J Neurol Sci. 1995;134:26–32. doi: 10.1016/0022-510x(95)00227-9. [DOI] [PubMed] [Google Scholar]
- 51.Zhang M, Olsson Y. Hematogenous metastases of the human brain-characteristics of peritumoral brain changes: a review. J Neurooncol. 1997;35:81–89. doi: 10.1023/a:1005799805335. [DOI] [PubMed] [Google Scholar]
- 52.Zwartkruis FJ, Wolthuis RM, Nabben NM, Franke B, Bos JL. Extracellular signal-regulated activation of Rap1 fails to interfere in Ras effector signalling. EMBO J. 1998;17:5905–5912. doi: 10.1093/emboj/17.20.5905. [DOI] [PMC free article] [PubMed] [Google Scholar]