

Abstract
Prostaglandins (PGs) are local mediators of several functions in the CNS. Both primary afferent neurons and intrinsic cells in the spinal cord produce PGs, with a marked upregulation during peripheral inflammation. Therefore, the significance of spinal PGs in the neuronal processing of mechanosensory information was herein investigated. In anesthetized rats, the discharges of spinal nociceptive neurons with input from the knee joint were extracellularly recorded. Topical administration of prostaglandin E2(PGE2) to the spinal cord facilitated the discharges and expanded the receptive field of dorsal horn neurons to innocuous and noxious pressure applied to the knee joint, the ankle, and the paw, thus mimicking inflammation-induced central sensitization. Conversely, topical administration of the PG synthesis inhibitor indomethacin to the spinal cord before and during development of knee joint inflammation attenuated the generation of inflammation-induced spinal neuronal hyperexcitability. However, after development of inflammation, the responses of spinal neurons to mechanical stimuli were only reduced by systemic indomethacin but not by indomethacin applied to the spinal cord. Thus, spinal PG synthesis is important for the induction and initial expression but not for the maintenance of spinal cord hyperexcitability. Spinal PGE2 application facilitated dorsal horn neuronal firing elicited by ionophoretic delivery of NMDA, suggesting that an interaction of PGs and NMDA receptors may contribute to inflammation-induced central sensitization. However, after development of inflammation, spinal indomethacin failed to reduce responses to ionophoretic delivery of NMDA or AMPA, suggesting that such an interaction is not required for the maintenance of central sensitization.
Keywords: central sensitization, glutamate, pain, nociception, NSAID, prostaglandin
Prostaglandins (PGs) are local mediators in a variety of functions in the CNS as well as in peripheral tissues. Considerable information has emerged in recent years regarding the involvement of spinal cord PGs in pain and central sensitization (Vanegas and Schaible, 2001). Indeed, cyclooxygenases 1 and 2 (COX-1 and COX-2), the enzymes that form prostaglandin E2 (PGE2) and other PGs from arachidonic acid, are expressed in both dorsal root ganglia (DRGs) and the spinal cord (Willingale et al., 1997; Inoue et al., 1999). Both isoforms of COX are expressed constitutively, but in particular COX-2 is markedly upregulated in the spinal cord during acute and chronic peripheral inflammation (Beiche et al., 1996, 1998a; Hay and de Belleroche, 1997; Hay et al., 1997; Goppelt-Struebe and Beiche, 1998;Ebersberger et al., 1999; Samad et al., 2001). In the spinal cord, there is a basal release of PGE2 as well as an increased release after noxious stimulation, such as electrical pulses (Ramwell et al., 1966), noxious heat (Coderre et al., 1990), subcutaneous formalin (Malmberg and Yaksh, 1995a,b; Hua et al., 1999;Muth-Selbach et al., 1999), and peripheral inflammation (Yang et al., 1996a; Hay et al., 1997; Hay and de Belleroche, 1998; Ebersberger et al., 1999; Gühring et al., 2000). Conversely, intrathecal administration of PGE2 causes allodynia and hyperalgesia in awake animals (Taiwo and Levine, 1988; Uda et al., 1990; Minami et al., 1994a, 1996; Malmberg et al., 1995a,b; Nishihara et al., 1995; Ferreira and Lorenzetti, 1996). Finally, antinociceptive nonsteroidal anti-inflammatory drugs (NSAIDs), such as indomethacin, inhibit PG synthesis not only in the inflamed peripheral tissues but also in the spinal cord, an effect that may contribute to their antinociceptive action (Vanegas and Schaible, 2001).
Curiously, the effects of PGs on spinal dorsal horn neuronal firing have until now received only scant attention (Vanegas and Schaible, 2001), although this activity is precisely the drive for segmental and suprasegmental reflexes as well as for the thalamocortical transmission that leads to the perception of pain. In the present experiments, we therefore determined the effects of spinally applied PGE2 on the responses of nociceptive dorsal horn neurons to natural mechanical innocuous and noxious stimulation of the knee and other peripheral tissues. We further investigated whether there is an interaction between PGE2 and glutamatergic synaptic transmission in the spinal dorsal horn. Finally, we tested whether spinal application of a COX inhibitor influences the development and maintenance of inflammation-induced hyperexcitability of nociceptive neurons to mechanosensory stimuli, because mechanical hyperalgesia (pressure hypersensitivity) is an important pain symptom of joint inflammation.
Parts of this work have been published previously in abstract form (Vasquez et al., 2000, 2001).
MATERIALS AND METHODS
Preparation
Experiments were performed on 75 male Wistar rats (200–350 gm; University of Jena, Jena, Germany) anesthetized with sodium thiopentone (Trapanal; initial dose of 85–115 mg/kg, i.p.; BYK Gulden Ltd., Konstanz, Germany). The trachea was cannulated, and catheters were inserted into the common carotid artery and the external jugular vein. The animals breathed spontaneously, and a gentle jet of oxygen was blown toward the opening of the tracheal cannula. Body temperature was kept at 37°C by means of a feedback-controlled system. Additional intraperitoneal injections of thiopentone (20–50 mg/kg) were given when necessary to achieve a sufficiently deep level of anesthesia as assessed by the absence of corneal or leg withdrawal reflexes. Mean arterial blood pressure was stable at 90–120 mmHg. Spinal cord segments L1–L4 were exposed by laminectomy. The dura mater was opened, and a thin-walled, elliptic rubber ring (∼3 × 5 mm) was tightly sealed with silicone gel onto the surface of the cord. This ring thus formed a trough with ∼30 μl capacity over the spinal segments in which the recordings were to be performed. This trough was immediately filled with Tyrode's solution. A solution of 3% agar in Tyrode's solution was poured around the trough to seal and stabilize the surgical area.
In 51 rats, an inflammation was induced in the left knee joint, either at the beginning of the experiment before recordings (n= 33 rats) or during the recording session while an ipsilateral neuron was being monitored (n = 18 rats). With this purpose, a 27 gauge needle was introduced through the patellar ligament, and 70 μl of a 4% kaolin suspension (Sigma, Deisenhofen, Germany) was slowly injected into the articular cavity. Then, the joint was slowly flexed and extended for 15 min. Thereafter, 70 μl of a 2% carrageenan solution (Sigma) was injected, and the joint was moved for another 5 min.
Recording and mechanical stimulation
Using glass-insulated carbon filaments for extracellular recording, individual neurons were identified by spike shape and height. The action potentials were continuously monitored on a digital oscilloscope to guarantee that the same neuron was recorded throughout. The signal was also fed into a window discriminator, the output of which was processed by an analog-to-digital interface and a personal computer so that peristimulus time histograms could be constructed. Action potentials were also stored on hard disk by means of the Spike/Spidi software (Forster and Handwerker, 1990). Final spike discrimination was performed off-line according to shape and size. Dorsal horn neurons were chosen for study if they responded to pressure applied to the ipsilateral, left knee but did not respond to brushing or squeezing of the skin over the knee. The size of, and the threshold within, receptive fields were determined using stimulation of the skin (brushing or squeezing of skin folds with forceps) and of the deep tissue (manual compression of joints and muscles). Mechanical test stimuli of two standard intensities were then applied to the knee, the ankle, and the paw. Each test stimulus lasted for 15 sec. A calibrated mechanical device (CORREX; Haag-Streit, Bern, Switzerland) was used for compression of the knee joint in the mediolateral axis; for innocuous intensity, a 1.9 N/40 mm2 holding pressure was applied, and for noxious intensity (felt painful when applied to the experimenter's fifth finger), the knee was compressed with 7.8 N/40 mm2 (or with 5.9 N/40 mm2 when the response was very strong). Two modified crocodile clips with teeth filed away and jaws wrapped in tape were used to apply mediolateral compression of the ankle joint and dorsoventral compression of the middle of the paw (1.1 N/20 mm2 for innocuous stimulation, and 5.8 N/20 mm2 for noxious stimulation).
Ionophoretic delivery of glutamatergic agonists
In one set of experiments, we used the ionophoretic administration of AMPA (Research Biochemicals, Deisenhofen, Germany) and NMDA (Research Biochemicals) close to the neurons. A carbon filament, glass-insulated microelectrode was glued onto a multibarrel glass micropipette so that their tips ended together. The carbon microelectrode was used for recording of extracellular unitary action potentials from nociceptive dorsal spinal neurons with input from the knee (see above). The multibarrel micropipette was used for ionophoresis. One barrel was filled with AMPA (10 mm in 155 mm NaCl, pH 7.5, adjusted with NaOH), and another one was filled with NMDA (50 mm in 115 mm NaCl, pH 7.5, adjusted with NaOH). Two other barrels were filled with NaCl; one of them (1 m) served for automatic current balancing, and the other (165 mm) served for control of current application. Ejection currents for NMDA and AMPA were negative, and positive retaining currents of 25 nA were used between ejection periods.
Experimental protocols
Effect of spinally applied PGE2 on responses to mechanical stimulation. In the first set of experiments (15 rats, one neuron per rat), we tested the effects of PGE2 on the responses of spinal cord neurons to mechanical stimulation of the left knee, ankle, and paw. Innocuous and noxious test stimuli were applied sequentially to the knee, the ankle, and the paw. This sequence was repeated every 5 min, even when the manipulations described below were being performed. The neuronal baseline responses were established with the vehicle solution (0.07% ethanol in Tyrode's solution) in the spinal trough, and, when the responses were stable, the last 25 min before application of PGE2 were taken as the predrug control period. After having established the baseline responses, the spinal trough was rinsed and filled with 30 μl of a solution containing PGE2 (100 ng/μl; Cayman Chemical, Ann Arbor, MI). This dose of PGE2 was chosen from behavioral experiments using intrathecal application of PGE2(Vanegas and Schaible, 2001). The mechanical stimulation of knee, ankle, and paw was continued, and, after 50 min, the PGE2 solution was replaced by the vehicle solution. The responses to mechanical stimulation were continued for another 25–50 min to observe recovery. In four experiments, the effect of the vehicle alone was tested and found to be nought.
In some experiments, PGE2 was applied a second time while the same cell (four experiments) or another cell (eight experiments) was being recorded. The same procedure was used with another eight rats, but in these experiments, the knee joint had been inflamed 5–10 (on average 7) hours before the recordings started.
Effect of the application of the COX inhibitor indomethacin.In the second set of experiments, we studied the effect of spinally administered indomethacin on the development and maintenance of inflammation-evoked mechanical hyperexcitability of dorsal horn neurons. To study its effect on development of hyperexcitability, indomethacin was applied before the inflammation was induced. In eight rats (one neuron per rat), the responses of the neurons to mechanical stimulation of knee, ankle, and paw (see above) were monitored for 25 min. Then, indomethacin (Calbiochem, Bad Soden, Germany) was administered at a dose of 8 mm (Vanegas and Schaible, 2001) into the spinal trough, and the responses to mechanical stimulation were tested for another 25 min. Thereafter, the inflammation was induced (see above), and the responses to stimulation of knee, ankle, and paw were followed for another 4 hr still in the presence of indomethacin. Another 10 rats were used to assess the development of hyperexcitability in the absence of indomethacin. In these animals, only the vehicle (10 mmNa2HPO4, 17.65 mm NaOH, and 63.4 mm NaCl, pH 7.5, adjusted with HCl) was administered to the spinal cord while the responses to mechanical stimulation of knee, ankle, and paw were monitored before (25 min) and after induction of inflammation (4 hr).
To study the effect of indomethacin on the maintenance of spinal cord hyperexcitability, an inflammation was induced first, and the recordings from neurons with knee input were started 5–10 hr thereafter (eight rats). The responses to mechanical stimulation of the knee, ankle, and paw were assessed for a period of 50 min, with vehicle (see above) on the spinal cord. Then, indomethacin (8 mm) was applied to the spinal cord surface, and the responses to the mechanical test stimuli were monitored for 100 min during which indomethacin was on the cord. Thereafter, indomethacin (4 mg/kg) was given intraperitoneally, and the protocol was continued for another 50 min.
Effect of PGE2 and indomethacin on glutamatergic responses. In the third set of experiments (nine rats with normal knee and 12 rats with inflamed knee, one neuron per rat), we tested whether the application of PGE2to the surface of the spinal cord would alter the responses to ionophoretic administration of AMPA or NMDA. Once a neuron had been characterized, ionophoretic application of AMPA or NMDA was started. After a variable number of seconds, the neuron began to fire, and ionophoretic application was then maintained for 10 sec after the first evoked spike. An ejection current was chosen such that it reproducibly induced clear responses that could be modulated in both directions (increase or decrease). Application of AMPA or NMDA began every 60 sec and was repeated until a stable baseline of 50 min duration was obtained. Then, PGE2 (100 ng/μl) was administered for 50 min, and the application of AMPA or NMDA was performed. Finally, the responses were monitored after washout of PGE2 for 50 min.
In another five rats with an inflammation in the knee joint, the effect of indomethacin on the responses to AMPA and NMDA was tested. AMPA and NMDA were administered ionophoretically for 50 min as described above, and then indomethacin (8 mm) was administered into the spinal trough and the responses to AMPA and NMDA were continued for 50 min. Thereafter, indomethacin was replaced by Tyrode's solution, and the responses to AMPA and NMDA were recorded for another 50 min.
Data analysis. The responses to pressure stimuli were calculated by subtracting the ongoing activity in the preceding 15 sec (if any) from the total activity during an innocuous or a noxious test stimulus. To evaluate the effects of PGE2 on the responses to mechanical stimulation, we averaged all of the test responses to each type of stimulus in the 25 min preceding drug application (baseline), as well as the test responses during the last 25 min of PGE2 application. Values are given in mean ± SE. Baseline and PGE2 values were compared by using the Wilcoxon matched-pairs signed rank test. A similar procedure was used for the evaluation of the effects of PGE2 on the responses to NMDA and AMPA. The responses in the last 25 min before PGE2(baseline) were averaged and normalized to 100%, and the values in the last 25 min of PGE2 application were expressed as percentage of baseline. A similar procedure was used to analyze effects of indomethacin.
In experiments in which we assessed the effects of indomethacin on the development of inflammation-induced hyperexcitability, the baseline responses before inflammation were averaged (mean ± SE). After injection of kaolin into the knee, the responses to each stimulus within every hour after kaolin were averaged, and the baseline responses were subtracted. For statistical analysis, we compared the increase of the responses in the nontreated and the indomethacin-treated animals at corresponding intervals of 1 hr using the Mann–Whitney U test. In all statistical tests, significance was acknowledged if p < 0.05.
RESULTS
All neurons with knee input were located in segments L1–L4 at depths of 700-1200 μm (mean of 954 μm), thus mainly in the deep dorsal horn. Typically, the receptive field also included deep tissues of the thigh and lower leg. In 42 rats with normal joints, 29 of 42 neurons were wide dynamic range neurons that responded to innocuous and noxious stimulation of the knee in a graded manner, and 13 of 42 neurons were nociceptive specific and responded only to noxious pressure applied to the knee and other tissues. In 20 of these 42 neurons, the receptive field extended to the ankle and, in eight neurons, further to the paw. Three of these neurons had cutaneous receptive fields located on the thigh and/or the lower leg. Spontaneous activity was usually absent. In the sample of 33 neurons with input from the inflamed knee joint, 26 neurons were wide dynamic range, and seven were nociceptive-specific neurons.
Effect of PGE2 on the responses of spinal cord neurons
The effects of spinally applied PGE2 on the responses of dorsal horn neurons to mechanical stimulation of the ipsilateral hindlimb were studied in 15 neurons. Figure1 shows the responses to innocuous and noxious pressure applied to the knee joint, the ankle, and the paw. During baseline stimulation the spinal cord trough was filled with vehicle solution. When this was replaced by the PGE2 solution (100 ng/μl), the responses to both innocuous and noxious pressure showed a gradual increase. When the PGE2-containing solution was replaced by the vehicle, the responses declined slowly. However, recovery was incomplete in most of the neurons (this is indicated by the large SE in the post-PGE2 period).
Fig. 1.
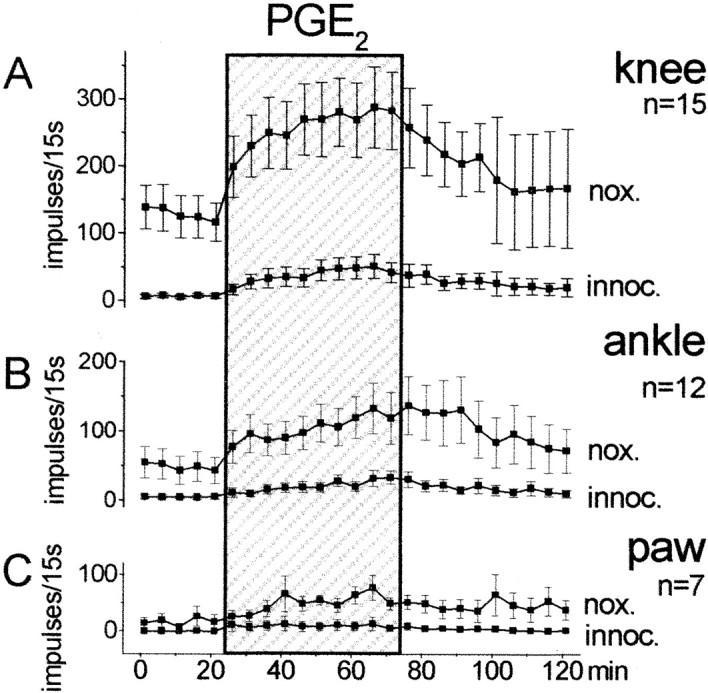
Effect of PGE2 on the responses of dorsal horn neurons to mechanical stimulation of the ipsilateral knee joint, ankle, and paw. Each symbol shows the mean ± SE of the responses. nox., Noxious pressure;innoc., innocuous pressure. During the first 25 min, only vehicle solution was in the spinal cord trough. This was replaced for 50 min (shaded area) by a solution containing PGE2 (100 ng/μl).
For statistical analysis, we compared the average responses in the last 25 min of PGE2 application versus the average baseline responses by using the Wilcoxon matched-pairs signed rank test. The increase of the responses was significant for innocuous (p < 0.005) and noxious (p < 0.001) pressure applied to the knee (n = 15 neurons), for innocuous (p < 0.005) and noxious (p < 0.01) pressure applied to the ankle (n = 12 neurons), and for innocuous (p < 0.05) and noxious (p < 0.05) pressure applied to the paw (n = 7 neurons). The number of neurons for knee was greater than for ankle or paw because their receptive fields did not always extend to these latter regions. Interestingly, however, PGE2 application caused an expansion of the receptive field in some neurons. Thus, under PGE2, three neurons that did not respond to ankle stimulation initially became responsive to noxious compression of the ankle, and three neurons whose receptive field did not include the paw initially became responsive to noxious stimulation of the paw. After PGE2, the responses increased in both nociceptive-specific and wide dynamic range neurons.
In four experiments, a second application of PGE2was made during recording from the same neuron, 2 hr 15 min after the first application. In eight additional experiments, a second neuron was identified ∼2 hr after the protocol with the first neuron had been completed, and the same protocol was performed with the new neuron. In contrast to the first application of PGE2, the second administration did not change the responses to mechanical stimulation (n = 12) (Fig.2).
Fig. 2.

Effect of PGE2 (shaded area) when spinally applied after a previous application of PGE2. The graph displays the responses of 12 spinal cord neurons to noxious (nox.) and innocuous (innoc.) pressure onto the knee joint. Same type of display as in Figure 1.
During inflammation of peripheral tissues, the milieu in the spinal cord is modified such that excitatory synaptic transmission becomes more efficient and spinal nociceptive neurons become hyperexcitable. Furthermore, endogenous release of PGE2 is increased (see introductory remarks). In view of the above results with a second PGE2 application, in eight neurons we tested the effects of PGE2 on the responses to mechanical stimulation when the knee joint had been inflamed for 5–10 (on average 7) hours. When applied during inflammation, PGE2 increased the neuronal responses to mechanical stimuli, but the effects were smaller than in non-inflamed animals (Fig. 3). However, this comparison reached statistical significance only for knee stimulation (p < 0.05; one-sided; Mann–WhitneyU test).
Fig. 3.

PGE2-induced changes in the responses to mechanical stimulation of knee, ankle, and paw in rats with non-inflamed knees and in rats with inflammation of the stimulated knee joint. The columns show the mean ± SE increase of the responses above baseline in the last 25 min of the PGE2application. *p < 0.05, significant difference between increase above baseline in rats with normal knees and rats with inflamed knees.
Effect of indomethacin on the development and maintenance of inflammation-induced hyperexcitability
In these experiments, we addressed the role of endogenous spinal PGs in the induction and maintenance of inflammation-induced hyperexcitability for mechanical stimuli. The PG synthesis inhibitor indomethacin that reduces stimulus-evoked PGE2release from spinal cord (Malmberg and Yaksh, 1994) was applied into the spinal trough. Figure 4 illustrates the experiments in which indomethacin was given before induction and during development of inflammation. First, we recorded the baseline responses, and then we induced the knee inflammation by injection of kaolin and carrageenan (K/C) and monitored the neurons for another 4 hr. Indomethacin was given 25 min before kaolin was injected and had no immediate effect on neuronal responses. Indeed, in five neurons, the average response to noxious pressure onto the knee joint before indomethacin was 273 ± 89 impulses/15 sec and after indomethacin was 260 ± 93 impulses/15 sec. However, indomethacin attenuated the inflammation-induced changes. Figure 4 shows the responses to noxious and innocuous pressure applied to the knee and ankle in nontreated control animals and in animals that received indomethacin in the spinal trough. In all graphs, the responses in the baseline period were set to zero, and the values after K/C express the increase of the responses above baseline. In the untreated rats, we observed a pronounced increase of the responses to mechanical stimulation of the injected knee (showing peripheral sensitization and ensuing spinal cord activation and sensitization) and of the non-inflamed ankle (showing central sensitization) within the 4 hr of knee inflammation (Fig. 4,open squares). The circumference of the injected knees increased on average from 43 to 50 mm. In contrast, when indomethacin was present in the spinal trough before and during development of knee inflammation, there was no increase in the responses to innocuous or noxious stimulation of the non-inflamed ankle or to innocuous stimulation of the injected knee. The responses to noxious pressure on the knee showed a small but significant increase. For statistical analysis, we compared for all stimuli the values obtained in each of the 4 hr after induction of inflammation (Mann–Whitney Utest). In neurons from rats without indomethacin, the responses to mechanical stimulation of the inflamed knee and of the non-inflamed ankle were significantly larger than in rats with indomethacin treatment (Fig. 4). In rats with spinal indomethacin, the circumference of the injected knees increased on average from 44 to 50 mm (same as in control experiments).
Fig. 4.

Effect of spinal application of indomethacin on the development of inflammation-induced hyperexcitability of spinal cord neurons. The graphs show the changes of the responses to noxious (nox.) and innocuous (innoc.) pressure ipsilaterally applied to the knee and to the ankle in the 4 hr after induction of inflammation in the knee by intra-articular injection of K/C. When the spinal trough was filled with vehicle solution (open squares) or with indomethacin (filled squares), the baseline (BL) values before K/C were set to zero, and the values after K/C injection show the mean ± SE change of all of the responses within each hourly interval compared with baseline. For stimulation of the knee joint, each value shows at least the averaged responses from seven animals and for stimulation of the ankle at least from four animals.
The inflammation induced by K/C and the resulting hyperexcitability of spinal cord neurons reach a plateau after 5–6 hr, i.e., the responses usually do not further increase after this time (Neugebauer et al., 1993). In additional experiments, we tested the effect of indomethacin when the compound was given after development of inflammation (Fig.5). Indomethacin (8 mm) was administered to the spinal cord 6–11 hr after induction of inflammation. Although indomethacin was applied to the spinal cord for 100 min, no reduction of the responses to knee stimulation was noted, nor were the responses to stimulation of the ankle and paw reduced. These data therefore suggest that inhibition of spinal PG synthesis after development of inflammation does not influence central sensitization. When indomethacin was given intraperitoneally at a later time, the responses to pressure onto the knee showed a progressive reduction. When the last three values after intraperitoneal injection of indomethacin were compared with the last three values before intraperitoneal injection of indomethacin, the reduction of the responses to noxious pressure was statistically significant for noxious pressure (p < 0.05; Wilcoxon matched-pairs signed rank test; n = 6 neurons). This is probably attributable to extraspinal effects of indomethacin.
Fig. 5.

Effects of indomethacin on neuronal responses after development of hyperexcitability. The recordings started 6–11 hr after induction of inflammation in the ipsilateral knee joint. Eachsymbol represents the mean ± SE of the responses of spinal cord neurons to innocuous (innoc.) and noxious (nox.) pressure onto the knee joint under spinal application and subsequent intraperitoneal administration of indomethacin.
Effect of PGE2 on the responses to AMPA and NMDA
Behavioral experiments have shown that the hyperalgesia evoked by PGE2 can be blocked by antagonists at NMDA and non-NMDA receptors (Minami et al., 1994b; Nishihara et al., 1995a). Furthermore, NMDA receptors are thought to be key players in the generation and maintenance of inflammation-induced hyperexcitability (Woolf and Thompson, 1991; Neugebauer et al., 1993). Thus, an interaction between PGE2 and glutamatergic synaptic transmission is of particular interest. We therefore studied whether the application of PGE2 would influence responses of spinal cord neurons to the application of AMPA and NMDA, the agonists at ionotropic glutamate receptors. Neurons that responded to mechanical stimulation of the knee joint were stimulated by the ionophoretic administration of AMPA or NMDA. The knee joint was either normal or inflamed. Figure 6 shows the effects of PGE2 on the responses of neurons to AMPA or NMDA. The line graphs show the average responses to each test application of AMPA or NMDA. The columns display the average responses in the 25 min before PGE2, the responses in the last 25 min of PGE2 application, and the responses after washout of PGE2, expressed as percentage of baseline. The baseline responses were obtained while Tyrode's solution was in the spinal cord trough. In animals with normal knees (Fig. 6, top), only the responses to NMDA showed a significant increase (p < 0.05; Wilcoxon matched-pairs signed rank test) during application of PGE2. However, when the knee was inflamed, both the responses to AMPA and to NMDA (Fig. 6, bottom) showed marked progressive increases (for AMPA, p < 0.005; for NMDA, p < 0.005; Wilcoxon matched-pairs signed rank test).
Fig. 6.
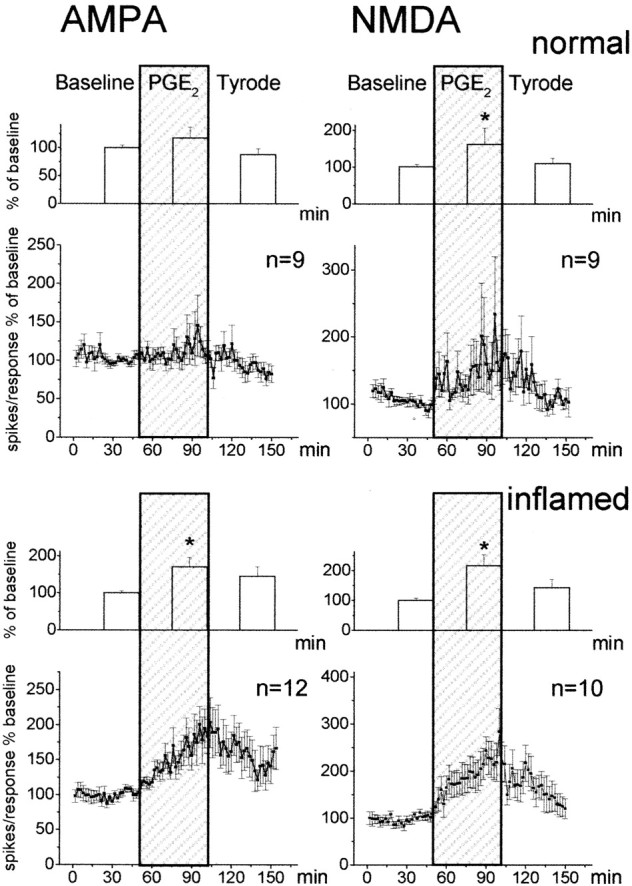
Effects of PGE2 on the responses of spinal cord neurons to the ionophoretic application of AMPA or NMDA. Neurons in the top panels were in animals with normal joints, and neurons in the bottom panels were in rats with inflammation of the ipsilateral knee joint. The line graphs below the column diagrams show the responses to each test application of AMPA or NMDA, averaged in the total sample of neurons. For each neuron, responses are expressed as percentage of the average of all its responses during baseline. Each squarerepresents the mean ± SE of the response of all neurons to one AMPA application (left) or one NMDA application (right). The column diagrams display the average response in the 25 min before PGE2, the average responses in the last 25 min of PGE2 application (shaded area), and the average responses after washout of PGE2, expressed as percentage of baseline. *p < 0.05 versus baseline.
To test whether a reduction of endogenous spinal PGs would alter the activation of glutamate receptors after inflammation is established, we tested the effect of spinal application of indomethacin on the responses to AMPA and NMDA. As with mechanical stimulation, indomethacin (8 mm) was administered 6–11 hr after induction of inflammation. Figure 7 shows that neither the responses to AMPA nor the responses to NMDA were significantly changed in the presence of indomethacin.
Fig. 7.

Effects of indomethacin on responses to AMPA or NMDA after development of hyperexcitability. The recordings started 6–11 hr after induction of inflammation in the ipsilateral knee joint.Graphs illustrate the effects of topical administration of indomethacin to the spinal cord on the responses of dorsal horn neurons to ionophoretically administered AMPA or NMDA. Eachsymbol displays one neuron.
DISCUSSION
In the present study, topical application of PGE2 to the spinal cord enhanced the responses of dorsal horn neurons to mechanical stimuli and expanded their receptive fields. These effects mimic the central sensitization that develops during knee joint inflammation. Spinal application of indomethacin before and during development of knee joint inflammation did not prevent the inflammation in the knee but markedly attenuated the generation of hyperexcitability in spinal cord neurons. Administration of indomethacin 6–11 hr after development of inflammation, however, did not reduce the enhanced responses. Thus, endogenous PG synthesis in primary afferent neurons and/or spinal cord seems necessary and sufficient for the induction of central sensitization but not for its maintenance. Topical application of PGE2 to the spinal cord enhanced the responses of nociceptive neurons to the ionophoretic application of NMDA, thus suggesting that PGE2 interaction with glutamatergic synaptic transmission contributes to the PG role in central sensitization. However, application of indomethacin after development of inflammation did not reduce responses to ionophoretically administered NMDA and AMPA, again suggesting that after establishment of inflammation spinal PG synthesis is not essential for glutamatergic sensitivity.
Prostaglandins and the increase in responsiveness of spinal cord neurons
The effects of PGE2 on neurons in vitro as well as on animal behavior have been studied extensively (Vanegas and Schaible, 2001). However, we have herein investigated the effects of PGE2 on nociceptive neuronal discharges, i.e., messages that may lead to perception and behavioral responses, elicited by “natural” innocuous and noxious stimuli. As mentioned, PGE2 induced a pattern of effects on spinal cord neurons similar to that of central sensitization in general and of inflammation-induced hyperexcitability in particular. After PGE2, like during peripheral inflammation (Woolf 1983; Hylden et al., 1989; Neugebauer and Schaible, 1990; Dougherty et al., 1992), and in agreement with behavioral experiments (see introductory remarks), the neurons showed an increased firing to noxious mechanical stimulation, which may lead to primary mechanical hyperalgesia, a decrease in the excitation threshold, and thus enhanced or novel responses to innocuous stimulation, which may lead to allodynia, and an expansion of the receptive field, which is typical of central sensitization and may lead to secondary mechanical hyperalgesia. Preliminary experiments show that butaprost, an EP2 agonist, produces similar effects as PGE2 in a dose-dependent manner.
We also studied herein the consequences of reducing the effect of PGs during the induction of inflammation, because in inflammation, PG synthesis and release in the spinal cord are upregulated (see introductory remarks). Local transmitters and modulators may contribute to this upregulation, because NMDA (Sorkin, 1993), kainic acid (Yang et al., 1996b), substance P (Hua et al., 1999), depolarization by K+ (Dirig et al., 1997), and capsaicin (Dirig and Yaksh, 1999) have been shown to stimulate PG synthesis and release in the spinal cord (Vanegas and Schaible, 2001). During inflammation, interleukin-1β-mediated induction of COX-2 may be particularly important (Samad et al., 2001). Because no specific antagonists are available for PG receptors, we applied indomethacin, a COX-1/COX-2 inhibitor, to the spinal cord before the inflammation began. This significantly attenuated the development of inflammation-induced neuronal hyperexcitability. Plausibly, the initial production of PGs in primary afferents and/or spinal cord intrinsic elements is essential for the full development of spinal cord plasticity. Indomethacin also reduces windup of dorsal horn neuronal discharges to electrical C-fiber stimulation (Willingale et al., 1997) and the discharges of dorsal horn neurons after subcutaneously applied formalin (Chapman and Dickenson, 1992). Although these and the present results are taken as attributable to COX inhibition, contribution by non-COX targets cannot be excluded (Vanegas and Schaible, 2001).
Effects of PGE2 on the responses to AMPA and NMDA
The results discussed above could arise from a PG effect on presynaptic elements, e.g., primary afferent terminals, or on postsynaptic elements, e.g., intrinsic spinal neurons. PGE2 acts on G-protein-coupled receptors (EP1–4), and DRG neurons express EP1 (Oida et al., 1995), EP3 (Sugimoto et al., 1994; Oida et al., 1995; Beiche et al., 1998b), and EP4 receptors (Oida et al., 1995), whereas spinal cord neurons express EP2 receptors (Kawamura et al., 1997). Thus, the spinal effects of naturally released or experimentally applied PGE2on nociception may be exerted on both the primary afferents and dorsal horn neurons.
The evidence for presynaptic targets is considerable. For example, PGE2 enhances the release of substance P and calcitonin gene-related peptide (CGRP) from cultured or native DRG neurons that is evoked by various chemical stimuli (Nicol et al., 1992; Andreeva and Rang, 1993; Vasko et al., 1993, 1994; Hingtgen and Vasko, 1994; Southall et al., 1998). Conversely, NSAIDs diminish the stimulus-induced release of substance P and CGRP from DRG neurons or spinal cord slices because this release is facilitated by PGs (Andreeva and Rang, 1993; Vasko et al., 1994). Furthermore, PGE2 application increases the frequency but not the amplitude of miniature EPSCs mediated by AMPA receptors in spinal cord neurons (Minami et al., 1999). Recently, however, Baba et al. (2001) showed that PGE2 excites spinal cord intrinsic neurons directly.
Here we show for the first time that PGE2increases firing of intrinsic neurons elicited by AMPA or NMDA. When the knees were normal, PGE2 caused a significant facilitation of firing to application of NMDA close to the neurons. This is of particular interest because the activation of NMDA receptors of dorsal horn neurons is thought to be a key mechanism in the induction of central sensitization (see above) and in particular the increase in mechanosensitivity (Neugebauer et al., 1993). When the joint was inflamed, PGE2 induced facilitated responses to AMPA and NMDA. This could result from presynaptic but also from postsynaptic effects of PGE2. For example, PGE2 might release compounds from primary afferents (e.g., substance P) that facilitate the responses to glutamate receptor agonists. Alternatively or additionally, the sensitivity of postsynaptic neurons to PGE2 might increase during inflammation attributable to, for example, EP receptor sensitization. However, because the responses to AMPA and NMDA were not reduced by indomethacin after inflammation was established, a facilitation of glutamatergic synapses by endogenous PGs does not seem to be the mechanism whereby central sensitization is maintained (see also below).
Role of prostaglandins after development of inflammation
Unexpectedly, when the inflammation was established, the responses of spinal cord neurons to mechanical stimulation (and the responses to AMPA or NMDA) were not reduced by spinal administration of indomethacin. However, the responses to mechanical stimuli were reduced after systemic administration of indomethacin. Because inhibition of PG synthesis by indomethacin is rapid, we should have seen a reduction of the responses after spinal application if the continuous production of PGs in the spinal cord were important for the maintenance of hyperexcitability to mechanical stimuli during inflammation. Interestingly, Dirig et al. (1998) observed no reduction of thermal hyperalgesia with intrathecal administration ofS(+)-ibuprofen or the specific COX-2 inhibitor SC58125 3 hr after induction of carrageenan inflammation. It thus seems that spinal PG synthesis is not required for the maintenance of central sensitization, at least in the time period studied, and that analgesic effects of NSAIDs in this period are exerted at sites other than spinal targets, at least with respect to mechanical stimulation. Such targets could be the inflamed knee joint (Heppelmann et al., 1986) and supraspinal structures (Vanegas et al., 1997).
The lack of effect of spinal indomethacin after establishment of inflammation could have several reasons. First, there might be no additional spinal PG synthesis after the initial PG production, and hence indomethacin would not act. This seems unlikely, because upregulation of COX-2 in the spinal cord begins in the first hours of inflammation and lasts for many hours or days (Vanegas and Schaible, 2001). Second, PGs and/or other spinal mediators might, as inflammation progresses, cause persistent changes, including a decrease in the sensitivity of spinal neurons to PGs. Indeed, the effects of PGE2 on the responses to mechanical stimulation were not fully reversible, a second application of PGE2 in the same experiment did not further enhance these responses, and the effects of PGE2on these responses were much less pronounced in rats with a knee inflammation. Thus, PGE2 could initiate cellular events that induce long-term changes in synaptic transmission that may contribute to the persistence of central sensitization. Because the activation of PG receptors sets in motion second messenger systems and enhances, for example cAMP (Vanegas and Schaible, 2001), it is likely that second messengers could induce these long-term changes. A desensitization of PG receptors or alterations in the transduction cascade may be part of these effects. It should be noted, however, that exogenous PGE2 enhanced the responses to AMPA and NMDA in the spinal cord of rats with inflamed knees, so that a total insensitivity of neurons to PGE2 is unlikely, and other explanations should be sought in the future.
In all, the present results highlight the involvement of PGs in the facilitation of somatosensory neuronal discharges, particularly during induction of central sensitization. A direct and/or indirect facilitation of glutamatergic synapses might underlie this effect, but other molecular players seem to be responsible for the sustained central hyperexcitability.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant Scha 404/11-1. E.V. held fellowships from the Deutscher Akademischer Austauschdienst and the Venezuelan Consejo Nacional de Investigaciones Científicas y Técnicas. We thank G. Cuny for skillful assistance with the laboratory work.
Correspondence should be addressed to Dr. Hans-Georg Schaible, Institut für Physiologie I der Universität Jena, Teichgraben 8, D-07740 Jena, Germany. E-mail: schaible@mti-n.uni-jena.de.
E.V. and H.V. are on leave from the Instituto Venezolano de Investigaciones Cientificas, Apartado 21827, Caracas 1020A, Venezuela.
REFERENCES
- 1.Andreeva L, Rang HP. Effect of bradykinin and prostaglandins on the release of calcitonin gene-related peptide-like immunoreactivity from the spinal cord in vitro. Br J Pharmacol. 1993;108:185–190. doi: 10.1111/j.1476-5381.1993.tb13460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baba H, Kohno T, Moore KA, Woolf CJ. Direct activation of rat spinal dorsal horn neurons by prostaglandin E2. J Neurosci. 2001;21:1750–1756. doi: 10.1523/JNEUROSCI.21-05-01750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beiche F, Scheuerer S, Brune K, Geisslinger G, Goppelt-Struebe M. Up-regulation of cyclooxygenase-2 mRNA in the rat spinal cord following peripheral inflammation. FEBS Lett. 1996;390:165–169. doi: 10.1016/0014-5793(96)00604-7. [DOI] [PubMed] [Google Scholar]
- 4.Beiche F, Brune K, Geisslinger G, Goppelt-Struebe M. Expression of cyclooxygenase isoforms in the rat spinal cord and their regulation during adjuvant-induced arthritis. Inflamm Res. 1998a;47:482–487. doi: 10.1007/s000110050362. [DOI] [PubMed] [Google Scholar]
- 5.Beiche F, Klein T, Nüsing R, Neuhuber W, Goppelt-Struebe M. Localization of cycloogygenase-2 and prostaglandin E2 receptor EP3 in the rat lumbar spinal cord. J Neuroimmunol. 1998b;89:26–34. doi: 10.1016/s0165-5728(98)00061-7. [DOI] [PubMed] [Google Scholar]
- 6.Chapman V, Dickenson AH. The spinal and peripheral roles of bradykinin and prostaglandins in nociceptive processing in the rat. Eur J Pharmacol. 1992;219:427–433. doi: 10.1016/0014-2999(92)90484-l. [DOI] [PubMed] [Google Scholar]
- 7.Coderre TJ, Gonzales R, Goldyne ME, West ME, Levine JD. Noxious stimulus-induced increase in spinal prostaglandin E2 is noradrenergic terminal-dependent. Neurosci Lett. 1990;115:253–258. doi: 10.1016/0304-3940(90)90464-k. [DOI] [PubMed] [Google Scholar]
- 8.Dirig DM, Konin GP, Isakson PC, Yaksh TL. Effect of spinal cyclooxygenase inhibitors in rat using the formalin test and in vitro prostaglandin E2 release. Eur J Pharmacol. 1997;331:155–160. doi: 10.1016/s0014-2999(97)01053-4. [DOI] [PubMed] [Google Scholar]
- 9.Dirig DM, Isakson PC, Yaksh TL. Effect of COX-1 and COX-2 inhibition on induction and maintenance of carageenan-evoked thermal hyperalgesia in rats. J Pharmacol Exp Ther. 1998;285:1031–1038. [PubMed] [Google Scholar]
- 10.Dirig DM, Yaksh TL. In vitro prostanoid release from spinal cord following peripheral inflammation: effects of substance P, NMDA and capsaicin. Br J Pharmacol. 1999;126:1333–1340. doi: 10.1038/sj.bjp.0702427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dougherty PM, Sluka KA, Sorkin LS, Westlund KN, Willis WD. Neural changes in acute arthritis in monkeys. I. Parallel enhancement of responses of spinothalamic tract neurons to mechanical stimulation and excitatory amino acids. Brain Res Rev. 1992;17:1–13. doi: 10.1016/0165-0173(92)90002-4. [DOI] [PubMed] [Google Scholar]
- 12.Ebersberger A, Grubb BD, Willingale HL, Gardiner NJ, Nebe J, Schaible H-G. The intraspinal release of prostaglandin E2 in a model of acute arthritis is accompanied by up-regulation of cyclo-oxygenase-2 in the spinal cord. Neuroscience. 1999;93:775–781. doi: 10.1016/s0306-4522(99)00164-5. [DOI] [PubMed] [Google Scholar]
- 13.Ferreira SH, Lorenzetti BB. Intrathecal administration of prostaglandin E2 causes sensitization of primary afferent neurons via the spinal release of glutamate. Inflamm Res. 1996;45:499–502. doi: 10.1007/BF02311085. [DOI] [PubMed] [Google Scholar]
- 14.Forster C, Handwerker HO. Automatic classification and analysis of microneurographic spike data using a PC/AT. J Neurosci Methods. 1990;31:109–118. doi: 10.1016/0165-0270(90)90155-9. [DOI] [PubMed] [Google Scholar]
- 15.Goppelt-Struebe M, Beiche F. Cyclooxygenase-2 in the spinal cord: localization and regulation after a peripheral inflammatory stimulus. Adv Exp Med Biol. 1998;433:213–216. doi: 10.1007/978-1-4899-1810-9_45. [DOI] [PubMed] [Google Scholar]
- 16.Gühring H, Görig M, Ates M, Coste O, Zeilhofer HU, Pahl A, Rehse K, Brune K. Suppressed injury-induced rise in spinal prostaglandin E2 production and reduced early thermal hyperalgesia in iNOS-deficient mice. J Neurosci. 2000;20:6714–6720. doi: 10.1523/JNEUROSCI.20-17-06714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hay CH, de Belleroche JS. Carrageenan-induced hyperalgesia is associated with increased cyclo-oxygenase-2 expression in spinal cord. NeuroReport. 1997;8:1249–1251. doi: 10.1097/00001756-199703240-00038. [DOI] [PubMed] [Google Scholar]
- 18.Hay CH, de Belleroche JS. Dexamethasone prevents the induction of COX-2 mRNA and prostaglandins in the lumbar spinal cord following intraplantar FCA in parallel with inhibition of oedema. Neuropharmacology. 1998;37:739–744. doi: 10.1016/s0028-3908(98)00073-2. [DOI] [PubMed] [Google Scholar]
- 19.Hay CH, Trevethick MA, Wheeldon A, Bowers JS, de Belleroche JS. The potential role of spinal cord cyclooxygenase-2 in the development of Freund's complete adjuvant-induced changes in hyperalgesia and allodynia. Neuroscience. 1997;78:843–850. doi: 10.1016/s0306-4522(96)00598-2. [DOI] [PubMed] [Google Scholar]
- 20.Heppelmann B, Pfeffer A, Schaible H-G, Schmidt RF. Effects of acetylsalicylic acid (ASA) and indomethacin on single groups III and IV units from acutely inflamed joints. Pain. 1986;26:337–351. doi: 10.1016/0304-3959(86)90062-X. [DOI] [PubMed] [Google Scholar]
- 21.Hingtgen CM, Vasko MR. Prostacyclin enhances the evoked-release of substance P and calcitonin gene-related peptide from rat sensory neurons. Brain Res. 1994;655:51–60. doi: 10.1016/0006-8993(94)91596-2. [DOI] [PubMed] [Google Scholar]
- 22.Hua X-Y, Chen P, Marsala M, Yaksh TL. Intrathecal substance P-induced thermal hyperalgesia and spinal release of prostaglandin E2 and amino acids. Neuroscience. 1999;89:525–534. doi: 10.1016/s0306-4522(98)00488-6. [DOI] [PubMed] [Google Scholar]
- 23.Hylden JLK, Nahin RL, Traub RJ, Dubner R. Expansion of receptive fields of spinal lamina I projection neurons in rats with unilateral adjuvant-induced inflammation: the contribution of dorsal horn mechanisms. Pain. 1989;37:229–243. doi: 10.1016/0304-3959(89)90135-8. [DOI] [PubMed] [Google Scholar]
- 24.Inoue A, Ikoma K, Morioka N, Kumagai K, Hashimoto T, Hide I, Nakata Y. Interleukin-1β induces substance P release from primary afferent neurons through the cyclooxygenase-2 system. J Neurochem. 1999;73:2206–2213. [PubMed] [Google Scholar]
- 25.Kawamura T, Yamauchi T, Koyama M, Maruyama T, Akira T, Nakamura N. Expression of prostaglandin EP2 receptor mRNA in the rat spinal cord. Life Sci. 1997;61:2111–2116. doi: 10.1016/s0024-3205(97)00884-9. [DOI] [PubMed] [Google Scholar]
- 26.Malmberg AB, Yaksh TL. Capsaicin-evoked prostaglandin E2 release in spinal cord slices: relative effect of cyclooxygenase inhibitors. Eur J Pharmacol. 1994;271:293–299. doi: 10.1016/0014-2999(94)90786-2. [DOI] [PubMed] [Google Scholar]
- 27.Malmberg AB, Yaksh TL. Cyclooxygenase inhibition and the spinal release of prostaglandin E2 and amino acids evoked by paw formalin injection: a microdialysis study in unanesthetized rats. J Neurosci. 1995a;15:2768–2776. doi: 10.1523/JNEUROSCI.15-04-02768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malmberg AB, Yaksh TL. The effect of morphine on formalin-evoked behaviour and spinal release of excitatory amino acids and prostaglandin E2 using microdialysis in conscious rats. Br J Pharmacol. 1995b;114:1069–1075. doi: 10.1111/j.1476-5381.1995.tb13315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minami T, Nishihara I, Uda R, Ito S, Hyodo M, Hayaishi O. Characterization of EP-receptor subtypes involved in allodynia and hyperalgesia induced by intrathecal administration of prostaglandin E2 to mice. Br J Pharmacol. 1994a;112:735–740. doi: 10.1111/j.1476-5381.1994.tb13139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minami T, Uda R, Horiguchi S, Ito S, Hyodo M, Hayashi O. Allodynia evoked by intrathecal administration of prostaglandin E2 to conscious mice. Pain. 1994b;57:217–223. doi: 10.1016/0304-3959(94)90226-7. [DOI] [PubMed] [Google Scholar]
- 31.Minami T, Okuda-Ashitaka E, Mori H, Ito S, Hayaishi O. Prostaglandin D2 inhibits prostaglandin E2-induced allodynia in conscious mice. J Pharmacol Exp Ther. 1996;278:1146–1152. [PubMed] [Google Scholar]
- 32.Minami T, Okuda-Ashitaka E, Hori Y, Sakuma S, Sugimoto T, Sakimura K, Mishina M, Ito S. Involvement of primary afferent C-fibres in touch-evoked pain (allodynia) induced by prostaglandin E2. Eur J Neurosci. 1999;11:1849–1856. doi: 10.1046/j.1460-9568.1999.00602.x. [DOI] [PubMed] [Google Scholar]
- 33.Muth-Selbach US, Tegeder I, Brune K, Geisslinger G. Acetaminophen inhibits spinal prostaglandin E2 release after peripheral noxious stimulation. Anesthesiology. 1999;91:231–239. doi: 10.1097/00000542-199907000-00032. [DOI] [PubMed] [Google Scholar]
- 34.Neugebauer V, Schaible H-G. Evidence for a central component in the sensitization of spinal neurons with joint input during development of acute arthritis in cat's knee. J Neurophysiol. 1990;64:299–311. doi: 10.1152/jn.1990.64.1.299. [DOI] [PubMed] [Google Scholar]
- 35.Neugebauer V, Lücke T, Schaible H-G. N-methyl-d-aspartate (NMDA) and non-NMDA receptor antagonists block the hyperexcitability of dorsal horn neurons during development of acute arthritis in rat's knee joint. J Neurophysiol. 1993;70:1365–1377. doi: 10.1152/jn.1993.70.4.1365. [DOI] [PubMed] [Google Scholar]
- 36.Nicol GD, Klingberg DK, Vasko MR. Prostaglandin E2 increases calcium conductance and stimulates release of substance P in avian sensory neurons. J Neurosci. 1992;12:1917–1927. doi: 10.1523/JNEUROSCI.12-05-01917.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishihara I, Minami T, Uda R, Ito S, Hyodo M, Hayaishi O. Effect of NMDA receptor antagonists on prostaglandin E2-induced hyperalgesia in conscious mice. Brain Res. 1995;677:138–144. doi: 10.1016/0006-8993(95)00133-b. [DOI] [PubMed] [Google Scholar]
- 38.Oida H, Namba T, Sugimoto Y, Ushikubi F, Ohishi H, Ichikawa A, Narumiya S. In situ hybridization studies of prostacyclin receptor mRNA expression in various mouse organs. Br J Pharmacol. 1995;116:2828–2837. doi: 10.1111/j.1476-5381.1995.tb15933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramwell PW, Shaw JE, Jessup R. Spontaneous and evoked release of prostaglandins from frog spinal cord. Am J Physiol. 1966;211:998–1004. doi: 10.1152/ajplegacy.1966.211.4.998. [DOI] [PubMed] [Google Scholar]
- 40.Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1β-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 41.Sorkin LS. IT ketorolac blocks NMDA-evoked spinal release of prostaglandin E2 (PGE2) and thromboxane B2 (TBXB2). Anesthesiology. 1993;79:A908. [Google Scholar]
- 42.Southall MD, Michael RL, Vasko MR. Intrathecal NSAIDS attenuate inflammation-induced neuropeptide release from rat spinal cord slices. Pain. 1998;78:39–48. doi: 10.1016/S0304-3959(98)00113-4. [DOI] [PubMed] [Google Scholar]
- 43.Sugimoto Y, Shigemoto R, Namba T, Negishi M, Mizuno N, Narumiya S, Ichikawa A. Distribution of the messenger RNA for the prostaglandin E receptor subtype EP3 in the mouse nervous system. Neuroscience. 1994;62:919–928. doi: 10.1016/0306-4522(94)90483-9. [DOI] [PubMed] [Google Scholar]
- 44.Taiwo YO, Levine JD. Prostaglandins inhibit endogenous pain control mechanisms by blocking transmission at spinal noradrenergic synapses. J Neurosci. 1988;8:1346–1349. doi: 10.1523/JNEUROSCI.08-04-01346.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uda R, Horiguchi S, Ito S, Hyodo M, Hayaishi O. Nociceptive effects induced by intrathecal administration of prostaglandin D2, E2, or F2alpha to conscious mice. Brain Res. 1990;510:26–32. doi: 10.1016/0006-8993(90)90723-o. [DOI] [PubMed] [Google Scholar]
- 46.Vanegas H, Schaible H-G. Prostaglandins and cyclooxygenases in the spinal cord. Prog Neurobiol. 2001;64:327–363. doi: 10.1016/s0301-0082(00)00063-0. [DOI] [PubMed] [Google Scholar]
- 47.Vanegas H, Tortorici V, Eblen-Zajjur A, Vasquez E. PAG-microinjected dipyrone (metamizol) inhibits responses of spinal dorsal horn neurons to natural noxious stimulation in rats. Brain Res. 1997;759:171–174. doi: 10.1016/s0006-8993(97)00360-0. [DOI] [PubMed] [Google Scholar]
- 48.Vasko MR, Zirkelbach SL, Waite KJ. Prostaglandins stimulate the release of substance P from rat spinal cord slices. Prog Pharmacol Clin Pharmacol. 1993;10:69–89. [Google Scholar]
- 49.Vasko MR, Campbell WB, Waite KJ. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. J Neurosci. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vasquez E, Bär K-J, Ebersberger A, Klein B, Vanegas H, Schaible H-G. Influence of PGE2 on spinal nociceptive processing in rats with normal and inflamed knee joints. Soc Neurosci Abstr. 2000;26:732.6. [Google Scholar]
- 51.Vasquez E, Bär K-J, Ebersberger A, Vanegas H, Schaible H-G. Spinal prostaglandin E2 modifies the mechanosensitivity of spinal cord neurons and the development of inflammation-evoked hyperexcitability. Pflügers Arch. 2001;441:R156. [Google Scholar]
- 52.Willingale HL, Gardiner NJ, McLymont N, Giblett S, Grubb BD. Prostanoids synthesized by cyclooxygenase isoforms in rat spinal cord and their contribution to the development of neuronal hyperexcitability. Br J Pharmacol. 1997;122:1593–1604. doi: 10.1038/sj.bjp.0701548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Woolf CJ. Evidence for a central component of post injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- 54.Woolf CJ, Thompson SWN. The induction and maintenance of central sensitization is dependent on N-methyl-d-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain. 1991;44:293–299. doi: 10.1016/0304-3959(91)90100-C. [DOI] [PubMed] [Google Scholar]
- 55.Yang LC, Marsala M, Yaksh TL. Characterization of time course of spinal aminoacids, citrulline and PGE2 release after carrageenan/kaolin-induced knee joint inflammation: a chronic microdialysis study. Pain. 1996a;67:345–354. doi: 10.1016/0304-3959(96)03106-5. [DOI] [PubMed] [Google Scholar]
- 56.Yang LC, Marsala M, Yaksh TL. Effect of spinal kainic acid receptor activation on spinal amino acid and prostaglandin E2 release in rat. Neuroscience. 1996b;75:453–461. doi: 10.1016/0306-4522(96)00294-1. [DOI] [PubMed] [Google Scholar]