

Abstract
Mutations in presenilin genes are linked to early onset familial Alzheimer's disease (FAD). Previous work in non-neuronal cells indicates that presenilin-1 (PS1) associates with cytoskeletal elements and that it facilitates Notch1 signaling. Because Notch1 participates in the control of neurite growth, cultured hippocampal neurons were used to investigate the cytoskeletal association of PS1 and its potential role during neuronal development. We found that PS1 associates with microtubules (MT) and microfilaments (MF) and that its cytoskeletal association increases dramatically during neuronal development. PS1 was detected associated with MT in the central region of neuronal growth cones and with MF in MF-rich areas extending into filopodia and lamellipodia. In differentiated neurons, PS1 mutations reduced the interaction of PS1 with cytoskeletal elements, diminished the nuclear translocation of the Notch1 intracellular domain (NICD), and promoted a marked increase in total neurite length. In developing neurons, PS1 overexpression increased the nuclear translocation of NICD and inhibited neurite growth, whereas PS1 mutations M146V, I143T, and deletion of exon 9 (D9) did not facilitate NICD nuclear translocation and had no effect on neurite growth. In cultures that were treated with amyloid β (Aβ), PS1 mutations significantly increased neuritic dystrophy and AD-like changes in tau such as hyperphosphorylation, release from MT, and increased tau protein levels. We conclude that PS1 participates in the regulation of neurite growth and stabilization in both developing and differentiated neurons. In the Alzheimer's brain PS1 mutations may promote neuritic dystrophy and tangle formation by interfering with Notch1 signaling and enhancing pathological changes in tau.
Keywords: Alzheimer's disease, presenilin, cytoskeleton, Notch1, amyloid β, tau, neuronal dystrophy
The most aggressive form of early onset familial Alzheimer's disease (FAD) is caused by mutations in two related genes, presenilin-1 and presenilin-2 (PS1 and PS2; Levy-Lahad et al., 1995; Sherrington et al., 1995). Both genes encode highly homologous integral membrane proteins spanning seven to eight transmembrane domains and a hydrophilic loop between transmembrane domains 6 and 7 (Doan et al., 1996; Li and Greenwald, 1998). In neurons the presenilins are localized in the endoplasmic reticulum (ER), intermediate compartment, and nuclear membranes (Busciglio et al., 1997; Capell et al., 1997; Lah et al., 1997; Annaert et al., 1999). Presenilins undergo endoproteolytic cleavage, generating stable N- and C-terminal fragments (NTF and CTF) that are regulated tightly in their levels and stoichiometry (Thinakaran et al., 1996, 1997). NTF and CTF interact with each other and with other proteins to form a high-molecular-weight heterodimeric complex (Capell et al., 1998; Yu et al., 1998).
Recent experiments suggest that PS1 is γ-secretase (Li et al., 2000) and that its function is related to the trafficking, turnover, and cleavage of amyloid precursor protein (APP; De Strooper et al., 1998;Wolfe et al., 1999) and other membrane proteins. Deficiency of PS1 inhibits the processing and transport of APP and the maturation of the TrkB receptor (De Strooper et al., 1998; Naruse et al., 1998); PS1 activity also is required for the cleavage of Notch1 at the plasma membrane and the release of the Notch1 intracellular domain (NICD; De Strooper et al., 1999; Song et al., 1999; Struhl and Greenwald, 1999;Ye et al., 1999).
Notch1 is a transmembrane receptor that mediates cell fate decisions during development (Artavanis-Tsakonas et al., 1995; Kopan et al., 1996; Weinmaster, 1997). Ligand binding of a member of the Delta-Serrate-LAG2 family induces the proteolytic cleavage of Notch1 and the nuclear translocation of NICD, where it activates the transcription of downstream genes (Jarriault et al., 1995; Schroeter et al., 1998; Struhl and Adachi, 1998). In differentiated neurons Notch1 signaling has been shown to inhibit neurite growth (Berezovska et al., 1999a; Franklin et al., 1999; Sestan et al., 1999; Redmond et al., 2000) and to promote dendritic branching (Redmond et al., 2000).
Previous results suggest that PS1 may interact with cytoskeletal elements, including the microtubule-associated protein tau (Takashima et al., 1998) and the actin-binding protein filamin (Zhang et al., 1998). In AD brains PS1 has been detected associated with neurofibrillary tangles (NFT) and dystrophic neurites (Busciglio et al., 1997; Giannakopoulos et al., 1997; Tomidokoro et al., 1999). In this report the cytoskeletal association of PS1 and its role in the control of neurite growth were analyzed in hippocampal cultures. We show that PS1 interacts with microtubules (MT) and microfilaments (MF) and that its association with cytoskeletal elements increases during neuronal differentiation. FAD-linked mutations M146V, I143T, and D9 reduce cytoskeletal binding, interfere with Notch1 signaling, and deregulate neurite growth. In addition, PS1 mutations potentiate amyloid β (Aβ)-induced neuronal dystrophy and pathological changes in tau. Our results provide evidence that PS1 mutations may enhance and accelerate neuritic dystrophy and tangle formation.
MATERIALS AND METHODS
Antibodies. PSN2 and PSN3 monoclonal antibodies were raised against a synthetic peptide corresponding to amino acid residues 31–56 of PS1 (Okochi et al., 1998). Antibody 2025 is a rabbit antiserum raised against a synthetic peptide corresponding to amino acid residues 1–20. The rabbit antiserum AD3L and the mouse antiserum mAD3L were raised against a synthetic peptide corresponding to residues 295–325 in the loop region of PS1 (Sahara et al., 1996; Okochi et al., 1998). Other antibodies that were used include a monoclonal antibody against β-tubulin isotype III (clone SDL.3D10; Sigma, St. Louis, MO), monoclonal anti-Aβ (clone 6E10; Senetek, Napa, CA), anti-Bip (clone 10c3; StressGen, Sidney, Canada), polyclonal anti-Notch1 (C-20; Santa Cruz Biotechnology, Santa Cruz, CA), anti-red fluorescent protein (RFP; D.s. peptide antibody, Clontech, Palo Alto, CA), and monoclonal antibodies to dephosphorylated tau at Ser-199/Ser-202 (Tau-1; Roche, Indianapolis, IN) and to phosphorylated tau at Ser-396/Ser-404 (PHF-1;Greenberg et al., 1992).
Cell culture. Rat hippocampal cultures were established from embryonic day 18 fetuses as described (Goslin, 1992). The cells were plated on poly-l-lysine-treated culture dishes or glass coverslips at a density of 20,000 cells/cm2 in DMEM plus 10% horse serum for 2–3 hr. After cell attachment the media were replaced with Neurobasal media supplemented with 2% B27 (Life Technologies, Grand Island, NY). To depolymerize MT selectively in neuronal growth cones, we incubated the cultures with 30 μmnocodazole for 30 min before fixation. To depolymerize MF, we treated the cultures with 10 μg/ml cytochalasin B for 1 hr.
Preparation of cytoskeletal fractions. Triton X-100 (TX-100)-resistant cytoskeletons were prepared as described (Busciglio et al., 1995). Briefly, the cultures were washed with prewarmed MT-stabilizing buffer (0.13 m HEPES, pH 6.9, 2 mm MgCl2, and 10 mm EGTA) and extracted in the same buffer plus 0.2% TX-100 for 5 min at 37°C. Then the cultures were fixed, and immunocytochemistry was performed on the TX-100-resistant material remaining on the coverslip. For Western blot the cultures that were grown in culture dishes were extracted as described above and harvested in cold RIPA lysis buffer (1% TX-100, 0.5% sodium deoxycholate, and 0.1% SDS plus 150 mm NaCl and 50 mm Tris-HCl, pH 7.2) supplemented with protease inhibitors (Complete, Roche, Indianapolis, IN) and phosphatase inhibitors (Busciglio et al., 1993). For some experiments a mild extraction protocol that preserves MF and cytoskeletal membrane interactions was used (Nakata and Hirokawa, 1987; Pigino et al., 1997). The cells were washed in extraction buffer PHEM [containing (in mm) 80 PIPES/KOH, pH 6.8, 1 MgCl2, 1 EGTA, and 1 GTP plus 30% glycerol], incubated for 1 min in 0.02% saponin/PHEM, washed, and fixed for immunocytochemistry. All steps were performed at 37°C.
Immunocytochemistry. Before or after detergent extraction the cells were fixed for 30 min at 37°C in 4% paraformaldehyde and 0.12 m sucrose in extraction buffer, washed with PBS, and, in the case of unextracted cultures, permeabilized with 0.1% TX-100/PBS for 10 min. The cultures were blocked for 1 hr in 5% BSA in PBS and incubated overnight at 4°C with primary antibody. For immunofluorescence (IF) Texas Red- or fluorescein-conjugated anti-rabbit, mouse, or goat secondary antibodies were used (Vector Laboratories, Burlingame, CA). For IF detection of endogenous Notch1, a highly sensitive tyramide signal amplification kit was used, following the vendor's protocol (NEN Life Science Products, Boston, MA). The specificity of Notch1 labeling was confirmed by abolishment of the IF signal by preabsorption of the primary antibody with the corresponding antigenic peptide. Fluorescence was visualized with a Zeiss LSM 410 confocal-scanning microscope or with an Olympus IX-70 inverted microscope.
Western blot and immunoprecipitation. For Western blots the unextracted cultures were washed with PBS at 37°C and harvested in cold RIPA. The lysates were centrifuged at 100,000 × gfor 60 min. Then the supernatants were collected, and the protein content was determined by a commercial kit (Bio-Rad, Hercules, CA). The samples were mixed with an equal volume of Laemmli sample buffer and maintained at room temperature for 15 min before loading onto gels. For immunoprecipitation, RIPA lysates were incubated overnight at 4°C with primary antibody and were immunoprecipitated with protein A–Sepharose (Pharmacia, Piscataway, NJ) for 2 hr. Then the beads were washed three times and resuspended in sample buffer containing protease inhibitors. The specificity of the immunoprecipitation was controlled by replacing the primary antibody for the corresponding nonimmune sera in the immunoprecipitate mixture. Proteins were separated by PAGE in 4–20% gradient gels and transferred to polyvinylidene difluoride; the membranes were incubated overnight at 4°C with primary antibody. After incubation with peroxidase-conjugated secondary antibody, the reaction was developed by enhanced chemiluminescence (ECL; Amersham, Arlington Heights, IL). Primary antibodies were used at the following dilutions: PSN2 and PSN3, 1:500; 2025, 1:1000; AD3L, 1:3000; mAD3L, 1:1500; anti-Bip, 1:500; anti-RFP, 1:1000; PHF-1, 1:2000; Tau-1, 1:1500. To control the specificity of PS1 antibodies on Western blot assays, we performed preabsorptions with the corresponding antigenic peptides as described (Busciglio et al., 1993). To control for protein loading and sample-to-sample variability, we stripped the immunoblots with 2% SDS in 100 mm β-mercaptoethanol for 2 hr at 37°C and restained them by using anti α-tubulin 1:4000 (clone DM1A; Sigma). The films were scanned with a FastScan densitometer (Molecular Dynamics, Sunnyvale, CA), and volume analysis was performed on the appropriate bands with NIH Image software. A standard curve of pixel values was constructed by immunoblotting a serial dilution of purified tubulin. All densitometric measurements that were used for analysis were within the linear range of pixel values.
Transfection. Full-length PS1 and PS1 mutations M146V, I143T, and D9 (Thinakaran et al., 1996) were cloned into pcDNA3 (Invitrogen, Carlsbad, CA). All constructs were confirmed by sequencing. Hippocampal cultures were transfected at 1 and 8 din vitro (DIV) by using calcium phosphate as previously described (Threadgill et al., 1997), with the following modification: the calcium phosphate/DNA precipitate was formed in HEPES-buffered saline, pH 7.05, incubated at room temperature in the dark for 1 min, and immediately added to the cultures. This protocol resulted in ∼30% transfection efficiency in 10 DIV cultures. Cells were cotransfected with red or green fluorescent protein expression vectors (RFP, GFP; Clontech) as transfection markers and one of the following constructs: empty vector pcDNA3, pcDNA3-PS1-wt, pcDNA3-PS1-M146V, pcDNA3-PS1-I143T, or pcDNA3-PS1-D9. The total cDNA concentration was 5 μg/500 μl of media. The ratio of RFP/pcDNA3-PS1 was 1:3. The efficiency of cotransfection was ∼95%. Neuronal cells were transfected in 24-well plates in quadruplicate wells. Expression of PS1 constructs was confirmed by IF. RFP fluorescence was distributed along the entire length of neuronal processes, allowing for the morphological analysis of transfected cells. The cultures were processed and analyzed 48 hr after transfection.
Morphometric analysis. To analyze the effect of PS1-wt and FAD-linked mutations on neurite growth, Notch1 IF ratio, and Aβ-induced neuronal dystrophy, we captured the images of cotransfected neurons with PS1 and RFP with a CCD camera (Spot, Diagnostic Instruments, Sterling Heights, MI) driven by Spot image acquisition software and analyzed with NIH Image software. Data were collected from at least five independent transfection experiments. The identity of transfected cultures was coded to avoid experimental bias. Data are expressed as the mean ± SE. Asterisks in the figures indicate significant statistical differences among groups as determined by Student's t test. To determine total neuritic length, we scored 50–100 transfected cells per well in quadruplicate wells. More than 200 cells were scored per experimental condition. To determine the Notch1 IF ratio of cytoplasm/nuclei, we measured the intensity of Notch1 IF in identical volumes of nuclear and cytoplasmic areas in the same cell in 80 individual cells per condition. To quantify the number of individual dystrophic events per neuron, we analyzed images of 30 neurons per condition, and we scored the number of dystrophic processes with NIH Image software.
Aβ peptide treatment. Synthetic Aβ1–42 (Bachem, King of Prussia, PA) was dissolved in double-distilled sterile H2O to 3 mg/ml, further diluted in sterile PBS to 1.5 mg/ml, and used after preincubation at 37°C for 3–4 d to preaggregate the peptide. Then Aβ aggregates were pelleted in a microfuge at 10,000 rpm for 5 min; the supernatant was discarded to eliminate soluble Aβ oligomers. The predominant form of Aβ in this preparation has been shown to be fibrillar by electron microscopy and Congo red birefringence (Lorenzo and Yankner, 1994). Fibrillar Aβ was added directly to the culture medium at a final concentration of 20 μm. Control cells were treated with reverse sequence Aβ or PBS alone. The peptides were added 2 hr after transfection to 8 DIV cultures and incubated for 48 hr before analysis.
RESULTS
PS1 is associated with cytoskeletal elements in cultured hippocampal neurons
The association of PS1 with components of the neuronal cytoskeleton was analyzed in growth cones where the MT and MF are less packed and accessible for subcellular localization studies at the light microscopy level. Significant expression of PS1 has been found in neuronal growth cones in culture (Busciglio et al., 1997; Capell et al., 1997; Levesque et al., 1999) and in growth cone-enriched fractions purified from rat brain homogenates (Beher et al., 1999). Confirming those reports, confocal microscopy studies showed a reticular pattern of PS1 IF in growth cones, in which a portion of it colocalized with the ER resident protein Bip (Fig.1A,B). PS1 IF extended to the periphery and into lamellipodia, whereas Bip IF was restricted to the neuritic shaft and central region of the growth cones (Fig.1A,B). PS1 IF was retained in cultures extracted with TX-100 under MT-stabilizing conditions before fixation (Fig.1C). In contrast, Bip immunostaining was lost completely in these preparations (Fig. 1D). In extracted cultures PS1 IF was detected in the central region of growth cones where it showed partial colocalization with MT (Fig. 1E,F,arrows). PS1 IF was negative in cytoskeletal preparations of cultures treated with the MT-depolymerizing drug nocodazole, further suggesting an association of PS1 with MT (Fig.1G,H).
Fig. 1.

PS1 associates with MT in neuronal growth cones. Confocal images show the distribution of PS1 (A) and Bip (B) in 3 DIV hippocampal growth cones. PS1 colocalizes with Bip at the central region of the growth cone. Note that PS1 IF extends to filopodia and lamellipodia (A). Cultures were stained sequentially with PSN2 and anti-Bip antibodies (see Materials and Methods). After TX-100 extraction, PS1 IF remains in the cytoskeletal preparation (C), whereas Bip IF is abolished completely (D). Shown is double IF for PS1 with antibody 2025 (E) and β-tubulin class III (F). PS1 IF appears closely associated with the microtubular network. Partial colocalization between PS1 and individual MT can be observed (arrows, E, F). A mild nocodazole treatment (see Materials and Methods) abolished PS1 (G) and β-tubulin class III (H) IF from neuronal growth cones of TX-100-extracted cultures. Note the presence of PS1 IF in the neuritic shaft where intact MT are still present (G, H). The perimeter of the growth cone before nocodazole treatment is shown in H. Scale bar, 10 μm.
As shown in Figure 1A, PS1 immunolabeling in nonextracted cells extended to the periphery of growth cones into lamellipodia. To determine whether PS1 localization in MF-enriched areas depends on the presence of intact MF, we used a mild extraction with saponin to preserve the actin cytoskeleton and its interaction with membrane-associated proteins (Nakata and Hirokawa, 1987; Pigino et al., 1997). In these preparations PS1 IF extended to the edges of lamellipodia and filopodia where it showed several points of contact with MF (Fig. 2A–C,arrows). Depolymerization of MF with cytochalasin B completely abolished actin and PS1 labeling from the periphery (Fig.2D,E), although PS1 IF remained in the central region of growth cones (Fig. 2D) where it is associated with ER membranes and MT (see Fig. 1). These results indicate that PS1 interacts with MT in the central region of growth cones and with MF at or near the cellular membrane of filopodia and lamellipodia.
Fig. 2.

PS1 associates with MF in growth cone filopodia and lamellipodia. Saponin extraction preserves the localization of PS1 (labeled with antibody AD3L) in lamellipodia (A), in which numerous MF are stained by phalloidin conjugated with Texas Red (B). Note several points of contact between PS1 and MF (arrows, A–C). Depolymerization of MF with cytochalasin B abolished PS1 (D) and phalloidin (E) IF from the periphery of saponin-extracted growth cones, whereas PS1 IF remained in the central region of the growth cones (D). Scale bar, 10 μm.
Western blot analysis of hippocampal culture homogenates confirmed the presence of PS1 in the insoluble fraction of TX-100-extracted cultures (Fig. 3A). The amount of PS1 present in this fraction increased dramatically during neuronal differentiation; by 10 DIV, most full-length (FL), CTF (Fig.3A), and NTF (data not shown) PS1 were recovered in the insoluble fraction. In contrast, after TX-100 extraction, Bip was solubilized completely (Fig. 3B). These results suggest that the interaction of PS1 with cytoskeletal elements increases during neuronal development and that this interaction is not a common feature of other ER resident proteins.
Fig. 3.

The cytoskeletal association of PS1 increases during neuronal development, and PS1 mutations reduce cytoskeletal binding. A, Western blot analysis of cytoskeletal preparations of 1, 6, and 10 DIV cultures. Shown are insoluble (Insol) and soluble (Sol) fractions prepared from triplicate cultures. All lanes were loaded with 10 μg of protein. The blots were developed with antibody AD3L that recognizes PS1 FL and CTF. Note the dramatic increase in the association of FL and CTF to the insoluble fraction during neuronal development. In contrast, the ER-resident protein Bip is solubilized completely by extraction with TX-100 (B).C, Cytoskeletal binding of RFP and PS1 expressed as the amount of protein in the insoluble-over-soluble fractions, determined by quantitative Western blot analysis with antibody AD3L to PS1 CTF and anti-RFP (see Materials and Methods). Cultures were transfected at 8 DIV and analyzed at 10 DIV. Similar results were obtained with antibodies PSN2 and 2025 recognizing PS1 NTF (data not shown). Note that PS1-wt is retained more efficiently in the cytoskeleton than PS1 mutations. Values are the mean ± SE; n = 6 independent experiments. *p < 0.001 relative to PS1-wt by Student's t test.
FAD-linked PS1 mutations reduce cytoskeletal binding, inhibit NICD nuclear translocation, and deregulate neurite growth
The ability of FAD-linked PS1 mutations M146V, I143T, and D9 to bind to the neuronal cytoskeleton was tested at 10 DIV, when most PS1 appears to be associated with the detergent-resistant fraction (Fig.3A). Hippocampal neurons were transfected at 8 DIV, and 48 hr later they were extracted and processed for Western blot. RFP, which is soluble, showed little retention in the cytoskeletal fraction (Fig.3C). The ratio of PS1 expression in the insoluble-over-soluble fractions revealed that the level of PS1 mutant proteins was diminished markedly in the insoluble fraction as compared with PS1-wt (Fig. 3C). Interestingly, this decrease in the cytoskeletal association of PS1 in 10 DIV neurons overexpressing PS1 mutations was correlated with a marked decrease in the levels of nuclear NICD (Fig. 4B) and a significant increase in total neuritic length as compared with RFP- or PS1-wt-transfected neurons (Fig. 4C). This result suggests that PS1 is involved in the regulation of Notch1-mediated neurite growth and that PS1 mutations alter this process. To investigate this possibility further, we also tested the effect of M146V, I143T, and D9 mutations in younger cultures plated at low density to minimize cell contact, which has been demonstrated to activate Notch1 inhibitory input on neurite growth (Sestan et al., 1999). Hippocampal neurons at 3 DIV were used because at this stage neuritic processes are actively growing, no cell contacts are present, and Notch1 signaling is not active. These cultures exhibited diffuse cytoplasmic IF for Notch1 and minimal Notch1 nuclear IF as compared with neurons at 10 DIV, by which time Notch1 nuclear IF is detected clearly (Fig. 4A). Overexpression of PS1-wt at 3 DIV significantly increased nuclear IF of Notch1 (Fig.4B) and reduced total neuritic length from 1574 ± 85 μm in cells transfected with RFP to 910 ± 36 μm (Fig.4C). These two events, increased nuclear Notch1 IF and reduced neuritic length, resemble a more advanced neurodevelopmental stage, e.g., 10 DIV neurons, in which cell contact activates Notch1 signaling and inhibits neuritic growth (Sestan et al., 1999), suggesting that PS1 overexpression in 3 DIV neurons inhibits neurite growth by increasing Notch1 signaling. All three PS1 mutations expressed at 3 DIV failed to enhance NICD nuclear translocation (Fig.4B) and have no effect on neurite extension (Fig.4C). Thus, FAD-linked mutations reduce the ability of PS1 to facilitate Notch1 signaling and deregulate neurite growth in both developing and differentiated neurons.
Fig. 4.
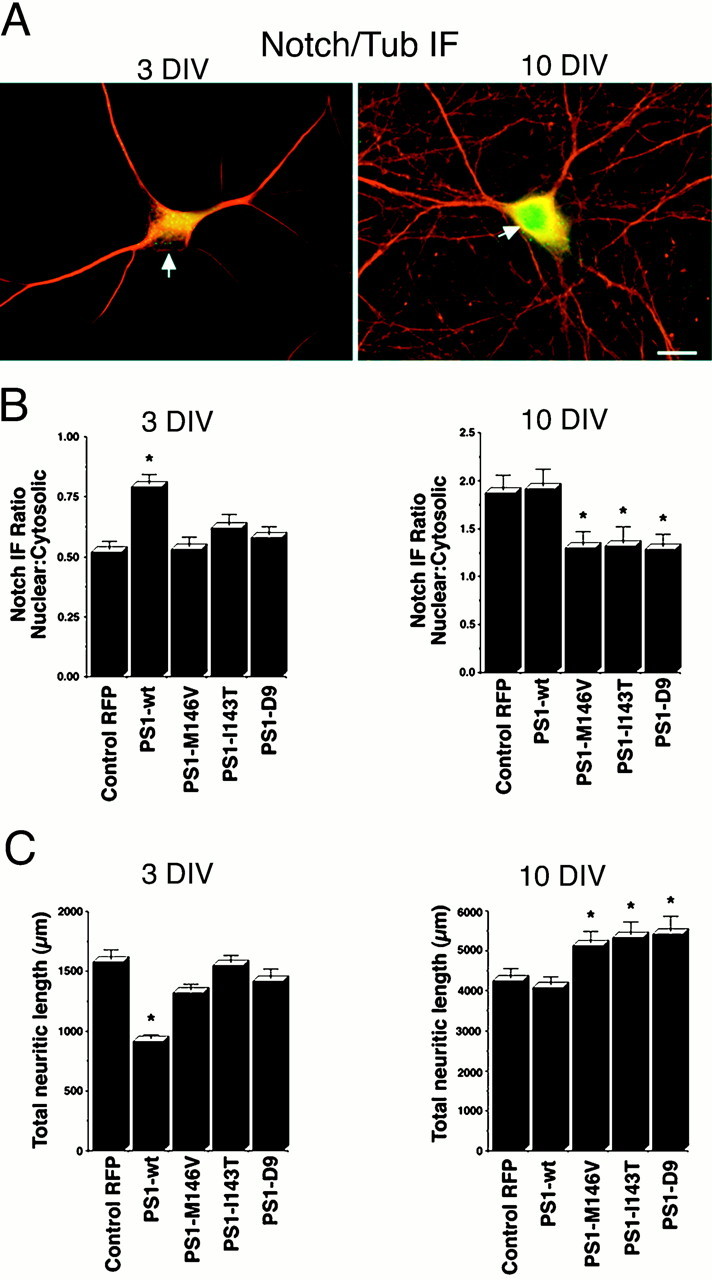
PS1 enhances nuclear translocation of NICD and reduces neurite growth, whereas PS1 mutations inhibit both effects.A, Confocal images of double IF labeling (merged images) showing the subcellular localization of endogenous Notch1 (green) and β-tubulin class III (red) in hippocampal neurons at 3 and 10 DIV. Diffuse Notch1 IF is detected in cell bodies and in the initial portion of neuritic processes but is excluded from neuronal nuclei (arrow) at 3 DIV; however, clear Notch1 nuclear staining is observed in the neurons by 10 DIV (arrow). Scale bar, 20 μm. B, Relative intensity of Notch1 IF in nuclear-over-cytoplasmic compartment in 3 and 10 DIV neurons transfected with the indicated plasmids at 1 and 8 DIV, respectively. IF ratios in individual cells were determined as described in Materials and Methods. At 3 DIV the PS1-wt-transfected cells showed a significant increase in nuclear IF as compared with RFP- or PS1 mutant-transfected cells. At 10 DIV the neurons expressing PS1 mutations exhibited significantly lower levels of nuclear IF than RFP- or PS1-wt-expressing cells. Values are the mean ± SE; n = 80 cells analyzed per condition in a representative experiment. Similar results were obtained in five independent experiments. *p< 0.01 relative to control RFP by Student's t test.C, Total neuritic length of 3 and 10 DIV neurons transfected with the indicated plasmids at 1 and 8 DIV, respectively. PS1 mutations failed to reduce neuritic growth in 3 DIV cultures, whereas at 10 DIV the neurons expressing PS1 mutations exhibited significantly longer processes than RFP- or PS1-wt-transfected cells. Morphometric analysis was performed as described in Materials and Methods. Values are the mean ± SE; n = 110 cells analyzed per condition in a representative experiment. Similar results were obtained in five independent experiments. *p < 0.01 relative to control RFP by Student'st test.
PS1 mutations potentiate Aβ-induced neuronal dystrophy
Exposure to Aβ fibrils causes major alterations in neuronal morphology that include shrinkage of cell bodies and neuritic dystrophy (Busciglio et al., 1992; Pike et al., 1992; Irizarry et al., 1997; Geula et al., 1998). In this context PS1 mutations may increase aberrant growth and neuritic dystrophy in neurons that are exposed to Aβ by suppressing the inhibitory input of Notch1 on neurite growth. To test this hypothesis, we analyzed neuritic dystrophy in 10 DIV neurons overexpressing PS1. At 2 hr after transfection the cells were treated with 20 μm fibrillar Aβ; 48 hr later the cultures were fixed, and neuronal dystrophy was analyzed. Trypan blue exclusion assays determined that most dystrophic neurons were alive after 48 hr (data not shown). In nontransfected cultures Aβ treatment induced dystrophy in 27% of the neurons (Fig.5A). A similar percentage of neurons expressing RFP (30%) and PS1-wt (32%) developed dystrophy as well (Fig. 5A). The percentage of dystrophic neurons expressing PS1 mutations increased to 48% (M146V), 52% (I143T), and 50% (D9) (Fig. 5A). The high degree of dystrophy observed in neurons expressing PS1 mutants (Fig. 5B) prompted us to determine whether PS1 mutations increased the number of individual dystrophic processes per cell. Dystrophic events that were scored included abnormal neuritic branches protruding from neuronal cell bodies (Fig. 6A) and neuritic processes with obvious aberrant morphology (Fig.6B). Interestingly, the majority of abnormal processes was found in physical contact with Aβ deposits (Fig.6A,B). The number of dystrophic events was elevated significantly in neurons expressing PS1 mutations as compared with RFP- or PS1-wt-transfected cells (Fig. 6C). This result indicates that FAD-linked PS1 mutations potentiate Aβ-induced dystrophy by increasing both the number of dystrophic processes per neuron and the total number of dystrophic neurons in the culture.
Fig. 5.

PS1 mutations potentiate neuronal dystrophy in cultures treated with fibrillar Aβ. A, Quantification of the number of dystrophic neurons in transfected cultures treated with Aβ. At 8 DIV the cells were treated with 20 μmfibrillar Aβ 2 hr after transfection and were analyzed at 10 DIV (see Materials and Methods). In nontransfected cultures (No Transf.) the number of dystrophic neurons is expressed as a percentage of the total number of neurons in the culture. In transfected cultures the number of transfected dystrophic neurons is expressed as a percentage of the total number of transfected cells. Note the significant increase of dystrophic neurons expressing PS1 mutations as compared with RFP-transfected and PS1-wt-transfected ones. Cultures treated with reverse Aβ peptide or vehicle alone did not develop neuritic dystrophy. Values are the mean ± SE;n = 80 cells scored per condition in a representative experiment. Similar results were obtained in five independent experiments. *p < 0.01 relative to control RFP by Student's t test. B, Morphology of 10 DIV hippocampal neurons expressing PS1 mutations treated with reverse sequence Aβ peptide (Control) or Aβ. Note the aberrant morphology developed by Aβ-treated neurons. Scale bar, 20 μm.
Fig. 6.

PS1 mutations increase the number of dystrophic neurites per cell in Aβ-treated cultures. A, Confocal images of neurons transfected with the indicated PS1 mutations and treated with Aβ. Red IF represents RFP autofluorescence, and the blue is Aβ IF detected with antibody 6E10. The images show examples of abnormal neuritic branches protruding from neuronal cell bodies in contact with Aβ deposits (arrows). Scale bar, 20 μm. B, Serial confocal sections (I, II, III) of cells transfected with PS1-I143T and PS1-D9 showing dystrophic processes in contact with Aβ. Dystrophic neurites grow upward, entering inside Aβ deposits and extending several micrometers above the initial focal plane. Scale bar, 5 μm. C, Quantification of the number of dystrophic events per neuron induced by 20 μmAβ in 10 DIV cells transfected with the indicated constructs. PS1 mutations markedly increased the number of dystrophic processes per neuron. Morphometric analysis was performed as described in Materials and Methods. Values are the mean ± SE; n = 30 individual cells scored per condition in a representative experiment. Similar results were obtained in four independent experiments. *p < 0.01 relative to control RFP by Student'st test.
PS1 mutations increase Aβ-induced tau phosphorylation
Treatment with Aβ increases tau phosphorylation in primary rat and human neurons (Busciglio et al., 1995; Ferreira et al., 1997), releasing phosphorylated tau from the cytoskeletal fraction (Busciglio et al., 1995). Previous work has shown that PS1 associates with tau and glycogen synthase kinase-3β (GSK3β) and that PS1 mutations increase GSK3β activity (Weihl et al., 1999) and tau phosphorylation when PS1 and tau are coexpressed by transient transfection in Cos-7 cells (Takashima et al., 1998). Double-labeling IF experiments showed colocalization of endogenous PS1 and tau in neuronal growth cones where, similar to PS1, tau IF extended beyond the MT-rich central region and entered into filopodia and lamellipodia (Fig.7A, arrows). To establish whether endogenous PS1 and tau interact in hippocampal neurons treated with Aβ, we performed coimmunoprecipitation experiments. Lysates of 10 DIV cultures treated with 20 μm Aβ for 48 hr were immunoprecipitated with PHF-1, a monoclonal antibody to phosphorylated tau at Ser-396/Ser-404 (Greenberg et al., 1992), which has been shown to recognize hyperphosphorylated soluble tau in Aβ-treated cultures (Busciglio et al., 1995). Western blot analysis of the immunoprecipitated samples showed that PS1 FL and NTF were coimmunoprecipitated by PHF-1 and recognized in the blots by specific antibodies against PS1 (Fig.7B). Preabsorption with antigenic peptides completely abolished PS1-specific immunoreactivity from the Western blots (Fig.7B). Conversely, hyperphosphorylated tau was recognized by PHF-1 in Western blots of samples immunoprecipitated with 2025, a polyclonal antibody that recognizes the NTF of PS1 (Fig.7B). These results suggest that endogenous PS1 interacts with hyperphosphorylated soluble tau in Aβ-treated neurons.
Fig. 7.
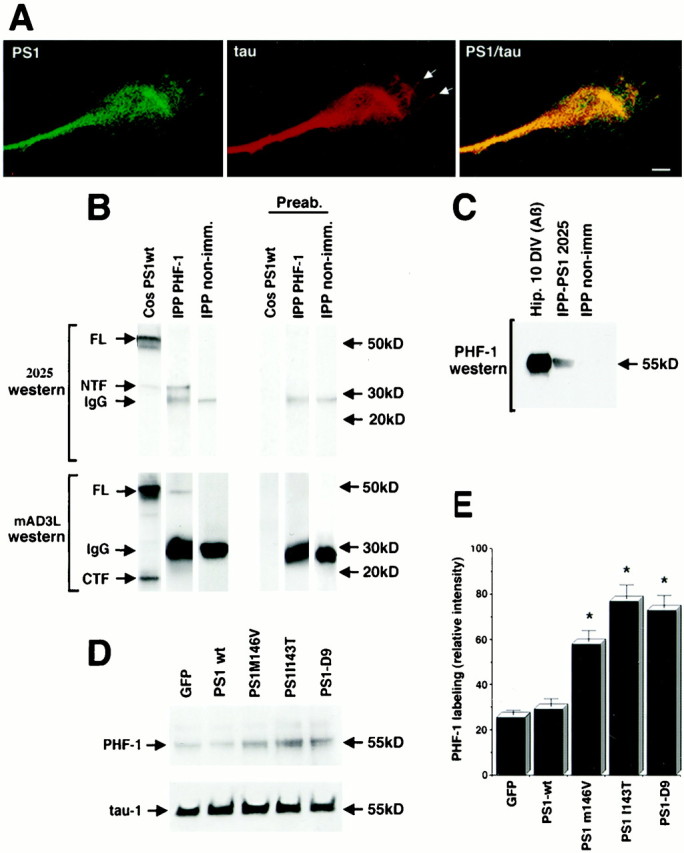
PS1 colocalizes and coimmunoprecipitates with tau, and PS1 mutations increase Aβ-induced tau phosphorylation.A, Double IF showing colocalization of endogenous PS1 (PSN2) and tau (Tau-1) in neuronal growth cones. Note the presence of tau IF in filopodia (arrows). Scale bar, 10 μm.B, Cell lysates of 10 DIV hippocampal neurons treated with Aβ were immunoprecipitated with PHF-1 (IPP PHF-1), blotted, and stained with antibodies 2025 and mAD3L. Antibody 2025 recognized PS1 NTF in PHF-1-immunoprecipitated material. Antibody 2025 also labeled PS1 FL and NTF in a homogenate of transfected Cos cells (Cos PS1wt) used as a positive control. Antibody mAD3L labeled PS1 FL and CTF in a homogenate of transfected Cos cells and recognized a band corresponding to PS1 FL in PHF-1-immunoprecipitated material. Cell lysate immunoprecipitated with nonimmune mouse sera (IPP non-imm) showed no reaction with 2025 and mAD3L antibodies. Preabsorption of both 2025 and mAD3L with the corresponding antigenic peptides completely abolished PS1-specific labeling in the Western blot (Preab). Secondary antibodies used to develop the immunoblots reacted with PHF-1 IgG in the immunoprecipitated material (IgG).C, Cell lysates of 10 DIV hippocampal neurons treated with Aβ were immunoprecipitated with anti-PS1 antibody 2025, blotted, and reacted with PHF-1, which recognizes phosphorylated tau at Ser 396/404. PHF-1 labeled a 55 kDa band in a homogenate of Aβ-treated neurons (Aβ) and a band of similar molecular weight in the material immunoprecipitated with 2025 (IPP-PS1 2025). Cell lysate immunoprecipitated with nonimmune rabbit sera (IPP non-imm) showed no reaction with PHF-1.D, Western blot analysis of transfected cultures treated with Aβ. Cultures transfected at 8 DIV were treated with 20 μm Aβ and harvested at 10 DIV. Protein (10 μg) was loaded in each lane. The blots were developed with PHF-1 and Tau-1 antibodies. Expression of green fluorescent protein (GFP) was used as a control. Note the increase in PHF-1 staining in cultures expressing PS1 mutations. No significant changes in the levels of nonphosphorylated tau were detected by Tau-1. E, Relative changes in the level of phosphorylated tau in Aβ-treated cultures expressing PS1 mutations detected by the PHF-1 antibody. Quantitative Western blot analysis was performed as described in Materials and Methods. Values are the mean ± SE; n = 5 independent experiments. *p < 0.01 relative to PS1-wt by Student's t test.
To determine the effect of PS1 mutations on tau hyperphosphorylation, we analyzed hippocampal cultures expressing wild-type or mutated PS1 after Aβ treatment. Western blot analysis of cellular homogenates revealed that the expression of PS1 mutations significantly increased PHF-1 immunoreactivity as compared with GFP or PS1-wt constructs (Fig.7D,E). In these cultures the amount of tau detected by Tau-1, which recognizes a nonphosphorylated epitope at Ser-199/Ser-202, did not change significantly, indicating that PS1 mutations increased total tau protein levels, that is, dephosphorylated plus phosphorylated tau in Aβ-treated neurons (Fig. 7D). The amount of tubulin in the cytoskeleton was not altered in cells expressing the mutations (data not shown), suggesting that the release of tau was not caused by disassembly of the microtubular network. These results indicate that endogenous tau and PS1 interact in hippocampal neurons and that PS1 mutations potentiate Aβ-induced hyperphosphorylation of tau and its release from the cytoskeleton.
DISCUSSION
These experiments indicate that PS1 regulates neuritic growth and that mutations linked to FAD destabilize the interaction of PS1 with the neuronal cytoskeleton, perturb Notch1 signaling, and enhance pathological changes in tau.
Immunocytochemical and biochemical studies revealed that PS1 associates with MT and MF in primary neurons (see Figs. 1-3). The presence of PS1 in lamellipodia and filopodia of neuronal growth cones (see Fig. 2) is not entirely surprising because PS1 has been detected at the cell surface and lamellipodia in non-neuronal cells (Dewji and Singer, 1997;Ray et al., 1999; Schwarzman et al., 1999). One possibility is that cytoskeletal interactions stabilize the heterodimeric complex containing PS1 at specific subcellular compartments, e.g., the tip of neuronal processes, where its presence may be critical to facilitate the activity of Notch1 that regulates neurite growth. The MT-associated protein tau and the MF-binding protein filamin have been identified previously as PS1-binding proteins (Takashima et al., 1998; Zhang et al., 1998), and both are enriched in growth cones, including filopodia and lamellipodia (Letourneau and Shattuck, 1989; Brandt et al., 1995). In this regard, tau and filamin are potential candidates to mediate the cytoskeletal association of PS1. In growth cones of hippocampal neurons, tau colocalizes with PS1 both in MT- and MF-rich areas (see Fig. 7A). Interestingly, previous studies have shown that the N-terminal projection domain of tau interacts with plasma membrane structures (Brandt et al., 1995), where it may contribute to the stabilization of the PS1 complex.
The amount of PS1 associated with the cytoskeleton increases dramatically during neuronal development (see Fig. 3A); by 10 DIV, when hippocampal neurons are fully differentiated and Notch1 signaling is activated by cell contact (Sestan et al., 1999) (see Fig.4A), most PS1 is found in the cytoskeletal fraction (see Fig. 3A). In 10 DIV cultures, PS1 mutations markedly decreased cytoskeletal binding (Fig. 3C) and Notch1 nuclear IF (see Fig. 4B) and increased total neuritic length (see Fig. 4C), suggesting that, in differentiated neurons, PS1 mutations deregulate cell contact-dependent inhibition of neurite growth mediated by Notch1.
Modulation of neurite growth by PS1 also was observed in 3 DIV neurons devoid of cell contacts and bearing fast-growing axonal processes. In these cultures the overexpression of PS1-wt significantly increased nuclear translocation of NICD and decreased axonal growth (see Fig.4B,C), although neither of these effects was observed in cultures expressing M146V, I143T, or D9 mutations (see Fig.4B,C). Similarly, neurite growth is not inhibited by the overexpression of a constitutively active NICD construct in cultured neurons derived from PS1-M146L transgenic mice (Berezovska et al., 1999b). Moreover, presenilin mutants induce morphological defects in Caenorhabditis eleganscholinergic interneurons by interfering with Notch1 signaling (Wittenburg et al., 2000). Taken collectively, these results suggest that (1) PS1 negatively regulates neuritic growth by facilitating the inhibitory input of Notch1 on neurite extension and (2) PS1 mutations reduce Notch1 signaling and alter neuritic growth and neuronal morphology. Future experiments will determine whether the cytoskeletal association of PS1 is relevant for the facilitation of Notch1 signaling in neuronal cells.
An important implication of these results is that, under pathological conditions, PS1 mutations may predispose differentiated neurons to develop dystrophic features by deregulating neurite growth. In this regard, different factors leading to excessive or maladaptive plasticity-related cellular events may play a key role in the development of AD pathology (Mesulam, 1999). To test this hypothesis, we evaluated neuritic dystrophy in Aβ-treated neurons expressing wild-type and mutant PS1. Most hippocampal neurons remained viable after a 48 hr exposure to 20 μm fibrillar Aβ but developed considerable dystrophic features (see Fig. 5B). Interestingly, the majority of dystrophic processes was found in contact with Aβ deposits (see Fig. 6A,B), suggesting that physical interaction between fibrils and neuronal membranes is required to trigger the dystrophic response. Neurons expressing PS1 mutations showed a dramatic increase in the number of dystrophic processes per cell (see Fig. 6C), the total number of dystrophic neurons in the culture (see Fig. 5A), and a marked acceleration in the appearance of dystrophic features (G. P. and J. B., unpublished observation) as compared with cultures expressing PS1-wt. These results suggest that PS1 mutations potentiate neuritic dystrophy by promoting abnormal neurite growth.
PS1 colocalizes with NFT in the AD brain (Busciglio et al., 1997;Giannakopoulos et al., 1997; Tomidokoro et al., 1999) and with tau in neuronal growth cones in culture (see Fig. 7A). Coimmunoprecipitation of endogenous PS1 and tau from cell lysates further suggests a molecular association between these two proteins in primary neurons (see Fig. 7B,C). PS1 mutations significantly increased tau phosphorylation, total tau protein level, and the release of tau from the cytoskeleton (see Fig. 7D,E). PS1 mutations may enhance tau hyperphosphorylation directly by increasing GSK3β tau kinase activity (Takashima et al., 1998; Weihl et al., 1999) and indirectly by destabilizing calcium homeostasis and increasing cytosolic Ca2+ levels (Begley et al., 1999), which have been shown to increase the activation of GSK3β and tau phosphorylation significantly (Hartigan and Johnson, 1999). In addition, the interaction of PS1 with MT-binding domains that are present in the tau molecule (Takashima et al., 1998) may contribute to the release of tau from the microtubular polymer. Thus, PS1 mutations promote tau hyperphosphorylation, reduced microtubular binding, and increased levels. All of these abnormal modifications in tau are present in AD brains and may be implicated directly in the development of cytoskeletal pathology and tangle formation (Khatoon et al., 1992;Goedert, 1993; Alonso et al., 1994).
In conclusion, our results indicate that PS1 regulates neuronal morphology, probably by facilitating Notch1 signaling, and that PS1 mutations potentiate neuritic dystrophy and enhance pathological changes in tau. It has been shown that FAD-linked presenilin mutations elevate the production and deposition of Aβ1–42 (Borchelt et al., 1996; Duff et al., 1996; Lemere et al., 1996; Xia et al., 1997). The finding that PS1 mutations promote aberrant neuritic growth and enhance AD-like changes in tau suggests a novel and unanticipated role for presenilins in the development of AD pathology. Thus, in FAD patients, PS1 mutations may advance the onset of neurodegeneration not only by accelerating Aβ deposition but also by promoting neuritic dystrophy and tangle formation.
Footnotes
This work was supported in part by National Institutes of Health Grant HD38466, a grant from the Alzheimer's Association, and funds from the University of Connecticut Health Center (J.B.) and by a grant-in-aid for Scientific Research from the Ministry of Education, Science, and Culture of Japan (H.M.) We thank Drs. David Papermaster and James Hewett for helpful comments.
Correspondence should be addressed to Dr. Jorge Busciglio, Department of Neuroscience, University of Connecticut Health Center, 263 Farmington Avenue, Farmington, CT 06030. E-mail:busciglio@nso1.uchc.edu.
REFERENCES
- 1.Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:5562–5566. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, George-Hyslop PS, Cordell B, Fraser P, De Strooper B. Presenilin-1 controls γ-secretase processing of amyloid precursor protein in pre-Golgi compartments of hippocampal neurons. J Cell Biol. 1999;147:277–294. doi: 10.1083/jcb.147.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- 4.Begley JG, Duan W, Chan S, Duff K, Mattson MP. Altered calcium homeostasis and mitochondrial dysfunction in cortical synaptic compartments of presenilin-1 mutant mice. J Neurochem. 1999;72:1030–1039. doi: 10.1046/j.1471-4159.1999.0721030.x. [DOI] [PubMed] [Google Scholar]
- 5.Beher D, Elle C, Underwood J, Davis JB, Ward R, Karran E, Masters CL, Beyreuther K, Multhaup G. Proteolytic fragments of Alzheimer's disease-associated presenilin-1 are present in synaptic organelles and growth cone membranes of rat brain. J Neurochem. 1999;72:1564–1573. doi: 10.1046/j.1471-4159.1999.721564.x. [DOI] [PubMed] [Google Scholar]
- 6.Berezovska O, McLean P, Knowles R, Frosch M, Lu FM, Lux SE, Hyman BT. Notch1 inhibits neurite outgrowth in postmitotic primary neurons. Neuroscience. 1999a;93:433–439. doi: 10.1016/s0306-4522(99)00157-8. [DOI] [PubMed] [Google Scholar]
- 7.Berezovska O, Frosch M, McLean P, Knowles R, Koo E, Kang D, Shen J, Lu FM, Lux SE, Tonegawa S, Hyman BT. The Alzheimer-related gene presenilin-1 facilitates Notch1 in primary mammalian neurons. Mol Brain Res. 1999b;69:273–280. doi: 10.1016/s0169-328x(99)00119-9. [DOI] [PubMed] [Google Scholar]
- 8.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin-1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 9.Brandt R, Leger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J Cell Biol. 1995;131:1327–1340. doi: 10.1083/jcb.131.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Busciglio J, Lorenzo A, Yankner BA. Methodological variables in the assessment of β-amyloid neurotoxicity. Neurobiol Aging. 1992;13:609–612. doi: 10.1016/0197-4580(92)90065-6. [DOI] [PubMed] [Google Scholar]
- 11.Busciglio J, Gabuzda DH, Matsudaira P, Yankner BA. Generation of β-amyloid in the secretory pathway in neuronal and non-neuronal cells. Proc Natl Acad Sci USA. 1993;90:2092–2096. doi: 10.1073/pnas.90.5.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busciglio J, Lorenzo A, Yeh J, Yankner BA. β-Amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14:879–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 13.Busciglio J, Hartmann H, Lorenzo A, Wong C, Baumann K, Sommer B, Staufenbiel M, Yankner BA. Neuronal localization of presenilin-1 and association with amyloid plaques and neurofibrillary tangles in Alzheimer's disease. J Neurosci. 1997;17:5101–5107. doi: 10.1523/JNEUROSCI.17-13-05101.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Capell A, Saffrich R, Olivo JC, Meyn L, Walter J, Grunberg J, Mathews P, Nixon R, Dotti C, Haass C. Cellular expression and proteolytic processing of presenilin proteins is developmentally regulated during neuronal differentiation. J Neurochem. 1997;69:2432–2440. doi: 10.1046/j.1471-4159.1997.69062432.x. [DOI] [PubMed] [Google Scholar]
- 15.Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, Beyreuther K, Selkoe DJ, Haass C. The proteolytic fragments of the Alzheimer's disease-associated presenilin-1 form heterodimers and occur as a 100–150 kDa molecular mass complex. J Biol Chem. 1998;273:3205–3211. doi: 10.1074/jbc.273.6.3205. [DOI] [PubMed] [Google Scholar]
- 16.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 17.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 18.Dewji NN, Singer SJ. Cell surface expression of the Alzheimer disease-related presenilin proteins. Proc Natl Acad Sci USA. 1997;94:9926–9931. doi: 10.1073/pnas.94.18.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doan A, Thinakaran G, Borchelt DR, Slunt HH, Ratovitsky T, Podlisny M, Selkoe DJ, Seeger M, Gandy SE, Price DL, Sisodia SS. Protein topology of presenilin-1. Neuron. 1996;17:1023–1030. doi: 10.1016/s0896-6273(00)80232-9. [DOI] [PubMed] [Google Scholar]
- 20.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid β42(43) in brains of mice expressing mutant presenilin-1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 21.Ferreira A, Lu Q, Orecchio L, Kosik KS. Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar Aβ. Mol Cell Neurosci. 1997;9:220–234. doi: 10.1006/mcne.1997.0615. [DOI] [PubMed] [Google Scholar]
- 22.Franklin JL, Berechid BE, Cutting FB, Presente A, Chambers CB, Foltz DR, Ferreira A, Nye JS. Autonomous and nonautonomous regulation of mammalian neurite development by Notch1 and Delta1. Curr Biol. 1999;9:1448–1457. doi: 10.1016/s0960-9822(00)80114-1. [DOI] [PubMed] [Google Scholar]
- 23.Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Aging renders the brain vulnerable to amyloid β protein neurotoxicity. Nat Med. 1998;4:827–831. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- 24.Giannakopoulos P, Bouras C, Kovari E, Shioi J, Tezapsidis N, Hof PR, Robakis NK. Presenilin-1-immunoreactive neurons are preserved in late-onset Alzheimer's disease. Am J Pathol. 1997;150:429–436. [PMC free article] [PubMed] [Google Scholar]
- 25.Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer's disease. Trends Neurosci. 1993;16:460–465. doi: 10.1016/0166-2236(93)90078-z. [DOI] [PubMed] [Google Scholar]
- 26.Goslin K, Banker G. Rat hippocampal neurons in low density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT; Cambridge, MA: 1992. pp. 252–281. [Google Scholar]
- 27.Greenberg SG, Davies P, Schein JD, Binder LI. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267:564–569. [PubMed] [Google Scholar]
- 28.Hartigan JA, Johnson GV. Transient increases in intracellular calcium result in prolonged site-selective increases in tau phosphorylation through a glycogen synthase kinase 3β-dependent pathway. J Biol Chem. 1999;274:21395–21401. doi: 10.1074/jbc.274.30.21395. [DOI] [PubMed] [Google Scholar]
- 29.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related Aβ deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signaling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- 31.Khatoon S, Grundke-Iqbal I, Iqbal K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer's disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem. 1992;59:750–753. doi: 10.1111/j.1471-4159.1992.tb09432.x. [DOI] [PubMed] [Google Scholar]
- 32.Kopan R, Schroeter EH, Weintraub H, Nye JS. Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci USA. 1996;93:1683–1688. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lah JJ, Heilman CJ, Nash NR, Rees HD, Yi H, Counts SE, Levey AI. Light and electron microscopic localization of presenilin-1 in primate brain. J Neurosci. 1997;17:1971–1980. doi: 10.1523/JNEUROSCI.17-06-01971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemere CA, Lopera F, Kosik KS, Lendon CL, Ossa J, Saido TC, Yamaguchi H, Ruiz A, Martinez A, Madrigal L, Hincapie L, Arango JC, Anthony DC, Koo EH, Goate AM, Selkoe DJ. The E280A presenilin-1 Alzheimer mutation produces increased Aβ42 deposition and severe cerebellar pathology. Nat Med. 1996;2:1146–1150. doi: 10.1038/nm1096-1146. [DOI] [PubMed] [Google Scholar]
- 35.Letourneau PC, Shattuck TA. Distribution and possible interactions of actin-associated proteins and cell adhesion molecules of nerve growth cones. Development. 1989;105:505–519. doi: 10.1242/dev.105.3.505. [DOI] [PubMed] [Google Scholar]
- 36.Levesque L, Annaert W, Craessaerts K, Mathews PM, Seeger M, Nixon RA, Van Leuven F, Gandy S, Westaway D, St. George-Hyslop P, De Strooper B, Fraser PE. Developmental expression of wild-type and mutant presenilin-1 in hippocampal neurons from transgenic mice: evidence for novel species-specific properties of human presenilin-1. Mol Med. 1999;5:542–554. [PMC free article] [PubMed] [Google Scholar]
- 37.Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, Bird TD, Schellenberg GD. A familial Alzheimer's disease locus on chromosome 1. Science. 1995;269:970–973. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- 38.Li X, Greenwald I. Additional evidence for an eight-transmembrane-domain topology for Caenorhabditis elegans and human presenilins. Proc Natl Acad Sci USA. 1998;95:7109–7114. doi: 10.1073/pnas.95.12.7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y-M, Xu M, Lai M-T, Huang Q, Castro J, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil J, Register R, Sardana MK, Shearman M, Smith A, Shi X-P, Yin K-C, Shafer JA, Gardell SJ. Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin-1. Nature. 2000;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 40.Lorenzo A, Yankner BA. β-Amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mesulam MM. Neuroplasticity failure in Alzheimer's disease: bridging the gap between plaques and tangles. Neuron. 1999;24:521–529. doi: 10.1016/s0896-6273(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 42.Nakata T, Hirokawa N. Cytoskeletal reorganization of human platelets after stimulation revealed by the quick-freeze deep-etch technique. J Cell Biol. 1987;105:1771–1780. doi: 10.1083/jcb.105.4.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- 44.Okochi M, Sahara N, Kametani F, Usami M, Arai T, Tanaka K, Ishii K, Yamamoto A, Mori H. Presenilin-1 cleavage is a universal event in human organs. Neurobiol Aging. 1998;19:S3–S10. doi: 10.1016/s0197-4580(98)00045-1. [DOI] [PubMed] [Google Scholar]
- 45.Pigino G, Paglini G, Ulloa L, Avila J, Caceres A. Analysis of the expression, distribution, and function of cyclin-dependent kinase 5 (cdk5) in developing cerebellar macroneurons. J Cell Sci. 1997;110:257–270. doi: 10.1242/jcs.110.2.257. [DOI] [PubMed] [Google Scholar]
- 46.Pike CJ, Cummings BJ, Cotman CW. β-Amyloid induces neuritic dystrophy in vitro: similarities with Alzheimer pathology. NeuroReport. 1992;3:769–772. doi: 10.1097/00001756-199209000-00012. [DOI] [PubMed] [Google Scholar]
- 47.Ray WJ, Yao M, Mumm J, Schroeter EH, Saftig P, Wolfe M, Selkoe DJ, Kopan R, Goate AM. Cell surface presenilin-1 participates in the γ-secretase-like proteolysis of Notch. J Biol Chem. 1999;274:36801–36807. doi: 10.1074/jbc.274.51.36801. [DOI] [PubMed] [Google Scholar]
- 48.Redmond L, Oh SR, Hicks C, Weinmaster G, Ghosh A. Nuclear Notch1 signaling and the regulation of dendritic development. Nat Neurosci. 2000;3:30–40. doi: 10.1038/71104. [DOI] [PubMed] [Google Scholar]
- 49.Sahara N, Yahagi Y, Takagi H, Kondo T, Okochi M, Usami M, Shirasawa T, Mori H. Identification and characterization of presenilin I-467, I-463, and I-374. FEBS Lett. 1996;381:7–11. doi: 10.1016/0014-5793(96)00054-3. [DOI] [PubMed] [Google Scholar]
- 50.Schroeter EH, Kisslinger JA, Kopan R. Notch1 signaling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 51.Schwarzman AL, Singh N, Tsiper M, Gregori L, Dranovsky A, Vitek MP, Glabe CG, St. George-Hyslop PH, Goldgaber D. Endogenous presenilin-1 redistributes to the surface of lamellipodia upon adhesion of Jurkat cells to a collagen matrix. Proc Natl Acad Sci USA. 1999;96:7932–7937. doi: 10.1073/pnas.96.14.7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sestan N, Artavanis-Tsakonas S, Rakic P. Contact-dependent inhibition of cortical neurite growth mediated by Notch signaling. Science. 1999;286:741–746. doi: 10.1126/science.286.5440.741. [DOI] [PubMed] [Google Scholar]
- 53.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K. Cloning of a gene bearing missense mutations in early onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 54.Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA. Proteolytic release and nuclear translocation of Notch1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci USA. 1999;96:6959–6963. doi: 10.1073/pnas.96.12.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Struhl G, Adachi A. Nuclear access and action of Notch in vivo. Cell. 1998;93:649–660. doi: 10.1016/s0092-8674(00)81193-9. [DOI] [PubMed] [Google Scholar]
- 56.Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- 57.Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, Nihonmatsu N, Mercken M, Yamaguchi H, Sugihara S, Wolozin B. Presenilin-1 associates with glycogen synthase kinase-3β and its substrate tau. Proc Natl Acad Sci USA. 1998;95:9637–9641. doi: 10.1073/pnas.95.16.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Endoproteolysis of presenilin-1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 59.Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, Sisodia SS. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J Biol Chem. 1997;272:28415–28422. doi: 10.1074/jbc.272.45.28415. [DOI] [PubMed] [Google Scholar]
- 60.Threadgill R, Bobb K, Ghosh A. Regulation of dendritic growth and remodeling by Rho, Rac, and Cdc42. Neuron. 1997;19:625–634. doi: 10.1016/s0896-6273(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 61.Tomidokoro Y, Ishiguro K, Igeta Y, Matsubara E, Kanai M, Shizuka M, Kawarabayashi T, Harigaya Y, Kawakatsu S, Ii K, Ikeda M, St. George-Hyslop PH, Hirai S, Okamoto K, Shoji M. Carboxyl-terminal fragments of presenilin-1 are closely related to cytoskeletal abnormalities in Alzheimer's brains. Biochem Biophys Res Commun. 1999;256:512–518. doi: 10.1006/bbrc.1998.0119. [DOI] [PubMed] [Google Scholar]
- 62.Weihl CC, Ghadge GD, Kennedy SG, Hay N, Miller RJ, Roos RP. Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J Neurosci. 1999;19:5360–5369. doi: 10.1523/JNEUROSCI.19-13-05360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weinmaster G. The ins and outs of Notch signaling. Mol Cell Neurosci. 1997;9:91–102. doi: 10.1006/mcne.1997.0612. [DOI] [PubMed] [Google Scholar]
- 64.Wittenburg N, Eimer S, Lakowski B, Rohrig S, Rudolph C, Baumeister R. Presenilin is required for proper morphology and function of neurons in C. elegans. Nature. 2000;406:306–309. doi: 10.1038/35018575. [DOI] [PubMed] [Google Scholar]
- 65.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 66.Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haass C, Seubert P, Koo EH, Selkoe DJ. Enhanced production and oligomerization of the 42-residue amyloid β protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem. 1997;272:7977–7982. doi: 10.1074/jbc.272.12.7977. [DOI] [PubMed] [Google Scholar]
- 67.Ye Y, Lukinova N, Fortini ME. Neurogenic phenotypes and altered Notch processing in Drosophila presenilin mutants. Nature. 1999;398:525–529. doi: 10.1038/19096. [DOI] [PubMed] [Google Scholar]
- 68.Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, St. George-Hyslop PH, Fraser PE. The presenilin-1 protein is a component of a high-molecular-weight intracellular complex that contains β-catenin. J Biol Chem. 1998;273:16470–16475. doi: 10.1074/jbc.273.26.16470. [DOI] [PubMed] [Google Scholar]
- 69.Zhang W, Han SW, McKeel DW, Goate A, Wu JY. Interaction of presenilins with the filamin family of actin-binding proteins. J Neurosci. 1998;18:914–922. doi: 10.1523/JNEUROSCI.18-03-00914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]