

Abstract
Wnt signaling has been implicated in the control of cell proliferation and in synapse formation during neural development, and these actions are presumed to be mediated byfrizzled receptors. In this paper we report the phenotype of mice carrying a targeted deletion of thefrizzled-4 (fz4) gene.fz4(−/−) mice exhibit three distinct defects: (1) progressive cerebellar degeneration associated with severe ataxia, (2) absence of a skeletal muscle sheath around the lower esophagus associated with progressive esophageal distension and dysfunction, and (3) progressive deafness caused by a defect in the peripheral auditory system unaccompanied by loss of hair cells or other auditory neurons. As assayed using a lacZ knock-in reporter,fz4 is widely expressed within the CNS. In particular,fz4 is expressed in cerebellar Purkinje cells, esophageal skeletal muscle, and cochlear inner hair cells, and the absence of Fz4 in these cells is presumed to account for thefz4(−/−) phenotype. In contrast to the early cell proliferation and patterning effects classically ascribed to Wnts, the auditory and cerebellar phenotypes of fz4(−/−) mice implicate Frizzled signaling in maintaining the viability and integrity of the nervous system in later life.
Keywords: frizzled-4, cerebellar degeneration, Purkinje cells, Wnt signaling, esophagus, progressive hearing loss
Members of the Wnt family of secreted signaling proteins are found throughout the animal kingdom and play important roles in the control of tissue patterning, cell fate, and cell proliferation (for review, see Cadigan and Nusse, 1997). Loss of function mutations in Wnt genes have been extensively characterized in Drosophila, Caenorhabditis elegans, and mice, and these mutations demonstrate that a single Wnt can have multiple and diverse functions in different developmental contexts. For example,Drosophila Wingless (Wg), the most extensively studied Wnt protein, plays an essential role in embryogenesis to maintain segment polarity in the epidermis, define the identities of a subset of developing neuroblasts, and pattern the midgut and heart; in imaginal disc development, Wg acts to pattern the wings, legs, and eyes (for review, see Klingensmith and Nusse, 1994). Gain of function mutations that lead to Wnt overexpression have also been characterized inDrosophila and mice. For example, overexpression of Wnt-1 or Wnt-3a in mice, first identified as a consequence of retroviral insertion, causes hyperproliferation and oncogenic transformation (Nusse and Varmus, 1982; Roelink et al., 1990). Consistent with this observation, recent work has implicated mutational activation of downstream effectors of Wnt signaling in the pathogenesis of various human cancers (Morin et al., 1997; Chan et al., 1999; Satoh et al., 2000). In general, the range of Wnt action is limited to the immediate vicinity of the site of synthesis and secretion, presumably as a consequence of the tight binding of Wnts to extracellular matrix molecules.
A role for Wnt signaling in the development of the vertebrate CNS has emerged from observations with knock-out mice: mutations inWnt-1 lead to a failure of midbrain development and a partial or complete failure of cerebellar development (McMahon and Bradley, 1990; Thomas and Capecchi, 1990), and mutations inWnt-3a (Lee et al., 2000) or Lef1 (Galceran et al., 2000), a downstream effector of Wnt signaling, block the normal expansion of hippocampal precursors. Moreover, the phenotype ofWnt-1/Wnt-3a double knock-out mice shows that these two Wnts act redundantly to expand the number of dorsal neural crest precursors along the length of the spinal cord (Ikeya et al., 1997). Consistent with these loss-of-function phenotypes, ectopic expression of Wnt-1 in the developing spinal cord causes a local increase in cell proliferation (Dickinson et al., 1994). These observations indicate that one role of Wnts in CNS development involves the stimulation of cell proliferation.
Wnts may also play a role in CNS synaptogenesis as indicated by the recent demonstration that Wnt-7a stimulates axonal growth and synaptogenesis by developing cerebellar mossy fibers as they find their granule cell targets (Lucas and Salinas, 1997; Hall et al., 2000). Interestingly, the defect in mossy fiber differentiation inWnt-7a knock-out mice is partial and transient, suggesting that other Wnts expressed in the cerebellum may largely compensate for the loss of Wnt-7a. At present there are 18 known mammalian Wnt genes, and many of them are expressed in the CNS (Salinas and Nusse, 1992;Parr et al., 1993; Grove et al., 1998; Lako et al., 1998), raising the possibility that additional aspects of CNS development may be controlled by Wnt signaling.
Wnt signals are transduced by the Frizzled family of cell surface receptors, the members of which are also found throughout the animal kingdom. In mammals there are currently nine known Frizzled genes, and for many of these there are highly conserved orthologs in fish, birds, and amphibia. In vitro studies of Wnt–Frizzled binding using soluble Wg or Xenopus Wnt-8 demonstrate that one Wnt can bind to any of a number of Frizzled proteins, although the affinity of binding varies among different ligand-receptor pairs (Hsieh et al., 1999; Rulifson et al., 2000). Several experiments have begun to address the biological consequences of this complex pattern of ligand-receptor specificity. Injection of Wnt and Frizzled RNAs into Xenopus embryos has demonstrated a specific interaction between Wnt-5A and Frizzled-5 but not between Wnt-5A and any of six other Frizzled receptors tested, indicating a high degree of ligand-receptor specificity in this experimental system (He et al., 1997). In contrast, duringDrosophila embryogenesis, Frizzled and Frizzled-2 appear to play largely redundant roles as Wg receptors, as judged by the development of nearly normal embryos when either receptor is absent, and the recapitulation of the full wg null phenotype when both are absent (Bhat, 1998; Kennerdell and Carthew, 1998; Bhanot et al., 1999; Chen and Struhl, 1999).
In the vertebrate CNS, several Frizzled genes are expressed during development and in the adult (Wang et al., 1996; Shi et al., 1998;Borello et al., 1999; Wheeler and Hoppler, 1999), but their roles in vertebrate nervous system development and function are largely unexplored. In this paper we report the phenotype of fz4 knock-out mice.
MATERIALS AND METHODS
Generation of knock-in mice. The fz4 knock-in construct was electroporated into R1 cells, and colonies were grown in medium containing G418 and gancyclovir. Colonies were picked 8 d after plating and screened by Southern blot hybridization, and positive ES clones were injected into C57/Bl6 blastocysts.
Auditory brainstem response. Auditory brainstem responses were recorded from adult fz4(−/−) and fz4(+/+) littermates without revealing the genotype to the experimenter. Mice were anesthetized with intraperitoneal injections of a 3–5 mg/kg mixture of xylazine hydrochloride (2.5 mg/ml) and ketamine (25 mg/ml). A subcutaneous injection of 1% lidocaine served to keep the animals quiet and helped reduce muscle noise during signal averaging. The absence of a withdrawal reflex to sharp paw pinches signaled adequate anesthesia. Electrodes were placed at the vertex of the skull, in the dorsum of the neck, and in the postauricular region of the ear closest to the speaker. Free field clicks (n = 1000) of 100 μsec duration were presented in 5 dB increments, starting at 5 dB and progressing to 100 dB sound pressure level (SPL). At each intensity level, recordings were collected for 20 msec and then averaged.
Immunohistochemistry and terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling. Immunostaining and terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling (TUNEL) were performed on brain sections that were fresh frozen, cryosectioned at 16 μm, and then post-fixed in 4% paraformaldehyde in PBS, except for calbindin immunostaining, which was performed with perfused tissues that were cryosectioned at 16 μm. Immunostaining of the esophagus and lower gastrointestinal (GI) tract was performed with tissue perfused with 4% paraformaldehyde and cryosectioned at 10 μm. Reagents were obtained from the following sources: anti-calbindin (Chemicon, Temecula, CA), anti-type 1 IP3 receptor (Affinity BioReagents, Golden, CO), anti-skeletal muscle myosin (Sigma, St. Louis, MO), anti-glial fibrillary acidic protein (GFAP) (Dako, Carpinteria, CA), anti-CGRP (Peninsula Laboratories, Belmont, CA), and fluorescein TUNEL reagents (Roche, Indianapolis, IN).
Histochemistry. Unfixed leg muscles were snap-frozen in isopentane and cryosectioned at 10 μm. Whole mounts of esophagus, stomach, and intestine were prepared by dissecting fresh tissues followed by fixation in fresh 4% paraformaldehyde in PBS. Acetylcholine esterase staining was performed at room temperature with minor modifications of the method of Karnovsky and Root (1964). The reaction mixture contained 0.1 m sodium phosphate, pH 6.0, 4 mm acetylcholine iodide, 5 mm sodium citrate, 3 mmcopper sulfate, 0.5 mm potassium ferricyanide, and 100 μm tetraisopropylpyrophosphoramide. NADH diaphorase staining was performed at room temperature for 30 min in 1 mm NADH, 0.2 mTris, pH 7.4, and 1 mg/ml nitro blue tetrazolium (NBT), as described inBancroft and Stevens (1982). NADPH diaphorase staining was performed at 37°C for 1–3 hr in PBS with 0.3% Triton X-100, 0.1 mm NADPH, and 0.25 mg/ml NBT, as described inBancroft and Stevens (1982). For semithin sections of the esophagus, isolated esophagi were fixed in 2% paraformaldehyde, 2% gluteraldehyde, in PBS with 1.5 mmMgCl2 and then post-fixed in osmium tetroxide, embedded in Epon, sectioned at 1 μm, and stained with toluidine blue.
For X-gal staining of temporal bones, esophagus, and embryos, or vibratome sections of fresh brain, tissues were fixed for 5–10 min at room temperature in 2.5% paraformaldehyde, 0.2% gluteraldehyde in PBS with 2 mm MgCl2 before incubation in X-gal overnight at 37°C. For inner ear staining, after the X-gal reaction temporal bones were decalcified for 2 weeks in 0.25m EDTA, 15% sucrose in PBS, equilibrated with OCT over 2 d, and cryosectioned. Retinas were X-gal stained after perfusion with 2.5% paraformaldehyde, 0.2% gluteraldehyde in PBS with 2 mm MgCl2, and cryosectioning of the eye.
The Sevier–Munger silver staining method, which highlights cerebellar basket cells and nerve fibers, was performed on deparaffinized sections of the CNS as described (Sevier and Munger, 1965; Luna, 1992). The development step was timed by visual inspection to maximize specificity and contrast before sections were rinsed, cleared, and mounted.
RESULTS
Expression pattern of a fz4–lacZknock-in allele
To investigate the function of fz4 in vivo, we constructed a targeted deletion of all but the last 70 nucleotides of the fz4 coding region (Fig.1). The targeting construct also incorporates a lacZ reporter beginning at the initiator methionine of fz4. As an initial guide to the analysis of the fz4(−/−) phenotype, we surveyed the pattern offz4 gene expression by assessing X-gal staining using thefz4–lacZ knock-in allele. In the midgestation embryo, the fz4–lacZ reporter is expressed widely (data not shown). In the adult, X-gal staining is observed in many locations throughout the CNS, including the olfactory bulb, cerebral cortex, midbrain, dorsal spinal cord, retina, and inner ear (Fig. 2). Within the adult cerebellum, X-gal staining is confined to Purkinje cells (Figs.2A) and is seen at postnatal day (P) 15 but not at P8 (data not shown). In most locations within the CNS, X-gal staining appears in a punctate pattern within the cell somata. This pattern presumably reflects a heterogeneous distribution of active tetrameric β-galactosidase, possibly as a consequence of low levels of expression. However, retinal photoreceptors, inner hair cells in the organ of Corti, and vestibular hair cells in the maculae and cristae show a more intense and uniform pattern of X-gal staining (Fig.2C–F), suggesting that the fz4 gene may be expressed at higher levels in these neurons. In all tissues examined thus far, fz4(+/−) and fz4(−/−) mice exhibit identical or very nearly identical patterns of X-gal staining.
Fig. 1.

Strategy for targeted replacement of thefz4 coding region by lacZ. A,Top, Map of the murine fz4 locus. Thefz4 coding exons reside within a region of 3 kb, indicated by a filled rectangle. B,BamHI; E, EcoRI;H, HindIII; N,NcoI. Center, Structure of thefz4 targeting construct. A 5′ flanking segment of 8.5 kb terminating at the initiator methionine of fz4 was joined to the initiator methionine codon of a β-galactosidase expression cassette. The β-galactosidase coding region is followed by an intron and poly(A) site from the mouse protamine-1 gene (lacZ-mp1) (Peschon et al., 1987) and by aPGK-neo selectable marker. A thymidine kinase selectable marker (MC1-TK) is located distal to the 3′ homology segment. Bottom, Structure of the targeted allele. The targeting event precisely deletes all but the 3′ 70 bp of the fz4 coding region. B, Genotyping of fz4(+/+), fz4(+/−), andfz4(−/−) mice by EcoRI digestion and Southern blotting with the 3′ flanking probe indicated inA. The wild-type and gene-targeted alleles generate fragments of 10.4 and 12.0 kb, respectively.
Fig. 2.

fz4 expression patterns as revealed by X-gal staining of the lacZ reporter infz4(+/−) and fz4(−/−) mice.A, B, X-gal staining of cerebellum (A) and a coronal section through the brain (B) from adult fz4(+/−) mice. In the cerebellum, X-gal staining is detected only in Purkinje cells.C, X-gal staining of the adult fz4(+/−) retina. The pigment epithelium is at the top, and the ganglion cell layer is at the bottom. Uniform filling of the photoreceptor soma by the X-gal reaction product contrasts with a punctate pattern of X-gal staining in the inner retina.D–F, X-gal staining pattern in the adultfz4(−/−) inner ear. X-gal staining is found in inner hair cells in the organ of Corti (D,arrowhead) and at very low levels in outer hair cells and in the primary sensory cells of the maculae (E) and cristae (F). As seen in the retina, the X-gal product exhibits uniform filling of primary sensory cells. fz4(+/−) andfz4(−/−) mice exhibited indistinguishable patterns of X-gal staining in each of these tissues. Scale bars: A, 200 μm; C–F, 40 μm.
Growth retardation, abnormal gait, and premature death infz4(−/−) mice
Despite the widespread expression of fz4 observed in the embryo and documented previously in multiple adult organs by RNase protection (Wang et al., 1996), fz4(−/−) mice are born at the expected Mendelian ratio in crosses between fz4(+/−) parents, and they grow at a normal rate during the first week of postnatal life. However, beginning in the second postnatal week,fz4(−/−) mice gain weight more slowly than their sex-matched fz4(+/+) and fz4(+/−) littermates, and ∼50% of fz4(−/−) mice die during the first several months of postnatal life (Fig. 3). No differences were observed between fz4(+/+) andfz4(+/−) mice with respect to weight gain, viability, or any of the other phenotypes examined in this study.
Fig. 3.
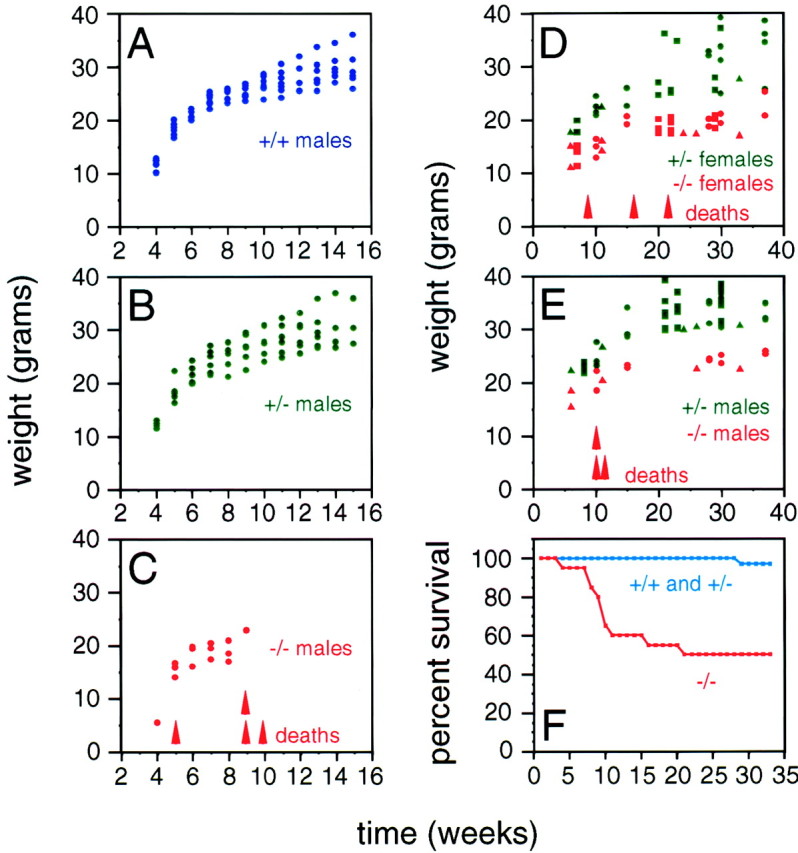
Growth retardation and early lethality in fz4(−/−) mice. A–C, Weight gain during early life in a cohort of sevenfz4(+/+), six fz4(+/−), and fourfz4(−/−) mice. No differences are observed betweenfz4(+/+) and fz4(+/−) mice. The fourfz4(−/−) mice in this cohort weighed less than theirfz4(+/+) and fz4(+/−) littermates at every time point examined and died between postnatal weeks 5 and 11.D, E, Weight gain over 35 weeks among 20fz4(+/−) and 13 fz4(−/−) mice.Squares, circles, andtriangles represent different cohorts. Long-livedfz4(−/−) mice remain underweight throughout life in comparison to their fz4(+/−) littermates.F, A Kaplan–Meyer survival curve shows ∼50% lethality among fz4(−/−) mice between 3 and 15 weeks of age. Many of the fz4(−/−) mice that survive beyond this early period die during the ensuing year.
The earliest outward phenotypic difference betweenfz4(−/−) mice and their fz4(+/+) andfz4(+/−) littermates is seen in coat color. In the 129/SVJ × C57BL/6J background of this line, fz4(+/+) and fz4(+/−) mice are either black or agouti, whereasfz4(−/−) mice are light black or silver, respectively. This phenotype suggests that fz4 may play a role in the expansion or migration of melanocyte precursors from the neural crest or the differentiation and survival of melanocytes. Other outward phenotypes that become increasingly apparent as fz4(−/−) animals age are a characteristic hunchback posture and an abnormal gait (Fig. 4). Older fz4(−/−) mice walk with a shortened stride, minimal alternation between the left and right feet, and frequent failures to fully lift each foot between steps. At rest, fz4(−/−) adults show a characteristically wide stance, as seen in the position of the hind feet in Figure4C.
Fig. 4.
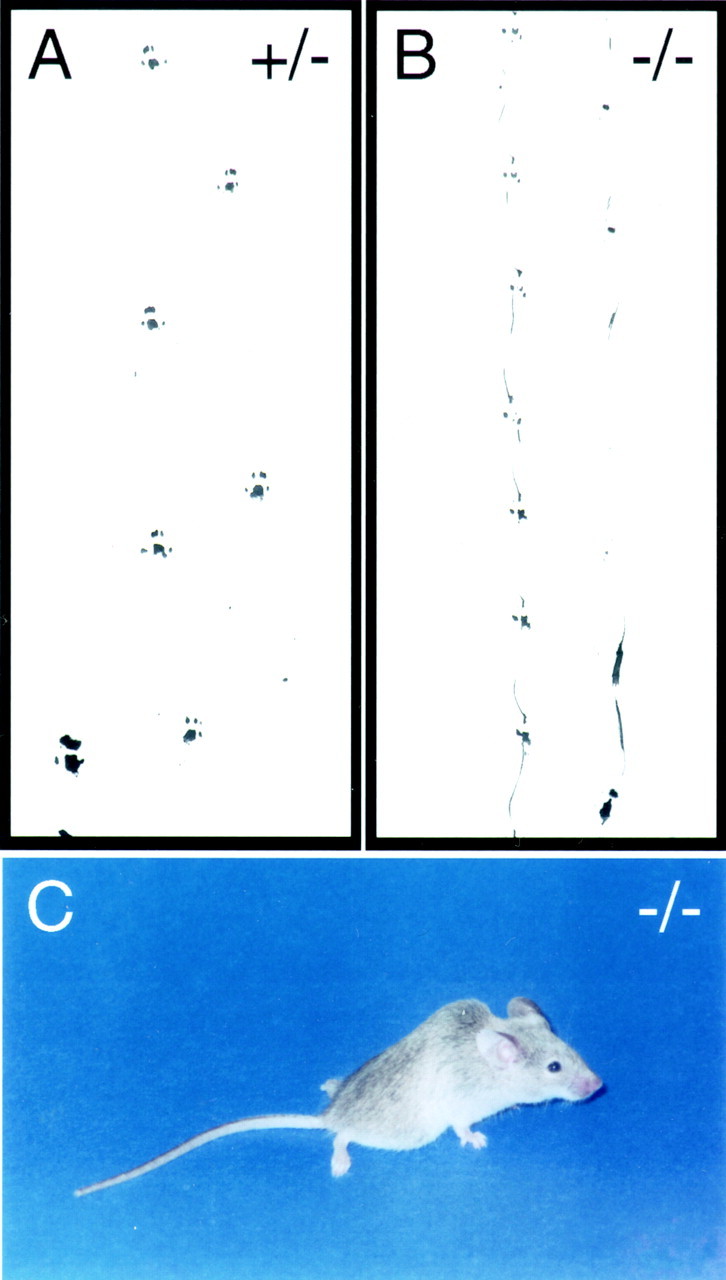
Ataxia in fz4(−/−) mice.A, B, Hind footprints from adultfz4(+/−) and fz4(−/−) mice.fz4(−/−) mice exhibit a shortened stride, minimal alternation between the left and right feet, and frequent failures to fully lift each foot between steps. C,fz4(−/−) adults shows a wide stance, as evident in the position of the hind feet. As seen here, older animals also have a characteristic hunchback posture.
Cerebellar degeneration in fz4(−/−) mice
The gait abnormalities noted above might be caused, at least in part, by a peripheral neuropathy or by abnormalities intrinsic to skeletal muscle. To test these possibilities, leg muscles fromfz4(+/+) and fz4(−/−) adults were examined by hematoxylin and eosin staining, the pattern of innervation was assessed by acetylcholine esterase histochemistry, and the distribution of type 1 and type 2 muscle fibers was assessed by NADH diaphorase staining (Fig. 5) [in this and all subsequent figures, paired photographs show the fz4(+/+) orfz4(+/−) section on the left and thefz4(−/−) section on the right]. This analysis shows that the average diameter of the fz4(−/−) muscle fibers is smaller than that of the fz4(+/+) fibers, a difference that we attribute to myocyte atrophy associated with the poor weight gain in fz4(−/−) mice. However, no other differences were observed between these muscle samples, and in particular, no evidence was found for denervation, loss, or regeneration of myofibrils. Cresyl violet staining of serial cross sections along the length of the fz4(+/+) andfz4(−/−) spinal cord shows little or no difference between the two (data not shown). Although we cannot rule out functional defects in spinal motor or sensory mechanisms not accompanied by cell loss, these data suggest that the gait abnormality is unlikely to arise from lower motor neuron loss, peripheral neuropathy, or myopathy.
Fig. 5.
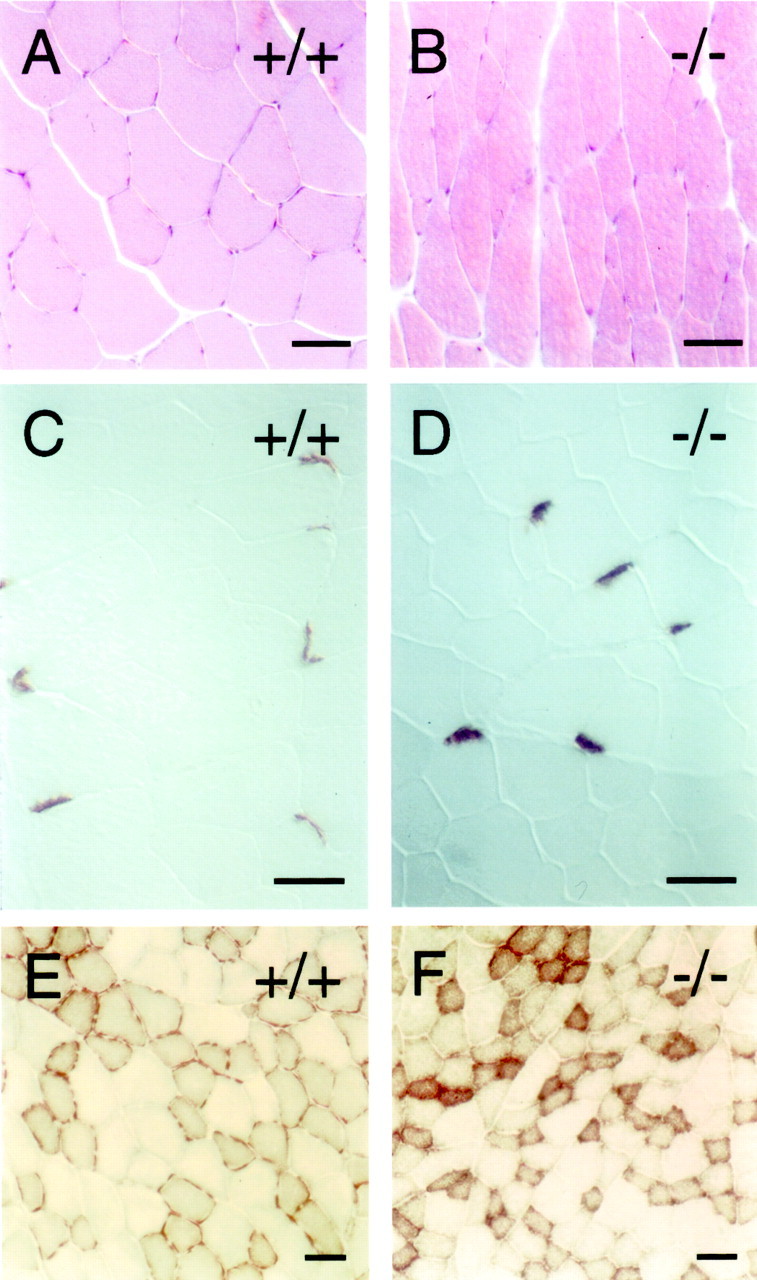
Histologic appearance of hindlimb skeletal muscle in fz4(−/−) mice. A, B, Hematoxylin and eosin staining. C, D, Acetylcholine esterase histochemistry to visualize neuromuscular junctions. E, F, NADH diaphorase histochemistry to distinguish type 1 fibers (strongly stained) from type 2 fibers (weakly stained). fz4(−/−) limb muscles show modest atrophy consistent with weight loss but no evidence of inflammation, fibrosis indicative of myopathy, or clustering of fiber types indicative of peripheral neuropathy. Scale bars, 40 μm.
To investigate the possibility of a central neurological basis for the gait abnormalities in fz4(−/−) mice, sections of adult brain were surveyed for evidence of developmental anomalies or cell loss using cresyl violet, hematoxylin and eosin, and the Sevier–Munger silver stain (Sevier and Munger, 1965), which highlights neuronal processes, and anti-calbindin immunostaining, which highlights cerebellar Purkinje cells (Fig. 6). This survey revealed a hypocellular cerebellum in the context of grossly normal cerebellar architecture (Fig.6A,B). Closer examination revealed a dramatic loss of cerebellar granule and Purkinje cells in a laminar pattern that followed the crests and sulci of the cerebellar folia with accompanying vacuolization that extended into the subjacent myelinated tracts (Fig.6C,D,G–N). Adult fz4(−/−) cerebella also exhibited astroglial activation and altered astroglial architecture as assayed by GFAP immunostaining (Fig. 6E,F). Other brain regions in adult fz4(−/−) mice appear to be unaltered. The disorganization of cerebellar structure infz4(−/−) mice suggests that the gait abnormalities infz4(−/−) mice reflect a progressive cerebellar ataxia.
Fig. 6.
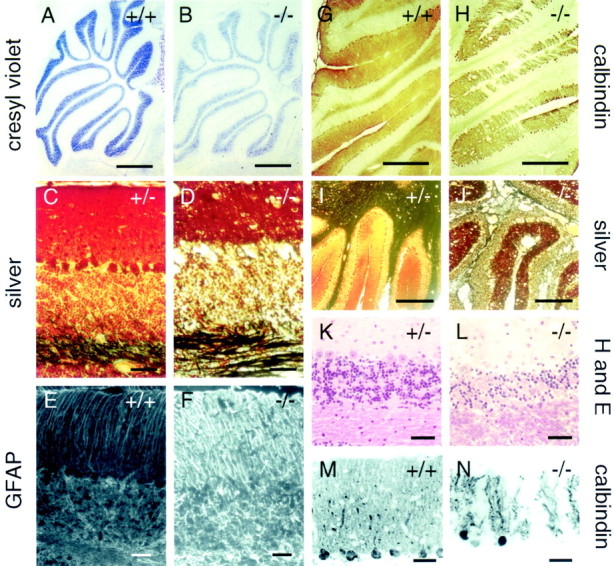
Cerebellar degeneration in adultfz4(−/−) mice. A, B, Cresyl violet staining shows hypocellularity in thefz4(−/−) cerebellum in the context of normal cerebellar architecture. C, D,I, J, Silver staining by the method ofSevier and Munger (1965), which visualizes neuronal processes (black), and hematoxylin and eosin staining (K, L) show vacuolation, loss of Purkinje and granule cells, and disorganization of the molecular and granule cell layers in adult fz4(−/−) cerebella.E, F, Increased activation of glia and disorganization of activated glial morphology in the adultfz4(−/−) cerebellum. G,H, M, N, Purkinje cell loss in the adult fz4(−/−) cerebellum as revealed by calbindin immunostaining. Scale bars: A,B, 1 mm;C–F, K–N, 40 μm; G, H, 0.5 mm;I, J, 400 μm.
Analysis of neuronal cell death in the cerebellum at different developmental times was performed using cresyl violet staining andin situ detection of DNA fragmentation by TUNEL labeling (Gavrieli et al., 1992). By cresyl violet staining the cerebella offz4(−/−) mice appear to develop normally up to P19 (Fig.7A,B). However, TUNEL analysis at P8, P14, P19, and P30 shows massive death of granule cells in the fz4(−/−) cerebellum beginning between P14 and P19 (Fig.7C–F,M). At P8 and P14, both fz4(−/−) and fz4(+/+) cerebella show small numbers of TUNEL+ cells in the external granule cell layer and in the developing inner granule cell layer. At P19, at least 50-fold more TUNEL+ cells are present in the fz4(−/−) cerebellum compared with the fz4(+/−) cerebellum, and > 95% of these are in the granule cell layer. By P30, the number of TUNEL+ cells in the fz4(−/−) cerebellum is reduced greatly. Elsewhere within the CNS, fz4(+/+), fz4(+/−), andfz4(−/−) littermates show differences of less than twofold in the number of TUNEL+ cells during postnatal development and in adulthood.
Fig. 7.
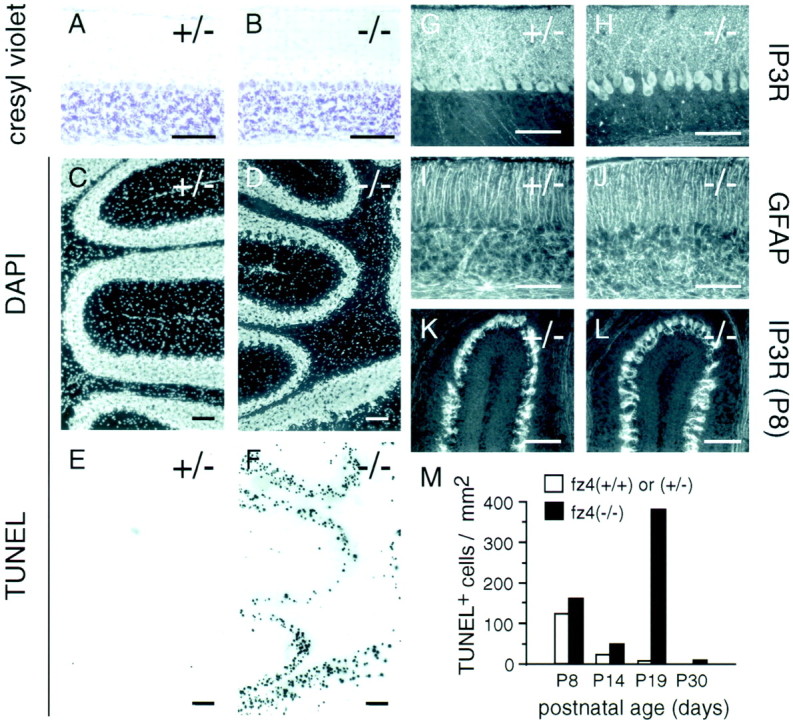
Extensive granule cell death between weeks 2 and 5 in the fz4(−/−) cerebellum.A–J, Cerebella fromfz4(+/−) and fz4(−/−) mice at P19.A, B, Cresyl violet staining reveals a grossly normal appearance including roughly normal numbers of Purkinje and granule cells. DAPI staining (C, D) and TUNEL labeling of DNA fragments (E,F) in the same sections reveal extensive granule cell death in the fz4(−/−) cerebellum (F) and minimal cell death in thefz4(+/−) control (E).G, H, Type I IP3 receptor immunostaining at P19 shows increased separation of Purkinje cell bodies and processes in the fz4(−/−) cerebellum.I, J, Activated glia, as revealed by GFAP immunostaining, show subtle disorganization in thefz4(−/−) granule cell layer. K,L, Type I IP3 receptor immunostaining at P8 shows that developing Purkinje cells of fz4(+/−) andfz4(−/−) mice have indistinguishable morphology.M, Histogram showing, at different ages, the number of TUNEL-positive nuclei per square millimeter of surface area in a series of randomly selected 16-μm-thick sections of cerebellum. Because cell sizes are smaller and cell densities are higher in younger animals, the total number of cells per square millimeter is higher in younger animals. Scale bars, 100 μm.
In fz4(−/−) mice, TUNEL labeling of cerebellar Purkinje cells was rarely seen, but this simply may reflect their paucity relative to granule cells. However, a progressive loss of Purkinje cells proceeds throughout adulthood in the fz4(−/−) cerebellum as judged by immunostaining for type-1 IP3 receptor and calbindin, both of which are specifically expressed in Purkinje cells (Figs.6M,N,7G,H,K,L). Interestingly, the peak of granule cell death occurs at a time (P19) when the vast majority of Purkinje cells are still present. At this time, the only Purkinje cell abnormality evident infz4(−/−) mice is a small but distinct separation of the Purkinje cell bodies from the network of processes in the molecular layer (Fig. 7G,H). The organization of activated astroglial cells is also subtly altered in the granule cell layer at P19 but not at earlier times, as determined by immunostaining with anti-GFAP (Fig. 7I,J). If the cerebellar phenotype results from a lack of fz4 activity in Purkinje cells, this temporal pattern implies that granule cell death in the fz4(−/−) cerebellum is not a consequence of Purkinje cell loss but is secondary to some alteration in the properties of the Purkinje cells.
Progressive hearing loss in fz4(−/−) mice
As part of a general screen for behavioral deficits, we observed that fz4(−/−) mice show a grossly normal auditory startle response at 1–2 months of age, whereas older fz4(−/−) mice show a diminished or absent startle response. Vestibular function in fz4(−/−) mice appears grossly intact as judged by their ability to remain balanced on a slowly rotating drum. Auditory brainstem responses recorded from adult fz4(−/−) mice and from age-matched fz4(+/+) or fz4(+/−) controls confirmed the presence of elevated auditory thresholds infz4(−/−) adults. In the example shown in Figure8, the auditory brainstem response threshold is at least 40 dB higher than the average control value of ∼60 dB. Some adult fz4(−/−) mice with detectable startle responses have auditory brainstem thresholds that are elevated but still below the 100 dB ceiling of the stimulus used here. The failure to observe the earliest component of the auditory brainstem response infz4(−/−) mice localizes the hearing defect to the peripheral auditory system, although this does not rule out the possibility of a coexisting central auditory defect.
Fig. 8.

Auditory brainstem responses in adultfz4(−/−) mice reveal a peripheral hearing defect. Representative ABRs in 11-month-old fz4(+/+) andfz4(−/−) mice are shown. The click stimulus at the indicated sound pressure levels (dB SPL) triggers the line sweep and occurs at the 0 msec position. Each traceis an average of 1000 responses. The fz4(+/+) mouse shows a threshold sensitivity of ∼60 dB SPL, whereas thefz4(−/−) littermate shows no response at any of the stimulus intensities tested. This fz4(−/−) mouse also shows no startle reflex when tested with a stimulus generated by striking two metal rods.
Strong expression of the fz4–lacZ knock-in reporter is evident in inner hair cells in the organ of Corti in both fz(+/−) and fz4(−/−) mice (Fig.2D). These results suggest that the peripheral auditory defect might be referable to defects in these cells. At the light microscope level, however, there is no morphological evidence of inner or outer hair cell loss in fz4(−/−) mice. Thus, in this instance, primary sensory cell loss is not responsible for the hearing loss. Similar analyses of vestibular sensory cells show strong expression of the fz4–lacZ knock-in reporter in both fz(+/−) and fz4(−/−) mice (Fig.2E,F) but no evidence of sensory cell loss. Thus, the mechanism of progressive hearing loss is presently unknown, and the question of whether there may also be a defect in the vestibular system remains open.
Esophageal dysfunction and distension infz4(−/−) mice
The only gross internal anatomic defect present infz4(−/−) mice is progressive enlargement of the esophagus (Fig.9A,B,E,F), an abnormality that is apparent at P8 but not at P0. A concomitant defect in esophageal peristalsis and gastric sphincter function is suggested by the observations that when fed ad libitumbefore sacrifice, adult fz4(−/−) mice typically have large quantities of food within the esophagus, whereas the esophagi fromfz4(+/+) and fz4(+/−) mice have little or no food. Moreover, in fz4(−/−) mice, large numbers of bacteria are found adhering to the esophageal epithelium, which also shows extensive desquamation (Fig. 9C,D). These observations suggest that the growth retardation infz4(−/−) mice may arise from a feeding defect caused by esophageal dysfunction.
Fig. 9.

Esophageal distension and absence of skeletal muscle in the lower esophagus in adult fz4(−/−) mice.A, B, Cross sections of the esophagus from fz4(+/−) and fz4(−/−) mice stained with hematoxylin and eosin. B shows one of the more extreme examples of esophageal distension seen infz4(−/−) mice. C, D, The esophageal wall in fz4(+/−) andfz4(−/−) mice; the lumenal face is at thetop. Epon sections (1 μm) were stained with toluidine blue. The outer layer of the fz4(+/+) esophagus is composed of skeletal muscle, whereas the outer layer of thefz4(−/−) esophagus is composed of smooth muscle. The lumenal face of the fz4(−/−) esophagus shows extensive desquamation of the epithelium and large numbers of bacteria, seen as clusters of darkly stained dots in the higher magnification inset. E, F, Cross sections of the esophagus from fz4(+/−) andfz4(−/−) mice immunostained with anti-skeletal muscle myosin (red). Cell nuclei are counterstained with DAPI (blue). Only the fz4(+/−) esophagus shows a ring of skeletal muscle. G, H, Acetylcholine esterase histochemistry of lower esophagus flat mounts from fz4(+/+) and fz4(−/−) mice. Large acetylcholine esterase-positive motor endplates characteristic of skeletal muscle are seen throughout the fz4(+/+) esophagus and in the upper esophagus of fz4(−/−) mice (data not shown). In the lower esophagus of fz4(−/−) mice, a fine network of acetylcholine esterase-stained fibers is seen instead of motor endplates. I, Whole-mount X-gal staining of an isolated fz4(−/−) esophagus at the junction between the rostral two-thirds (left), which is ensheathed in skeletal muscle, and the caudal one-third (right), which is ensheathed in smooth muscle. ThelacZ reporter is strongly expressed in the esophageal skeletal muscle but not in the smooth muscle. The location of this transition zone varies among fz4(−/−) mice and in extreme cases can be found within 1–2 mm of the rostral end of the esophagus. The fz4(+/−) esophagus shows a pattern of X-gal staining along its entire length, which is less intense but otherwise resembles that seen in the skeletal muscle of thefz4(−/−) esophagus. J,K, Absence of significant differences in the lower gastrointestinal tract of fz4(+/+) andfz4(−/−) mice as seen in the indistinguishable patterns of NADPH diaphorase-stained enteric neurons in flat mounts of duodenum. Scale bars: A, B, 400 μm;C, D, G, H,J, K, 40 μm; E,F, 200 μm.
A potential mechanism for defective esophageal peristalsis and sphincter function is suggested by the observation that, amongfz4(−/−) mice, a variable length of the lower esophagus, ranging from the lower one-fourth to nearly the entire length of the esophagus, is devoid of the normal sheath of skeletal muscle. Toluidine blue staining of 1 μm plastic sections and immunostaining with anti-skeletal muscle myosin show the expected ring of skeletal muscle encircling the fz4(+/−) esophagus (Fig.9C,E). By contrast, a ring of smooth muscle encircles the fz4(−/−) esophagus (Fig.9D,F). Acetylcholine esterase staining of whole mounts of a fz4(+/+) esophagus reveals the expected pattern of large motor endplates terminating on skeletal muscle fibers along the entire length of the esophagus (Fig.9G), and a similar pattern is seen for variable distances in the upper esophagi of fz4(−/−) mice (data not shown). However, acetylcholine esterase staining of the lower esophagus offz4(−/−) mice shows no motor endplates and reveals instead a fine network of neuronal processes (Fig. 9H). In both fz4(+/−) and fz4(−/−) mice, thefz4–lacZ reporter is expressed in the skeletal muscle fibers that ensheath the upper esophagus, but the reporter is not expressed in the smooth muscle that lines the lower esophagus offz4(−/−) mice (Fig. 9I).
Gross and microscopic examination of the stomach and lower gastrointestinal tract by hematoxylin and eosin staining and microscopic examination of the associated enteric neurons by NADPH diaphorase and acetylcholine esterase histochemistry and by anti-CGRP immunostaining reveal few if any differences betweenfz4(−/−) and fz4(+/+) or fz4(+/−) mice (Fig. 9J,K).
DISCUSSION
The experiments reported here establish a role for Fz4 in maintaining the structure and function of the cerebellum, inner ear, and esophagus during postnatal life. Although the discussion that follows assumes that the involvement of Fz4 in these processes reflects its inferred role as a Wnt receptor, we note that for Frizzled-mediated tissue polarity in Drosophila there is no evidence at present for Wnt involvement, suggesting the possibility that in some contexts Frizzled proteins may function independently of Wnts.
Earlier studies of Wnt and lef-1 mutant mice revealed a role for Wnts and Wnt signaling in the proliferation of neural precursors, the patterned expression of cell adhesion molecules (Shimamura et al., 1994), and synaptogenesis. In each of these cases, Wnt activity was manifest during prenatal or immediately postnatal life. By contrast, the progressive cerebellar degeneration and auditory dysfunction seen in older fz4(−/−) mice suggest that defects in Wnt signaling may also play a role in progressive neurodegenerative processes in later life. In keeping with this general hypothesis, recent work has implicated Wnt signaling in the suppression of apoptosis in cultured fibroblasts (Chen et al., 2001) and has suggested β-catenin destabilization by mutant presenilin-1 as a mechanism that enhances apoptosis in the brain in patients with Alzheimer's disease (Zhang et al., 1998). Whether the progressive esophageal enlargement in older fz4(−/−) mice simply represents the late sequela of an early defect in esophageal skeletal muscle development remains to be determined. In the paragraphs that follow we consider each of thefz4(−/−) phenotypes in the context of previous work with genetically modified mice and related human diseases.
Esophageal enlargement and dysfunction
Under normal circumstances, the esophagus propels food by peristalsis in a rostrocaudal direction, selectively relaxes the lower esophageal sphincter to allow passage of food into the stomach, and maintains lower esophageal sphincter tone to block reflux of gastric contents (Pelot, 1995). In humans, esophageal enlargement is found in the context of achalasia, a disorder of esophageal motility caused by diminished peristalsis of the esophageal body and impaired relaxation of the lower esophageal sphincter (Ouyang and Cohen, 1995). Primary achalasia is thought to arise from defects in esophageal innervation, involving either the enteric neurons within the myenteric plexus or extrinsic innervation from the dorsal motor nucleus via the vagus nerve. A similar etiology has been described for achalasia secondary to Chagas's disease, in which loss of enteric neurons follows infection by Trypanosoma cruzi. Although the expression of thefz4–lacZ reporter in skeletal muscle within the esophagus suggests that the progressive esophageal dilatation seen infz4(−/−) mice may be of myogenic origin, the data do not rule out a neurogenic contribution.
The finding of smooth rather than skeletal muscle in the lower esophagus of fz4(−/−) mice is of interest because in normal development the mouse esophagus is initially ensheathed in smooth muscle. During late fetal and early postnatal life the esophageal smooth muscle transdifferentiates into skeletal muscle, a process that proceeds in a rostrocaudal direction until the entire esophagus is ensheathed in skeletal muscle (Patapoutian et al., 1995). It is possible that loss of Fz4 directly or indirectly causes a developmental arrest in the process of esophageal transdifferentiation. Interestingly, in humans the lower half of the esophagus is ensheathed exclusively in smooth muscle, whereas the upper half of the esophagus contains a mixture of smooth and striated muscle (Pelot, 1995). Thus, the absence of skeletal muscle per se in the lower esophagus offz4(−/−) mice is not necessarily the cause of esophageal enlargement and dysfunction.
It is of interest to compare the esophageal phenotype infz4(−/−) mice with that seen in MASH1(−/−) mice (Guillemot et al., 1993; Sang et al., 1999). NeonatalMASH1(−/−) mice do not show evidence of milk in their stomachs, indicating that either they fail to nurse or having nursed fail to ingest the milk. In the neonatal MASH1(−/−) esophagus, enteric neurons are missing almost completely, but transdifferentiation of esophageal muscle and vagal innervation appear to be undisturbed. Sang et al. (1999) have hypothesized that the failure to find milk in the stomachs of MASH1(−/−) pups reflects a failure to relax the lower esophageal sphincter secondary to the nearly complete absence of nitric oxide synthase-containing enteric neurons. The esophageal dilatation in fz4(−/−) mice could also reflect a failure of lower esophageal relaxation. Becausefz4(−/−) mice have no difficulty nursing, the severity of any esophageal sphincter defect is presumably less than that hypothesized for MASH1(−/−) mice.
Progressive hearing loss
In the human population, ∼0.1% of individuals suffer from some form of early onset hereditary deafness (Keats and Berlin, 1999). The incidence of late onset deafness, whether hereditary or acquired, increases with age, such that 30% of individuals over 65 years of age suffer from hearing impairment (Weinstein, 2000). Numerous mouse models of early onset Mendelian hearing disorders have been identified as naturally occurring mutants or have been constructed by targeted gene disruption (Holme and Steele, 1999). Most of these mouse models exhibit severe early onset defects in auditory or vestibular function, or both, and their phenotypes closely resemble the corresponding Mendelian disorders in humans.
The complex etiology of late onset hearing loss has made it more difficult to study. Environmental factors that confound genetic analysis include chronic exposure to loud sounds and ototoxic compounds. In a few cases, a genetic etiology for late onset hearing loss in humans has been identified as arising from partial defects in genes that are essential for inner ear function in the mouse. For example, heterozygosity for an 8 bp deletion in the human POU3F4/Brn-3c/Brn3.0 transcription factor gene produces late onset hearing loss (Vahava et al., 1998), whereas homozygous deletion of the orthologous gene in mice results in a complete loss of auditory and vestibular hair cells early in development (Erkman et al., 1996; Xiang et al., 1997, 1998). The expression of fz4 in auditory and vestibular hair cells together with the late onset hearing loss exhibited by fz4(−/−) mice in the absence of auditory or vestibular hair cell death suggests that Fz4 plays a role in maintaining hair cell function, but that it is not essential for the initial development and functioning of the inner ear. These observations suggest the general possibility that derangements in Wnt signaling might play a role in human hearing loss.
Cerebellar degeneration
The size, cellularity, and overall structure of thefz4(−/−) cerebellum appear normal before the third week of postnatal life. Thus, cell proliferation, migration, and arborization occur with little or no disruption. Cerebellar degeneration infz4(−/−) mice begins in the third postnatal week with extensive granule cell death, and over the ensuing months Purkinje cells also die. The time window for the initial wave of granule cell death overlaps with the peak of synaptogenesis during cerebellar development, roughly P10–P20, suggesting that granule cell death could arise from a defect in some aspect of synaptogenesis.
Among the various mouse cerebellar mutants, theleaner mutant most closely approximates thefz4(−/−) mice with respect to the time course and pattern of cell loss (Herrup and Wilczynski, 1982; Fletcher et al., 1996). In leaner mice, granule cell death begins at P10, occurs mostly over the ensuing several months, and is characterized by large numbers of TUNEL-positive granule cells. Purkinje cell death begins several weeks later and continues for at least 6 months.Leaner, and its alleles tottering androlling mouse Nagoya, are caused by mutations in the gene encoding the α1A calcium channel (Fletcher et al., 1996). Despite the widespread expression of this gene in the CNS, the cell death phenotype is largely confined to the cerebellum. This similarity in phenotype suggests that cell death in the fz4(−/−) cerebellum could involve misregulation of intracellular calcium or of a common effector that is regulated by both calcium and Wnt signaling.
On the basis of the expression of thefz4–lacZ reporter in Purkinje cells and our inability to detect reporter expression in cerebellar granule cells, granule cell death in fz4(−/−) mice may arise as a secondary consequence of Purkinje cell dysfunction. However, we note that fz4 is widely expressed in the CNS, including in brainstem nuclei that project to the cerebellum (e.g., the dorsal cochlear nuclei). Thus, granule cell loss could also arise from a defect in some fz4-dependent property of cells that project to the cerebellum.
Clear precedents exist for developmental signaling from Purkinje cells to granule cells in the context of granule cell proliferation in the external granule layer and granule cell survival in the immediate postmitotic period (Goldowitz and Hamre, 1999; Heintz and Zoghbi, 2000). In staggerer mice, a deletion in the gene encoding ROR-α, a nuclear hormone receptor expressed specifically in Purkinje cells (Hamilton et al., 1996), leads to a block in Purkinje cell development and a secondary decrease in granule cell proliferation and survival (Herrup, 1983; Sonmez and Herrup, 1984). A similar loss of granule cells secondary to Purkinje cell loss is observed in heterozygous Lurcher mice (Caddy and Biscoe, 1979) and after targeted ablation of cerebellar Purkinje cells using transgenic diphtheria toxin (Smeyne et al., 1995). InLurcher heterozygotes a point mutation in theGrid2 ionotropic glutamate receptor gene produces a constitutively active channel that causes excitotoxic Purkinje cell death beginning in the second postnatal week (Wetts and Herrup, 1982;Zuo et al., 1997). By contrast, in the Purkinje cell degeneration mouse, Purkinje cell death begins in the third postnatal week and is associated with minimal granule cell death (Mullen et al., 1976).
Signaling between Purkinje and granule cell precursors appears to be mediated, at least in part, by Sonic hedgehog (Shh). Purkinje cells produce Shh, granule cells express the Shh receptor Patched, and Shh acts to promote proliferation and block differentiation of granule cell precursors (Dahmane and Ruiz-i-Altaba, 1999; Wechsler-Reya and Scott, 1999). The late granule cell death in fz4(−/−) mice suggests that communication between Purkinje and granule cells continues beyond the proliferative phase of cerebellar development and that this communication remains essential for granule cell viability. It will be of interest to determine whether cell–cell communication is similarly implicated in any of the human cerebellar degenerations.
Diversity and redundancy in frizzled function
The complex and widespread patterns of expression exhibited by many of the mammalian frizzled genes suggest that each Frizzled receptor is likely to play multiple roles in different tissues and at different developmental times. Moreover, the significant overlap among different frizzled family members in the time and place of expression suggests that partial redundancy of Frizzled function may be the rule rather than the exception. The present work reveals a role for fz4 in three unrelated processes. As noted in the introductory remarks, earlier work on Frizzled and Frizzled-2 in Drosophila revealed their nearly complete functional redundancy as Wingless receptors in the embryo (Bhat, 1998;Kennerdell and Carthew, 1998; Bhanot et al., 1999; Chen and Struhl, 1999). Therefore it will be of great interest to determine whether the pattern of frizzled redundancy seen in Drosophilaalso holds in mammals. If so, then we can anticipate that mice harboring mutations in fz4 together with mutations in otherfrizzled genes will reveal a range of phenotypes significantly richer than those revealed by the corresponding single gene mutations.
Footnotes
This work was supported by the Howard Hughes Medical Institute (Y.W., J.N.) and National Institutes of Health Grant DC00232 (H.C., D.R.). We thank Se-jin Lee for the 129/SVJ genomic library; Andras Nagy and Janet Rossant for ES cells; Richard Behringer for the knock-out vector; Jen-Chih Hsieh for assistance with mapping of genomic clones; Phil Smallwood for help with animal husbandry; Mitra Cowan, Diane Blesh, and Chip Hawkins for blastocyst injection; Anthony Wynshaw-Boris and Amy Chen for advice on ES cell methodology; David Linden, Mark Molliver, Elizabeth O'Hearn, Randy Reed, and Amir Rattner for helpful discussions; and Tudor Badea for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Jeremy Nathans, 805 PCTB, 725 North Wolfe Street, Johns Hopkins University School of Medicine, Baltimore, MD 21205. E-mail: jnathans@jhmi.edu.
REFERENCES
- 1.Bancroft JD, Stevens A. Theory and practice of histological techniques. Churchill Livingstone; Edinburgh: 1982. [Google Scholar]
- 2.Bhanot P, Fish M, Jemison J, Nusse R, Nathans J, Cadigan KM. Frizzled and frizzled-2 function as redundant receptors for Wingless during Drosophila embryonic development. Development. 1999;126:4175–4186. doi: 10.1242/dev.126.18.4175. [DOI] [PubMed] [Google Scholar]
- 3.Bhat KM. Frizzled and frizzled2 play a partially redundant role in wingless signaling and have similar requirements to wingless in neurogenesis. Cell. 1998;95:1027–1036. doi: 10.1016/s0092-8674(00)81726-2. [DOI] [PubMed] [Google Scholar]
- 4.Borello U, Buffa V, Sonnino C, Melchionna R, Vivarelli E, Cossu G. Differential expression of the Wnt putative receptors Frizzled during mouse somitogenesis. Mech Dev. 1999;89:173–177. doi: 10.1016/s0925-4773(99)00205-1. [DOI] [PubMed] [Google Scholar]
- 5.Caddy KW, Biscoe TJ. Structural and quantitative studies on the normal C3H and Lurcher mutant mouse. Philos Trans R Soc Lond B Biol Sci. 1979;287:167–201. doi: 10.1098/rstb.1979.0055. [DOI] [PubMed] [Google Scholar]
- 6.Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- 7.Chan EF, Gat U, McNiff JM, Fuchs E. A common human skin tumour is caused by activating mutations in beta-catenin. Nat Genet. 1999;21:410–413. doi: 10.1038/7747. [DOI] [PubMed] [Google Scholar]
- 8.Chen C, Struhl G. Wingless transduction by the Frizzled and Frizzled2 proteins of Drosophila. Development. 1999;126:5441–5452. doi: 10.1242/dev.126.23.5441. [DOI] [PubMed] [Google Scholar]
- 9.Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J, Wang CY. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J Cell Biol. 2001;152:87–96. doi: 10.1083/jcb.152.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dahmane N, Ruiz-i-Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;14:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 11.Dickinson ME, Krumlauf R, McMahon AP. Evidence for a mitogenic effect of Wnt-1 in the developing central nervous system. Development. 1994;120:1453–1471. doi: 10.1242/dev.120.6.1453. [DOI] [PubMed] [Google Scholar]
- 12.Erkman L, McEvilly RJ, Luo L, Ryan AK, Hooshmand F, O'Connell SM, Keithley EM, Rapaport DH, Ryan AF, Rosenfeld MG. Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature. 1996;381:603–606. doi: 10.1038/381603a0. [DOI] [PubMed] [Google Scholar]
- 13.Fletcher CF, Lutz CM, O'Sullivan TN, Shaughnessy JD, Jr, Hawkes R, Frankel WN, Copeland NG, Jenkins NA. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- 14.Galceran J, Miyashita-Lin EM, Devaney E, Rubenstein JL, Grosschedl R. Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development. 2000;3:469–482. doi: 10.1242/dev.127.3.469. [DOI] [PubMed] [Google Scholar]
- 15.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trends Neurosci. 1999;21:375–382. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- 17.Guillemot F, Lo LC, Johnson JE, Auerbach A, Anderson DJ, Joyner AL. Mammalian achaete-scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell. 1993;75:463–476. doi: 10.1016/0092-8674(93)90381-y. [DOI] [PubMed] [Google Scholar]
- 18.Grove EA, Tole S, Limon J, Yip Y, Ragsdale CW. The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development. 1998;125:2315–2325. doi: 10.1242/dev.125.12.2315. [DOI] [PubMed] [Google Scholar]
- 19.Hall AC, Lucas FR, Salinas PC. Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell. 2000;100:525–535. doi: 10.1016/s0092-8674(00)80689-3. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton BA, Frankel WN, Kerrebrock AW, Hawkins TL, FitzHugh W, Kusumi K, Russell LB, Mueller KL, van Berkel V, Birren BW, Kruglyak L, Lander ES. Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature. 1996;379:736–739. doi: 10.1038/379736a0. [DOI] [PubMed] [Google Scholar]
- 21.He X, Saint-Jeannet J-P, Wang Y, Nathans J, Dawid I, Varmus R. A member of the Frizzled protein family mediating axis induction by Wnt-5A. Science. 1997;275:1652–1654. doi: 10.1126/science.275.5306.1652. [DOI] [PubMed] [Google Scholar]
- 22.Heintz N, Zoghbi H. Insights from mouse models into the molecular basis of neurodegeneration. Annu Rev Physiol. 2000;62:779–802. doi: 10.1146/annurev.physiol.62.1.779. [DOI] [PubMed] [Google Scholar]
- 23.Herrup K. Role of staggerer gene in determining cell number in cerebellar cortex I granule cell death is an indirect consequence of staggerer gene action. Dev Brain Res. 1983;11:267–274. doi: 10.1016/0165-3806(83)90225-0. [DOI] [PubMed] [Google Scholar]
- 24.Herrup K, Wilczynski SL. Cerebellar cell degeneration in the leaner mutant mouse. Neuroscience. 1982;7:2185–2196. doi: 10.1016/0306-4522(82)90129-4. [DOI] [PubMed] [Google Scholar]
- 25.Hsieh J-C, Rattner A, Smallwood PM, Nathans J. Biochemical characterization of Wnt-Frizzled interactions using a soluble, biologically active vertebrate Wnt protein. Proc Natl Acad Sci USA. 1999;96:3546–3551. doi: 10.1073/pnas.96.7.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holme RH, Steele KP. Genes involved in deafness. Curr Opin Genet Dev. 1999;9:309–314. doi: 10.1016/s0959-437x(99)80046-x. [DOI] [PubMed] [Google Scholar]
- 27.Ikeya M, Lee SMK, Johnson JE, McMahon AP, Takada S. Wnt signaling required for expansion of neural crest and CNS progenitors. Nature. 1997;389:966–970. doi: 10.1038/40146. [DOI] [PubMed] [Google Scholar]
- 28.Karnovsky MJ, Root L. A “direct coloring” thiocholine method for choline esterases. J Histochem Cytochem. 1964;12:219–221. doi: 10.1177/12.3.219. [DOI] [PubMed] [Google Scholar]
- 29.Keats BJB, Berlin CI. Genomics and hearing impairment. Genome Res. 1999;9:7–16. [PubMed] [Google Scholar]
- 30.Kennerdell JR, Carthew RW. Use of dsRNA-mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell. 1998;95:1017–1026. doi: 10.1016/s0092-8674(00)81725-0. [DOI] [PubMed] [Google Scholar]
- 31.Klingensmith J, Nusse R. Signaling by wingless in Drosophila. Dev Biol. 1994;166:396–414. doi: 10.1006/dbio.1994.1325. [DOI] [PubMed] [Google Scholar]
- 32.Lako M, Lindsay S, Bullen P, Wilson DI, Robson SC, Strachen T. A novel mammalian wnt gene, WNT8B, shows brain-restricted expression in early development, with sharply delimited expression boundaries in the developing forebrain. Hum Mol Genet. 1998;7:813–822. doi: 10.1093/hmg/7.5.813. [DOI] [PubMed] [Google Scholar]
- 33.Lee SMK, Tole S, Grove E, McMahon AP. A local Wnt-3a signal is required for development of the mammalian hippocampus. Development. 2000;127:457–467. doi: 10.1242/dev.127.3.457. [DOI] [PubMed] [Google Scholar]
- 34.Lucas FR, Salinas PC. WNT-7a induces axonal remodeling and increases synapsin I levels in cerebellar neurons. Dev Biol. 1997;192:31–44. doi: 10.1006/dbio.1997.8734. [DOI] [PubMed] [Google Scholar]
- 35.Luna L. Histopathologic methods and color atlas of special stains and tissue artifacts. American Histolabs; Gaithersburg, MD: 1992. [Google Scholar]
- 36.McMahon AP, Bradley A. The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell. 1990;62:1073–1085. doi: 10.1016/0092-8674(90)90385-r. [DOI] [PubMed] [Google Scholar]
- 37.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 38.Mullen RJ, Eicher EM, Sidman RL. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc Natl Acad Sci USA. 1976;73:208–212. doi: 10.1073/pnas.73.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nusse R, Varmus H. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31:99–109. doi: 10.1016/0092-8674(82)90409-3. [DOI] [PubMed] [Google Scholar]
- 40.Ouyang A, Cohen S. Motility disorders of the esophagus. In: Haubrich WS, Schaffner F, Berk JE, editors. Gastroenterology. W. B. Saunders; Philadelphia: 1995. pp. 418–435. [Google Scholar]
- 41.Parr BA, Shea MJ, Vassileva G, McMahon AP. Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development. 1993;119:247–261. doi: 10.1242/dev.119.1.247. [DOI] [PubMed] [Google Scholar]
- 42.Patapoutian A, Wold BJ, Wagner RA. Evidence for developmentally programmed transdifferentiation in mouse esophageal muscle. Science. 1995;270:1818–1821. doi: 10.1126/science.270.5243.1818. [DOI] [PubMed] [Google Scholar]
- 43.Pelot D. Anatomy, anomalies, and physiology of the esophagus: applied anatomy and anomalies. In: Haubrich WS, Schaffner F, Berk JE, editors. Gastroenterology. W. B. Saunders; Philadelphia: 1995. pp. 397–411. [Google Scholar]
- 44.Peschon JJ, Behringer RR, Brinster RL, Palmiter RD. Spermatid-specific expression of protamine 1 in transgenic mice. Proc Natl Acad Sci USA. 1987;84:5316–5319. doi: 10.1073/pnas.84.15.5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roelink H, Wagenaar E, Lopes da Silva S, Nusse R. Wnt-3, a gene activated by proviral insertion in mouse mammary tumors, is homologous to int-1/Wnt-1 and is normally expressed in mouse embryos and adult brain. Proc Natl Acad Sci USA. 1990;87:4519–4523. doi: 10.1073/pnas.87.12.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rulifson EJ, Wu CH, Nusse R. Pathway specificity by the bifunctional receptor frizzled is determined by affinity for wingless. Mol Cell. 2000;6:117–126. [PubMed] [Google Scholar]
- 47.Salinas PC, Nusse R. Regional expression of the Wnt-3 gene in the developing mouse forebrain in relationship to diencephalic neuromeres. Mech Dev. 1992;39:151–160. doi: 10.1016/0925-4773(92)90042-i. [DOI] [PubMed] [Google Scholar]
- 48.Sang Q, Ciampoli D, Greferath U, Sommer L, Young HM. Innervation of the esophagus in mice that lack MASH1. J Comp Neurol. 1999;408:1–10. doi: 10.1002/(sici)1096-9861(19990524)408:1<1::aid-cne1>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 49.Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, Sasaki Y, Imaoka S, Murata M, Shimano T, Yamaoka Y, Nakamura Y. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24:245–250. doi: 10.1038/73448. [DOI] [PubMed] [Google Scholar]
- 50.Sevier AC, Munger BL. Sevier–Munger method for neural tissues. J Neuropathol Exp Neurol. 1965;23:130–135. doi: 10.1097/00005072-196501000-00012. [DOI] [PubMed] [Google Scholar]
- 51.Shi DL, Goisset C, Boucaut JC. Expression of Xfz3, a Xenopus frizzled family member, is restricted to the early nervous system. Mech Dev. 1998;70:35–47. doi: 10.1016/s0925-4773(97)00166-4. [DOI] [PubMed] [Google Scholar]
- 52.Shimamura K, Hirano S, McMahon AP, Takeichi M. Wnt-1-dependent regulation of local E-cadherin and alpha N-catenin expression in the embryonic mouse brain. Development. 1994;8:2225–2234. doi: 10.1242/dev.120.8.2225. [DOI] [PubMed] [Google Scholar]
- 53.Smeyne RJ, Chu T, Lewin A, Bian F, Crisman S, Kunsch C, Lira SA, Oberdick J. Local control of granule cell generation by cerebellar Purkinje cells. Mol Cell Neurosci. 1995;6:230–251. doi: 10.1006/mcne.1995.1019. [DOI] [PubMed] [Google Scholar]
- 54.Sonmez E, Herrup K. Role of staggerer gene in determining cell number in cerebellar cortex: granule cell death and persistence of the external granule cell layer in young mouse chimeras. Dev Brain Res. 1984;12:271–283. doi: 10.1016/0165-3806(84)90049-x. [DOI] [PubMed] [Google Scholar]
- 55.Thomas KR, Capecchi MR. Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nature. 1990;346:847–850. doi: 10.1038/346847a0. [DOI] [PubMed] [Google Scholar]
- 56.Vahava O, Morell R, Lynch ED, Weiss S, Kagan ME, Ahituv N, Morrow JE, Lee MK, Skvorak AB, Morton CC, Blumenfeld A, Frydman M, Friedman TB, King MC, Avraham KB. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science. 1998;279:1950–1954. doi: 10.1126/science.279.5358.1950. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y, Macke JP, Abella BS, Andreasson K, Worley P, Gilbert DJ, Copeland NG, Jenkins NA, Nathans J. A large family of putative transmembrane receptors homologous to the product of the Drosophila tissue polarity gene frizzled. J Biol Chem. 1996;271:4468–4476. doi: 10.1074/jbc.271.8.4468. [DOI] [PubMed] [Google Scholar]
- 58.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 59.Weinstein BE. Geriatric audiology. Thieme; New York: 2000. [Google Scholar]
- 60.Wetts R, Herrup K. Interaction of granule, Purkinje, and inferior olivary neurons in lurcher chimeric mice: granule cell death. Brain Res. 1982;250:358–362. doi: 10.1016/0006-8993(82)90431-0. [DOI] [PubMed] [Google Scholar]
- 61.Wheeler GN, Hoppler S. Two novel Xenopus frizzled genes expressed in developing heart and brain. Mech Dev. 1999;86:203–207. doi: 10.1016/s0925-4773(99)00117-3. [DOI] [PubMed] [Google Scholar]
- 62.Xiang M, Gan L, Li D, Chen Z-Y, Zhou L, O'Malley BW, Klein W, Nathans J. Essential role of POU-domain factor Brn-3c in auditory and vestibular hair cell development. Proc Natl Acad Sci USA. 1997;94:9445–9450. doi: 10.1073/pnas.94.17.9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiang M, Gao W-Q, Hasson T, Shin JJ. Requirement for Brn-3c in maturation and survival, but not in fate determination of inner hair cells. Development. 1998;125:3935–3946. doi: 10.1242/dev.125.20.3935. [DOI] [PubMed] [Google Scholar]
- 64.Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, van de Wetering M, Clevers H, Saftig P, De Strooper B, He X, Yankner BA. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998;395:698–702. doi: 10.1038/27208. [DOI] [PubMed] [Google Scholar]
- 65.Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N. Neurodegeneration in Lurcher mice caused by mutation in delta-2 glutamate receptor gene. Nature. 1997;388:769–733. doi: 10.1038/42009. [DOI] [PubMed] [Google Scholar]