

Abstract
Injury to the CNS is often followed by a spread of damage (secondary degeneration), resulting in neuronal losses that are substantially greater than might have been predicted from the severity of the primary insult. Studies in our laboratory have shown that injured CNS neurons can benefit from active or passive immunization with CNS myelin-associated antigens. The fact that autoimmune T-cells can be both beneficial and destructive, taken together with the established phenomenon of genetic predisposition to autoimmune diseases, raises the question: will genetic predisposition to autoimmune diseases affect the outcome of traumatic insult to the CNS? Here we show that the survival rate of retinal ganglion cells in adult mice or rats after crush injury of the optic nerve or intravitreal injection of a toxic dosage of glutamate is up to twofold higher in strains that are resistant to the CNS autoimmune disease experimental autoimmune encephalomyelitis (EAE) than in susceptible strains. The difference was found to be attributed, at least in part, to a beneficial T-cell response that was spontaneously evoked after CNS insult in the resistant but not in the susceptible strains. In animals of EAE-resistant but not of EAE-susceptible strains devoid of mature T-cells (as a result of having undergone thymectomy at birth), the numbers of surviving neurons after optic nerve injury were significantly lower (by 60%) than in the corresponding normal animals. Moreover, the rate of retinal ganglion cell survival was higher when the optic nerve injury was preceded by an unrelated CNS (spinal cord) injury in the resistant strains but not in the susceptible strains. It thus seems that, in normal animals of EAE-resistant strains (but not of susceptible strains), the injury evokes an endogenous protective response that is T-cell dependent. These findings imply that a protective T-cell-dependent response and resistance to autoimmune disease are regulated by a common mechanism. The results of this study compel us to modify our understanding of autoimmunity and autoimmune diseases, as well as the role of autoimmunity in non-autoimmune CNS disorders. They also obviously have far-reaching clinical implications in terms of prognosis and individual therapy.
Keywords: protective autoimmunity, encephalitogenicity, neuroprotection, autoimmune disease, CNS, EAE
Any CNS insult (e.g., trauma, stroke, or ischemia) inevitably results in a loss of neurons that is substantially greater than might be expected from the severity of the injury. This is because the insult triggers a cascade of events, starting with degeneration of the directly injured neurons and leading to the eventual degeneration (secondary degeneration) of neurons that escaped the initial injury. This spread of damage involves compounds that are essential for the survival and function of neurons yet become toxic when their physiological concentrations are exceeded. Among the injury-related mechanisms that might underlie the posttraumatic spread of damage are biochemical and metabolic changes in oxygen and glucose use, energy state, lipid-dependent enzymes, free radicals, eicosanoids, tissue ions, biogenic amines, endogenous opioids, and excitatory amino acids (Hovda et al., 1991; Yoshino et al., 1991; Yoles et al., 1992;Faden, 1993; Liu et al., 1994). These changes cause alterations in cellular homeostasis, excitotoxicity, local production of agents harmful to nerve cells, and a loss of trophic support from targets, all of which result in secondary neuronal loss. The presumed mechanisms of secondary degeneration have served as a basis for the development and evaluation of various pharmacological interventions for the treatment of CNS injuries. The therapeutic approach of preventing or decreasing the secondary degeneration accompanying CNS trauma is termed neuroprotection (Faden and Salzman, 1992; McIntosh, 1993; Smith et al., 1995).
T-cells are important players in the adaptive arm of the immune system. They respond to antigens through interactions of their specific antigen receptor with the antigen presented by major histocompatibility complex molecules and a group of costimulatory molecules. When activated, they can kill their target cells or produce cytokines that activate or suppress the growth, or differentiation of other cells. Thus, T-cells play a central role in the protection of tissues against foreign invaders, as well as in tissue maintenance.
Immune responses in the CNS are relatively restricted, resulting in the status of the CNS as an immune-privileged site (Streilein, 1995). The unique nature of the communication between the CNS and the immune system can be observed, for example, in the dialog between the CNS and T-cells. In the CNS, under normal conditions, activated T-cells can cross the blood–brain barrier and enter the CNS parenchyma. However, only T-cells capable of reacting with a CNS antigens seem to persist there (Hickey et al., 1991). Comparative studies of the T-cell response at sites of axotomy in the CNS and the peripheral nervous system (PNS), using T-cell immunocytochemistry, revealed a significantly greater accumulation of endogenous T-cells in injured PNS axons than in injured CNS axons (Moalem et al., 1999a). Moreover, in cases of inflammation, the CNS showed a high potential for elimination of T-cells via apoptosis, whereas such elimination was less effective in the PNS and was almost absent in other tissues, such as muscle and skin (Gold et al., 1997). These findings, suggesting that the T-cell response to CNS injury is relatively limited, prompted us to examine how boosting of the T-cell response at a site of CNS injury affects the outcome of secondary degeneration.
Recent studies in our laboratory showed that systemic injection of activated T-cells directed against CNS myelin-associated self-proteins, or peptides derived from them, reduces the secondary degeneration of neurons after crush injury of the rat optic nerve (Moalem et al., 1999b; Kipnis et al., 2000). It was further shown that passive transfer of T-cells reactive to myelin proteins or active immunization with myelin-associated antigens promoted recovery from spinal cord contusion by protection of spared neurons (Hauben et al., 2000a,b). These findings prompted the suggestion that autoimmunity, usually considered detrimental, can under certain circumstances be beneficial (Schwartz et al., 1999a,b; Schwartz and Cohen, 2000). Our additional studies demonstrated that beneficial autoimmunity is not merely a reflection of immune manipulation but is a physiological response to trauma (Yoles et al., 2001).
These findings raised some key questions, which are addressed in the present study. Can a beneficial autoimmune response be manifested by all individuals? If not, is the ability to manifest a beneficial autoimmunity determined by an individual's genetic predisposition to the development of autoimmune disease? In other words, will the outcome of identical CNS insults (mechanical or biochemical) differ in individuals who differ in their susceptibility to autoimmune disease?
Susceptibility to the development of autoimmune diseases is a complicated phenomenon. Animals of a particular strain are defined as “resistant” to autoimmune disease development if they do not develop a disease after active immunization with any of the myelin-associated self-proteins and “susceptible” if they develop the disease after experimental challenge with one of the several known myelin-associated proteins that can cause the disease, such as proteolipid protein (PLP) (Bebo et al., 1998), myelin basic protein (MBP) (Kerlero de Rosbo et al., 1995), or myelin oligodendrocyte glycoprotein (MOG) (Mendel et al., 1995). Regulation of the immune response in general, and of the autoimmune response in particular, may take place on more than one physiological level. Accordingly, malfunction of control mechanisms in susceptible strains or individuals may occur at different levels in the hierarchy of autoimmune regulation. Mechanisms of control might include (1) appropriate presentation of the antigen in a complex with major histocompatibility complex (MHC) molecules, (2) the ability to evoke regulatory T-cells, and (3) neuroendocrine effects on immune cell regulation and activation.
In the present study, using injured optic nerves of rats and mice as models, we show that the extent of degeneration that inevitably follows any CNS insult of given severity is controlled by the immune system and is manifested by the ability of individuals that are resistant to autoimmune disease (and the inability of those that are susceptible) to display a beneficial T-cell-dependent response. We show that the survival rate of retinal ganglion cells (RGCs) in adult mice or rats, after crush injury of the optic nerve or intravitreal injection of glutamate at a toxic dosage, was up to twofold higher in strains that are resistant to CNS autoimmune diseases than in susceptible strains. The difference was found to be attributable to a beneficial T-cell-dependent response, which was spontaneously evoked after CNS insult in the resistant but not in the susceptible strains. These results suggest the existence of a close association between resistance to autoimmune diseases and ability to evoke protective T-cell-dependent immunity.
MATERIALS AND METHODS
Animals. Inbred female adult Lewis (Lew), Sprague Dawley (SPD), and Fisher (F344) rats and adult mice of the BALB/c/OLA, BALB/c/OLA nu/nu, C57BL/6J, C57BL/6J nu/nu, SJL/J, B10.PL, F1(SJL/J×BALB/c/OLA), C3H.SW, and C3H/HeJ strains (8–12 weeks old) were supplied by the Animal Breeding Center of The Weizmann Institute of Science under germ-free conditions. Note that not all the resistant strains used in this work have been classified as absolutely experimental autoimmune encephalomyelitis (EAE)-resistant but showed varying degree of resistance. The rats and mice were housed in a light- and temperature-controlled room and matched for age in each experiment. Animals were handled according to the regulations formulated by the Institutional Animal Care and Use Committee.
Crush injury of the optic nerve in rats and mice. Animals were deeply anesthetized by intraperitoneal injection of 2% XYL-M (xylazine, 10 mg/kg; VMD, Arendonk, Belgium) and Ketaset (ketamine, 50 mg/kg; Fort Dodge Laboratories, Fort Dodge, IA). The rat optic nerve was exposed using a binocular operating microscope, and calibrated cross-action forceps were used to inflict a crush injury on the nerve 1–2 mm from the eye. Mice were subjected to severe crush injury of the intraorbital portion of the optic nerve. The uninjured contralateral nerve was left undisturbed.
Intravitreal glutamate injection. The right eye of an anesthetized animal was punctured with a 27 gauge needle in the upper part of the sclera, and a 10 μl Hamilton syringe with a 30 gauge needle was inserted as far as the vitreal body. A total volume of 1 μl of l-glutamate (200 nmol) dissolved in saline was injected.
Measurement of secondary degeneration in rats. Secondary degeneration of optic nerve axons and their attached RGCs was measured by retrograde labeling of RGCs after postinjury application of the fluorescent lipophilic dye 4-(4-(didecylamino)styryl)-N-methylpyridinium iodide (4-Di-10-Asp) (Molecular Probes Europe BV, Leiden, The Netherlands), distal to the lesion site, 2 weeks after crush injury. Because only axons that are intact can transport the dye back to their cell bodies, application of the dye distal to the lesion site after 2 weeks ensures that only axons that survived both the primary damage and the secondary degeneration will be counted. This approach enabled us to differentiate between neurons that are still functionally intact and neurons in which the axons are injured but the cell bodies are still viable, because only those neurons whose fibers are morphologically intact can take up dye applied distally to the site of injury and transport it to their cell bodies. Using this method, the number of labeled RGCs reliably reflects the number of still-functioning neurons. Labeling and measurement were performed as follows. The right optic nerve was exposed for the second time, again without damaging the retinal blood supply. Complete axotomy was performed 1–2 mm from the distal border of the injury site, and solid crystals (0.2- to 0.4-mm-diameter) of 4-Di-10-Asp were deposited at the site of the newly formed axotomy. Five days after dye application, the rats were killed. The retina was detached from the eye, prepared as a flattened whole mount in 4% paraformaldehyde solution, and examined for labeled RGCs by fluorescence microscopy.
Retrograde labeling of retinal ganglion cells in mice. Mice were anesthetized and placed in a stereotactic device. The skull was exposed and kept dry and clean. The bregma was identified and marked. The designated point of injection was at a depth of 2 mm from the brain surface, 2.92 mm behind the bregma in the anteroposterior axis and 0.5 mm lateral to the midline. A window was drilled in the scalp above the designated coordinates in the right and left hemispheres. The neurotracer dye FluoroGold (5% solution in saline; Fluorochrome, Denver, CO) was then applied (1 μl, at a rate of 0.5 μl/min in each hemisphere) using a Hamilton syringe, and the skin over the wound was sutured.
Assessment of retinal ganglion cell survival in mice. Mice were given a lethal dose of pentobarbitone (170 mg/kg). Their eyes were enucleated, and the retinas were detached and prepared as flattened whole mounts in 4% paraformaldehyde solution. Labeled cells from four to six selected fields of identical size (0.7 mm2) were counted. The selected fields were located at approximately the same distance from the optic disk (0.3 mm) to overcome the variation in RGC density as a function of distance from the optic disk. Fields were counted under the fluorescence microscope (magnification of 800×) by observers blinded to the identity of the retinas. The average number of RGCs per field in each retina was calculated.
Spinal cord contusion in rats. Rats were anesthetized, and their spinal cords were exposed by laminectomy at the level of T8. A 10 gm rod was dropped onto the laminectomized cord from a height of 50 mm, using the NYU impactor (Basso et al., 1996a,b; Hauben et al., 2000a). Sham-operated rats were laminectomized but not contused.
RESULTS
Rate of retinal ganglion cell survival after optic nerve insult in mice correlates with resistance to autoimmune disease induction
We initially selected strains of rats or mice that differ in their susceptibility to EAE, a CNS autoimmune disease associated with CNS myelin proteins. The mice used in the following experiments were susceptible to the development of EAE after challenge with different CNS self-proteins emulsified in complete Freund's adjuvant. As discussed below, the susceptibility of each of the selected strains to EAE is similar to their susceptibility to other autoimmune diseases. We selected strains that are susceptible or resistant not only to one myelin-associated protein but to several. Optic nerves were subjected to severe crush injury in mice of five strains [B10.PL, C57BL/6J, SJL/J, BALB/c/OLA, and F1(SJL/J×BALB/c/OLA); n = 6–8 in each strain] known to differ in their resistance to EAE. Three days before injury, the RGCs of these mice had been stereotactically labeled with a neurotracer dye injected into the superior colliculus. Significantly more surviving RGCs (mean ± SE per square millimeter for all values recorded here) were found 1 week after axonal injury in BALB/c/OLA mice (a strain resistant to EAE; 1645 ± 26) than in mice of two EAE-susceptible strains: B10.PL mice (1239 ± 51) and C57BL/6J mice (1251 ± 117) (susceptible strains that develop EAE when challenged with MBP and MOG, respectively) (Fig. 1a). Similarly, the number of surviving RGCs measured 2 weeks after optic nerve injury in the mouse strains SJL/J and F1(SJL/J×BALB/c/OLA) (susceptible strains that develop EAE when challenged with PLP and MBP, respectively) were 439 ± 46 and 447 ± 25 and were significantly lower than those obtained in EAE-resistant BALB/c/OLA mice (638 ± 31) (Fig. 1b). There were no differences in the total numbers of RGCs in the retinas of undamaged nerves of any of the tested strains (3500 ± 100). The above results suggested that a genetic predisposition to autoimmune disease, regardless of the identity of the myelin-associated antigen that triggers it, is correlated with a relatively more severe outcome of CNS injury. To verify this tentative conclusion, we used congenic mice (C3H.SW and C3H/HeJ) that differ only in their H-2 haplotype and are known to respond differently to challenge by MOG peptides (only the C3H.SW mice develop EAE) (Duong et al., 1994; Mendel et al., 1995). Two weeks after optic nerve injury, significantly more surviving RGCs were found in the C3H/HeJ mice (738 ± 48) than in the C3H.SW mice (545 ± 23) (Fig. 1c).
Fig. 1.
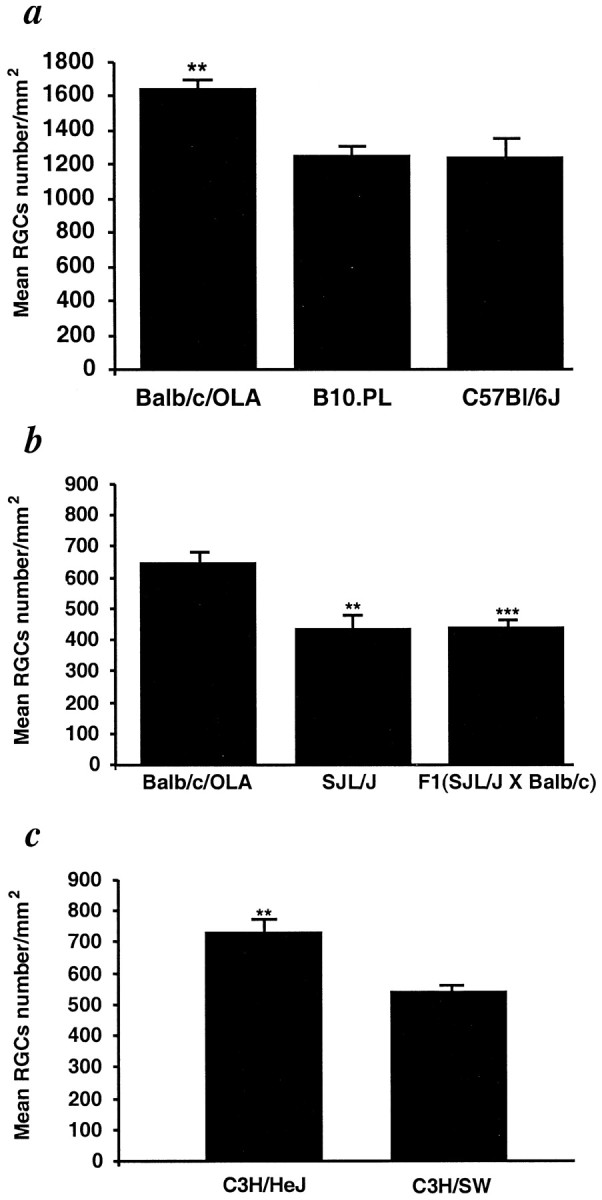
Survival rate of retinal ganglion cells after optic nerve injury is associated with resistance to the autoimmune disease EAE. The RGCs of adult C57BL/6J, B10.PL, BALB/c/OLA (a), BALB/c/OLA, SJL/J, F1(SJL/J×BALB/c/OLA) (b), and C3H.SW and C3H/HeJ (c) mice were retrogradely labeled with the neurotracer dye FluoroGold injected stereotactically into the superior colliculus. Three days later, the mice were subjected to severe crush injury of the intraorbital portion of the optic nerve. One (a) or 2 (b, c) weeks after optic nerve crush injury, the retina was detached from the eye, prepared as a flattened whole mount, and examined for labeled RGCs by fluorescence microscopy. One week after the injury, survival rates were significantly lower in B10.PL and C57BL/6J mice than in resistant BALB/c/OLA mice (p < 0.01) (a). Similar results were obtained in SJL/J (p < 0.01) and F1(SJL/J×BALB/c/OLA) mice (p < 0.001), in which RGC survival rates were significantly lower than in the resistant BALB/c/OLA mice (b). RGC survival rates were significantly higher in C3H/HeJ mice (EAE-resistant) than in congenic (EAE-susceptible) C3H.SW mice (p < 0.001) (c). The average numbers of RGCs on the uninjured side were similar (∼3500 RGCs per square millimeter) in all mouse strains. **p < 0.01; ***p < 0.001; Student'st test.)
Secondary degeneration in injured rat optic nerve correlates with susceptibility to experimental autoimmune encephalomyelitis
In the mouse model used for the above experiments, the severe crush injury damages all of the axons at the same time. Cell body death is then inevitable, and differences in the physiological response to trauma in this model are therefore assessed in terms of the rate of cell body death. This explains why the differences among the groups, although significant, were small. To examine the link between secondary degeneration (degeneration of initially spared neurons because of the hostility of their extracellular environment) and susceptibility to autoimmune diseases, we used the model of partial crush injury of the rat optic nerve, in which some of the neurons are intentionally left undamaged and, in the absence of any intervention, will undergo secondary degeneration (Yoles and Schwartz, 1998). The injury was inflicted in rats of three strains: F344, SPD (both resistant to EAE), and Lew (susceptible to EAE after MBP challenge). Lew and F344 rats possess the same alleles of the rat MHC RT1.B locus, except for a single allele in the nonclassical region corresponding to the mouse Qa-Tla (Kunz et al., 1989). These two strains differ in their neuroendocrine balance. However, it was shown recently that their differences with respect to susceptibility to autoimmune disease development, after active immunization with CNS self-proteins, can be attributed to differences in the immune regulatory network that is upregulated to a much greater extent in F344 rats than in Lew rats (Sun et al., 1999). Interstrain differences in hypothalamus pituitary axis, which might also contribute to the differences in the outcome of CNS injury, have not yet been investigated in these two strains. The outcome of the injury, in terms of the survival of neurons that had escaped the primary injury, was significantly better in the two EAE-resistant strains (F344, 43 ± 3; SPD, 60 ± 4) than in the susceptible strain (Lew, 28 ± 2 or 32 ± 3; tested in two different experiments; n = 6 in each) (Fig.2a,b).
Fig. 2.

Survival of neurons after partial crush injury of the optic nerve in Lew, SPD, and F344 rats. The optic nerves of adult Lew, SPD, and F344 rats were subjected to a partial crush injury 1–2 mm from the eye, using calibrated cross-action forceps. Two weeks later, the optic nerves were exposed for the second time, and the fluorescent dye 4-Di-10-Asp was applied distally to the injury site. Five days after dye application, the retinas were detached from the eyes and prepared for fluorescence microscopy as described in Figure 1. The amount of endogenous neuroprotection was significantly greater in F344 and SPD rats (p < 0.01 andp < 0.001, respectively), known to be resistant to EAE induction. **p < 0.01; ***p < 0.001; Student's t test.
Our recent studies in Lew and SPD male rats after contusive injury of the spinal cord, a model that allows repeated noninvasive assessment of recovery by examination of functional activity, showed that the response to trauma is profoundly affected by strain differences. A 3 month follow-up of the spontaneous recovery from identical contusions at the level of T7 revealed that, in the Lew rats, known to be susceptible to autoimmune disease development, there was almost complete paralysis of the hind limbs (measured by mobility in an open field) (Hauben et al., 2000), whereas significant walking ability was observed in the EAE-resistant SPD rats (U. Hauben, E. Agranov, and M. Schwartz, unpublished observations).
EAE-resistant rats show better postinjury neuronal survival than susceptible rats because of their beneficial T-cell-dependent response
The better postinjury neuronal survival in the resistant animals, if it indeed reflects a T-cell-dependent response, could be the result of either an injury-evoked beneficial T-cell-dependent response that occurs in the resistant but not in the susceptible strains or an injury-evoked destructive immunity that occurs in the susceptible but not in the resistant strains. To examine these possibilities, we measured secondary degeneration after optic nerve injury in adult EAE-susceptible and EAE-resistant rats that were devoid of mature T-cells as a result of having undergone thymectomy at birth. When compared with their respective nonthymectomized controls, the number of surviving neurons in the resistant (SPD) rats was significantly lower (by 60 ± 7%), whereas in the susceptible (Lew) rats it was not significantly affected (Fig.3a). These findings imply that, in normal SPD rats, the spontaneous T-cell-dependent response to injury is beneficial for CNS recovery and thus, in the absence of such a response, i.e., after thymectomy, recovery is worse. In contrast, in the susceptible Lew rats, this beneficial T-cell-mediated response is apparently missing and might even be replaced by a response that is deleterious to nerve survival; thus, in the absence of T-cells, i.e., after thymectomy, recovery is either unaffected or slightly improved (Fig. 3a). This interpretation was further substantiated by comparing the survival rate of RGCs in Lew rats with that in SPD rats in response to optic nerve crush sustained 2 weeks after contusive injury to the spinal cord. This experiment was designed on the basis of a previous study in our laboratory showing that, in SPD (resistant) rats, the number of surviving RGCs after optic nerve injury inflicted 7–17 d after spinal cord contusion is significantly higher than after optic nerve injury in sham-operated rats (Yoles et al., 2001). It was further shown that this conditioning effect is mediated by T-cells. The ratios (expressed as a percentage) of the RGC survival rate in spinally contused rats to that in rats without previous spinal cord injury were compared between the two strains. The comparison showed that, in contrast to the finding in the resistant SPD rats in which the number of surviving RGCs after optic nerve injury in the preconditioned group was significantly better (by 80 ± 15%) than in controls, in the susceptible Lew rats the effect of previous injury of the spinal cord on recovery from optic nerve injury was slightly worse, but the difference was not significant (Fig. 3b).
Fig. 3.
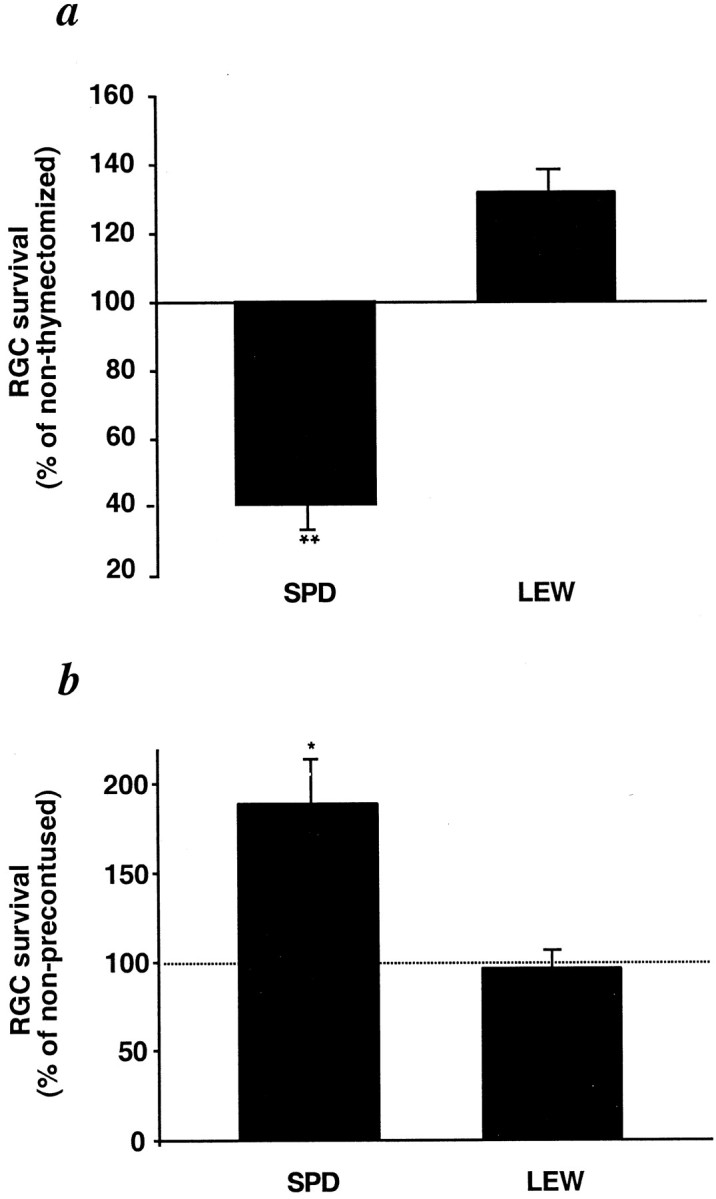
T-cell dependence of physiological differences in the response to optic nerve crush injury in EAE-resistant and EAE-susceptible rats. a, Adult Lew and SPD rats, which had undergone thymectomy 1 d after birth, and normal adult control rats were subjected to optic nerve crush, and their RGCs were counted 2 weeks later, as described in Figure 2. Significantly fewer labeled RGCs were seen in the thymectomized SPD rats than in the normal SPD rats (p < 0.01). In Lew rats, the opposite was seen, but the effect was not significant (p> 0.1). b, Lew and SPD rats, thymectomized 1 d after birth, were subjected to contusive injury of the spinal cord at the level of T7 or T9 using the NYU impactor. Sham-operated rats were laminectomized but not contused. After 2 weeks, both sham-operated and contused rats were subjected to optic nerve crush and counting of surviving RGCs, as described in Figure 2. Significantly more labeled RGCs were seen in the preinjured SPD rats than in the sham-operated SPD controls (p < 0.05). In Lew rats, differences in the number of labeled RGCs between the preinjured and the sham-operated rats after optic nerve injury were not significant (p > 0.1). *p < 0.05; **p < 0.01; Student's t test.
Survival of neurons after injection of a toxic dose of glutamate correlates with resistance to autoimmune disease induction
The finding that the neuronal survival rate in EAE-resistant mice was higher than in EAE-susceptible mice after mechanical injury prompted us to examine whether the same applies after biochemical insult produced by direct exposure of the neuronal cell bodies to glutamate, the main mediator of neurotoxicity in CNS trauma or stroke (Juurlink et al., 1998). We found that RGC survival, measured by retrogradely labeling the RGCs and then counting the labeled cells 1 week after intraocular glutamate injection, was significantly higher in the resistant BALB/c/OLA mice (2170 ± 48) than in susceptible C57BL/6J mice (1408 ± 129; n = 12 in each group) (Fig. 4a). The association between neuronal survival after glutamate toxicity and susceptibility to autoimmune disease was further strengthened by our findings in the congenic mice C3H/HeJ and C3H.SW, which differ only in their H-2 haplotype. The number of RGCs that survived an intravitreal injection of a toxic dose of glutamate was higher in the resistant C3H/HeJ mice (1090 ± 74) than in the susceptible C3H.SW mice (807 ± 65;n = 6 in each group) (Fig. 4b). Interestingly, intraocular injection of glutamate into rats yielded a similar correlation between RGC loss and EAE susceptibility (F344 rats, 1627 ± 40; Lew rats, 1220 ± 70) (Fig. 4c) to that seen in the mice (Fig. 4a,b).
Fig. 4.
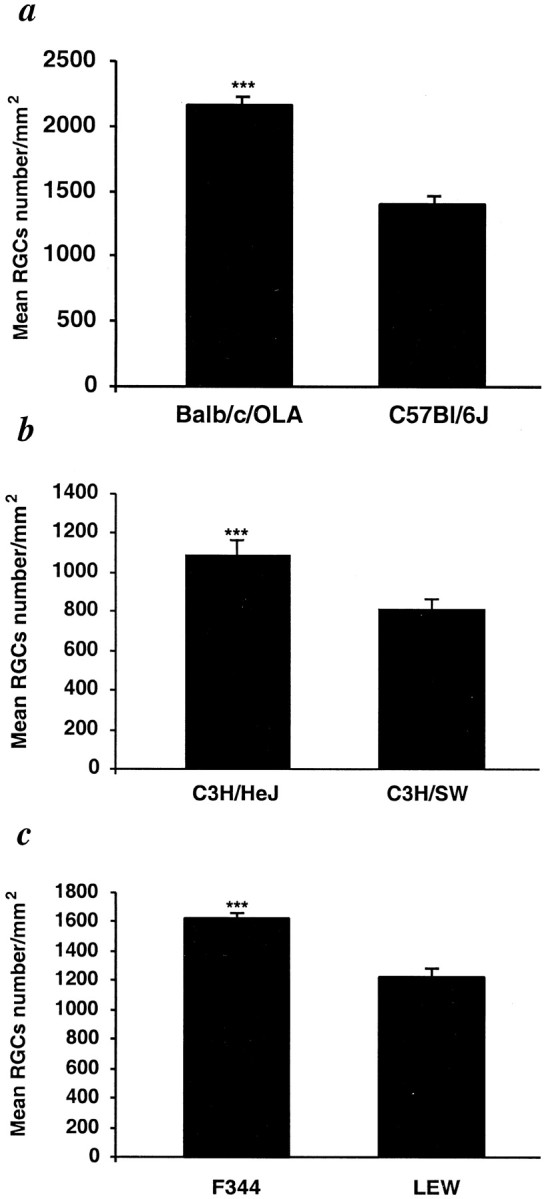
Glutamate toxicity is correlated with susceptibility to autoimmune disease. Glutamate (200 nmol) was injected intravitreally into EAE-susceptible and EAE-resistant strains of mice (C57BL/6J and BALB/c/OLA, respectively), the congenic mice C3H.SW and C3H/HeJ (differing in H-2 haplotype), and rats (Lew and F344). One week after glutamate injection, more surviving RGCs were found in mice of EAE-resistant strains (C3H/HeJ and BALB/c/OLA) than in susceptible strains (C3H.SW and C57BL/6J) (p < 0.001 in both a and b). As with the mice, significantly more RGCs were seen in the retinas of EAE-resistant rats (F344) than in the retinas of EAE-susceptible rats (Lew) (c, p < 0.001). No differences in the numbers of RGCs between susceptible and resistant strains were observed on the uninjured side in either rats or mice. ***p< 0.001; Student's t test.
These results demonstrated that recovery from CNS trauma, regardless of the nature of the insult, is better in rats and mice that are endowed with a higher resistance to induction of CNS-associated autoimmune disease. The higher rate of RGC survival observed in EAE-resistant rats than in EAE-susceptible rats after glutamate insult may result, as shown in the case of axonal injury (Fig. 3), from an insult-induced T-cell-dependent beneficial response that is present in the former and absent in the latter. To verify this possibility, we compared the effect of glutamate injection in normal adult SPD rats with that in adult SPD rats lacking mature T-cells as a result of having undergone thymectomy at birth. The number of surviving RGCs was significantly lower in the rats devoid of T-cells (1221 ± 66) than in the normal rats (1627 ± 39) (Fig.5a). Additional confirmation came from experiments using transgenic mice with defective development of the cortical epithelium of the thymus (nude mice), leading to a deficiency of mature T-cells. We compared the effects of toxic concentrations of glutamate in the nude mice and their matched wild-type controls from two strains (Fig. 5b). In the genetically resistant BALB/c/OLA strain, the number of surviving RGCs after exposure to glutamate toxicity was significantly lower in the nude mice than in the normal mice [1188 ± 51 (n= 6) compared with 2170 ± 48 (n = 13)]. In contrast, there were no significant differences in RGC survival between nude and normal mice of the EAE-susceptible C57BL/6J strain [1408 ± 129 (n = 17) compared with 1259 ± 45 (n = 12)].
Fig. 5.

Glutamate evokes a self-protective autoimmune response in mice and rats genetically resistant to EAE induction. Normal adult SPD rats and adult SPD rats that had undergone thymectomy at birth were injected intravitreally with glutamate (200 nmol). One week later, the numbers of surviving RGCs were determined by retrograde labeling with 4-Di-10-Asp. Significantly fewer surviving RGCs were seen in the thymectomized rats than in the normal controls (a, p < 0.01). After glutamate toxicity, nude mice of a genetically susceptible strain (C57BL/6J) showed no differences in RGC survival rates compared with their wild-type counterparts, whereas RGC survival rates in nude mice of a genetically resistant strain were significantly decreased (p < 0.001) (b). *p < 0.05; ***p < 0.001; Student'st test.
DISCUSSION
The results of this study demonstrate a link between the rate of neuronal survival after CNS insult and the resistance to autoimmune disease development. This link was found to be mediated by an injury-induced beneficial T-cell response that was present in the genetically resistant animals but not in the susceptible ones (defined in terms of the development of EAE after challenge with any myelin-associated antigen). The beneficial T-cell-dependent response evoked by the insult was found to be independent of the type of the insult (mechanical or biochemical) or the location (axon or cell body) of the primary insult.
Different strains are known to respond differently to myelin-associated antigens and to show different susceptibilities to EAE (Arnon, 1981). The source of the differences has been located in both the MHC class II antigens and elsewhere. Congenic mice (C3H.SW, H-2b, and C3H/HeJ, H-2k), differing only in their H-2 haplotype, were found to differ in their susceptibility to EAE (Duong et al., 1994). Mice that share the same H-2 haplotype (SJL/J and B10.S) were also found to have different susceptibilities to EAE, and the source of this difference is located outside of the MHC class II antigens (Livingstone et al., 1995; Kuchroo and Weiner, 1998). Similarly, Lew and F344 rats were found to differ in their susceptibilities to autoimmune diseases not associated with H-2. Although these two strains differ in their neuroendocrine interactions, difference in regulatory T-cells could explain the difference in susceptibility (Sun et al., 1999).
All strains examined in this study exhibited the same relationship between outcome of injury and resistance to autoimmune disease, regardless of whether the insult was mechanical (primarily affecting the axons and involving myelin-associated antigens) or biochemical (directly affecting the cell bodies and presumably involving antigens other than those associated with myelin). Moreover, the outcome of both mechanical and biochemical CNS injury was worse in transgenic nude mice or animals devoid of T-cells because of neonatal thymectomy than in the corresponding wild type. In contrast, among susceptible strains, the outcome of the injury in animals without T-cells did not differ from that in their wild-type counterparts. Because T-cell-mediated mechanisms disappeared in thymectomized and nude animals, it is suggestive that the differences in the outcome of CNS injury in strains of different susceptibilities are T-cell dependent. It further suggests that a beneficial T-cell-dependent response is present in resistant but not in susceptible strains.
In animal studies of traumatic CNS injury worldwide, no effort was made to find a link between neuronal recovery and the resistance or susceptibility of a particular strain to autoimmune disease. Studies have attempted to link the outcome of neuronal trauma to genetic (Friedman et al., 1999; Mattson et al., 2000) or anatomic (Yang et al., 1997) differences between strains, but no attention was directed to the possibility that the response to trauma is controlled by the immune system. Indeed, the traditional view was that if CNS insult evokes any immune response at all, it could only be detrimental for neuronal survival. Our results strongly suggest that animals of different strains differ in the amount of beneficial (neuroprotective) T-cell-dependent autoimmunity that they develop in response to CNS insult. Thus, a genetic background determining susceptibility to autoimmune CNS diseases will predispose the individual to a severe outcome from CNS insult.
One of the major factors determining whether or not an autoimmune disease will develop in a particular individual appears to be the presence or absence, not of autoreactive T-cells (which are found in patients with the autoimmune disease multiple sclerosis, as well as in healthy individuals), but of mechanisms that regulate the proper functioning of the autoreactive T-cells (for review, see Shevach, 2000). It is not clear, however, from the present study whether the same T-cells, depending on the regulatory conditions and the tissue context, can either beneficially affect the neurons or cause myelin damage. It is possible that the autoimmune T-cell response evoked by a traumatic insult is common to all individuals, and the nature of the subsequent T-cell regulatory response, determining whether the effect is beneficial or harmful, differs in resistant and susceptible individuals. Preliminary data from our laboratory (J. Kipnis, I. Shaked, T. Mizrachi, and M. Schwartz, unpublished observations) (Schwartz and Kipnis, 2001) show that protective autoimmunity is a multicellular response exhibited by a network of autoimmune T-cells, including the classical CD4+ cells (considered to be the T-cells that mediate autoimmune disease development) and regulatory T-cells (evoked in response to the CD4+ cells). The presence of the CD4+ T-cells without the regulatory T-cells is not neuroprotective and may even be destructive to myelin. Such a regulatory T-cell response may be related to the T-cell response described recently as “the third function of the thymus” (Seddon and Mason, 2000). In addition, it may be affected by neuroendocrine balance, as appeared to be the case in F344 rats. Indeed, it was shown recently that the resistance of F344 rats to autoimmune disease development, after active immunization with CNS self-proteins, can be attributed to the presence of an immune regulatory network (Sun et al., 1999). It seems that such T-cell-dependent regulatory mechanisms exist in resistant individuals or strains but are lacking or malfunctioning in susceptible ones. This further supports our early proposal that the spontaneous autoimmune response is beneficial and becomes destructive (causing an autoimmune disease) only when the regulatory mechanism breaks down (Cohen and Schwartz, 1999; Schwartz et al., 1999a; Schwartz and Cohen, 2000). The existence of a regulatory mechanism that inhibits the development of an autoimmune disease, although not of an autoimmune response, may explain the high incidence of autoimmune T-cells often found in healthy individuals. It may also explain the observed occurrence of multiple autoimmune diseases in a single, probably susceptible, individual when the resistance breaks down (Bellone et al., 1991; Oshima et al., 1998; Zhu et al., 1998; Ligier and Sternberg, 1999; Yocum, 1999; Goebels et al., 2000). Such a mechanism can also explain why non-autoimmune CNS degenerative diseases progress differently in different individuals.
Our studies may suggest that an upstream mechanism(s) regulates the response to stress signals associated with pathogen-free tissue damage (i.e., to self-antigens) in such a way as to derive the beneficial T-cell-dependent response and not an autoimmune disease. Several hypotheses have been proposed for controlling the response to self (Burnet, 1959; Jerne, 1974; Cohen, 1992; Matzinger, 1994). Our results point to an immune response to trauma that operates not by distinguishing between self and non-self antigens but rather according to how the response to self is regulated and what characterizes the phenotype of the beneficial anti-self response.
The results of this study strongly suggest that an individual's genetically determined predisposition to autoimmune diseases seems to be crucial not only for prognosis of damage after CNS injury but also for planning therapy, because what is applicable to resistant individuals or strains might not be helpful in those that are susceptible. An understanding of the genetic basis of the mechanisms that control the beneficial T-cell-dependent response evoked by CNS trauma may lead to the discovery of ways to boost the immunological conditions needed for recovery from CNS acute insults, for arresting the progression of chronic degenerative CNS disorders, and for avoidance of autoimmune disease.
Footnotes
This work was supported by Proneuron Ltd. (Ness-Ziona, Israel) and in part by grants from the Glaucoma Research Foundation and the Alan Brown Foundation for Spinal Cord Injury awarded to M.S. We thank S. Smith for editing this manuscript and A. Shapira for animal maintenance. M.S. holds the Maurice and Ilse Katz Professorial Chair in Neuroimmunology.
J.K. and E.Y. contributed equally to this work.
Correspondence should be addressed to Dr. Michal Schwartz, Department of Neurobiology, The Weizmann Institute of Science, 76100 Rehovot, Israel. E-mail: michal.schwartz@weizmann.ac.il.
REFERENCES
- 1.Arnon R. Experimental allergic encephalomyelitis—susceptibility and suppression. Immunol Rev. 1981;55:5–30. doi: 10.1111/j.1600-065x.1981.tb00337.x. [DOI] [PubMed] [Google Scholar]
- 2.Basso DM, Beattie MS, Bresnahan JC. Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Exp Neurol. 1996a;139:244–256. doi: 10.1006/exnr.1996.0098. [DOI] [PubMed] [Google Scholar]
- 3.Basso DM, Beattie MS, Bresnahan JC, Anderson DK, Faden AI, Gruner JA, Holford TR, Hsu CY, Noble LJ, Nockels R, Perot PL, Salzman SK, Young W. MASCIS evaluation of open field locomotor scores: effects of experience and teamwork on reliability. Multicenter Animal Spinal Cord Injury Study. J Neurotrauma. 1996b;13:343–359. doi: 10.1089/neu.1996.13.343. [DOI] [PubMed] [Google Scholar]
- 4.Bebo BF, Zelinka-Vincent E, Adamus G, Amundson D, Vandenbark AA, Offner H. Gonadal hormones influence the immune response to PLP 139–151 and the clinical course of relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. 1998;84:122–130. doi: 10.1016/s0165-5728(97)00214-2. [DOI] [PubMed] [Google Scholar]
- 5.Bellone M, Ostlie N, Lei S, Conti-Tronconi BM. Experimental myasthenia gravis in congenic mice. Sequence mapping and H-2 restriction of T helper epitopes on the alpha subunits of Torpedo californica and murine acetylcholine receptors. Eur J Immunol. 1991;21:2303–2310. doi: 10.1002/eji.1830211003. [DOI] [PubMed] [Google Scholar]
- 6.Burnet FM. The clonal selection theory of acquired immunity. Cambridge UP; Cambridge, UK: 1959. [Google Scholar]
- 7.Cohen IR. The cognitive paradigm and the immunological homunculus. Immunol Today. 1992;13:490–494. doi: 10.1016/0167-5699(92)90024-2. [DOI] [PubMed] [Google Scholar]
- 8.Cohen IR, Schwartz M. Autoimmune maintenance and neuroprotection of the central nervous system. J Neuroimmunol. 1999;100:111–114. doi: 10.1016/s0165-5728(99)00190-3. [DOI] [PubMed] [Google Scholar]
- 9.Duong TT, Finkelman FD, Singh B, Strejan GH. Effect of anti-interferon-gamma monoclonal antibody treatment on the development of experimental allergic encephalomyelitis in resistant mouse strains. J Neuroimmunol. 1994;53:101–107. doi: 10.1016/0165-5728(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 10.Faden AI. Experimental neurobiology of central nervous system trauma. Crit Rev Neurobiol. 1993;7:175–186. [PubMed] [Google Scholar]
- 11.Faden AI, Salzman S. Pharmacological strategies in CNS trauma. Trends Pharmacol Sci. 1992;13:29–35. doi: 10.1016/0165-6147(92)90013-v. [DOI] [PubMed] [Google Scholar]
- 12.Friedman G, Froom P, Sazbon L, Grinblatt I, Shochina M, Tsenter J, Babaey S, Yehuda B, Groswasser Z. Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology. 1999;52:244–248. doi: 10.1212/wnl.52.2.244. [DOI] [PubMed] [Google Scholar]
- 13.Goebels N, Hofstetter H, Schmidt S, Brunner C, Wekerle H, Hohlfeld R. Repertoire dynamics of autoreactive T cells in multiple sclerosis patients and healthy subjects: epitope spreading versus clonal persistence. Brain. 2000;123:508–518. doi: 10.1093/brain/123.3.508. [DOI] [PubMed] [Google Scholar]
- 14.Gold R, Hartung HP, Lassmann H. T-cell apoptosis in autoimmune diseases: termination of inflammation in the nervous system and other sites with specialized immune-defense mechanisms. Trends Neurosci. 1997;20:399–404. doi: 10.1016/S0166-2236(97)01079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hauben E, Butovsky O, Nevo U, Yoles E, Moalem G, Agranov G, Mor F, Leibowitz-Amit R, Pevsner E, Akselrod S, Neeman M, Cohen IR, Schwartz M. Passive or active immunization with myelin basic protein promotes recovery from spinal cord contusion. J Neurosci. 2000a;20:6421–6430. doi: 10.1523/JNEUROSCI.20-17-06421.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hauben E, Nevo U, Yoles E, Moalem G, Agranov E, Mor F, Akselrod S, Neeman M, Cohen IR, Schwartz M. Autoimmune T cells as potential neuroprotective therapy for spinal cord injury. Lancet. 2000b;355:286–287. doi: 10.1016/s0140-6736(99)05140-5. [DOI] [PubMed] [Google Scholar]
- 17.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 18.Hovda DA, Yoshino A, Kawamata T, Katayama Y, Becker DP. Diffuse prolonged depression of cerebral oxidative metabolism following concussive brain injury in the rat: a cytochrome oxidase histochemistry study. Brain Res. 1991;567:1–10. doi: 10.1016/0006-8993(91)91429-5. [DOI] [PubMed] [Google Scholar]
- 19.Jerne NK. Towards a network theory of the immune system. Ann Immunol (Paris) 1974;125C:373–389. [PubMed] [Google Scholar]
- 20.Juurlink BH, Paterson PG. Review of oxidative stress in brain and spinal cord injury: suggestions for pharmacological and nutritional management strategies. J Spinal Cord Med. 1998;21:309–334. doi: 10.1080/10790268.1998.11719540. [DOI] [PubMed] [Google Scholar]
- 21.Kerlero de Rosbo N, Mendel I, Ben-Nun A. Chronic relapsing experimental autoimmune encephalomyelitis with a delayed onset and an atypical clinical course, induced in PL/J mice by myelin oligodendrocyte glycoprotein (MOG)-derived peptide: preliminary analysis of MOG T cell epitopes. Eur J Immunol. 1995;25:985–993. doi: 10.1002/eji.1830250419. [DOI] [PubMed] [Google Scholar]
- 22.Kipnis J, Yoles E, Porat Z, Cohen A, Mor F, Sela M, Cohen IR, Schwartz M. T cell immunity to copolymer 1 confers neuroprotection on the damaged optic nerve: therapy for optic neuropathies. Proc Natl Acad Sci USA. 2000;97:7446–7451. doi: 10.1073/pnas.97.13.7446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuchroo VK, Weiner HL. Antigen-driven regulation of experimental autoimmune encephalomyelitis. Res Immunol 149 1998. 759 771; discussion 842–843, 855–860. [DOI] [PubMed] [Google Scholar]
- 24.Kunz HW, Cortese Hassett AL, Inomata T, Misra DN, Gill TJD. The RT1.G locus in the rat encodes a Qa/TL-like antigen. Immunogenetics. 1989;30:181–187. doi: 10.1007/BF02421204. [DOI] [PubMed] [Google Scholar]
- 25.Ligier S, Sternberg EM. Neuroendocrine host factors and inflammatory disease susceptibility. Environ Health Perspect [Suppl 107] 1999;5:701–707. doi: 10.1289/ehp.99107s5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu D, Yang R, Yan X, McAdoo DJ. Hydroxyl radicals generated in vivo kill neurons in the rat spinal cord: electrophysiological, histological, and neurochemical results. J Neurochem. 1994;62:37–44. doi: 10.1046/j.1471-4159.1994.62010037.x. [DOI] [PubMed] [Google Scholar]
- 27.Livingstone KD, Sudweeks JD, Blankenhorn EP, Hickey WF, Teuscher C. Susceptibility to actively-induced murine experimental allergic encephalomyelitis is not linked to genes of the T cell receptor or CD3 complexes. Autoimmunity. 1995;21:195–201. doi: 10.3109/08916939509008016. [DOI] [PubMed] [Google Scholar]
- 28.Mattson MP, Zhu H, Yu J, Kindy MS. Presenilin-1 mutation increases neuronal vulnerability to focal ischemia in vivo and to hypoxia and glucose deprivation in cell culture: involvement of perturbed calcium homeostasis. J Neurosci. 2000;20:1358–1364. doi: 10.1523/JNEUROSCI.20-04-01358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 30.McIntosh TK. Novel pharmacologic therapies in the treatment of experimental traumatic brain injury: a review. J Neurotrauma. 1993;10:215–261. doi: 10.1089/neu.1993.10.215. [DOI] [PubMed] [Google Scholar]
- 31.Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. 1995;25:1951–1959. doi: 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- 32.Moalem G, Monsonego A, Shani Y, Cohen IR, Schwartz M. Differential T cell response in central and peripheral nerve injury: connection with immune privilege. FASEB J. 1999a;13:1207–1217. doi: 10.1096/fasebj.13.10.1207. [DOI] [PubMed] [Google Scholar]
- 33.Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med. 1999b;5:49–55. doi: 10.1038/4734. [DOI] [PubMed] [Google Scholar]
- 34.Oshima M, Yokoi T, Deitiker P, Atassi MZ. T cell responses in EAMG-susceptible and non-susceptible mouse strains after immunization with overlapping peptides encompassing the extracellular part of Torpedo californica acetylcholine receptor alpha chain. Implication to role in myasthenia gravis of autoimmune T-cell responses against receptor degradation products. Autoimmunity. 1998;27:79–90. doi: 10.3109/08916939809008038. [DOI] [PubMed] [Google Scholar]
- 35.Schwartz M, Cohen IR. Autoimmunity can benefit self-maintenance. Immunol Today. 2000;21:265–268. doi: 10.1016/s0167-5699(00)01633-9. [DOI] [PubMed] [Google Scholar]
- 36.Schwartz M, Kipnis J. Protective autoimmunity: regulation and prospects for vaccination after brain and spinal cord injuries. Trends Mol Med. 2001;7:252–258. doi: 10.1016/s1471-4914(01)01993-1. [DOI] [PubMed] [Google Scholar]
- 37.Schwartz M, Moalem G, Leibowitz-Amit R, Cohen IR. Innate and adaptive immune responses can be beneficial for CNS repair. Trends Neurosci. 1999a;22:295–299. doi: 10.1016/s0166-2236(99)01405-8. [DOI] [PubMed] [Google Scholar]
- 38.Schwartz M, Yoles E, Levin LA. “Axogenic” and “somagenic” neurodegenerative diseases: definitions and therapeutic implications. Mol Med Today. 1999b;5:470–473. doi: 10.1016/s1357-4310(99)01592-0. [DOI] [PubMed] [Google Scholar]
- 39.Seddon B, Mason D. The third function of the thymus. Immunol Today. 2000;21:95–99. doi: 10.1016/s0167-5699(99)01559-5. [DOI] [PubMed] [Google Scholar]
- 40.Shevach EM. Regulatory T cells in autoimmunity. Annu Rev Immunol. 2000;18:423–449. doi: 10.1146/annurev.immunol.18.1.423. [DOI] [PubMed] [Google Scholar]
- 41.Smith DH, Casey K, McIntosh TK. Pharmacologic therapy for traumatic brain injury: experimental approaches. New Horiz. 1995;3:562–572. [PubMed] [Google Scholar]
- 42.Streilein JW. Unraveling immune privilege. Science. 1995;270:1158–1159. doi: 10.1126/science.270.5239.1158. [DOI] [PubMed] [Google Scholar]
- 43.Sun D, Whitaker JN, Wilson DB. Regulatory T cells in experimental allergic encephalomyelitis. III. Comparison of disease resistance in Lewis and Fischer 344 rats. Eur J Immunol. 1999;29:1101–1106. doi: 10.1002/(SICI)1521-4141(199904)29:04<1101::AID-IMMU1101>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 44.Yang G, Kitagawa K, Matsushita K, Mabuchi T, Yagita Y, Yanagihara T, Matsumoto M. C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: selective neuronal death in the murine transient forebrain ischemia. Brain Res. 1997;752:209–218. doi: 10.1016/s0006-8993(96)01453-9. [DOI] [PubMed] [Google Scholar]
- 45.Yocum DE. T cells: pathogenic cells and therapeutic targets in rheumatoid arthritis. Semin Arthritis Rheum. 1999;29:27–35. doi: 10.1016/s0049-0172(99)80035-3. [DOI] [PubMed] [Google Scholar]
- 46.Yoles E, Schwartz M. Degeneration of spared axons following partial white matter lesion: implications for optic nerve neuropathies. Exp Neurol. 1998;153:1–7. doi: 10.1006/exnr.1998.6811. [DOI] [PubMed] [Google Scholar]
- 47.Yoles E, Zalish M, Lavie V, Duvdevani R, Ben Bassat S, Schwartz M. GM1 reduces injury-induced metabolic deficits and degeneration in the rat optic nerve. Invest Ophthalmol Vis Sci. 1992;33:3586–3591. [PubMed] [Google Scholar]
- 48.Yoles E, Hauben E, Palgi O, Agranov E, Gothilf A, Cohen A, Kochroo V, Cohen IR, Weiner H, Schwartz M. Protective autoimmunity is a physiological response to CNS trauma. J Neurosci. 2001;21:3740–3748. doi: 10.1523/JNEUROSCI.21-11-03740.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshino A, Hovda DA, Kawamata T, Katayama Y, Becker DP. Dynamic changes in local cerebral glucose utilization following cerebral concussion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991;561:106–119. doi: 10.1016/0006-8993(91)90755-k. [DOI] [PubMed] [Google Scholar]
- 50.Zhu J, Zou LP, Bakhiet M, Mix E. Resistance and susceptibility to experimental autoimmune neuritis in Sprague-Dawley and Lewis rats correlate with different levels of autoreactive T and B cell responses to myelin antigens. J Neurosci Res. 1998;54:373–381. doi: 10.1002/(SICI)1097-4547(19981101)54:3<373::AID-JNR8>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]