

Abstract
Exposure of rat hippocampal neurons or human D283 medulloblastoma cells to the apoptosis-inducing kinase inhibitor staurosporine induced rapid cytochrome c release from mitochondria and activation of the executioner caspase-3. Measurements of cellular tetramethylrhodamine ethyl ester fluorescence and subsequent simulation of fluorescence changes based on Nernst calculations of fluorescence in the extracellular, cytoplasmic, and mitochondrial compartments revealed that the release of cytochrome c was preceded by mitochondrial hyperpolarization. Overexpression of the anti-apoptotic protein Bcl-xL, but not pharmacological blockade of outward potassium currents, inhibited staurosporine-induced hyperpolarization and apoptosis. Dissipation of mitochondrial potassium and proton gradients by valinomycin or carbonyl cyanidep-trifluoromethoxy-phenylhydrazone also potently inhibited staurosporine-induced hyperpolarization, cytochrome c release, and caspase activation. This effect was not attributable to changes in cellular ATP levels. Prolonged exposure to valinomycin induced significant matrix swelling, and per se also caused release of cytochrome c from mitochondria. In contrast to staurosporine, however, valinomycin-induced cytochrome c release and cell death were not associated with caspase-3 activation and insensitive to Bcl-xL overexpression. Our data suggest two distinct mechanisms for mitochondrial cytochrome c release: (1) active cytochrome c release associated with early mitochondrial hyperpolarization, leading to neuronal apoptosis, and (2) passive cytochrome c release secondary to mitochondrial depolarization and matrix swelling.
Keywords: mitochondrial membrane potential, staurosporine, valinomycin, potassium ionophore, proton ionophore, Bcl-2, necrosis, hippocampal neurons, medulloblastoma cells
Mitochondria play a central role in both necrotic and apoptotic neuron death by controlling cellular energetics, increasing the production of reactive oxygen species, and releasing pro-apoptotic factors into the cytosol (Murphy et al., 1999;Beal, 2000). The most prominent pro-apoptotic factor released from mitochondria is cytochrome c (Liu et al., 1996). It is electrostatically bound to the outer surface of the inner mitochondrial membrane. After its release into the cytosol, cytochrome c is capable of binding to the apoptotic protease-activating factor 1 (Apaf-1). This complex activates procaspase-9 in the presence of deoxy-ATP (dATP), resulting in the activation of the caspase cascade (Li et al., 1997; Zou et al., 1997). The release of cytochrome c in apoptosis is controlled by Bcl-2 family proteins (Kim et al., 1997;Kluck et al., 1997; Yang et al., 1997; Desagher and Martinou, 2000).
An increase in mitochondrial outer membrane permeability is required to trigger the release of cytochrome c. Several routes for mitochondrial cytochrome c release have been proposed that cause a selective outer membrane permeability increase (Schendel et al., 1998; Kluck et al., 1999; Shimizu et al., 1999). Another theory proposes that the opening of the mitochondrial permeability transition pore (PTP) stimulates the release of pro-apoptotic factors (Zamzami et al., 1996; Lemasters et al., 1998; Marzo et al., 1998; Vande Velde et al., 2000). The PTP complex is composed of several proteins of the outer and inner mitochondrial membrane, including the voltage-dependent anion channel (VDAC) and the adenine nucleotide translocator (Zoratti and Szabo, 1995; Bernardi and Petronilli, 1996; Beutner et al., 1996; Nicolli et al., 1996). PTP opening could trigger cytochrome c release indirectly by causing mitochondrial swelling and a subsequent rupture of the outer mitochondrial membrane.
Because PTP opening also results in inner membrane permeability to protons, it is associated with a decrease in mitochondrial transmembrane potential (ΔΨm) (Zoratti and Szabo, 1995). Decreases in ΔΨm or sensitivity to PTP inhibitors have been reported in several models of excitotoxic and apoptotic neuron death (Nieminen et al., 1996; White and Reynolds, 1996; Wadia et al., 1998; Heiskanen et al., 1999; Luetjens et al., 2000). However, in many neuronal and non-neuronal apoptotic systems, cytochrome c release occurred before a loss of ΔΨm and proceeded in the presence of PTP inhibitors (Bossy-Wetzel et al., 1998; Yoshida et al., 1998; Krohn et al., 1999;Stefanis et al., 1999; Deshmukh et al., 2000; Goldstein et al., 2000). It has also been reported that plasma membrane (Yu et al., 1996; Krohn et al., 1999) and/or mitochondrial membrane hyperpolarization (Vander Heiden et al., 1997; Kennedy et al., 1999; Scarlett et al., 2000) may precede cytochrome c release and caspase activation during survival factor withdrawal-, UV irradiation-, and staurosporine-induced apoptosis. The present study demonstrates the existence of two separate pathways of mitochondrial cytochrome c release in neural cells that differ with respect to changes in ΔΨm, Bcl-xL sensitivity, and their ability to activate the caspase cascade.
MATERIALS AND METHODS
Materials. Mitotracker Red (CMXRos) and tetramethylrhodamine ethyl ester (TMRE) were purchased from Molecular Probes (Leiden, The Netherlands). Rhodamine 123 (R123), valinomycin, and carbonyl cyanide p-trifluoromethoxy-phenylhydrazone (FCCP) were from Sigma (Deisenhofen, Germany). Clofilium tosylate and tetraethylammonium chloride (TEA) were from RBI Biotrend (Cologne, Germany). Staurosporine, benzyloxycarbonyl-IETD-7-amido-4-trifluoromethylcoumarin (z-IETD-AFC), and z-VDVAD-AFC were purchased from Alexis (Grünstetten, Germany). Acetyl-DEVD-7-amido-4-methylcoumarin (Ac-DEVD-AMC) and Ac-LEHD-AMC substrates were from Bachem (Heidelberg, Germany). All other chemicals were purchased in analytical grade purity from Merck (Darmstadt, Germany).
Cell culture and transfection. Primary cultures of hippocampal neurons were prepared from neonatal [postnatal day 1 (P1)] Fischer 344 rats as described (Krohn et al., 1998). Dissected hippocampi were incubated for 20 min at 37°C in Leibovitz L-15 medium (Life Technologies, Karlsruhe, Germany) containing 0.1% papain. Subsequently, the medium was removed, and the cells were suspended by gentle trituration in MEM medium supplemented with 10% NUR-serum, 2% B-27 supplement (50× concentrate), 2 mml-glutamine, 20 mmd-glucose, 26.2 mm sodium bicarbonate, 100 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies). The suspension was layered over medium containing 10 mg/ml trypsin inhibitor and centrifuged for 10 min at 600 rpm. The cells were then resuspended, plated, and maintained in the above described MEM culture medium at 37°C in an atmosphere of 95% air and 5% carbon dioxide. For imaging studies, cells were grown on poly-l-lysine-coated glass coverslips that had been placed into 35 mm Petri dishes (Falcon Becton Dickinson, Heidelberg, Germany). For immunofluorescence microscopy experiments, cells were plated onto eight-well tissue culture slides (Falcon Becton Dickinson). After 24 hr in vitro, cultures were treated with the antiproliferation agent cytosine β-arabinofuranoside (1 μm; Sigma). Experiments were performed on 8- to 10-d-old cultures. Animal care followed official governmental guidelines.
Human medulloblastoma D283 cells and breast adenocarcinoma MCF-7/Casp-3 cells stably transfected with caspase-3 (Jänicke et al., 1998) were cultured in RMPI 1640 medium (Life Technologies) supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), and 10% fetal calf serum (PAA, Cölbe, Germany). D283 cells originate from a human cerebellar medulloblastoma and are positive for neurofibrillary proteins, glutamine synthetase, and neuron-specific enolase (Vinoris et al., 1994). D283 cells deficient in mitochondrial DNA were established and cultured as described (Luetjens et al., 2000). For confocal laser-scanning microscopy, cells were cultivated at least overnight in 150 μl of medium on 35 mm glass-bottomed dishes (Willco BV, Amsterdam, The Netherlands) coated with poly-l-lysine. For immunofluorescence microscopy experiments, cells were plated onto eight-well tissue culture slides. In all other cases, cells were plated onto 6-, 24-,or 96-well tissue culture plates (Nunc, Hamburg, Germany) or tissue culture flasks (Falcon Becton Dickinson).
For transfections, D283 and MCF-7/Casp-3 cells were plated onto 12.5 cm2 culture flasks. One day later, cells were transfected with plasmids cytochrome c-enhanced green fluorescent protein (EGFP) (Heiskanen et al., 1999), pSFFV-Neo-Bcl-xL, or empty plasmid pSFFV-Neo (Boise et al., 1993) using the F2 transfection reagent (Targeting Systems, Santee, CA). Five micrograms of DNA and 5 μl of F2 reagent were diluted in 2.5 ml RPMI medium under serum-free conditions and incubated at 37°C for 20 min. Then cultures were incubated with the DNA-F2-transfection mixture at 37°C for 2 hr. Cells were cultured overnight in RPMI medium containing 10% fetal calf serum. For generation of stable cell lines, transfected MCF-7/Casp-3 cells containing cytochrome c–EGFP were selected in the presence of 1 mg/ml G418 for 2 weeks, and clones expressing mitochondrial cytochrome-EGFP were enriched. Expression of cytochrome c–EGFP was verified by Western blot analysis as described below using antibodies against GFP (Clontech, Heidelberg, Germany) and cytochrome c (Heiskanen et al., 1999). Cytochrome c–EGFP-positive cells were routinely checked for colocalization of cytochrome c with 50 nm CMXRos as mitochondrial marker. D283 cells stably overexpressing Bcl-xL were selected in the presence of 500 μg/ml G418 for 2 weeks. Expression of Bcl-xL was analyzed by Western blot analysis as described below.
TMRE-based detection of changes in mitochondrial transmembrane potential (ΔΨm). TMRE is a cationic, membrane-permeant dye that accumulates in the negatively charged mitochondrial matrix according to the Nernst equation potential (Ehrenberg et al., 1988). Estimation of ΔΨmin cultured rat hippocampal neurons was performed essentially as described previously (Krohn et al., 1999). Briefly, cells were treated with staurosporine (300 nm) or vehicle (dimethylsulfoxide) for the indicated period of time and incubated with 100 nm TMRE for 15 min at room temperature in HEPES-buffered saline (HBS) (in mm: 144 NaCl, 10 HEPES, 2 CaCl2, 1 MgCl2, 5 KCl, and 10 d-glucose; 320 mOsm, pH 7.4). The dye was present in the buffer during data collection. TMRE fluorescence was measured with a fluorescence microscope (Eclipse TE 300 inverted-stage microscope; Nikon, Düsseldorf, Germany) and a 40× S-fluorescence objective. Optics were as follows: excitation, 540–580 nm; dichroic mirror, 595 nm; and emission, 600–660 nm. Digital images of equal exposure were acquired using a 12-bit digital CCD camera (Visicam; Visitron, Munich, Germany) and Metamorph software (Universal Imaging Cooperation, West Chester, PA). Fluorescence data, which reflect the average pixel intensity obtained from the neuronal soma excluding the nucleus, are expressed in arbitrary fluorescence units (AU). Background fluorescence was subtracted from the values.
In the experiments shown in Figure1b, hippocampal neurons were treated with 300 nm staurosporine (STS) or vehicle for 6 hr. Subsequently, cells were incubated for 15 min in HBS or modified HBS containing 25 mm TEA, 1 μm clofilium tosylate, 120 mm NaCl, 10 mm HEPES, 2 mm CaCl2, 1 mm MgCl2, 5 mm KCl, and 10 mmd-glucose. Cells were loaded with 100 nm TMRE for 10 min, and TMRE fluorescence was acquired and analyzed as described above.
Fig. 1.
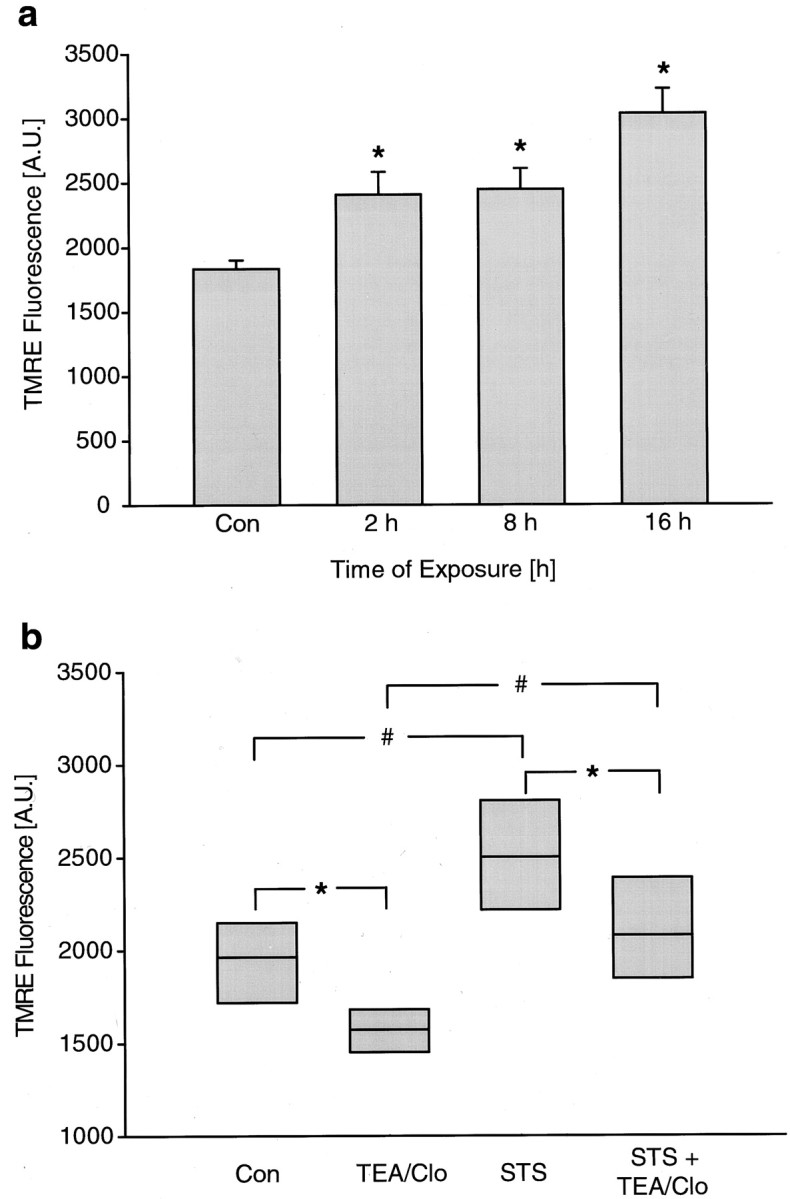
Staurosporine induces hyperpolarization of rat hippocampal neurons. a, Cultured rat hippocampal neurons exposed to STS for the indicated period of time were loaded with TMRE (100 nm, 15 min), and fluorescence was quantified. The TMRE uptake increased significantly at all time points measured. Data are means ± SEM from n = 27–150 neurons fromn = 5–20 separate experiments per time point. Different from controls *p < 0.05.b, STS-induced increase in TMRE fluorescence was not inhibited by blocking outward potassium currents. Hippocampal neurons were treated with 300 nm STS for 6 hr. Subsequently, cultures were exposed to HBS or HBS + TEA + clofilium tosylate (Clo) for a further 15 min. After a 10 min loading period, cellular TMRE fluorescence was acquired. Data were not expressed as an SD and therefore were presented in median/quartile form and represent values from n = 82–185 cells. *,#p < 0.05 compared with respective controls.
TMRE uptake of human D283 medulloblastoma cells in response to STS was quantified by confocal laser-scanning microscopy using an inverted Olympus IX70 microscope equipped with a confocal laser-scanning unit and a 60× oil immersion objective (Fluoview, Olympus, Hamburg) as described previously (Luetjens et al., 2000). After detection of autofluorescence, 10 or 100 nm TMRE was added to the cultures. TMRE was excited with the 488 nm line, and emission was detected from a 0.63-μm-thick optical section with a 565 nm long-pass filter. Whole cellular fluorescence after background subtraction was calculated using the UTHSCSA ImageTool program (developed at the University of Texas Health Science Center at San Antonio, TX; available from the Internet by anonymous FTP from www.maxrad6.uthscsa.edu).
Simulation of changes in cellular TMRE fluorescence based on Nernst calculations of fluorescence in the extracellular, cytoplasmic, and mitochondrial compartments was performed as described previously (Ward et al. 2000). The following constants were used: (1) mitochondrial volume fraction: 2%. To estimate the mitochondrial volume, areas defined as mitochondria by TMRE fluorescence intensity were integrated from 40 scans with 0.47-μm-thick confocal sections and 0.5 μm intervals in TMRE-stained D283 cells. Mitochondria in neuronal cells are rod-shaped with an average diameter of 0.25 μm and an average length of 1 μm (Jacobson, 1972; Trimmer et al. 2000). Blurring of an artificial image of mitochondria-like structures using the microscope point spread function revealed a threefold overestimation. The mitochondrial volume fraction used for the simulation was therefore less than the confocally measured mitochondrial volume fraction (6%). The volume of the somata were integrated from optical sections of the same cells after addition of 5 μm oligomycin and 2 μm FCCP. (2) Permeability constant for the equilibration across the plasma membrane: 1 ± 0.1% sec−1. The constant was calculated from the slope of the fluorescence intensity decay in the somata of TMRE-stained cells after addition of 2 μm FCCP and 5 μm oligomycin. (3) Saturation constant: 700 μm. The saturation concentration of TMRE was evaluated by adding 10, 30, 50, 100, 150, 250, and 500 nm TMRE to D283 cells. The measured fluorescence intensity of mitochondria-rich regions after cytoplasmic background subtraction at each concentration after equilibration was fitted to a sigmoidal Boltzmann equation to calculate the saturation concentration. The calculated saturation concentration of D283 medulloblastoma cells is higher than the published saturation concentration of primary neurons (Ward et al., 2000), suggesting an increased quench threshold and/or the existence of additional cellular processes that regulate TMRE distribution in transformed cells. (4) External probe concentration: 10 or 100 nm TMRE. Plasma and mitochondrial membrane potential changes were simulated as indicated.
Use of alternative probes to detect changes in ΔΨm. R123, like TMRE, is a cationic, voltage-sensitive probe that reversibly accumulates in mitochondria (Emaus et al., 1986) and that was used in the present study as a second probe to detect changes in ΔΨm. Cells were loaded with 100 nm–2 μm R123 in medium for 30 min at 37°C. Subsequently, cultures were washed with R123-free HBS, and R123 fluorescence was monitored by epifluorescence microscopy as described above.
For immunocytochemistry experiments and as a third probe to detect changes in ΔΨm, we used the voltage-sensitive probe CMXRos (Zamzami et al., 1996). In contrast to TMRE and R123, CMXRos forms stable thiol conjugates with mitochondrial proteins, inhibiting release of the dye from mitochondria and enabling fixation of cells while retaining the dye (Krohn et al., 1999). Cells were treated with valinomycin, staurosporine, or vehicle. For fluorescence microscopy, cells were incubated with 50 nm CMXRos during the last 15 min of the exposure. CMXRos-stained cells were fixed using methanol–acetic acid (3:1) for 15 min at −20°C and washed with PBS. Fluorescence was observed using an Eclipse TE 300 inverted microscope and a 40× dry or 100× oil immersion objective with the following optics: excitation, 510–560 nm; dichroic mirror, 575 nm; and emission, >590 nm. Digital images of equal exposure were acquired with a SPOT-2 camera (Diagnostic Instruments, Sterling Heights, MI) using Spot software version 2.21 (Diagnostic Instruments).
To exclude the possibility that fluorescence signal changes were caused by alterations in cell morphology, TMRE and CMXRos uptakes were also quantified in cell lysates. In the case of TMRE, cells were treated with 100 nm TMRE at 37°C for 10 min after the exposure to STS. Medium was discarded, and cells were lysed in lysis buffer (2% SDS, 0.1 m Tris, pH 7.5). Fluorescence was measured using an HTS fluorescence plate reader (Perkin-Elmer, Langen, Germany) (excitation, 577 nm; emission, 595 nm). In the case of CMXRos, D283 cells were loaded with 100 nm CMXRos during the last 15 min of exposure, washed with HBS, and fluorescence was measured using the above-mentioned fluorescence plate reader (excitation 590 nm, emission 635 nm). Protein content per well was determined with the Pierce Micro-BCA Protein assay Kit (KMF, Cologne, Germany). TMRE or CMXRos fluorescence were expressed as fluorescence units per microgram of protein. As a positive control, cultures were exposed to the protonophore FCCP (10 μm). Exposure to FCCP decreased CMXRos uptake from 55.8 ± 2.5 AU/μg of protein in vehicle-treated controls to 37.4 ± 4.2 AU/μg of protein in cultures treated for 30 min with FCCP (p < 0.05; n = 8 cultures per treatment).
Determination of the cell viability and Hoechst staining.Cells were simultaneously stained with 1 μmcalcein AM and 2 μm ethidium homodimer (EthD-1) in serum-free medium using the LIVE/DEAD Viability/Cytotoxicity Kit (Molecular Probes). Calcein AM is converted to intensely green fluorescent calcein in metabolically active cells through the activation of intracellular esterases. EthD-1 is a DNA-binding dye that enters dead cells through damaged membranes. Calcein and EthD-1 fluorescence were observed using the above-mentioned inverted microscope and a 20× dry immersion objective with the following optics: for calcein, excitation, 465–495 nm; dichroic mirror, 505 nm; emission, 515–555 nm; for EthD-1, excitation, 510–560 nm; dichroic mirror, 575 nm; emission, >590 nm. Digital images were acquired as described above. A total of 430–3000 cells were counted per culture. Chromatin condensation and fragmentation were visualized using the DNA-binding fluorescent dye Hoechst 33258 (Sigma). Cells cultured on coverslips were fixed in methanol–acetic acid (3:1) for 15 min at −20°C. Cells were washed in PBS and incubated in PBS containing 1 μg/ml Hoechst 33258 for 20 min at room temperature. Nuclei were observed using the Eclipse TE 300 inverted microscope and a 20× dry immersion objective with the following optics: excitation, 340–380 nm; dichroic mirror, 400 nm; and emission, 435–485 nm. A total of 700–800 nuclei were counted per culture.
Immunofluorescence analysis. For immunofluorescence analysis, cells were fixed on eight-well tissue culture slides, washed three times with PBS, permeabilized at 4°C for 3 min in PBS containing 0.1% Triton X-100, and then incubated with blocking solution (PBS with 5% horse serum and 0.3% Triton X-100) for 30 min at room temperature. Cytochrome c was detected using a monoclonal anti-cytochrome c antibody (clone 6H2.B4; PharMingen Becton Dickinson) that recognizes the native form of cytochrome c. The antibody was used at a concentration of 1:1000 in PBS containing 1% horse serum and 0.3% Triton X-100. After incubation at room temperature for 2 hr, cells were washed twice with PBS and incubated with biotin-conjugated anti-mouse IgG antibody (Vector Laboratories, Burlingame, CA) diluted 1:500. The secondary antibody was detected using Oregon green-conjugated streptavidin (Molecular Probes) diluted 1:1000 in PBS for 20 min at room temperature. Epifluorescence microscopy was performed as described above.
Digital imaging of EGFP-tagged cytochrome c. Cytochrome c–EGFP expressing cells were cultivated for at least 1 d in 150 μl of medium on 35 mm glass-bottomed dishes (Willco BV) coated with poly-d-lysine to let them attach firmly. EGFP fluorescence was observed using the Eclipse TE 300 inverted microscope and a 100× oil immersion objective equipped with the appropriate filter set. Digital images were acquired with the SPOT-2 camera described above. For time-lapse images, dishes were mounted onto the microscope stage. In control experiments, the cytochrome c signal could be monitored for up to 24 hr. The cells were stimulated with 3 μm staurosporine or 10 μm valinomycin or the combination of both directly on the stage after acquiring the first image. The media was enriched with 10 mm HEPES and thoroughly mixed to ensure a proper distribution of the drugs. To prevent evaporation, the media was covered with embryo-tested paraffin-oil (Sigma).
Measurement of caspase-3-like protease activity. After treatment with valinomycin, staurosporine, FCCP, or vehicle, cells were lysed in 200 μl of lysis buffer [10 mm HEPES, pH 7.4, 42 mm KCl, 5 mmMgCl2, 1 mmphenylmethylsulfonyl fluoride (PMSF), 0.1 mmEDTA, 0.1 mm EGTA, 1 mmdithiothreitol (DTT), 1 μg/ml pepstatin A, 1 μg/ml leupeptin, 5 μg/ml aprotinin, and 0.5% 3-(3-cholamidopropyldimethylammonio)-1-propane sulfonate (CHAPS)]. Fifty microliters of this lysate were added to 150 μl of reaction buffer [25 mm HEPES, 1 mmEDTA, 0.1% CHAPS, 10% sucrose, 3 mm DTT, pH 7.5 and 10 μm of the respective caspase substrate (Ac-DEVD-AMC, Ac-LEHD-AMC, z-IETD-AFC, or z-VDVAD-AFC)]. Accumulation of AMC or AFC fluorescence was monitored over 120 min using an HTS fluorescent plate reader (excitation, 380 nm; emission, 465 nm). Fluorescence of blanks containing no cell lysate were subtracted from the values. Protein content was determined using the Pierce Coomassie Plus Protein Assay Reagent (KMF). Caspase activity is expressed as change in fluorescent units per microgram of protein per hour.
Subcellular fractionation and immunoblotting. D283 cells from one 175 cm2 flask were collected at 200 × g for 5 min and washed with PBS. The cell pellet was resuspended in 100 μl of buffer A (20 mmHEPES–KOH, pH 7.5, 10 mm KCl, 1.5 mm MgCl2, 1 mm EGTA, 1 mm DTT, 250 mm sucrose, 100 mm PMSF, 1 μg/ml pepstatin A, 2 μg/ml leupeptin, and 2 μg/ml aprotinin). Cells were homogenized using a glass dounce and a “tight” pestle (10 strokes). Cell homogenates were centrifuged at 15,000 ×g for 15 min at 4°C. The obtained pellet represented the mitochondria-containing nuclear–heavy membrane fraction. The supernatant was respun for a further 15 min at 20,000 ×g at 4°C. This second supernatant represented the cytosol (including the light membrane fraction; Ellerby et al., 1997). Thirty micrograms of protein was loaded onto a 15% SDS-polyacrylamide gel. Proteins were separated for 1 hr at 120 V and then blotted to nitrocellulose membranes (Protean BA 83; 2 μm; Schleicher & Schuell, Dassel, Germany) in Towbin-buffer [25 mm Tris, 192 mm glycine, 20% methanol (v/v) and 0.01% SDS] at 15 V for 45 min. The blots were blocked with 5% nonfat milk in TBST (15 mm Tris-HCl, pH 7.5, 200 mm NaCl, and 0.1% Tween 20) for 2 hr at room temperature. Membranes were incubated with a mouse monoclonal anti-cytochrome c antibody (clone 7H8.2C12; 1:1000; PharMingen Becton Dickinson), a mouse monoclonal anti-VDAC antibody (31HL; 1:1000; Calbiochem, Bad Soden, Germany) to exclude contamination of cytoplasmic extracts with mitochondria, or a mouse monoclonal anti-α-tubulin antibody (clone DM 1A; 1:1000; Sigma) to prove equal loading of the samples. For caspase-3 immunoblots, 30 μg of protein of whole-cell lysate (lysis buffer: 68.5 mm Tris/HCl, pH 6.8, 2% SDS, and 10% glycerol) were loaded onto a 15% SDS-polyacrylamide gel. After blotting, membranes were probed with a rabbit polyclonal anti-caspase 3 antibody (H277; 1:500; Santa Cruz, Heidelberg, Germany). For detection of Bcl-xL expression, membranes were probed with a rabbit polyclonal anti-Bcl-x antibody diluted 1:1000 (kindly provided by Prof. Craig B. Thompson, University of Pennsylvania). Antibodies were diluted in blocking solution, and blots were incubated overnight at 4°C. Primary antibodies were detected using horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit antibodies (Promega, Heidelberg, Germany) used 1:5000 in blocking solution for 1 hr at room temperature. The blots were developed using the Pierce SuperSignal substrate chemiluminescence reagent.
Determination of cellular ATP. ATP was determined using the ATP Bioluminescence Assay Kit CLS II (Boehringer Mannheim, Mannheim, Germany). Cells cultivated on six-well tissue culture plates were collected at 10,000 rpm for 2 min at 4°C and washed with PBS. The cell pellet was resuspended in 50 μl of ice-cold ATP-lysis buffer (100 mm Tris and 4 mm EDTA, pH 7.75), 150 μl of boiling ATP-lysis buffer was added, and samples were incubated for 2 min at 99°C. Cell lysates were centrifuged at 10,000 rpm for 1 min at 4°C, and supernatants were collected. ATP measurement was performed using 50 μl of supernatant and 50 μl of luciferase reagent. After a 20 sec delay the chemiluminescence was measured with 2 sec integration time in a lumi-imager (Boehringer Mannheim). Protein content was determined using the Roti-Quant protein assay (Roth, Karlsruhe, Germany). Luciferase activity was expressed as fluorescent units per microgram of protein. Cultures treated with vehicle were set to 100% activity.
Statistics. Data are given as means ± SEM. For statistical comparison, t test or one-way ANOVA followed by Tukey test were used. Data that were not measured as SDs are presented as medians/quartiles and were analyzed by Kruskal–WallisH test and subsequent Bonferroni-corrected Mann–WhitneyU test. P values <0.05 were considered to be statistically significant.
RESULTS
Exposure of cultured rat hippocampal neurons to staurosporine induces early membrane hyperpolarization
Exposure to the protein kinase inhibitor staurosporine induces apoptosis in cultured rat hippocampal neurons (Prehn et al., 1997). We have previously demonstrated that mitochondrial depolarization is not required for cytochrome c release and caspase activation in this model (Krohn et al., 1999). Mitochondria remained polarized and could be depolarized by addition of the protonophore FCCP at a time point when cytochrome c was released and the caspase cascade was maximally activated. On the contrary, an increased uptake of TMRE into mitochondria could be observed during the staurosporine exposure. We exposed cultured rat hippocampal neurons to 300 nmstaurosporine and detected a significant increase in TMRE uptake 2 hr after onset of the treatment (Fig. 1a). By 8 hr, 73 ± 2.7% of the STS-treated hippocampal neurons had released their cytochrome c, as evidenced by a transition from a punctate to a diffuse cytochrome c immunofluorescence (n = 3 experiments). At this time point, mitochondrial TMRE uptake remained at a high level and even increased with time (Fig. 1a).
We have previously demonstrated that exposure of cultured rat hippocampal neurons to a high extracellular K+ concentration (50 mm) greatly abolished the increase in TMRE uptake during the STS exposure (Krohn et al., 1999). However, a strong plasma membrane depolarization may inhibit the entry of TMRE into the cytosolic compartment and may thus limit mitochondrial TMRE uptake (Nicholls and Ward, 2000). Outward potassium currents have been shown to mediate plasma membrane hyperpolarization during neuronal apoptosis (Yu et al., 1996). Treatment of cultured rat hippocampal neurons with selective inhibitors of outward potassium currents (25 mm TEA plus 1 μm clofilin) caused a significant decrease in cellular TMRE uptake in control cells, suggesting that these currents are involved in the regulation of plasma membrane potential (ΔΨp) under physiological conditions. Interestingly, staurosporine was still able to increase TMRE uptake in cultures treated with TEA plus clofilin, suggesting that the increase in TMRE uptake during staurosporine-induced apoptosis was not attributable to activation of outward potassium currents and subsequent plasma membrane hyperpolarization (Fig. 1b).
Mitochondrial depolarization inhibits staurosporine-induced cytochrome c release in rat hippocampal neurons
To address the issue regarding whether mitochondria have to remain polarized for staurosporine-induced cytochrome c release to occur, we treated cultured rat hippocampal neurons with the potassium ionophore valinomycin (10 nm) or the protonophore FCCP (10 μm). Both treatments induced a prominent mitochondrial depolarization, as evidenced by decreased uptake of the voltage-sensitive probes TMRE, R123, and CMXRos into mitochondria (data not shown). Hippocampal neurons were exposed to 100 nmstaurosporine, valinomycin, FCCP, or a combination of staurosporine and the ionophores. After 6 hr, cytochrome c release was evaluated by immunofluorescence analysis. Exposure to staurosporine induced a significant cytochrome c release, as shown by a diffuse cytochrome c immunofluorescence compared with vehicle-treated controls (Fig.2). Valinomycin and FCCP alone induced little cytochrome c release at this time point. In contrast to cultures treated with staurosporine alone, cotreatment with valinomycin or FCCP inhibited staurosporine-induced cytochrome c release.
Fig. 2.

Inhibition of cytochrome c release in rat hippocampal neurons by dissipation of mitochondrial potassium and proton gradients. Cultured rat hippocampal neurons were exposed to vehicle (a), 10 nm Val (b), 10 μm FCCP (c), 300 nm STS (d), Val + STS (e), or FCCP + STS (f) for 6 hr. Distribution of cytochrome c is shown by immunofluorescence analysis. Only cultures exposed to 300 nm STS showed a diffuse cytochrome c staining pattern (d). Scale bar, 50 μm.
Staurosporine induces early mitochondrial, but not plasma membrane hyperpolarization in D283 medulloblastoma cells
Increased uptake of TMRE was also observed in the D283 medulloblastoma cells in response to 3 μm staurosporine (Fig. 3). However, mitochondrial TMRE fluorescence may change in response to alterations in either ΔΨm or ΔΨp (Nicholls and Ward, 2000). To distinguish whether the increased uptake of TMRE was attributable to mitochondrial or plasma membrane hyperpolarization, we simulated single-cell fluorescence changes in D283 medulloblastoma cells in response to alterations in either ΔΨm or ΔΨp and compared these simulations with the traces we obtained during an exposure to staurosporine. For this purpose, we used a simulation program that is based on Nernst calculations of the distribution of voltage-sensitive cationic probes in the extracellular, cytoplasmic, or mitochondrial compartments (Ward et al., 2000; kindly provided by Prof. David G. Nicholls, The Buck Institute, Novato, CA). Constants for mitochondrial volume fraction, plasma membrane permeability rate for TMRE, as well as TMRE saturation concentration were experimentally determined as described in Materials and Methods. To validate the simulation, D283 medulloblastoma cells were incubated with 10 nm TMRE (Fig. 3a, dotted line). After equilibration of the TMRE signal, cells were exposed to 5 μm oligomycin, which hyperpolarizes mitochondria. This was followed by an exposure to 2 μm FCCP plus 5 μmoligomycin to depolarize mitochondria. The solid line in Figure3a shows the simulation of the experiment, with an immediate change of ΔΨm after addition of oligomycin (from −150 to −190 mV), followed by an immediate mitochondrial depolarization after addition of FCCP plus oligomycin (from −190 to 0 mV). We next recorded TMRE traces of D283 medulloblastoma cells exposed to 3 μm staurosporine using two different TMRE concentrations, 10 nm (Fig. 3b,c) and 100 nm (Fig. 3d,e). After 200 min of staurosporine treatment, mitochondria were depolarized by the addition of 2 μm FCCP plus 5 μmoligomycin. The dotted lines in Figure 3 show the respective time lapse of total cellular fluorescence in percentage of baseline after equilibration with 10 nm (b, c) and 100 nm (d, e) TMRE. In either case, TMRE fluorescence increased significantly after exposure to staurosporine, and a sharp drop in TMRE fluorescence was observed in response to FCCP plus oligomycin. Next, simulations were calculated for each TMRE concentration with an immediate change in either ΔΨm (Fig. 3b,d, solid lines) (hyperpolarization from −150 to −190 mV, representing the best fit) or ΔΨp (Fig. 3c,e, solid lines) (hyperpolarization from −60 to −90 mV, representing the best fit) after addition of staurosporine. This was followed by a simulation of the depolarization of mitochondria from −190 to 0 mV after addition of FCCP plus oligomycin. With either TMRE concentration, the simulations based on ΔΨp changes were less convincing than those based on ΔΨm changes. Moreover, the peak in TMRE fluorescence occurring in 100 nm TMRE-loaded D283 medulloblastoma cells after the addition of FCCP plus oligomycin (“unquenching” of TMRE fluorescence) is not well represented using the plasma membrane simulation.
Fig. 3.

Effect of oligomycin (a) and STS (b–e) on cellular TMRE uptake in D283 medulloblastoma cells and simulation of TMRE fluorescence changes according to Ward et al. (2000). The dotted linesrepresent the time lapse of total cellular fluorescence in percentage of baseline after equilibration with 10 nm(a–c) or 100 nm (d, e) TMRE. After equilibration of the TMRE signal, cells were exposed to 5 μm oligomycin (a) or 3 μm STS (b–e). The onset of treatment is indicated by the arrows on the left. After the respective treatments, mitochondria were depolarized by the addition of 2 μm FCCP plus 5 μm oligomycin (arrows on the right). Simulations based on the Nernst equation were calculated with immediate changes in ΔΨM (solid lines in a,b, and d; left arrow,hyperpolarization from −150 to −190 mV) or ΔΨP(solid lines in c and e;left arrow, hyperpolarization from −60 to −90 mV). In each case, this was followed by a simulation of mitochondrial depolarization after addition of FCCP plus oligomycin (right arrows, depolarization from −190 to 0 mV). Note that the time lapse of the oligomycin and STS treatments have the same shape. Simulations based on changes in ΔΨP are less convincing, and the peak after FCCP plus oligomycin in cells loaded with 100 nm TMRE in d does not occur ine (solid lines). Traces are means from all cells within the microscope field in a typical experiment (n = 18–26 cells). The experiments were performed in duplicate (10 nm TMRE experiments) and triplicate (100 nm TMRE experiment) with similar results.
Both staurosporine and valinomycin induce cytochrome c release in human D283 medulloblastoma cells
The above data indicated that mitochondrial hyperpolarization may precede cytochrome c release in neuronal apoptosis. However, mitochondrial depolarization has also been suggested to trigger cytochrome c release and caspase activation in neural cells (Wadia et al., 1998; Heiskanen et al., 1999; Luetjens et al., 2000). We were therefore interested to investigate in more detail the influence of ΔΨm, overexpression of anti-apoptotic proteins (Bcl-xL), and mitochondrial respiratory chain activity on cytochrome c release in human D283 medulloblastoma cells. To this end, D283 cells were exposed to staurosporine or the potassium ionophore valinomycin, which depolarizes mitochondria (Holmuhamedov et al., 1998; Rottenberg and Wu, 1998). Subcellular fractionation experiments revealed that treatment of D283 medulloblastoma cells with staurosporine (3 μm) induced a significant release of cytochrome c from mitochondria into the cytosol already after 4 hr of treatment. The appearance of cytochrome c in the cytosolic fraction was associated with a corresponding decrease in the mitochondria-containing nuclear–heavy membrane fraction (Fig. 4a). Exposure to valinomycin (10 μm) also induced a release of cytochrome c into the cytosol after 6 and 16 hr of exposure (Fig. 4b), albeit the magnitude of this release was smaller. As with staurosporine, the increased accumulation of cytochrome c in the cytosol was associated with a corresponding decrease in the cytochrome c content of the mitochondria-containing nuclear–heavy membrane fraction.
Fig. 4.

Cytochrome c release induced by staurosporine and valinomycin in human medulloblastoma D283 cells. Cultures were incubated for the indicated periods of time with staurosporine (STS) (a), valinomycin (Val) (b), or the vehicle. Cells were fractionated into a cytosolic and a mitochondria-containing nuclear–heavy membrane fraction (Pellet). Immunoblot analysis was performed using an anti-cytochrome c antibody. Blots were incubated with an anti-VDAC antibody to exclude contaminations of the cytosolic fraction with mitochondria. An anti-α-tubulin antibody was used as loading control. The experiment was repeated three times with similar results. c, Immunoblot analysis of whole-cell lysates using an anti-cytochrome c and an anti-α-tubulin antibody after exposure to STS, Val, or vehicle for the indicated periods of time. The experiment was repeated twice with similar results.
Immunoblot analysis of whole-cell lysates exposed to staurosporine for 8 hr showed a reduction in total cytochrome c content compared with vehicle-treated control cells (Fig. 4c), likely because of the reported degradation of cytochrome c during apoptosis (Bobba et al., 1999). In contrast, exposure to valinomycin for 6 hr did not alter total cellular cytochrome c content. Interestingly, prolonged exposure to valinomycin (16 hr) increased cytochrome c content, presumably a consequence of increased cytochrome c transcription during conditions of metabolic stress (Dey and Moraes, 2000).
Overexpression of Bcl-xL inhibits staurosporine-induced mitochondrial hyperpolarization and cytochrome c release
To relate cytochrome c release with changes in ΔΨm during apoptosis, we performed experiments using the voltage-sensitive probe CMXRos in combination with immunofluorescence microscopy. Controls treated with vehicle and loaded with CMXRos showed a rod-like staining pattern characteristic of mitochondria (Fig.5a). In these cells, CMXRos fluorescence colocalized with cytochrome c immunofluorescence (Fig.5c). In agreement with our TMRE confocal imaging studies, cultures exposed to staurosporine showed an increased CMXRos uptake after 30 min of treatment (Fig. 5a,b). At this time, cytochrome c was still localized to mitochondria (Fig. 5d). Interestingly, cytochrome c immunofluorescence concentrated around the nucleus, indicative of perinuclear clustering of mitochondria early during apoptosis (De Vos et al., 1998). Quantification of CMXRos (Fig.5e) as well as TMRE (Fig. 5f) fluorescence in cell lysates confirmed the increased uptake of voltage-sensitive probes during staurosporine-induced apoptosis and demonstrated that the increase in CMXRos and TMRE fluorescence was not an artifact caused by staurosporine-induced morphological alterations.
Fig. 5.
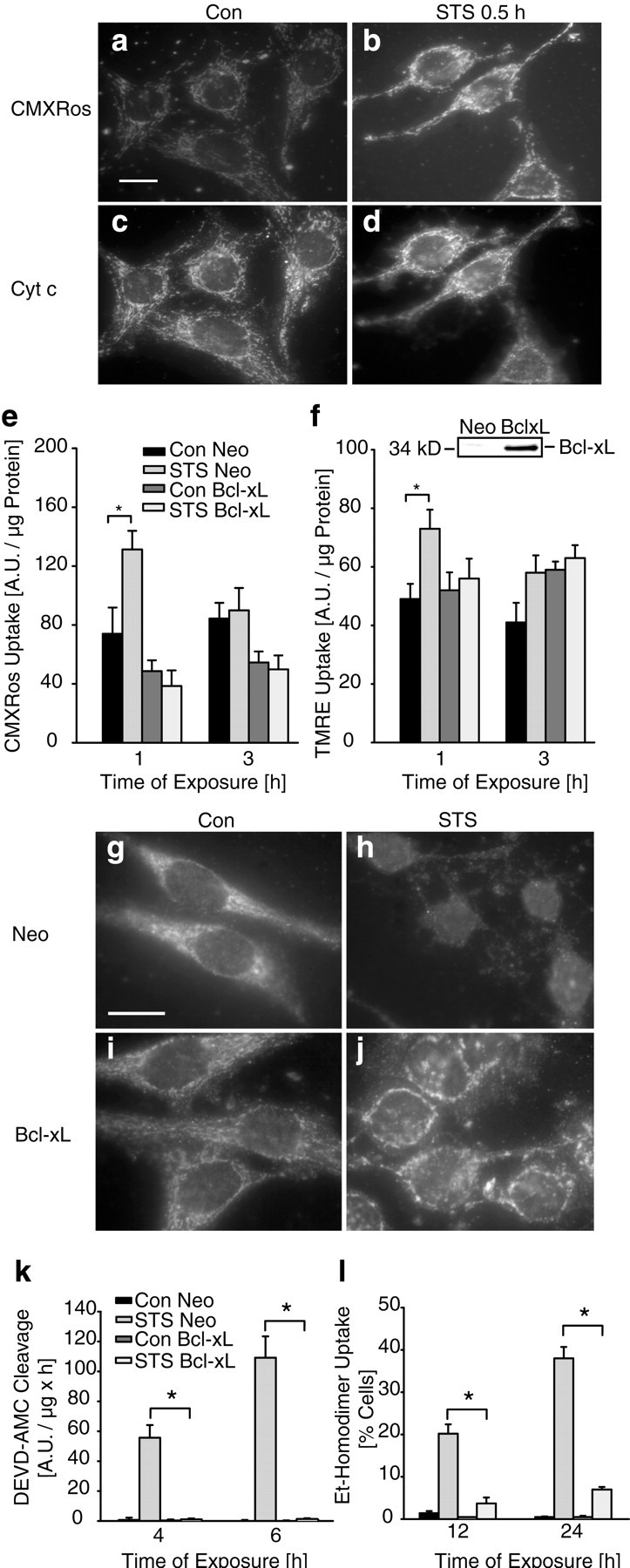
Overexpression of Bcl-xL inhibits staurosporine-induced mitochondrial hyperpolarization and cytochrome c release. a–d, D283 cells were exposed to vehicle or STS. After 30 min exposure to STS, cytochrome c immunofluorescence remained mitochondrial (d), whereas CMXRos uptake increased significantly (b). Scale bar, 10 μm. Experiments were performed three times with comparable results.e–l, D283 medulloblastoma cells were stably transfected with pSFFV-Neo-Bcl-xL (D283/Bcl-xL) or empty plasmid pSFFV-Neo (D283/Neo). Overexpres- sion of Bcl-xL was confirmed by immunoblotting using an anti-Bcl-x antibody (f, inset).e, Quantification of CMXRos fluorescence in D283/Neo cultures confirmed an early increase in CMXRos uptake after the exposure to STS. In contrast, the increase of CMXRos uptake was inhibited in D283/Bcl-xL cells. Data are means ± SEM fromn = 8 cultures; *p < 0.05 with respect to control. Experiment was performed in triplicate and yielded comparable results. f, Quantification of TMRE uptake in D283/Neo cultures confirmed the early increase of fluorescence obtained with CMXRos. The increase of TMRE uptake was inhibited in D283/Bcl-xL cells. Data are means ± SEM from n = 8 cultures; *p < 0.05 with respect to control. Experiment was repeated twice with comparable results.g–j, Cytochrome c distribution was visualized by immunofluorescence analysis after 3 hr exposure to STS or vehicle. Note that the STS-induced cytochrome c release was significantly reduced in D283/Bcl-xL cells (j) compared with D283/SFFV cells (h). Scale bar, 10 μm (g). k, Control cells and Bcl-xL-overexpressing cells were treated with vehicle (Con) or STS for 4 and 6 hr. Caspase-3-like protease activity was measured by cleavage of the fluorigenic substrate Ac-DEVD-AMC. Activities are presented as increase in AMC fluorescence (in arbitrary fluorescence units) over 60 min per microgram of protein. Data are means ± SEM from n = 8 cultures. The experiment was repeated three times with similar results. Different from controls, *p < 0.05.l, Quantification of cell death after exposure to STS. D283 cells were incubated with STS or vehicle for up to 24 hr. Cells were stained simultaneously with 1 μm calcein AM and 2 μm ethidium homodimer (EthD-1). Live (green) and dead (red) cells were counted. n = 4 cultures (430–3000 cells) per time point. Different from controls, *p < 0.05.
The ability of the anti-apoptotic protein Bcl-xL to inhibit staurosporine-induced mitochondrial hyperpolarization and cytochrome c release was investigated in human D283 medulloblastoma cells stably transfected with plasmid pSFFV-Neo-Bcl-xL (D283/Bcl-xL) or empty vector pSFFV-Neo (D283/neo). Overexpression of Bcl-xL in D283/Bcl-xL cells was confirmed by Western blot analysis (Fig. 5f, inset). We observed decreased CMXRos uptake in Bcl-xL-overexpressing cells under control conditions. This was not observed in TMRE-loaded cells, suggesting that CMXRos does not solely report membrane potential changes. Interestingly, the staurosporine-induced increase in CMXRos and TMRE uptake was significantly inhibited in Bcl-xL-overexpressing cultures (Fig. 5e,f).
After 3 hr of exposure, cytochrome c was released from mitochondria in the majority of D283/neo cells, resulting in a weak, diffuse cytochrome c staining in the cytosol (Fig. 5g,h). In cells overexpressing Bcl-xL, staurosporine-induced cytochrome c release was significantly inhibited (Fig. 5i,j). However, overexpression of Bcl-xL did not prevent the perinuclear clustering of mitochondria during apoptosis. Bcl-xL overexpression also significantly inhibited staurosporine-induced caspase-3 like protease activity (Fig.5k) determined in cytosolic extracts by measuring the cleavage of Ac-DEVD-AMC, a fluorigenic substrate preferentially cleaved by caspase-3, -7, and -8, but also by caspase-1, -6, -9, and -10. Moreover, Bcl-xL significantly inhibited the staurosporine-induced decrease in cell viability determined by staining of cultures with calcein AM and EthD-1 (Fig. 5l). EthD-1 is a DNA binding dye that enters dead cells through damaged membranes and thus indicates (secondary) necrosis.
Bcl-xL overexpression does not inhibit valinomycin-induced cytochrome c release and mitochondrial depolarization
D283 medulloblastoma cells exposed to valinomycin showed a dramatically altered CMXRos uptake (Fig.6a,b) (15 min of exposure). Mitochondria failed to accumulate CMXRos, and instead the entire cell was diffusely stained. The increased uptake of CMXRos into the cytosolic compartment is likely attributable to plasma membrane hyperpolarization occurring additionally at micromolar concentrations of valinomycin (Holmuhamedov et al., 1998; Rottenberg and Wu, 1998). An immediate mitochondrial depolarization after addition of valinomycin was also observed using the probes TMRE and R123 (data not shown). After 15 min exposure to valinomycin, cytochrome c was retained in mitochondria (Fig. 6c,d). To test whether valinomycin, like staurosporine, was toxic to D283 cells, the effect of valinomycin on viability of D283 cells was determined by simultaneous staining of cultures with calcein AM and EthD-1. Exposure to valinomycin for 24 hr induced a significant increase in the number of EthD-1-positive cells. Interestingly, overexpression of Bcl-xL failed to provide a significant protection against valinomycin-induced cell death (Fig. 6e). Moreover, overexpression of Bcl-xL did not influence the valinomycin-induced loss of cytochrome c (Fig.6f,h,j,l). The decreased CMXRos uptake into mitochondria induced by valinomycin was also not prevented in Bcl-xL-overexpressing cells (Fig. 6g,i,k,m).
Fig. 6.

Overexpression of Bcl-xL does not inhibit valinomycin-induced mitochondrial depolarization and cytochrome c release. a–d, Representative images of vehicle-treated (a, c) and Val-treated (b, d) D283 medulloblastoma cells fixed after 15 min of exposure. a,b, Changes of mitochondrial membrane potential were determined by CMXRos uptake. Note that the mitochondrial staining pattern was lost after treatment with Val (b).c, d, Distribution of cytochrome c was determined by immunofluorescence analysis. Cytochrome c immunofluorescence remained intact after exposure to Val (d). Scale bar, 10 μm. e, Quantification of cell death after exposure to Val. D283/Neo and D283/Bcl-xL cells were exposed to vehicle or Val for 12 and 24 hr and stained simultaneously with 1 μm calcein AM and 2 μm ethidium homodimer. The percentage of dead cells was determined. n = 4 cultures (600–3000 cells) per time point. Different from controls, *p < 0.05.n.s., Not statistically significant.f–m, Cells were treated with Val or vehicle for 6 hr. Cytochrome c distribution was visualized by immunofluorescence analysis. Val-induced cytochrome c release was not inhibited in Bcl-xL-overexpressing cells (l, arrowheads) compared with control cells (h). Mitochondria appeared swollen in control and Bcl-xL-overexpressing cells (h, l; arrows). Overexpression of Bcl-xL could also not inhibit loss of mitochondrial membrane potential (i, m). Scale bar (in f): f–m, 10 μm.n–p, High magnification of mitochondria and mitochondria-rich regions in vehicle- and Val-treated cultures (6 hr) stained with the cytochrome c antibody. Overexpression of Bcl-xL does not inhibit large-scale mitochondrial swelling induced by Val. Mitochondria of vehicle-treated D283/Neo and D283/Bcl-xL cells were indistinguishable, therefore only mitochondria of a D283/Bcl-xL cell are shown. Images were deconvoluted using No Neighbor Deblurring software, which applies the algorithm of Monck et al. (1992) to reduce image background haze attributable to light originating from unsharp areas of the specimen. Scale bar (in n): n–p; 5 μm. N, Nucleus. All experiments were repeated twice with comparable results. q–r, Images of mitochondria in vehicle- and Val-treated (6 hr) rat primary astrocytes stained with the anti-cytochrome c antibody. Images were deconvoluted using the above-mentioned software. Scale bar, 5 μm (q).
High magnification of cytochrome c immunofluorescence revealed that exposure to valinomycin induced mitochondrial alterations indicative of mitochondrial swelling (Fig. 6n,o). These alterations were not sensitive to overexpression of Bcl-xL (Fig. 6p). Mitochondrial swelling in response to valinomycin could be more clearly detected in rat primary astrocytes (Fig. 6q,r). The low density of mitochondria in the somata of astrocytes allowed a quantification of mitochondrial swelling after a 6 hr exposure to 10 μm valinomycin. Measurement of the mitochondrial area using Metamorph software revealed a mean increase of 209 ± 80% compared with vehicle-treated controls (n = 50 and 75 mitochondria, respectively;p < 0.05).
Valinomycin-induced cytochrome c release occurs without significant activation of the caspase cascade
We next compared the effects of valinomycin and staurosporine on caspase activation in D283 medulloblastoma cells. Caspase-3-like protease activity determined by measuring the cleavage of Ac-DEVD-AMC after exposure to 10 μm valinomycin was not significantly different from control cultures treated with the vehicle at all time points investigated (Fig. 7a). Cleavage of z-IETD-AFC and Ac-LEHD-AMC (substrates for caspase-6, -8, -9 and -10), and z-VDVAD-AFC (a substrate for caspase-2 and -3) did also not increase after exposure to valinomycin (data not shown). In contrast, cultures treated with staurosporine showed significant caspase-3-like protease activity after 6 hr of treatment (Fig.7a).
Fig. 7.

Valinomycin does not activate neural apoptosis.a, D283 medulloblastoma cells were treated with vehicle (Con), Val, or STS for up to 24 hr. Caspase-3 like activity was measured by cleavage of the fluorigenic substrate Ac-DEVD-AMC. Activities are presented as increase in AMC fluorescence (in arbitrary fluorescence units) over 60 min per microgram of protein. Data are means ± SEM from n = 8 cultures, and experiments were repeated three times with similar results. Different from controls, *p < 0.05. b, Immunoblot probed with an antibody recognizing procaspase-3 and active caspase-3. Cultures were exposed to vehicle (Con), Val, and STS for 6 hr. Experiment was repeated twice with similar results.c–e, Hoechst staining of D283 cells treated with vehicle (c) or Val (d) for 20 hr. Exposure to Val induced no chromatin condensation in contrast to cells treated with STS for 6 hr (e). Scale bar, 50 μm.
Lack of caspase-3 activation was also confirmed by immunoblot analysis. Caspase-3 is proteolytically activated by cleavage of its precursor pro-caspase-3 (32 kDa) into active subunits. Staurosporine-treated cultures accumulated the active caspase-3 subunit (p17) after 6 hr of treatment (Fig. 7b). In contrast, active caspase-3 could not be detected in cultures treated with valinomycin for 6 hr (Fig.7b). Similar results were obtained in cultures treated for 24 hr with valinomycin (data not shown). Chromatin condensation and fragmentation are considered to be one of the hallmarks of apoptotic cells. To observe nuclear changes, D283 cells were stained with the chromatin-specific dye Hoechst 33258. Exposure to valinomycin for 20 hr induced little chromatin condensation or fragmentation (Fig.7c–e). In contrast, nuclei of cells treated with 3 μm staurosporine for 6 hr showed a prominent apoptotic morphology (Fig. 7e). Interestingly, the toxicity of valinomycin was associated with the appearance of multiple cytoplasmic vacuoles, but cell shrinkage was not observed (data not shown). Therefore, valinomycin induced a necrotic, rather than apoptotic cell death in D283 medulloblastoma cells.
Exposure to valinomycin inhibits staurosporine-induced mitochondrial hyperpolarization and cytochrome c release
Similar to the results obtained with TMRE and CMXRos (Figs. 4, 5), mitochondria of staurosporine-treated cells accumulated more R123 fluorescence compared with vehicle-treated controls (Fig.8a,b). In contrast, cells treated with valinomycin failed to accumulate significant amounts of R123 in their mitochondria (Fig. 8c). Of note, the combined exposure to staurosporine and valinomycin reversed the increase in R123 uptake induced by staurosporine (Fig. 8d). This effect was independent of the concentration of R123 used, because similar uptake patterns were observed in cultures incubated with 100 nm, 300 nm, or 2 μm R123. Moreover, this effect was independent of the probe used, because the staurosporine-induced increase in TMRE or CMXRos uptake was also inhibited in cultures treated with valinomycin (data not shown).
Fig. 8.

Valinomycin inhibits staurosporine-induced hyperpolarization and cytochrome c release. a–d, Cultures were exposed for 30 min to vehicle (Con) (a), STS (b), Val (c), or to a combination of STS and Val (d), loaded with 2 μm R123 for 30 min and washed with HBS. STS induced an increased uptake of R123 into mitochondria (b), whereas treatment with Val decreased R123 uptake into mitochondria (c). Combined exposure to STS and Val reduced the STS-induced increase in R123 uptake (d). Scale bar, 25 μm. Experiments were repeated four times with comparable results. e, Cultures were incubated for 4 hr with vehicle, Val, STS, or a combination of Val and STS. Cells were fractionated into a cytosolic and a mitochondria-containing nuclear–heavy membrane fraction (Pellet). Immunoblot analysis of cytosolic fractions was performed using an anti-cytochrome c antibody. Blots were incubated with an anti-VDAC antibody to exclude contaminations of the cytosolic fractions with mitochondria and with an anti-α-tubulin antibody to confirm equal loading of each sample. Control pellet of vehicle-treated cells is shown. The experiment was performed in triplicate and yielded comparable results.
To examine the effect of valinomycin-induced mitochondrial depolarization on staurosporine-induced cytochrome c release, we performed an immunoblot analysis of cytosolic fractions of D283 medulloblastoma cells treated for 4 hr with staurosporine (3 μm) or valinomycin (10 μm). Exposure to staurosporine or valinomycin alone showed a significant release of cytochrome c into the cytosol after both treatments (Fig.8e), thus confirming our previous immunofluorescence and immunoblot observations (Figs. 4-6). Interestingly, in cultures treated simultaneously with valinomycin and staurosporine, release of cytochrome c was significantly decreased. Similar results were obtained in cells analyzed by cytochrome c immunofluorescence microscopy (data not shown).
Valinomycin inhibits staurosporine-induced cytochrome c release in MCF-7/Casp-3 cells transfected with cytochrome c–EGFP
The results obtained in D283 medulloblastoma cells confirmed our previous observation of an inhibition of staurosporine-induced cytochrome c release by valinomycin or FCCP in cultured rat hippocampal neurons. The interaction of staurosporine and valinomycin was further investigated in MCF-7/Casp-3 cells stably transfected with cytochrome c–EGFP. Cells exposed to vehicle for 6 hr maintained their mitochondrial cytochrome c–EGFP fluorescence (Fig.9a,e). As reported previously (Heiskanen et al., 1999), exposure to 3 μmstaurosporine induced a significant release of cytochrome c–EGFP from mitochondria, resulting in a diffuse staining of the cytosol and the nucleus (Fig. 9b,f). Six hours of exposure to 10 μm valinomycin induced prominent mitochondrial swelling, and the cytochrome c–EGFP signal was diffuse in a subpopulation of the cells (Fig. 9c,g). Of note, exposure to valinomycin greatly inhibited cytochrome c–EGFP release induced by staurosporine because mitochondrial cytochrome c signal was still clearly visible after 6 hr (Fig. 9d,h).
Fig. 9.

Valinomycin induces mitochondrial swelling and inhibits staurosporine induced cytochrome c release in MCF-7/Casp-3 cells stably transfected with cytochrome c–EGFP. Cultures were treated with vehicle (Con; a,e), STS (b, f), Val (c,g), and STS and Val in combination (d, h). Digital images were acquired before (a–d) and 6 hr after (e–h) treatment. Exposure to STS induced a significant cytochrome c release resulting in a diffuse staining of cytoplasm and nucleus (f). Val alone induced cytochrome c release (arrowheads), as well as mitochondrial swelling (arrows) (g). Exposure to Val inhibited STS-induced cytochrome c release (h). Scale bar, 10 μm. Experiments were repeated six times with similar results.
Dissipation of mitochondrial potassium and proton gradients inhibits staurosporine-induced activation of the caspase cascade
We next examined the ability of valinomycin to inhibit the activation of the caspase cascade in staurosporine-exposed D283 cells. After 6 hr treatment with 3 μm staurosporine, caspase cleavage activity increased significantly in the cultures (Fig.10a). In cultures treated simultaneously with staurosporine and valinomycin, DEVD-cleavage was significantly reduced (Fig. 10a). This result was confirmed by immunoblot analysis using an anti-caspase-3 antibody (Fig.10b). The staurosporine-induced appearance of the active p17 subunit was significantly reduced in cultures treated simultaneously with valinomycin. Cultures exposed to staurosporine plus valinomycin also demonstrated a significant decrease in the percentage of apoptotic nuclei compared with cultures exposed to staurosporine alone (Fig.10c). FCCP also inhibited staurosporine-induced caspase-3-like protease activity shown by the reduced cleavage of the Ac-DEVD-AMC substrate (Fig. 10d). Finally, cellular ATP levels were determined in D283 cells after exposure to staurosporine and valinomycin. Treatment with either staurosporine or valinomycin induced a decrease in cellular ATP levels. However, there was no difference in the reduction of cellular ATP levels between staurosporine-treated cultures and cultures treated simultaneously with staurosporine and valinomycin (Fig. 10e). Moreover, the ability of valinomycin to inhibit staurosporine-induced caspase-3-like protease activity was preserved in D283 ρ− cells deficient in mitochondrial respiration (51 ± 5% decrease in DEVDase activity after 8 hr of treatment; n = 8 cultures; p < 0.05).
Fig. 10.

Valinomycin reduces staurosporine-induced increase in caspase-3 like protease activity and apoptotic nuclear morphology. a, Apoptotic cell death was induced in D283 cells by an exposure to STS for 6 hr. Val or vehicle was added as indicated. Caspase-3 like protease activity was measured by cleavage of the Ac-DEVD-AMC substrate. Activities are represented as increase in AMC fluorescence over 60 min per microgram of protein. Val significantly reduced STS-induced Ac-DEVD-AMC cleavage. Data are means from n = 8 cultures; experiment was repeated twice with comparable results, *p < 0.05 different from controls.b, Immunoblot analysis of cultures treated with STS, Val, STS + Val or vehicle for 6 hr using an anti-caspase-3 antibody. Addition of Val to cultures treated with STS reduced cleavage of pro-caspase-3 compared with cultures treated with STS alone. Experiment was repeated twice with similar results. c, D283 cells were exposed to STS for 4 hr. Morphology of nuclei was visualized by Hoechst 33258 staining. Percentage of nuclei with fragmented chromatin was determined. n = 4 cultures (700–800 nuclei). Different from controls, *p < 0.05.d, Cell death was induced by exposure to STS for 6 hr. Vehicle or FCCP were added as indicated. Caspase-3 like activity was measured by cleavage of the Ac-DEVD-AMC substrate. Data are means from n = 8 cultures; experiments were done in duplicate and yielded similar results, *p < 0.05 different from controls. e, Determination of cellular ATP content in D283 cells exposed to vehicle, STS, Val, or STS + Val for 3 and 6 hr. Luciferase activity of cultures treated with vehicle was set to 100% activity. Treatment with STS, Val, and STS + Val resulted in a decrease in luciferase activity. Data are means fromn = 4–6 cultures per treatment.
DISCUSSION
One proposed mechanism for apoptosis induction by trophic factor withdrawal, UV irradiation, or staurosporine exposure is a disturbance of mitochondrial ion and volume homeostasis, with several theories arguing for mitochondrial depolarization, hyperpolarization, swelling, or shrinkage (Nieminen et al., 1996; White and Reynolds, 1996;Bossy-Wetzel et al., 1998; Wadia et al., 1998; Yoshida et al., 1998;Heiskanen et al., 1999; Krohn et al., 1999; Stefanis et al., 1999;Deshmukh et al., 2000; Goldstein et al., 2000; Luetjens et al., 2000;Matsuyama et al., 2000). We observed an increased uptake of the voltage-sensitive probes TMRE, R123, and CMXRos into mitochondria during staurosporine-induced apoptosis of rat hippocampal neurons and human D283 medulloblastoma cells that occurred before cytochrome c release and caspase activation. Increased uptake of voltage-sensitive probes could be detected using dyes that are taken up reversibly or irreversibly by mitochondria. However, the Nernstian behavior of these probes predicts an increase in uptake as either ΔΨm or ΔΨp increases (Nicholls and Ward, 2000). Moreover, increased uptake of voltage-sensitive probes during apoptosis could be caused by an increase in mitochondrial matrix volume (Vander Heiden et al., 1997;Buckman et al., 2001).
To distinguish between increases in ΔΨm and ΔΨp as a cause for the increased TMRE uptake during staurosporine-induced apoptosis, we have remodelled changes in cellular TMRE fluorescence based on Nernst calculations of TMRE fluorescence distribution in the extracellular, cytoplasmic, and mitochondrial compartment (Ward et al., 2000). Simulation of single-cell fluorescence changes demonstrated that the staurosporine-induced increase in TMRE fluorescence could be precisely remodelled by increasing ΔΨm, but not ΔΨp. However, it should be noted that the simulation of plasma membrane hyperpolarization in a “virtual” cell produced traces that were not identical, but similar in shape to those recorded after addition of staurosporine, or obtained with the simulation of mitochondrial membrane hyperpolarization (Fig. 3). This indicates that simple quantifications of fluorescence of membrane potential probes should be interpreted with caution (see also Ward et al., 2000). It has been reported that apoptosis of mouse neocortical neurons induced by staurosporine is associated with plasma membrane hyperpolarization because of the enhancement of a delayed rectifierIK current (Yu et al., 1996). In the latter study, however, plasma membrane hyperpolarization became evident only 9–11 hr after addition of staurosporine, and theIK current actually tended to decrease during the early stage of apoptosis (30 min after onset of the staurosporine treatment). Two other lines of evidence suggest that the increased mitochondrial uptake of voltage-sensitive probes during apoptosis was indeed caused by mitochondrial membrane hyperpolarization: (1) Blocking of outward potassium currents did not inhibit staurosporine-induced hyperpolarization. (2) The staurosporine-induced increase in TMRE and CMXRos uptake was inhibited in cells overexpressing Bcl-xL, an anti-apoptotic protein with a mainly mitochondrial site of action (Vander Heiden et al., 1997).
It is possible that the mitochondrial hyperpolarization observed during the early stage of staurosporine-induced apoptosis is caused by a Ca2+-dependent stimulation of mitochondrial energetics. Staurosporine induces a Ca2+-dependent cell death in neurons (Prehn et al., 1997), and cytosolic and mitochondrial Ca2+ overloading have been detected early during staurosporine-induced apoptosis (Kruman and Mattson, 1999). Mitochondrial Ca2+ uptake is known to stimulate the activity of several NADH-producing dehydrogenases of the mitochondrial matrix and could thereby increase the mitochondrial proton motive force. However, because this is a physiological, regulated process, it is likely that other mechanisms operate in cells undergoing stress-induced apoptosis. One potential mechanism that could cause a pathophysiological increase in ΔΨm is a shift from state 3 to state 4 respiration during apoptosis. This could be achieved by inhibiting mitochondrial ADP–ATP exchange (Vander Heiden et al., 1999).
Interestingly, dissipation of mitochondrial potassium or proton gradients by valinomycin or FCCP not only inhibited mitochondrial hyperpolarization, but also staurosporine-induced cytochrome c release. The inhibitory effect of valinomycin on staurosporine-induced cytochrome c release (or vice versa) was confirmed in three cell types using three independent techniques. It has been demonstrated that sufficient ATP levels are required to trigger apoptosis, in particular the activation of the caspase cascade (Eguchi et al., 1997; Leist et al., 1997; Lee and Shacter, 1999). Valinomycin depolarizes mitochondria and thereby reduces mitochondrial ATP production. However, the inhibitory effect of valinomycin on apoptosis occurred upstream of the caspase cascade, i.e., at the level of mitochondrial cytochrome c release. Moreover, measurements of cellular ATP revealed that the inhibitory effect of valinomycin on staurosporine-induced cytochrome c release was not attributable to a reduction of cellular ATP content. Furthermore, the ability of valinomycin to inhibit staurosporine-induced caspase-3-like protease activity was preserved in D283 ρ− cells deficient in mitochondrial respiration. Valinomycin also induces plasma membrane hyperpolarization at micromolar concentrations, as well as cytosolic acidification (Furlong et al., 1998; Holmuhamedov et al., 1998;Rottenberg and Wu, 1998). Both processes should promote, rather than inhibit apoptosis (Yu et al., 1996; Matsuyama et al., 2000).
It is therefore conceivable that valinomycin inhibited cytochrome c release and apoptosis activation by inducing mitochondrial depolarization or by counteracting a mitochondrial ion–volume homeostasis disregulation during staurosporine-induced apoptosis. Valinomycin and FCCP dissipate ΔΨm. However, the proton gradient across the inner mitochondrial membrane can be maintained in the presence of valinomycin and can increase significantly. Our findings therefore suggest that an electrical gradient across the inner membrane is required for apoptotic signaling or that the protonmotive force must be of a certain threshold. A mitochondrial membrane potential may be required for the insertion of pro-apoptotic proteins that trigger the release of cytochrome c. Depolarization of the mitochondrial membrane could also inhibit cytochrome c release by reducing Ca2+ influx into the matrix via the mitochondrial Ca2+ uniporter (Nicholls and Akerman, 1982; Murphy et al., 1996; Andreyev et al., 1998). In primary neuron cultures exposed to glutamate receptor agonists, mitochondrial depolarization has been shown to inhibit mitochondrial Ca2+ overloading, free radical production, and cell death (Castilho et al., 1998; Sengpiel et al., 1998; Stout et al., 1998).
It has also been suggested that cytochrome c release during apoptosis could be triggered by mitochondrial swelling and a subsequent rupture of the outer mitochondrial membrane (Vander Heiden et al., 1997). Large-scale mitochondrial swelling could not be detected in D283 cells treated with staurosporine. Moreover, valinomycin per se induced a prominent swelling of mitochondria but inhibited staurosporine-induced cytochrome c release. Previous studies have shown that mitochondria do not enlarge during apoptosis (Bossy-Wetzel et al., 1998; Kluck et al., 1999; Matsuyama et al., 2000; Scarlett et al., 2000), but may actually condense (Kluck et al., 1999; Martinou et al., 1999). Mitochondrial condensation could facilitate cytochrome c release, e.g., by loosening outer–inner membrane contact sites. However, it remains to be shown whether changes in mitochondrial morphology play an active role in cytochrome c release during apoptosis.
Exposure of human medulloblastoma D283 cells to the potassium-ionophore valinomycin caused significant cytochrome c release from mitochondria. In contrast to staurosporine, valinomycin decreased ΔΨm, induced mitochondrial swelling, and the subsequent cell death was necrotic rather than apoptotic. No caspase activity was detectable, and overexpression of Bcl-xL failed to reverse valinomycin-induced mitochondrial alterations. The valinomycin-induced cytochrome c release may be secondary to mitochondrial matrix swelling. Of note, our data suggest that mitochondria have to swell significantly and over a prolonged period of time to physically release their cytochrome c (Minamikawa et al., 1999). Valinomycin triggers mitochondrial swelling by selectively increasing the transport of potassium ions into the mitochondrial matrix, which then triggers the influx of anions and water. Several groups have reported that valinomycin induces PTP opening (Furlong et al., 1998; Dallaporta et al., 1999). However, it should be noted that the mechanism of valinomycin-induced mitochondrial swelling may be different from that induced by a Ca2+-induced PTP. Moreover, a Ca2+-induced PTP could be sensitive to Bcl-xL overexpression (Marzo et al., 1998).
In conclusion, the present study suggests the existence of two mechanisms of mitochondrial cytochrome c release during stress-induced cell death: (1) passive cytochrome c release secondary to strong mitochondrial depolarization and matrix swelling that is not inhibited by Bcl-xL overexpression and that predominantly leads to cell necrosis and (2) active cytochrome c release that is inhibited by Bcl-xL overexpression as well as by a dissipation of mitochondrial potassium and proton gradients.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Pr338/9–1), the Interdisciplinary Center for Clinical Research, University of Münster (Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie Grant 01 KS 9604/0), and Stiftung VerUm. We thank Gudrun Münstermann and Christiane Schettler for technical assistance, Dr. Reiner Jänicke for gift of the MCF-7/Casp-3 cell line, Prof. Craig B. Thompson for providing plasmid pSFFV-Neo-Bcl-xL and Bcl-x antiserum, and Prof. David G. Nicholls for providing the simulation program for single-cell fluorescence of voltage-sensitive probes and helpful discussions.
Correspondence should be addressed to Dr. Jochen H. M. Prehn, Interdisciplinary Center for Clinical Research, Research Group “Apoptosis and Cell Death,” Faculty of Medicine, Westphalian Wilhelms-University, Röntgenstrasse 21, D-48149 Münster, Germany. E-mail: prehn@uni-muenster.de.
REFERENCES
- 1.Andreyev AY, Fahy B, Fiskum G. Cytochrome c release from brain mitochondria is independent of the mitochondrial permeability transition. FEBS Lett. 1998;20:373–376. doi: 10.1016/s0014-5793(98)01394-5. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000;23:298–304. doi: 10.1016/s0166-2236(00)01584-8. [DOI] [PubMed] [Google Scholar]
- 3.Bernardi P, Petronilli The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr. 1996;28:131–138. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- 4.Beutner G, Ruck A, Riede B, Welte W, Brdiczka D. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett. 1996;396:189–195. doi: 10.1016/0014-5793(96)01092-7. [DOI] [PubMed] [Google Scholar]
- 5.Bobba A, Atlante A, Giannattasio S, Sgaramella G, Calissano P, Marra E. Early release and subsequent caspase-mediated degradation of cytochrome c in apoptotic granule cells. FEBS Lett. 1999;457:126–130. doi: 10.1016/s0014-5793(99)01018-2. [DOI] [PubMed] [Google Scholar]
- 6.Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 7.Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buckman JF, Hernandez H, Kress GJ, Votyakova TV, Pal S, Reynolds IJ. MitoTracker labeling in primary neuronal and astrocytic cultures: influence of mitochondrial membrane potential and oxidants. J Neurosci Methods. 2001;104:165–176. doi: 10.1016/s0165-0270(00)00340-x. [DOI] [PubMed] [Google Scholar]
- 9.Castilho RF, Hansson O, Ward MW, Budd SL, Nicholls DG. Mitochondrial control of acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 1998;18:10277–10286. doi: 10.1523/JNEUROSCI.18-24-10277.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dallaporta B, Marchetti P, de Pablo MA, Maisse C, Duc HT, Metivier D, Zamzami N, Geuskens M, Kroemer G. Plasma membrane potential in thymocyte apoptosis. J Immunol. 1999;162:6534–6542. [PubMed] [Google Scholar]
- 11.Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–377. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- 12.Deshmukh M, Kuida K, Johnson EM., Jr Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol. 2000;150:131–143. doi: 10.1083/jcb.150.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Vos K, Goossens V, Boone E, Vercammen D, Vancompernolle K, Vandenabeele P, Haegeman G, Fiers W, Grooten J. The 55-kDa tumor necrosis factor receptor induces clustering of mitochondria through its membrane-proximal region. J Biol Chem. 1998;273:9673–9680. doi: 10.1074/jbc.273.16.9673. [DOI] [PubMed] [Google Scholar]
- 14.Dey R, Moraes CT. Lack of oxidative phosphorylation and low mitochondrial membrane potential decrease susceptibility to apoptosis and do not modulate the protective effect of Bcl-xL in osteosarcoma cells. J Biol Chem. 2000;275:7087–7094. doi: 10.1074/jbc.275.10.7087. [DOI] [PubMed] [Google Scholar]
- 15.Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–1840. [PubMed] [Google Scholar]
- 16.Ehrenberg B, Montana V, Wie MD, Wuskell JP, Loew LM. Membrane potential can be determined in individual cells from the Nernstian distribution of cationic dyes. Biophys J. 1988;53:785–794. doi: 10.1016/S0006-3495(88)83158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellerby HM, Martin SJ, Ellerby LM, Naiem SS, Rabizadeh S, Salvesen GS, Casiano CA, Cashman NR, Green DR, Bredesen DE. Establishment of a cell-free system of neuronal apoptosis: comparison of premitochondrial, mitochondrial, and postmitochondrial phases. J Neurosci. 1997;17:6165–6178. [PMC free article] [PubMed] [Google Scholar]
- 18.Emaus RK, Grunwald R, Lemasters JJ. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties. Biochim Biophys Acta. 1986;850:436–448. doi: 10.1016/0005-2728(86)90112-x. [DOI] [PubMed] [Google Scholar]
- 19.Furlong IJ, Lopez Mediavilla C, Ascaso R, Lopez Rivas A, Collins MK. Induction of apoptosis by valinomycin: mitochondrial permeability transition causes intracellular acidification. Cell Death Differ. 1998;5:214–221. doi: 10.1038/sj.cdd.4400335. [DOI] [PubMed] [Google Scholar]
- 20.Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 21.Heiskanen KM, Bhat MB, Wang HW, Ma J, Nieminen AL. Mitochondrial depolarization accompanies cytochrome c release during apoptosis in PC6 cells. J Biol Chem. 1999;274:5654–5658. doi: 10.1074/jbc.274.9.5654. [DOI] [PubMed] [Google Scholar]
- 22.Holmuhamedov EL, Jovanovic S, Dzeja PP, Jovanovic A, Terzic A. Mitochondrial ATP-sensitive K+ channels modulate cardiac mitochondrial function. Am J Physiol. 1998;275:H1567–H1576. doi: 10.1152/ajpheart.1998.275.5.H1567. [DOI] [PubMed] [Google Scholar]
- 23.Jacobson S. Neurocytology. In: Curtis BA, Jacobson S, Marcus EM, editors. An introduction to the neurosciences. Saunders; Philadelphia: 1972. pp. 36–71. [Google Scholar]
- 24.Jänicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 25.Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19:5800–5810. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim CN, Wang X, Huang Y, Ibrado AM, Liu L, Fang G, Bhalla K. Overexpression of Bcl-X(L) inhibits Ara-C-induced mitochondrial loss of cytochrome c and other perturbations that activate the molecular cascade of apoptosis. Cancer Res. 1997;57:3115–3120. [PubMed] [Google Scholar]
- 27.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 28.Kluck RM, Esposti MD, Perkins G, Renken C, Kuwana T, Bossy-Wetzel E, Goldberg M, Allen T, Barber MJ, Green DR, Newmeyer DD. The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J Cell Biol. 1999;147:809–822. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krohn AJ, Preis E, Prehn JHM. Staurosporine-induced apoptosis of cultured rat hippocampal neurons involves caspase-1-like proteases as upstream initiators and increased production of superoxide as a main downstream effector. J Neurosci. 1998;18:8186–8197. doi: 10.1523/JNEUROSCI.18-20-08186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krohn AJ, Wahlbrink T, Prehn JHM. Mitochondrial depolarization is not required for neuronal apoptosis. J Neurosci. 1999;19:7394–7404. doi: 10.1523/JNEUROSCI.19-17-07394.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kruman II, Mattson MP. Pivotal role of mitochondrial calcium uptake in neural cell apoptosis and necrosis. J Neurochem. 1999;72:529–540. doi: 10.1046/j.1471-4159.1999.0720529.x. [DOI] [PubMed] [Google Scholar]
- 32.Lee YJ, Shacter E. Oxidative stress inhibits apoptosis in human lymphoma cells. J Biol Chem. 1999;274:19792–19798. doi: 10.1074/jbc.274.28.19792. [DOI] [PubMed] [Google Scholar]
- 33.Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. 1998;1366:177–196. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- 35.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 36.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 37.Luetjens CM, Bui NT, Sengpiel B, Münstermann G, Poppe M, Krohn AJ, Bauerbach E, Krieglstein J, Prehn JHM. Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J Neurosci. 2000;20:5715–5723. doi: 10.1523/JNEUROSCI.20-15-05715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinou I, Desagher S, Eskes R, Antonsson B, Andre E, Fakan S, Martinou JC. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J Cell Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 40.Matsuyama S, Llopis J, Deveraux QL, Tsien RY, Reed JC. Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nat Cell Biol. 2000;2:318–324. doi: 10.1038/35014006. [DOI] [PubMed] [Google Scholar]
- 41.Minamikawa T, Williams DA, Bowser DN, Nagley P. Mitochondrial permeability transition and swelling can occur reversibly without inducing cell death in intact human cells. Exp Cell Res. 1999;246:26–37. doi: 10.1006/excr.1998.4290. [DOI] [PubMed] [Google Scholar]
- 42.Monck JR, Oberhauser AF, Keating TJ, Fernandez JM. Thin-section ratiometric Ca2+ images obtained by optical sectioning of fura-2 loaded mast cells. J Cell Biol. 1992;116:745–759. doi: 10.1083/jcb.116.3.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc Natl Acad Sci USA. 1996;93:9893–9898. doi: 10.1073/pnas.93.18.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murphy AN, Fiskum G, Beal MF. Mitochondria in neurodegeneration: bioenergetic function in cell life and death. J Cereb Blood Flow Metab. 1999;19:231–245. doi: 10.1097/00004647-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 45.Nicholls D, Akerman K. Mitochondrial calcium transport. Biochim Biophys Acta. 1982;683:57–88. doi: 10.1016/0304-4173(82)90013-1. [DOI] [PubMed] [Google Scholar]
- 46.Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- 47.Nicolli A, Basso E, Petronilli V, Wenger RM, Bernardi P. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, and cyclosporin A-sensitive channel. J Biol Chem. 1996;271:2185–2192. doi: 10.1074/jbc.271.4.2185. [DOI] [PubMed] [Google Scholar]
- 48.Nieminen AL, Petrie TG, Lemasters JJ, Selman WR. Cyclosporin A delays mitochondrial depolarization induced by N-methyl-d-aspartate in cortical neurons: evidence of the mitochondrial permeability transition. Neuroscience. 1996;75:993–997. doi: 10.1016/0306-4522(96)00378-8. [DOI] [PubMed] [Google Scholar]
- 49.Prehn JHM, Jordan J, Ghadge GD, Preis E, Galindo MF, Roos RP, Krieglstein J, Miller RJ. Ca2+ and reactive oxygen species in staurosporine-induced neuronal apoptosis. J Neurochem. 1997;68:1679–1685. doi: 10.1046/j.1471-4159.1997.68041679.x. [DOI] [PubMed] [Google Scholar]
- 50.Rottenberg H, Wu S. Quantitative assay by flow cytometry of the mitochondrial membrane potential in intact cells. Biochim Biophys Acta. 1998;1404:393–404. doi: 10.1016/s0167-4889(98)00088-3. [DOI] [PubMed] [Google Scholar]
- 51.Scarlett JL, Sheard PW, Hughes G, Ledgerwood EC, Ku HH, Murphy MP. Changes in mitochondrial membrane potential during staurosporine-induced apoptosis in Jurkat cells. FEBS Lett. 2000;475:267–272. doi: 10.1016/s0014-5793(00)01681-1. [DOI] [PubMed] [Google Scholar]
- 52.Schendel SL, Montal M, Reed JC. Bcl-2 family proteins as ion channels. Cell Death Differ. 1998;5:372–380. doi: 10.1038/sj.cdd.4400365. [DOI] [PubMed] [Google Scholar]
- 53.Sengpiel B, Preis E, Krieglstein J, Prehn JHM. NMDA-induced superoxide production and neurotoxicity in cultured rat hippocampal neurons: role of mitochondria. Eur J Neurosci. 1998;10:1903–1910. doi: 10.1046/j.1460-9568.1998.00202.x. [DOI] [PubMed] [Google Scholar]
- 54.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 55.Stefanis L, Park DW, Friedman WJ, Greene LA. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- 57.Trimmer PA, Swerdlow RH, Parks JK, Keeney P, Bennett JP, Jr, Miller SW, Davis RE, Parker WD., Jr Abnormal mitochondrial morphology in sporadic Parkinson's and Alzheimer's disease cybrid cell lines. Exp Neurol. 2000;162:37–50. doi: 10.1006/exnr.2000.7333. [DOI] [PubMed] [Google Scholar]
- 58.Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- 60.Vander Heiden MG, Chandel NS, Schumacker PT, Thompson CB. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol Cell. 1999;3:159–167. doi: 10.1016/s1097-2765(00)80307-x. [DOI] [PubMed] [Google Scholar]
- 61.Vinoris SA, Herman MM, Katsetos CD, May EE, Frankfurter A. Neuron-associated class III tubulin, tau and MAP2 in the D-283 Med cell line and in primary explants of human medulloblastoma. Histochem J. 1994;26:678–685. doi: 10.1007/BF00158293. [DOI] [PubMed] [Google Scholar]
- 62.Wadia JS, Chalmers-Redman RME, Ju WJH, Carlile GW, Phillips JL, Fraser AD, Tatton WG. Mitochondrial membrane potential and nuclear changes in apoptosis caused by serum and nerve growth factor withdrawal: time course and modification by (−)-deprenyl. J Neurosci. 1998;18:932–947. doi: 10.1523/JNEUROSCI.18-03-00932.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ward MA, Rego AC, Frenguelli BG, Nicholls DG. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 2000;20:7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 66.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 67.Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LMT, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1996;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- 68.Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 70.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]