

Abstract
AMPA receptor (AMPAR)-mediated ionic currents that govern gene expression, synaptic strength, and plasticity also can trigger excitotoxicity. However, native AMPARs exhibit heterogeneous pharmacological, biochemical, and ionic permeability characteristics, which are governed partly by receptor subunit composition. Consequently, the mechanisms governing AMPAR-mediated excitotoxicity have been difficult to elucidate. The GluR2 subunit is of particular interest because it influences AMPAR pharmacology, Ca2+ permeability, and AMPAR interactions with intracellular proteins. In this paper we used mutant mice lacking the AMPAR subunit GluR2 to study AMPAR-mediated excitotoxicity in cultured cortical neurons and in hippocampal neurons in vivo. We examined the hypothesis that in these mice the level of GluR2 expression governs the vulnerability of neurons to excitotoxicity and further examined the ionic mechanisms that are involved. In cortical neuronal cultures AMPAR-mediated neurotoxicity paralleled the magnitude of kainate-evoked AMPAR-mediated currents, which were increased in neurons lacking GluR2. Ca2+ permeability, although elevated in GluR2-deficient neurons, did not correlate with excitotoxicity. However, toxicity was reduced by removal of extracellular Na+, the main charge carrier of AMPAR-mediated currents. In vivo, the vulnerability of CA1 hippocampal neurons to stereotactic kainate injections and of CA3 neurons to intraperitoneal kainate administration was independent of GluR2 level. Neurons lacking the GluR2 subunit did not demonstrate compensatory changes in the distribution, expression, or function of AMPARs or of Ca2+-buffering proteins. Thus GluR2 level may influence excitotoxicity by effects additional to those on Ca2+ permeability, such as effects on agonist potency, ionic currents, and synaptic reorganization.
Keywords: AMPA receptors, kainate, excitotoxicity, GluR2 subunit, calcium permeability, cortical neurons
Ionic flux via NMDA and AMPA glutamate receptors (NMDARs and AMPARs, respectively) can trigger neuronal death after excitotoxic and hypoxic/ischemic insults (Choi, 1988; Tymianski, 1996; Ying et al., 1997). NMDAR activity exhibits relatively homogeneous macroscopic ionic currents characterized by a high permeability to Na+, K+, and Ca2+ions for which the roles in NMDAR-mediated excitotoxicity are established (Choi, 1988; Tymianski, 1996; Sattler et al., 1999). Compared with NMDARs, native neuronal AMPARs exhibit more heterogeneous macroscopic ionic current properties and ionic permeability characteristics. Their biophysical and pharmacological properties are governed by four genes (GluR1 to GluR4 or GluR-A to GluR-D) that encode heteromeric receptors with high AMPA affinities that are permeable to Na+ and K+ions (Hollmann and Heinemann, 1994). However, the relative expression of these genes, as well as the splicing and editing of their mRNAs, imparts a diversity of pharmacological properties, gating characteristics, and Ca2+ permeability between cells (Geiger et al., 1995). Specifically, impermeability to Ca2+ is determined by the presence of the GluR2 subunit, which has a positively charged arginine at position 586 of transmembrane segment 2 (Q/R site) instead of a neutral glutamine (Hume et al., 1991; Burnashev et al., 1992). Thus permeability to Ca2+ ions is highest in AMPARs that lack GluR2.
However, the GluR2 subunit governs more than just Ca2+ permeability. GluR subunits display sequence divergence within the C-terminal (CT) cytoplasmic tail, and this region has been shown to mediate subunit-specific interactions with various cytoplasmic proteins (Dong et al., 1997; Lin and Sheng, 1998; Osten et al., 1998; Xia et al., 1999). These AMPAR CT–protein interactions may govern the pharmacological properties of the receptor (Mainen et al., 1998; Cotton and Partin, 2000), receptor turnover at synapses (Man et al., 2000), clustering (Matsuda et al., 2000), synaptic transmission, efficacy, and plasticity (Jia et al., 1996;Nishimune et al., 1998; Luthi et al., 1999). Thus the influence of GluR2 subunits on neuronal function and vulnerability to excitotoxicity may occur by mechanisms other than solely those attributable to the effects of GluR2 on ionic permeability profiles.
GluR2 is expressed widely in mammalian neurons. For example, in cultured dissociated cortical neurons, a preparation that commonly is used to study excitotoxicity, only 8–15% of neurons express AMPA channels lacking GluR2 (Pruss et al., 1991; Turetsky et al., 1994; Lu et al., 1996). In vivo, GluR2 is expressed widely in hippocampal pyramidal and granule neurons (Hollmann and Heinemann, 1994) and in cortical neurons (Kondo et al., 1997) that frequently are damaged by ischemia. Thus the relative abundance and, yet, heterogeneity of GluR2 expression have made it more difficult to define its role in AMPAR-mediated excitotoxicity.
Previous studies already have examined ionic mechanisms of AMPAR-mediated excitotoxicity. Despite generally low calcium permeability, AMPAR toxicity is likely to be, at least in part, mediated by Ca2+ ions (Pellegrini-Giampietro et al., 1992; Brorson et al., 1994; Turetsky et al., 1994; Lu et al., 1996; Gorter et al., 1997; Carriedo et al., 1998). However, difficulties arise in determining how GluR2 level and Ca2+ permeability relate to AMPAR-mediated toxicity because neurons that express GluR2 exhibit at least some Ca2+ permeability (Brorson et al., 1999), and measurements of whole-cell relative Ca2+ permeability and GluR2 levels in single cultured neurons do not necessarily correlate with their overall vulnerability to AMPAR overactivation (Vandenberghe et al., 2000). Further indication that Ca2+ permeability alone may not be the sole predictor of vulnerability arose from studies that used mice with GluR2 mutations producing AMPARs with high Ca2+ permeability. These animals displayed adverse changes in behavior and phenotype, underscoring the importance of the GluR2 subunit. However, they did not exhibit neuropathological lesions suggestive of excitotoxicity (Jia et al., 1996; Kask et al., 1998; Feldmeyer et al., 1999). These raise the possibility that GluR2 is involved in governing neurological development and function by subtler mechanisms than those related only to Ca2+ permeability. We wondered whether similar factors also could contribute to AMPAR-mediated neurotoxicity.
Because of the importance of AMPARs, the GluR2 subunit, and Ca2+ ions in neuronal function and excitotoxicity (Turetsky et al., 1994; Lu et al., 1996; Tymianski, 1996; Carriedo et al., 1998), we examined the hypothesis that the level of GluR2 expression governs the vulnerability of neurons to AMPAR-mediated neuronal damage. Additional experiments also were performed to determine whether Ca2+permeability alone or additional GluR2-related factors participate in governing AMPAR-mediated excitotoxicity. To this end, we studied mice deficient in the GluR2 AMPAR subunit (Jia et al., 1996). AMPARs in neurons of homozygous mice [GluR2(−/−)] are uniformly Ca2+-permeable, providing an unprecedented opportunity to examine the effect of Ca2+ permeability on AMPAR function as compared with wild-type [GluR2(+/+)] and heterozygous [GluR2(+/−)] controls. By controlling for GluR2 level, we eliminated the confounding effects of uncertain Ca2+ permeability and were able to determine its impact on excitotoxicity. Here we demonstrate that AMPAR-mediated excitotoxicity cannot be attributed solely to increased Ca2+ permeability. AMPAR-mediated excitotoxicity is affected by GluR2 level because of the influence of the GluR2 subunit on agonist affinity and the amplitude of macroscopic ionic currents.
MATERIALS AND METHODS
Experimental animals. GluR2 mutant mice were generated as described in Jia et al. (1996). In brief, for disruption of the GluR2 locus, an isogenic targeting vector was designed to delete transmembrane region 1 and the pore loop, which are essential for receptor function (Hollmann and Heinemann, 1994). R1 embryonic stem (ES) cells (strain 129) were electroporated with this vector and selected in G418 and ganciclovir (Nagy et al., 1993). Double-resistant clones were screened for the desired homologous recombination by Southern blotting, using a probe 5′ to exon 10. Four ES clones contained the targeting events and were used to produce aggregation chimeras with CD1 morulae (Wood et al., 1993). Only one ES clone transmitted the GluR2 mutation through the germline. Heterozygous mice from a CD1 × 129 cross were intercrossed to produce 477 F2 offspring in a 1:2:1 Mendelian ratio of GluR2(+/+):GluR2(+/−):GluR2(−/−), suggesting no embryonic lethality in the mutants. F2 littermates from the same cross were used throughout.
Mixed cortical cell cultures. Cultures containing both neurons and glia were prepared separately from each 1- to 2-d-old postnatal mouse pup born of GluR2(+/−)parents. Otherwise, the cultures were prepared as previously described (Sattler et al., 1997, 1998). In brief, cerebral cortices from each pup were incubated for 10–12 min in 0.05% trypsin in EDTA, dissociated by trituration, and plated on poly-l-ornithine-coated 24-well plates (Corning, Corning, NY) or glass coverslips at a density of 0.43 × 106 cells/well or 0.9 × 106 cells/coverslip. Plating medium consisted of Eagle's minimum essential medium (MEM, Earle's salt) supplemented with 10% heat-inactivated horse serum (ICN Biochemicals, Montr—al, Canada) and (in mm) 2 glutamine, 25 glucose, and 26 bicarbonate. The cultures were maintained at 37°C in a humidified 5% CO2 atmosphere. After 3–5 d in vitro the growth of non-neuronal cells was halted by a 24–48 hr exposure to 10 μm FDU solution [5 μm uridine and 5 μm(+)-5-fluor-2′-deoxyuridine]. The cultures were used for experiments on day 11 (12–13 d postnatal). Embryonic cortical neuronal cultures (used for Fig. 6A) were produced as above from embryonic Swiss mice at 15 d of gestation and used on days 12–14in vitro.
Fig. 6.

Effects of ion substitution and of GluR2 levels on kainate toxicity. A, Effect of Na+ removal, K+ supplementation, and protein synthesis inhibition on kainate toxicity in wild-type neurons. For low Na+, NMDG was substituted for NaCl in the control solution. For high-K+, 15 mm KCl was substituted for 15 mm NaCl, for a total of 20 mm K+. Cycloheximide (CHX) was applied at 1 μg/ml. *t(14) = 6.06; p < 0.0001. **t(22) = 5.83;p < 0.0001. n = 8–12 cultures per condition. B, Effect of Na+removal on the toxicity of equi-effective kainate concentrations in GluR2 mutant neurons. Data for controls are from Figure3Cii. Na+ removal had equal effects on all mutant groups (ANOVA, F = 0.16;p = 0.85) and reduced kainate toxicity by ∼50%. *Difference from same GluR2 group control.t16(+/+) = 2.83;p = 0.01.t15(+/−)= 2.30; p = 0.04.t15(−/−)= 3.59; p = 0.002. C, Effect of Na+ removal on the toxicity of 100 μmkainate in GluR2 mutants. Data for controls are from Figure3Ciii. GluR(−/−)neurons were more vulnerable to kainate toxicity than Glu(+/+) neurons both in the presence of Na+ (see Fig. 3) and in its absence (ANOVA,F = 6.4; p = 0.007 for NMDG group). *Difference from GluR(+/+), Bonferronit test; p < 0.01. Na+ removal was protective in GluR2(−/−)neurons. **Difference from controls,t22(−/−)= 3.88; p = 0.0008. D, Effect of Ca2+ removal on the toxicity of 100 μmkainate in GluR2 mutants. The control solution was modified by omitting CaCl2 and by adding 100 μm EGTA, a Ca2+ chelator. The rank order of vulnerability to kainate toxicity was proportional to the GluR2 level, with GluR(−/−)remaining more vulnerable to kainate toxicity than Glu(+/+) neurons (ANOVA, F = 3.7; p = 0.039). *Difference from GluR(+/+), Bonferroni t test;p < 0.05. N.S., No significant difference from controls (t13(−/−)= 1.55; p = 0.144).
Because experiments were performed on cultures grown from pups born of two GluR2(+/−)parents, each data set was obtained from sister cultures that included same-generation GluR2(+/+), GluR2(+/−), and GluR2(−/−)cultures.
Electrophysiology. Whole-cell patch-clamp recordings were performed in the cultured neurons at room temperature (RT), as previously described (Jia et al., 1996). The extracellular solution contained (in mm): 140 NaCl, 5.4 KCl, 1.0 CaCl2, 25 HEPES, 33 glucose, and 0.0003 tetrodotoxin, pH 7.3–7.4, at 320–335 mOsm. A multi-barrel perfusion system was used to exchange kainate-containing solutions rapidly. The pipette solution contained (in mm): 140 CsF, 35 CsOH, 10 HEPES, 11 EGTA, 2 tetraethylammonium chloride (TEA), 1 CaCl2, and 4 MgATP, pH 7.3, at 300 mOsm. The neurons were patch-clamped at a holding potential of −60 mV. ThePCa2+/PCs+permeability ratios for mutant and wild-type neurons, acutely isolated from hippocampal slices, were calculated previously in our laboratory by studying the reversal potential of currents recorded in low or high Ca2+ solutions. These solutions contained the following (in mm): 140 NaCl, 0.2 or 20 CaCl2, 5.4 KCl, 25 HEPES, 33 or 13 glucose, and 0.0005–0.0001 TTX. A Na+-free solution consisted of 10 mm CaCl2and 25 mm HEPES with equi-osmotic glucose or sucrose substituted for NaCl. The relative permeability ratios were determined to bePCa2+/PCs+= 3.51 and 0.41 for mutant and wild-type neurons, respectively, as calculated from the reversal potential of the constant field equation:
where E is the reversal potential, F,R, and room temperature T are standard thermodynamic parameters, and PCa2+and PCs+ represent permeability coefficients for Ca2+ and Cs+ (Lewis, 1979; Burnashev et al., 1995). To determine whether the mutant cultured cortical neurons were also relatively more permeable to Ca2+, we changed the extracellular Ca2+concentration from 1 to 20 mm, and we examined the shift in reversal potential.
Histological techniques. Kainate-activated cobalt labeling (see Fig. 1) was performed as previously described (Pruss et al., 1991;Turetsky et al., 1994). In brief, the cells were exposed to 100 μm kainate plus 5 mmCoCl2 in uptake buffer (in mm): 139 sucrose, 57.7 NaCl, 5 KCl, 2 MgCl2, 1 CaCl2, 12 glucose, and 10 HEPES, pH 7.6, for 30 min at RT. Then the cultures were washed in uptake buffer containing 5 mm EDTA to chelate any extracellular cobalt. After a 5 min incubation in 0.12% (NH4)2S, the cells were washed three times in uptake buffer and finally fixed in 4% paraformaldehyde for 30 min at RT. Enhancement of the CoS precipitation was performed by washing the fixed cells three times in development buffer (in mm): 292 sucrose, 15.5 hydroquinone, and 42 citric acid and then by incubating them in 0.1% AgNO3 in development buffer at 50°C. This solution was changed every 15 min until the silver enhancement was complete (usually four changes). The reaction was terminated by washing the cultures three times with development buffer.
Fig. 1.
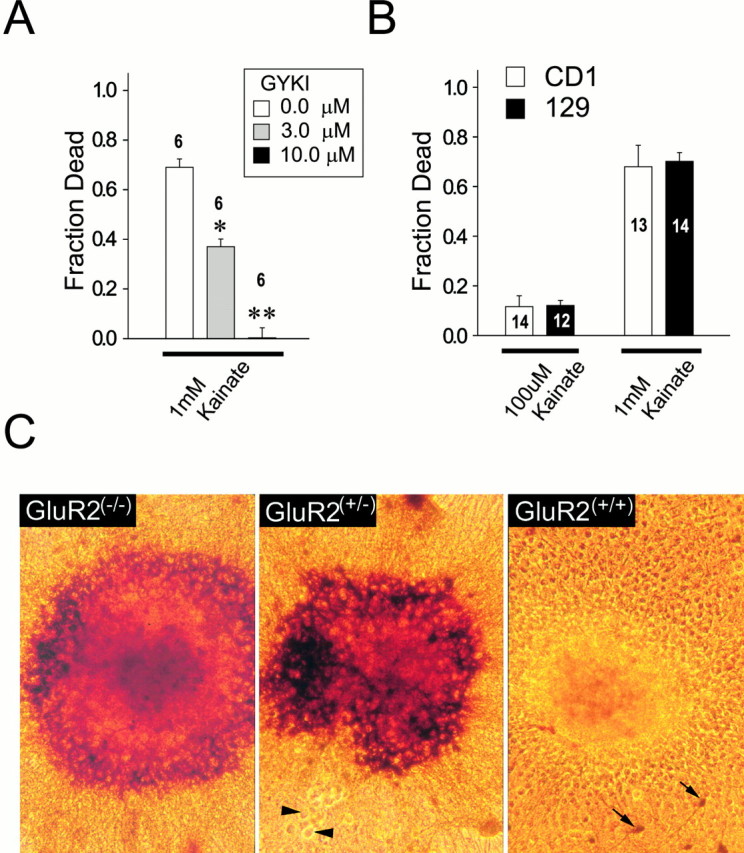
Characterization of kainate toxicity in GluR2 mutant cultures. A, Kainate toxicity is mediated by AMPARs. Wild-type neurons were exposed to 1 mm kainate for 24 hr in solution containing 10 μm MK-801, 2 μm nimodipine, and (in mm): 121 NaCl, 5 KCl, 1 Na-pyruvate, 1.8 CaCl2, 25 NaHCO3, and 20 d-glucose, pH 7.4. Kainate toxicity was abolished by 10 μm (−)-GYKI 53784, a selective AMPAR antagonist (ANOVA, F = 223;p < 0.0001; n = 6 cultures per condition). *p < 0.05, **p < 0.01 differences from controls. B, No differences in vulnerability to kainate toxicity between CD1 and 129 strains at 100 μm kainate (t24 = 0.52;p = 0.61) and at 1 mm kainate (t25 = 0.26; p = 0.80). Cultures were exposed to kainate (0.1–1 mm) as above. Numbers in bars indicaten cultures per condition. C, Representative staining for kainate-activated cobalt uptake in neuronal clusters from GluR2 mutant mice. Although clusters were uncommon in the cultures, these pictures are provided to illustrate the striking differences between GluR2(+/+) and GluR2(−/−)cultures. Arrowheads, Cobalt-positive neurons in GluR2(+/+) cultures. Scale bar, 100 μm.
Immunolabeling for GluR1 was performed in the cultured cells as described previously (Allison et al., 1998; Sattler et al., 2000). In brief, the cells were fixed first with 4% paraformaldehyde in PBS plus 4% sucrose for 20 min at 4°C. Cultures subsequently were fixed in ice-cold 100% methanol for 10 min at 4°C. After repeated washing, they were permeabilized with 0.02% Triton X-100 in PBS for 10 min at 4°C, blocked in 10% goat serum in PBS for 45 min at RT, followed by incubation with a rabbit affinity-purified anti-rat GluR1 IgG (1:3000 dilution; Upstate Biotechnology, Lake Placid, NY) primary antibody in 10% goat serum in PBS for 3 hr at RT or 37°C. Then the cultures were washed and incubated with secondary antibody (Cy5.5-tagged goat anti-rabbit IgG; 1:500 dilution; Jackson ImmunoResearch, West Grove, PA) for 1.5 hr at RT. Immunostaining was visualized with a laser-scanning confocal microscope (Bio-Rad MRC 1000, Hercules, CA) through a 60× oil immersion lens.
For calbindin staining, paraformaldehyde-fixed brain sections (30 μm) from four 1-month-old GluR2 mutant mice were labeled with a monoclonal anti-calbindin-D mouse IgG1 (1:200 dilution; Sigma, St. Louis, MO) and then with the Vectastatin elite kit (Vector Labs, Burlingame, CA), using diaminobenzidine as the chromogen.
Immunoblotting. Immunoblotting was done as described (Jia et al., 1996; Sattler et al., 1999, 2000) by using cells harvested from two cultures per genotype per lane or from three brains of each GluR2 genotype. The blotted proteins were probed with a rabbit affinity-purified anti-rat GluR1 IgG (1:3000 dilution; Upstate Biotechnology) or a monoclonal anti-calbindin-D mouse IgG1 (1:200 dilution; Sigma). Then the blots were probed with sheep anti-mouse or donkey anti-rabbit Ig conjugated to horseradish peroxidase (Amersham, Arlington Heights, IL), and the proteins were detected by enhanced chemiluminescence (Amersham).
Neuronal cell death measurements. These measurements were performed by serial quantitative measurements of propidium iodide (PI) fluorescence, using a multiwell plate fluorescence scanner (Cytofluor II, PerSeptive Biosytems, Framingham, MA) as described and previously validated (Sattler et al., 1997, 1998). In brief, the culture medium in each tissue culture well was replaced with control solution containing 50 μg/ml PI, and a baseline fluorescence reading was taken. Then sequential readings were taken up to 24 hr after the experimental manipulations. The fraction of dead neurons in each culture at a given time was calculated as:
where Ft = PI fluorescence at time t, Fc = PI fluorescence of controls at 24 hr, andFNMDA = background-subtracted PI fluorescence of identical cultures from the same dissection and plating 24 hr after a 60 min exposure to 100 μm NMDA at 37°C. Based on manual observations made at the time of validation of this technique, this NMDA exposure routinely produced near-complete neuronal death in each culture but had no effect on surrounding glia (also see Bruno et al., 1994; David et al., 1996; Sattler et al., 1997). The control solution consisted of MEM supplemented with 50 μg/ml PI. For kainate exposures the solution also contained MK-801 (10 μm; Research Biochemicals, Natick, MA) and nimodipine (2 μm; Miles Pharmaceuticals, Elkhart, IN), whereas for NMDA exposures the solution contained 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μm; Research Biochemicals) and nimodipine. All experiments were performed at 37°C.
In vivo kainate injections. For intrathecal injection studies, kainic acid was injected stereotactically into mice 7–9 weeks of age. Kainate was administered through a pulled glass capillary needle (60 μm diameter) that was inserted halfway between the bregma and lambda sutures, 2 mm lateral to the midline, at a depth of 1.5 mm (see Fig. 4C). Kainic acid or saline (200 nl) was introduced over 2 min. The needle was withdrawn after an additional 1 min wait. Intraperitoneal kainate injections (15–25 mg/kg) were performed by using kainate dissolved in 200–300 μl of saline. Animals were monitored for 2 hr for the onset of seizures, and the extent of injury was determined after 48 hr or 7 d from 10 μm sections taken over a 250 μm interval between 200 and 450 μm rostral to the needle tract. Estimates of cell death estimates were obtained by manually counting Nissl-stained sections from the central portion of the CA1 or CA3 sector from every fifth section.
Fig. 4.

Increased Ca2+ entry into GluR2(−/−)neurons. Kainate was applied in the presence of MK-801 and nimodipine.A–C, Experiments with fura-2. Kainate was applied for 10 min. A, Representative time course of [Ca2+]i averaged fromn = 4 GluR2(+/+) neurons.B, Representative time course of [Ca2+]i averaged fromn = 5 GluR2(−/−)neurons. C, Pooled baseline and peak [Ca2+]i measurements from three separate cultures per group. *p < 0.05 between wild-type and homozygous neurons. D, E, Confocal imaging of [Ca2+]i with fluo-3. Kainate (100 μm) was applied for 25 sec.D, Representative time course of [Ca2+]i averaged fromn = 4 GluR2(+/+) neurons.E, Representative time course of [Ca2+]i averaged fromn = 4 GluR2(−/−)neurons. F, Peak [Ca2+]i transients in the soma and dendrites of neurons measured with fluo-3 and evoked by 30 sec applications of NMDA (100 μm) in the presence of CNQX and nimodipine. Data were pooled from 13–18 cultures from two dissections.G, Peak [Ca2+]itransients measured with fluo-3 and evoked by 25 sec applications of kainate, using equipotent (i, ii) and nonequipotent (iii, iv) concentrations. i, EC 50(+/+); ii, EC 90(+/+); iii, 100 μm for all groups; iv, 1 mmfor all groups. *p < 0.01; Bonferronit test indicating differences from GluR2(+/−) and GluR2(−/−).Numbers in legends indicate numbers of cultures per group. Data were pooled from at least two dissections. Dendritic [Ca2+]i was measured 50–100 μm from the cell soma.
Fluorescence imaging. All experiments were performed on dissociated cultures grown on glass coverslips. Immunostained GluR1 clusters were visualized with the 647 nm laser line of a confocal microscope (Bio-Rad MRC 1000) through a 60× oil immersion lens [numerical aperture (NA) 1.4; Nikon]. Fluorescent clusters were counted by two independent observers in randomly selected dendrites in imaged neurons and were expressed as the numbers of clusters per unit of dendrite length (Allison et al., 1998; Sattler et al., 2000).
Fura-2 [Ca2+]iimaging was performed in the neuronal cultures identically to methods previously described (Tymianski et al., 1993; Sattler et al., 1998). In brief, neurons were loaded with fura-2 AM (2 μm; Molecular Probes, Eugene, OR) and viewed with an inverted microscope (Nikon Diaphot, xenon epifluorescence optics) through a fluorite oil immersion lens (Nikon CF UV-F 40×, NA = 1.3). Fura-2 excitation was evoked through narrow bandpass filters (340 ± 5/380 ± 6.5 nm), and fluorescence emissions >510 nm were recorded with an intensified CCD array camera (Quantex Model QX-100) interfaced to a PC-based personal computer. Four to eight images were averaged at each excitation wavelength and corrected for background fluorescence and camera dark current by subtracting a frame taken at the beginning of each experiment at each excitation wavelength from an area of the coverslip devoid of cells. Changes in [Ca2+]i were expressed as the background-subtracted 340/380 nm fura-2 fluorescence ratio. Fluo-3 [Ca2+]i imaging was performed with the confocal microscope in cultures loaded with fluo-3 AM (5 μm) with a 40× oil immersion lens (Nikon, 1.3 NA), using identical settings for all experiments (excitation 488 nm; emission 515 LP; iris 6.7 mm; gain 1440; laser intensity 3%; zoom 3.0), and a multi-barrel perfusion system to exchange solutions. Kainate solutions contained 10 μm MK-801, 2 μm nimodipine, and (in mm): 121 NaCl, 5 KCl, 20 d-glucose, 10 HEPES acid, 7 HEPES-Na salt, 3 NaHCO3, and 1.8 CaCl2.
Data analysis. All data were analyzed by ANOVA, with apost hoc Student's t test, using the Bonferroni correction for multiple comparisons. All means are presented with their standard errors. Cell death measurements reported in all figures are baseline-subtracted to reflect only the cell death produced by the experimental insult (e.g., kainate or NMDA application). Baseline cell death in the absence of an insult ranged from 13 to 28% of the neurons at the 24 hr observation time point.
RESULTS
We first performed a series of control experiments that identified the glutamate receptor subtype (AMPA vs kainate) responsible for kainate-induced injury, the effects of genetic background on kainate toxicity, and the calcium permeability characteristics of mutant AMPA receptors in cortical neurons maintained in dissociated cultures (Figs.1, 2).
Fig. 2.

Enhanced Ca2+permeability and increased kainate potency in GluR2-deficient cortical pyramidal neurons. A–C, Representative kainate-evoked (100 μm) whole-cell currents and the I–Vrelationships for averaged steady-state currents in GluR2 mutant neurons recorded in low (1 mm, open symbols) and high (20 mm, filled symbols) extracellular Ca2+. Curves were fit by a fourth order polynomial equation from which interpolated reversal potentials were calculated.Erev(+/+), +1.1 ± 0.9 and +0.6 ± 1.1 mV;Erev(+/−), −0.4 ± 1.2 and −0.3 ± 1.0 mV;Erev(−/−), +4.5 ± 2.5 and +11.8 ± 2.3 mV, for low and high Ca2+, respectively. D, Representative kainate-evoked whole-cell currents and concentration–response relationships for peak kainate-evoked currents recorded in GluR2 mutant cortical pyramidal neurons. Concentration–response curves at 10, 30, 100, 300, 1000, and 3000 μm kainate were constructed and normalized to the maximal response in GluR2(+/+)(▪), GluR2(+/−) (■), and GluR2(−/−) (▵) neurons. The potencies of kainate (EC50) and Hill coefficients (nH) were determined by fitting the curves to the equation: I =Imax × 1/(1 + (EC50/[kainate])n), whereImax in the response at 3 mm kainate. GluR2(+/+)EC50, 142.252 ± 15.672 μm;nH, 1.330 ± 0.027 (n = 19). GluR2(+/−)EC50, 131.286 ± 26.692 μm;nH, 1.303 ± 0.062 (n = 11). GluR2(−/−)EC50, 56.511 ± 7.480 μm;nH, 1.159 ± 0.048 (n = 15). *Differences from GluR2(+/−) and GluR2(+/+), one-way ANOVA (F = 8.155; p = 0.001) with post hocBonferroni t tests; p < 0.05.E, Currents from D plotted without normalization to Imax.
Characterization of kainate toxicity in cortical cultures of wild-type and parental strains
Kainic acid often is used for studies of AMPAR-mediated excitotoxicity (Brorson et al., 1994; Turetsky et al., 1994; Bindonkas and Miller, 1995; Lu et al., 1996; Carriedo et al., 1998). It activates both kainate- and AMPA-preferring receptors, the latter as an incompletely desensitizing agonist (Burnashev et al., 1992). We first examined which receptor subtype (AMPA or kainate) mediated kainic acid neurotoxicity in this study. Kainate was applied to the cultured cortical neurons for 24 hr in the presence of 10 μmMK-801 and 2 μm nimodipine, antagonists of NMDA and Ca2+ channels, respectively, to prevent Ca2+ entry through these alternative pathways (Brorson et al., 1994; Sattler et al., 1998; Jensen et al., 1999). This approach isolates ionic influx to AMPA/kainate channels (Sattler et al., 1998) and causes minimal Ca2+ accumulation in cortical neurons from nonmutant mouse strains that express the GluR2 subunit (Sattler et al., 1999). Kainate was applied throughout the 24 hr observation period in all studies. This treatment protocol maximizes the ionic disturbance produced by the activation of kainate-sensitive receptors to reduce the proportional impact of other potentially toxic events initiated after terminating the stimulus, such as the activation of the reverse operation of the Na+/Ca2+exchanger (Yu and Choi, 1997). In wild-type cultures the kainate (0.3–1.0 mm) produced rapid swelling and the death of >70% of neurons by 24 hr (Fig. 1A). A selective noncompetitive AMPAR antagonist (−)-GYKI 53784 (3–10 μm;Bleakman et al., 1996) completely blocked neuronal damage (Fig. 1A), indicating thatkainate neurotoxicity in these cultures is predominantly AMPAR-mediated (Ohno et al., 1997; Jensen et al., 1999).
Next, we examined the influence of genetic background on kainate toxicity. Mutant mice used in the experiments that are described below were generated by GluR2 gene targeting in ES cells of the 129 strain origin. Chimeric offspring were mated with the CD1 strain to obtain offspring that were tested for the presence or absence of the GluR2 null allele. F2 littermates from the same cross were used throughout. It is possible that the different offspring littermates tested here might contain a different complement of 129 genes linked to the GluR2 locus (Gerlai, 1996; Stryker et al., 1997). To determine whether this might affect excitotoxic vulnerability, we obtained cultures from each parental strain (129 and CD1) and tested them separately for their sensitivity to kainate toxicity. Neuronal death in response to kainate (at both 100 μm and 1 mm concentrations) was the same in neurons derived from the 129 or CD1 strain. Therefore, any anticipated differences in the excitotoxic response in GluR2(+/+), GluR2(+/−), and GluR2(−/−)mice could be attributed to the absence of the GluR2 receptor subunit and not to any differential inheritance of background genes.
Ionic currents and Ca2+ permeability of AMPARs in cultured GluR2 mutant neurons
Kainate-activated cobalt uptake is a staining technique that identifies neurons bearing Ca2+-permeable AMPARs (Pruss et al., 1991; Turetsky et al., 1994; Yin et al., 1999). We first stained cortical neuronal cultures from GluR2 mutants by this method to confirm that our cultures maintained Ca2+-permeable AMPARs after 2 weeksin vitro. No attempt was made to quantify the intensity of staining, because the significance of this measure to the physiological function of neurons is controversial. Figure 1C illustrates cobalt staining of neuronal clusters containing hundreds of neurons from the three mutant groups. Large clusters were uncommon in these cultures, and all other experiments in this paper, including the counts of Co2+-positive cells, were performed in dispersed (nonclustered) cultures. However, the many neurons in each cluster illustrate the striking paucity of cobalt staining in GluR2(+/+) neurons as compared with GluR2(+/−)and GluR2(−/−). We also counted cobalt-positive cells in three cultures per mutant group. All GluR2(−/−)neurons (100%) were stained intensely, as compared with only 4.47 ± 1.76% of GluR2(+/+) neurons (n = 3 cultures per group;t(4) = 54.13; p < 0.0001). Most (>80%) GluR2(+/−)neurons also were cobalt-stained, showing that this method also detects neurons expressing a heterogeneous population of AMPARs, of which only a fraction may be Ca2+-permeable.
The potency of kainate in activating inward currents and calcium permeability in GluR2(+/+) and GluR2(−/−)neurons has been examined in acutely isolated CA1 pyramidal cells (Jia et al., 1996; Joo et al., 1999), but not in cultured hippocampal nor in cortical cells. Therefore, we examined whether AMPAR currents with high Ca2+ permeability were maintained when cortical neurons were cultured from GluR2 mutant mice. The current–voltage relationships for GluR2(+/+) and GluR2(+/−)neurons exhibited little or no inward rectification, and their reversal potentials were insensitive to change from low (1 mm) to high (20 mm) extracellular Ca2+ (Fig.2A,B). This suggests that most AMPARs in GluR2(+/+) neurons contain the edited GluR2 subunit that confers a low permeability to Ca2+. Furthermore, GluR2(+/−)neurons also must express sufficient numbers of GluR2 subunits so that their macroscopic currents also exhibit linear I–Vrelationships insensitive to extracellular Ca2+. Currents from GluR2(−/−)neurons exhibited both an enhanced inward rectification and a Ca2+-dependent shift of the reversal potential (Fig. 2C), as predicted for the loss of the GluR2 subunit (Hollmann et al., 1991; Jonas et al., 1994; Burnashev et al., 1995). The reversal potential for current recorded from mutant neurons was shifted to the right when currents were recorded with low (1 mm) versus high (20 mm) extracellular concentrations of Ca2+:Erev(−/−), +4.5 ± 2.5 and +11.8 ± 2.3 mV (p < 0.05). Thus similar to neurons acutely dissociated from mouse brains, cortical neurons cultured from GluR2 mutant mice retain robust AMPAR-mediated currents and characteristic Ca2+ permeability that renders them suitable for examining the effect of GluR2 level on AMPAR-mediated excitotoxicity.
GluR2(−/−)neurons exhibit increased kainate potency and macroscopic currents
The subunit composition of ligand-gated receptors influences the EC50 value of the receptor for agonists as well as their sensitivity to pharmacological agents. Therefore, to determine the equi-effective concentration of kainate that could be used for eliciting excitotoxicity, we first examined the magnitudes and the concentration–response relationships for kainate-evoked ionic currents in GluR2(+/+), GluR2(+/−), and GluR2(−/−)neurons. Applications of kainate (>10 μm) activated an inward current in all of the neurons that were tested. Recordings revealed a higher potency of kainate in GluR2(−/−)neurons as compared with GluR2(+/−)and GluR2(+/+) neurons (Fig.2D). The potency of kainate (EC50) in GluR2(−/−)neurons was approximately threefold higher than in the GluR2(+/+) controls (EC50(−/−), 57 ± 7.5 μm versus EC50(+/+), 142 ± 16 μm; Bonferroni t test,p < 0.01).
Our previous studies in acutely dissociated neurons revealed that membrane capacitance and the maximum current evoked by a saturating concentration of kainate (Imax) was unaffected by the presence of the GluR2 subunit (Joo et al., 1999). However, the acute dissociation obliterates dendritic arbors to which functional AMPARs are localized and which are highly developed in cultured neurons (Sattler et al., 1998, 2000). In the cultured neurons used in the present study, GluR2(−/−)cells exhibited increased peak kainate currents as compared with GluR2(+/−)and GluR2(+/+) (Table1). Although this is consistent with the influence of the GluR2 subunit on increasing the single channel conductance of AMPA channels (Swanson et al., 1997), it also may indicate an influence of GluR2 on dendritic development (Feldmeyer et al., 1999) or on the subcellular distribution or functionality of AMPARs in dendrites. Also, GluR2(−/−)neurons exhibited a reduced membrane capacitance, resulting in a significantly higher current density as compared with their GluR2(+/+) and GluR2(+/−)counterparts (Table 1). The reduced membrane capacitance implies smaller neurons in GluR2(−/−)cultures, although this was not apparent on light microscopic examination (Fig. 3B).
Table 1.
Current density measurements in GluR2 mutant neurons
| C(pF) | Imax(pA) | Imax/C (pA/pF) | |
|---|---|---|---|
| GluR2(+/+), n = 8 | 45 ± 10 | 1878 ± 371 | 43 ± 4 |
| GluR2(+/−), n = 6 | 51 ± 8 | 2160 ± 287* | 45 ± 6 |
| GluR2(−/−),n = 6 | 34 ± 6 | 3027 ± 534* | 88 ± 5* |
Kainate current density was calculated by normalizing the peak current evoked by 3 mm kainate to cell capacitance calculated by integrating the capacitive transient evoked during each experiment by applying a voltage step of −10 mV from holding potential.
One-way ANOVA with post hoc Bonferronit tests for statistical difference from GluR2(+/+) at p < 0.01.
Fig. 3.

Kainate toxicity in vitro.A, Use of the concentration–response data from Figure2E for selecting kainate concentrations for excitotoxicity experiments (i–iv). B, Appearance of GluR2(−/−)and GluR2(+/+) cultures at baseline (0 hr) and at the end (24 hr) of a challenge with 1 mm kainate, using phase contrast (top) and PI fluorescence optics (bottom). C, Effects of kainate insults on 24 hr neuronal survival, using equipotent (i, ii) and nonequipotent (iii, iv) kainate concentrations.i, EC 50 (+/+);ii, EC 90 (+/+);iii, 100 μm for all groups,iv, 1 mm for all groups; v, toxicity of NMDA (100 μm × 60 min) at 24 hr. *p < 0.05, **p < 0.01; Bonferroni t test indicating differences from GluR2(+/+). The fraction of dead neurons ini–v was obtained by averaging measurements from four cultures per mutant mouse. The digits on eachbar indicate the number of mice per group. Data for each bar were replicated from at least two litters of mutant pups.
Vulnerability to AMPAR toxicity in vitro
We next studied AMPAR-mediated excitotoxicity in the GluR2 mutant cortical cultures. GluR2(−/−)neurons appeared morphologically similar to GluR2(+/+) controls and exhibited a low basal propidium iodide (PI) fluorescence (Fig. 3B,leftmost two panels). First, it was our goal to use equi-effective kainate concentrations that would expose neurons in the different mutant groups to similar ionic loads. To this end, it was necessary to take into account that GluR2(+/+), GluR2(+/−), and GluR2(−/−)neurons exhibited differences both in EC50 (see Fig. 2D) and in peak currents (Table 1). Thus equi-effective concentrations were calculated from the absolute rather than from normalized kainate-evoked currents (see Fig.2E), because the former represent the actual ionic current incurred in the cell. Using this approach, we determined the kainate concentration needed to elicit, in each GluR2 group, currents measuring 50 and 90% of the maximum current attainable in GluR2(+/+) neurons [Imax(+/+)]. The effective concentrations are termed EC50(+/+) and EC90(+/+), respectively, and are as listed in Table 2 and illustrated in Figure 3A, i and ii. In addition to these two equi-effective concentrations, toxicity was assessed by using 100 μm and 1 mm kainate, which evoke different inward currents in the different groups (Fig. 3Aiii, Aiv).
Table 2.
Equipotent kainate concentrations for Figure 3C
| Percentage of maximal current elicited in GluR2(+/+)neurons | Kainate concentration (μm) | ||
|---|---|---|---|
| GluR2(+/+) | GluR2(+/−) | GluR2(−/−) | |
| 50 | 142 | 107 | 28 |
| 90 | 742 | 351 | 69 |
Kainate concentrations for experiments in Figure 3Cwere calculated as illustrated in Figure 3A to achieve similar currents via GluR2(+/+), GluR2(+/−), and GluR2(−/−) neurons. Concentrations were calculated from the equation Ieq = Imax × 1/(1 + (EC50/[kainate])nH), where Ieq is the equipotent current, Imax is the maximum current elicited in the group in the response to 3 mm kainate (see Table 1), EC50 is the kainate concentration required to reach half of Imax, and nH is the Hill coefficient. Values of EC50 and nHfor each group are as calculated in Figure 2 (see legend). Experiments were performed at concentrations aimed at eliciting 50% (Fig.3Ci) and 90% (Fig. 3Cii) of the peak current that can be elicited in GluR2(+/+) neurons, as determined in Table 1.
The cultures were exposed to kainate for 24 hr in MK-801 (10 μm) and nimodipine (2 μm), antagonists of NMDARs and voltage-sensitive Ca2+ channels (VSCCs), respectively (Sattler et al., 1998, 1999). Sister cultures were exposed to NMDA (100 μm) in the presence of nimodipine (2 μm) and CNQX (10 μm), an AMPA/kainate antagonist, to isolate Ca2+influx to NMDA receptors (Sattler et al., 1998).
The use of equi-effective kainate concentrations is anticipated to control for confounding effects of kainate potency between mutant groups, thus leaving the GluR2 level as the only variable. We first exposed the cells to kainate concentrations that evoked 50% ofImax(+/+)[EC50(+/+); Table 2; Fig. 3Ai]. These insults produced 10–20% cell death in all groups but revealed no effect of the GluR2 level on AMPAR-mediated excitotoxic vulnerability (Fig. 3Ci; one-way ANOVA,F = 0.24; p = 0.79). These experiments produced relatively low neuronal mortality. Consequently, they were repeated by using equi-effective kainate concentrations that evoked 90% ofImax(+/+)[EC90(+/+); Table 2; Fig. 3Aii]. Although these higher kainate concentrations caused ∼50% of the neurons to die, there was no apparent effect of GluR2 level on excitotoxic vulnerability (Fig. 3Cii; one-way ANOVA, F = 0.02; p = 0.98).
Next, we used 100 μm kainate, an intermediate agonist concentration that evokes larger kainate-activated currents in GluR2(−/−)as compared with GluR2(+/+) neurons (see Fig. 2E). As anticipated, this challenge triggered more toxicity in neurons lacking GluR2 than in wild-type controls (Fig.3Aiii; one-way ANOVA, F = 25.7;p < 0.0001). We then treated the cultures with 1 mm kainate (Fig. 3Aiv). This is a near-saturating agonist concentration that produced near-maximal currents in all mutant groups (see Fig. 2D), although the actual current remained highest in the GluR2(−/−)group (see Fig. 2E). Kainate (1 mm) caused neuronal swelling and a rise in PI fluorescence that peaked at 24 hr (Fig. 3B, rightmost two panels). However, even at this highly toxic kainate concentration, GluR2(−/−)neurons were no more vulnerable than the GluR2(+/−)or GluR2(+/+) controls (Fig.3Aiii; one-way ANOVA, F = 0.11;p = 0.90).
The data in Figure 3, Ci–Civ, suggest that vulnerability to AMPAR-mediated toxicity parallels the magnitude of the kainate-evoked ionic current. When equipotent kainate concentrations were used, mortality was similar between the mutant groups and rose with increasing kainate concentration [compare EC50(+/+) versus EC90(+/+)]. When nonequipotent concentrations were used, neuronal loss evoked by 100 μm kainate also paralleled the size of the anticipated current, with the highest mortality in GluR2(−/−). This effect disappeared at 1 mm, a near-saturating kainate insult (see Fig. 2D) that causes 70–80% neuronal death, the maximum achievable with kainic acid in these cultures. This suggests that with 1 mmkainate the current that triggers excitotoxicity had reached a threshold level sufficient to trigger maximal neurotoxicity.
As a control we treated the cultures with 100 μm NMDA for 60 min, an insult that is highly toxic to cortical neurons in culture (Sattler et al., 1999, 2000). GluR2(−/−), GluR2(+/−), and GluR2(+/+) neurons were equally vulnerable to NMDA toxicity at these concentrations (Fig.3Cv; one-way ANOVA, F = 0.04;p = 0.96), indicating that mechanisms of NMDA-mediated toxicity remain in these cells.
Calcium dynamics in GluR2 mutant neurons
To probe further the impact of increased Ca2+ permeability on Ca2+ homeostasis and excitotoxicity in GluR2-deficient cells, we measured kainate-evoked changes in free intracellular Ca2+ concentration ([Ca2+]i). As Ca2+ ions are sequestered into intracellular organelles, buffered by Ca2+-binding proteins, or extruded through membrane pumps and exchangers (Blaustein, 1988; Pozzan et al., 1994), the kainate-evoked [Ca2+]i rise reflects the result of AMPAR-mediated Ca2+influx, efflux, and buffering. All kainate applications were performed in the presence of NMDAR and Ca2+ channel blockers (Sattler et al., 1998).
Baseline [Ca2+]iwas similar in GluR2(−/−)and GluR2(+/+) neurons loaded with the ratiometric Ca2+ indicator fura-2 (see Fig. 4A–C; t(22) = 0.59; p = 0.56). However, kainate exposure (100 μm) elicited significantly greater [Ca2+]i elevations in GluR2(−/−)neurons as compared with GluR2(+/+)controls (Fig. 4A–C;t(22) = 3.3; p = 0.003). The kainate-evoked change in [Ca2+]i was only detectable in GluR2(−/−), but not in GluR2(+/+), neurons loaded with fura-2 FF, a low Ca2+-affinity indicator (KD ∼35 μm) (Golovina and Blaustein, 1997; Carriedo et al., 1998) (data not shown). Thus fura-2 (KD ∼224 nm) may be more sensitive to the relatively small [Ca2+]i changes evoked in the controls. The rise and persistence of higher [Ca2+]i levels in GluR2(−/−)neurons during kainate exposure (Fig. 4B,C) suggest that [Ca2+]i-lowering mechanisms did not compensate for increased Ca2+ permeability in GluR2(−/−)neurons.
Our excitotoxicity studies (see Fig. 3) indicated a dependence of AMPAR-mediated toxicity on the magnitude of the anticipated ionic current but did not indicate which ions were responsible. Given that GluR2(−/−)cells are highly permeable to Ca2+, it was surprising that equipotent kainate insults failed to demonstrate increased toxicity in this group, because these cells are expected to incur larger Ca2+ loads. We therefore examined kainate-evoked changes in [Ca2+]i in the GluR2 mutant neurons under the same conditions as in the excitotoxicity experiments. Because kainate excitotoxicity also may depend on dendritic AMPARs (Bindonkas and Miller, 1995), we studied kainate-evoked [Ca2+]i changes in the neurons by confocal imaging of both soma and dendrites (Fig.5). On the basis of our experience with fura-2 (KD for Ca2+ ∼224 nm) versus fura-2 FF (KD for Ca2+ ∼35 μm; see above), we used fluo-3, an indicator with a Ca2+ affinity more similar to that of fura-2 (KD for Ca2+ ∼500 nm). Thus we anticipated to better resolve [Ca2+]i changes in GluR2(+/+) neurons versus GluR2(+/−)and GluR2(−/−)cells.
Fig. 5.
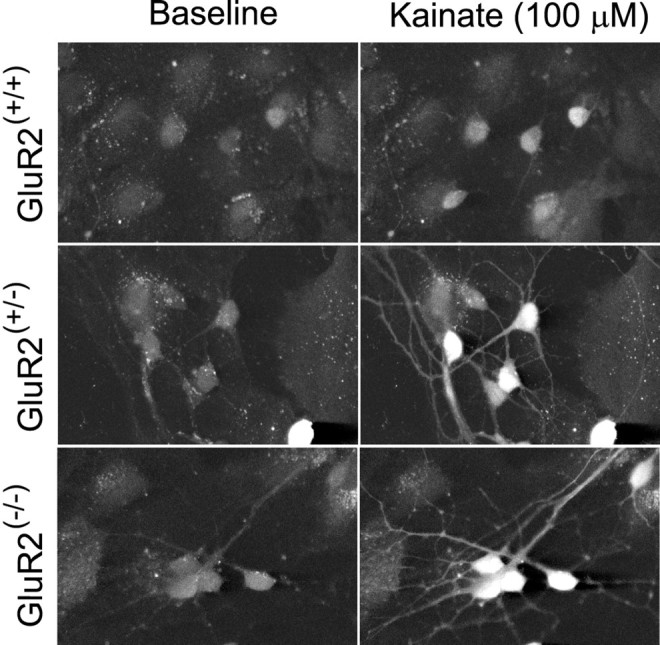
Representative confocal images of fluo-3 fluorescence from GluR2 mutant neurons at baseline and at the peak of a [Ca2+]i transient evoked with 100 μm kainate. Scale bar, 50 μm.
[Ca2+]i changes were measured in the soma and dendrites of neurons during 25–30 sec applications of kainic acid in the presence of MK-801 and nimodipine. Representative experiments (Fig. 4D,E) that used 100 μm kainate produced results similar to those obtained with fura-2 (Fig. 4A,B), with larger [Ca2+]i changes occurring in GluR2(−/−)cells as compared with GluR2(+/+)controls.
Next we used equi-effective kainate concentrations at EC50(+/+) and EC90(+/+) (Table 2), which produce similar degrees of AMPAR-mediated cell death (see Fig.3Ci,Cii). Consistent with the results obtained with cobalt staining (see Fig. 1C) and electrophysiology (see Fig. 2A–C), GluR2(+/+) cells, having AMPARs with low Ca2+ permeability, exhibited significantly smaller increases in [Ca2+]i in both soma and dendrites as compared with GluR2(+/−)and GluR2(−/−)mutants (Fig. 4Gi,Gii). Thus when the anticipated overall ionic currents are similar between groups, neurons that incur higher increases in [Ca2+]i (Fig.4Gi,Gii) do not necessarily exhibit increased mortality (see Fig. 3Ci,Cii). Exposing the cultures to 100 μm and to 1 mm kainate also revealed that GluR2(+/+) cells exhibited significantly smaller increases in [Ca2+]i in both soma and dendrites as compared with GluR2(+/−)and GluR2(−/−)mutants (Fig. 4Giii,Giv). These [Ca2+]i imaging data indicate that the overall size of the anticipated ionic current, but not necessarily the Ca2+ permeability, is the property of AMPARs that more closely predicts excitotoxic vulnerability.
Next, to determine whether GluR2 mutant neurons maintain normal [Ca2+]i responses via pathways other than AMPARs, we examined the effects of 30 sec applications of NMDA (100 μm) in the presence of CNQX and nimodipine to isolate Ca2+ influx to NMDARs (Sattler et al., 1998). All three groups exhibited similar changes in [Ca2+]iboth in the soma (ANOVA, F = 0.06; p = 0.94) and the dendrites (ANOVA, F = 0.22;p = 0.80; Fig. 4F). This indicates that the differences in the [Ca2+]i responses observed after AMPAR stimulation were attributable to the GluR2 level, not to differences in Ca2+ buffering or extrusion, because these factors also would have influenced the NMDAR-mediated responses. Furthermore, because [Ca2+]i transients were significantly larger in both soma and dendrites of GluR2(+/−)and GluR2(−/−)neurons as compared with GluR2(+/+)controls, it is unlikely that GluR2-deficient neurons had upregulated their [Ca2+]i-lowering mechanisms to compensate for increased Ca2+ permeability.
Ionic dependence of AMPAR-mediated excitotoxicity
The majority of the AMPAR-mediated ionic current is carried by Na+ ions. Because excitotoxic vulnerability was predicted by the size of the ionic current (see Fig.3), not Ca2+ permeability (see Fig. 4), we investigated further the ionic dependence of kainate-evoked AMPAR neurotoxicity. AMPAR-evoked neurotoxicity might be mediated by Na+ influx (Kato et al., 1991; Bindonkas and Miller, 1995; Itoh et al., 1998) or by K+ efflux, because K+ depletion promotes both necrosis and apoptosis (Miller and Johnson, 1996; Villalba et al., 1997; Yu et al., 1997) and K+ supplementation promotes neuronal survival (Gallo et al., 1987; Tymianski et al., 1994).
To examine further the ionic mechanisms of kainate toxicity, we performed experiments according to the same protocols as in Figure 3. In wild-type cortical neuronal cultures grown from Swiss mice, kainate toxicity was abolished completely by substituting extracellular Na+ withN-methyl-d-glucamine (NMDG) to prevent Na+ influx (Fig.6A). However, toxicity was unaffected by increasing extracellular K+ to 20 mm (Fig.6A) or to 50 mm (data not shown) to reduce K+ depletion (Yu et al., 1997) or by cycloheximide (Fig. 6A), a protein synthesis inhibitor that inhibits neuronal apoptosis caused by potassium depletion (Yu et al., 1997). Thus in wild-type neurons from the Swiss mouse strain, kainate toxicity was determined primarily by Na+ ions.
We next examined the ionic dependence of kainate toxicity in the GluR2 mutants. Concurrently with the AMPAR-mediated excitotoxicity experiments shown in Figure 3C, we also studied the effect of Na+ removal. In recombinant AMPARs Na+ ions are the main charge carriers and are responsible for >95% of the charge transfer regardless of GluR2 level (Burnashev et al., 1995). Thus we examined whether Na+ removal affects AMPAR-mediated excitotoxicity. First, we applied kainate at the equi-effective concentration that produces 90% ofImax(+/+)(Table 2) and that kills approximately one-half of the neurons in each GluR2 group (see Fig. 3Cii). Removing Na+ reduced the mortality of neurons by >50% in each group challenged with kainate (Fig.6B). Next, we applied kainate at 100 μm (a nonequipotent concentration) to each group. Removing Na+ also reduced neuronal mortality in each group as compared with the same group controls (Fig.6C). However, cell death in the GluR2(−/−)group was not abolished completely by removing Na+ under any conditions (Fig.6B,C), suggesting either that AMPAR-mediated neurotoxicity may depend on factors additional to the Na+ component of the ionic current or that Na+ removal has inherent deleterious effects on GluR2 mutant neurons.
Because Na+ removal did not abolish kainate-mediated AMPAR toxicity completely (Fig.6B,C), we investigated the effect of removing extracellular Ca2+ ions on kainate toxicity in the GluR2 mutants. Experiments by other authors already have suggested that this maneuver reduces kainate-mediated toxicity (Brorson et al., 1994), thus causally implicating Ca2+ ions in the process. Figure6D shows that removing Ca2+ was well tolerated by the cultures in the absence of a kainate challenge. Kainate (100 μm) still produced toxicity in the absence of Ca2+ influx, apparently to a lesser but not statistically significant degree than in the presence of Ca2+ (compare with Fig. 6C;t21 = 1.89; p = 0.073). The rank order of vulnerability to kainate toxicity was proportional to GluR2 level, with GluR(−/−)remaining more vulnerable to kainate toxicity than GluR(+/+) neurons. This experiment suggests, as have previous studies, that Ca2+ ions may be implicated, at least in part, in mediating cell death in AMPAR-mediated excitotoxicity. However, consistent with the other findings in this paper, the data also indicate that the GluR2 level may dictate excitotoxic vulnerability by mechanisms other than those dependent solely on the magnitude of Ca2+ loading.
AMPAR localization in GluR2 mutant neurons
The AMPAR GluR2 subunit interacts via its cytoplasmic C terminus with PDZ domain-containing proteins such as GRIP (glutamate receptor-interacting protein) that serve to cluster AMPARs at excitatory synapses (Dong et al., 1997; Matsuda et al., 2000). Thus the GluR2 level could impact receptor targeting and localization, which may affect function. For example, if GluR2 deficiency were to decrease the trafficking of AMPARs to dendrites, this could result in a larger number of receptors at the cell soma and could be reflected in our finding of larger kainate-evoked whole-cell currents (Imax) and current density in the GluR2(−/−)cultures (see Table 1). Alternately, these same findings could be explained by an effect of GluR2 level on receptor expression levels. Thus we examined the expression, localization, and clustering of AMPARs in our cultures.
Western blot analyses of whole brain homogenates from GluR2 mutant mice have already revealed no alterations in the levels of GluR1 and GluR4 AMPAR subunits, nor in GluR6 and 7 kainate subunits, nor in NR1, NR2A, and NR2B NMDAR subunits (Jia et al., 1996). We performed Western blot measurements of GluR1 protein levels in the GluR2 mutant cultures and also found no differences in protein expression among the groups (Fig.7B, insets). However, this does not exclude the possibility that neurons might compensate for the lack of GluR2 by modifying the function or the subcellular distribution of AMPARs. Thus we examined the distribution of AMPARs in the cultured cortical neurons. GluR1 immunostaining revealed a punctate staining pattern in the soma and dendrites of neurons, with no obvious qualitative differences among GluR2(−/−), GluR2(+/−)(data not shown), and GluR2(+/+) cells (Fig. 7A). We then counted GluR1 clusters along dendrites of randomly selected neurons (Allison et al., 1998; Sattler et al., 2000). This revealed no quantitative differences in the numbers of GluR1 clusters per unit of dendrite length (see Fig. 5B; one-way ANOVA, F = 1.33; p = 0.27). Our observations suggest that differences in the efficacy of kainate in evoking ionic currents and excitotoxicity are not explained by compensatory changes in the expression, clustering, or distribution of AMPARs, as observed by our methods. However, these findings do not rule out more subtle changes, such as an increased recruitment of AMPARs to the synapse, an increase in functional AMPARs at synapses, or an increase in AMPAR conductance.
Fig. 7.
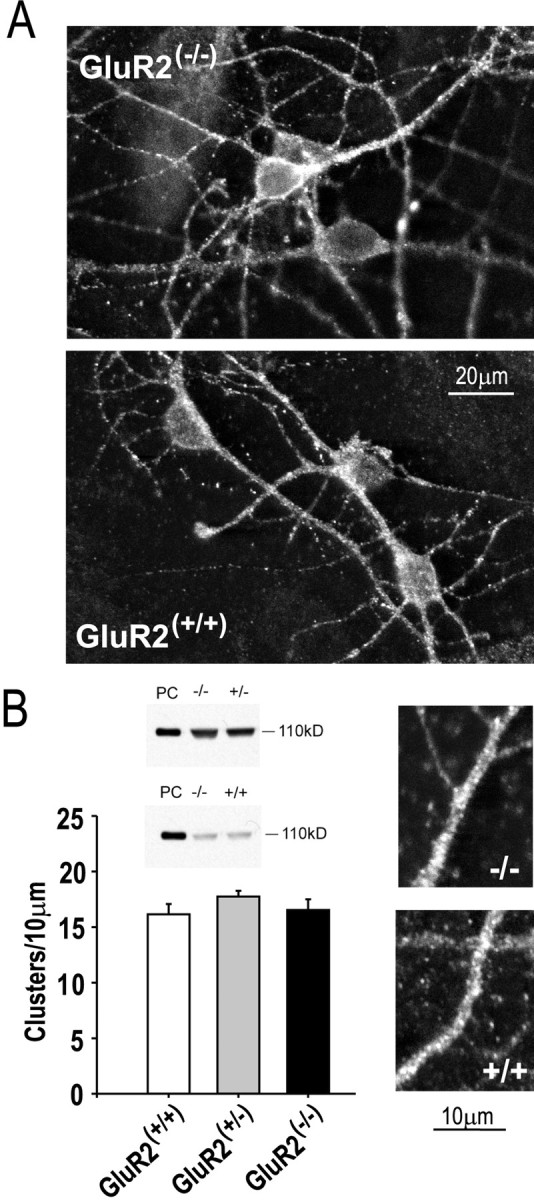
Unchanged AMPAR expression and distribution in GluR2 mutant neurons. A, Representative punctate GluR1 immunostaining in cultured cortical neurons from GluR2 mutants.B, Quantitation of the expression and numbers of GluR1 clusters per dendrite length. Immunoblots reveal equal GluR1 levels in GluR2(−/−)and GluR2(+/−) cultures (top) and in GluR2(−/−)and GluR2(+/+) cultures (bottom) Each immunoblot is representative of three experiments. PC, Positive control, using protein from isolated rat brain membranes. Counts of GluR1 clusters were obtained from 40 randomly selected dendrite segments per group from neurons in four separate cultures. Data were averaged from counts obtained by two independent observers.Insets, Representative dendrite segments from GluR2(−/−)(top) and GluR2(+/+)(bottom) neurons imaged from cultures as inA at higher magnification.
Vulnerability to AMPAR toxicity in vivo
Our experiments in vitro have demonstrated that neuronal vulnerability to AMPAR-mediated neurotoxicity increased with a reduced GluR2 level because of an effect on kainate potency. The importance of the GluR2 subunit in governing vulnerability to excitotoxicity is consistent with reports in nonmutant animals that a potentially neurotoxic challenge can cause a relative reduction in neuronal GluR2 expression that correlates with delayed neuronal death in epilepsy and global cerebral ischemia (Pellegrini-Giampietro et al., 1992, 1997; Friedman, 1998; Friedman and Veliskova, 1998). In our mutant mice we have noted that the GluR2(−/−)animals exhibit developmental delays and behavioral abnormalities (Jia et al., 1996; Gerlai et al., 1998) and that other mice with Q/R site-editing mutations and/or expressing GluR2 (GluR-B) subunits at different levels develop neurological deficits, seizures, or increased mortality (Brusa et al., 1995; Feldmeyer et al., 1999). However, despite these altered neurological phenotypes, none of these animals is reported to exhibit obvious changes in gross brain structure or in neuronal cell counts in relevant areas, particularly in areas in which selective neuronal death might be attributable to a loss of GluR2. This is unusual, given the role of GluR2 in governing excitotoxic vulnerability. Thus it was possible that in these studies the mutant animals showed no sign of neuronal loss because they were not challenged with a neurotoxic insult. Therefore, we studied the effects of kainate-triggered excitotoxicity in the GluR2 mutant mice in vivo, because kainate administration to rodents causes seizures and excitotoxic neurodegeneration in selected hippocampal neurons (Nadler et al., 1978; Ben-Ari, 1985).
We first studied neurons in the hippocampal CA1 sector because previously we have characterized kainate-evoked currents and Ca2+ permeability in these cells (Jia et al., 1996). They are affected in cerebral ischemia (Pellegrini-Giampietro et al., 1992), and they are less vulnerable than CA3 neurons to kainate-evoked damage caused by the secondary activation of hippocampal mossy fiber projections (Okazaki and Nadler, 1988). Intrathecal kainate injections (0.75, 2, or 5 nmol) were made directly into the CA1 sector (see Fig. 4A,C–F), and the survival of CA1 neurons was examined at 48 hr. Kainate produced CA1 pyramidal cell death in a dose-dependent manner (Fig.8A), ranging from 20–26% cell death with 0.75 nmol to >95% cell death at 5 nmol. However, there were no observable differences in kainate-induced toxicity to CA1 neurons of GluR2(−/−), GluR2(+/−), and GluR2(+/+) mice (Fig.8A,E,F).
Fig. 8.
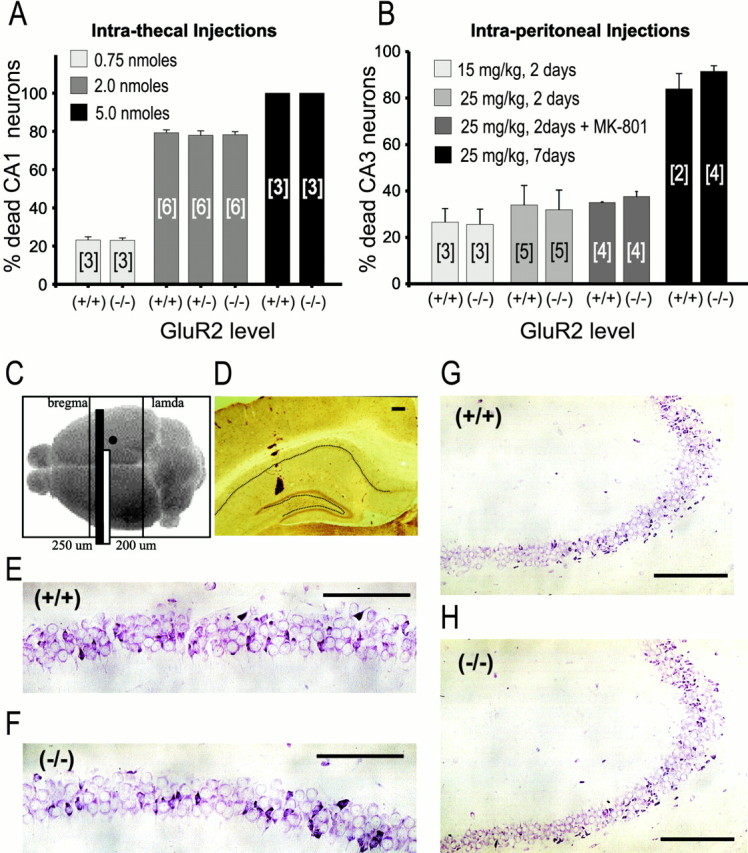
Lack of increased kainate toxicity in GluR2(−/−)neurons in vivo. Experiments were performed in 7- to 9-week-old mice. A, Effects of intrathecal kainate injections. B, Effects of intraperitoneal injections. Neurons in A and B were counted 48 hr after injection except where marked. [n], Number of mice per group. C, Schematic of region of analysis and site of kainic acid deposition for intrathecal and intraperitoneal studies. Black circle, Site of intrahippocampal kainic acid deposition. Black bar, Hippocampal segment used in analyses. D, Position of microcapillary within the hippocampus for deposition of kainic acid. Section was stained for calretinin. Dotted line, Position of pyramidal and granule cell layers. E, F, Examples of CA1 pyramidal cell loss in animals receiving an intrahippocampal dose of 0.75 nmol of kainic acid at 48 d after injection (distance from deposition site, 325 μm); E, GluR2(+/+) animal. F, GluR2(−/−)animal. G, H, Examples of CA3 pyramidal cell loss in animals receiving an intraperitoneal kainate injection, 25 mg/kg, at 2 d. G, GluR2(+/+)animal. H, GluR2(−/−)animal. Scale bars: E, F, 100 μm;G, H, 200 μm.
Next, intraperitoneal kainic acid injections were used to elicit neuronal damage in the hippocampal CA3 sector (Ben-Ari, 1985; Strain and Tasker, 1991). One group of animals also received MK-801 (5 mg/kg) at the time of kainic acid injection, and again at 8 hr, to minimize the possibility of obtaining neurotoxicity caused by indirect NMDAR activation by seizure activity (Okazaki and Nadler, 1988) (Fig, 8B). Seizures occurred in all of the mice that were injected with kainate alone with a latency of 15–18 min, consisting of behavior characterized by motion arrest, staring, and myoclonic jerks. The resultant mortality of CA3 neurons was dose-dependent (Fig.8B), ranging from 15–35% at 48 hr in mice given 15 mg/kg kainate to 20–45% at 48 hr and >90% at 7 d in mice given 25 mg/kg kainate. However, as in the intrathecal injection experiments on CA1 neurons (above), kainate-induced toxicity to CA3 neurons was similar in GluR2(−/−)and GluR2(+/+) mice (Fig.8B,G,H).
Given the lack of increased vulnerability in GluR2-deficient animals, we examined whether Ca2+-binding proteins (CaBPs) were altered in the GluR2 mutant animals to compensate for increased Ca2+ permeability. Kondo and colleagues (1997) examined the relationship between AMPAR subunit expression in individual neurons of the rat cortex and the expression of CaBPs. They found that parvalbumin-positive neurons were mainly GluR2-negative, whereas calbindin D28k-positive cells were mainly GluR2-expressing. Because both proteins bind Ca2+ with similar affinities (Van Eldik et al., 1982), the functional significance of these differences is unclear. However, in our animals the distribution of calbindin immunoreactivity was unchanged in the hippocampus (Fig.9A,B) and cortex (Fig.9C) of GluR2-deficient mice. Calbindin expression by Western blot analysis was unchanged also (Fig. 9D). Similar results were obtained with other Ca2+-BPs (parvalbumin, calmodulin, and calretinin; data not shown). Thus a compensatory change in CaBP expression is unlikely to have occurred.
Fig. 9.
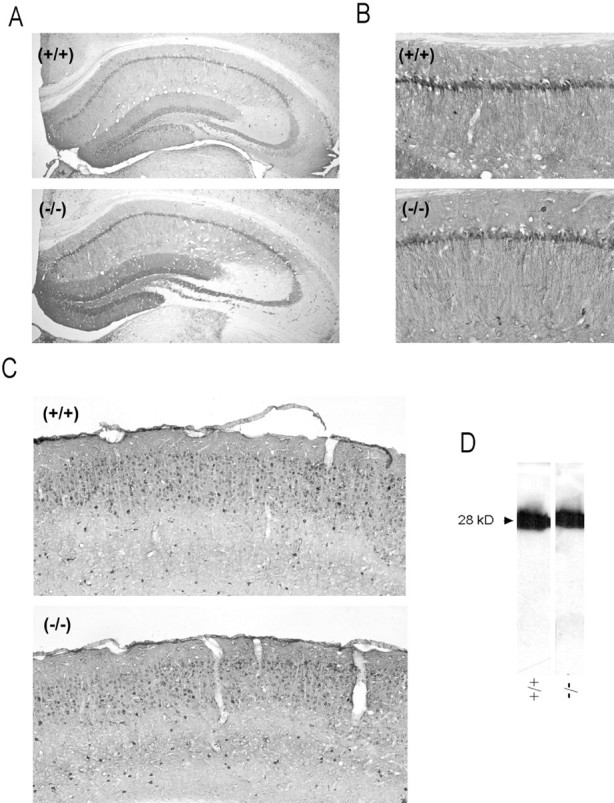
Unchanged calbindin levels in the brain of GluR2 mutant mice. A, B, Representative calbindin immunostaining in the hippocampus.A, Low magnification. B, CA1 region.C, Calbindin staining in the cerebral cortex.D, Calbindin expression by Western blot analysis.
Contrary to our in vitro data, our experiments in vivo failed to demonstrate an effect of GluR2 level on vulnerability to kainate-evoked neurotoxicity. Particularly, they do not demonstrate an increased vulnerability to toxicity in neurons lacking GluR2, as suggested by the GluR2 hypothesis (Bennett et al., 1996) and by our own findings in vitro. Thus it is likely that, in the whole animal, neurons and/or neuronal circuits have compensated for the alterations in GluR2 level in a manner that was not observed readily, such as a reduction in functional AMPARs. Notably, a recent report has shown that increased synaptic activity induces a reduction in AMPAR Ca2+ permeability via the incorporation of GluR2 subunits into Ca2+-permeable AMPARs (Liu and Cull-Candy, 2000). However, this mechanism is unlikely in our GluR2(−/−)animals because they lack GluR2 altogether.
Despite a lack of increased neuronal lethality in vivo in GluR2(−/−)animals, diverse GluR2 mutant animals express obvious GluR2-dependent differences in developmental, behavioral, neurological, and survival phenotypes (Brusa et al., 1995; Jia et al., 1996; Gerlai et al., 1998;Kask et al., 1998; Feldmeyer et al., 1999). This reinforces the notion that the GluR2 subunit may be involved in regulating aspects of neuronal function by mechanisms other than those governing Ca2+ permeability.
DISCUSSION
The availability of mutant mice that lack GluR2 has made it possible to examine the influence of GluR2 level on AMPAR-mediated excitotoxicity while controlling for the influence of GluR2 on Ca2+ permeability. Kainic acid was used to evoke AMPAR-mediated excitotoxicity (see Fig. 1A) in neuronal cultures expressing different levels of GluR2 as confirmed by cobalt staining (see Fig. 1C), electrophysiology (see Fig.2A–C), and fluorescent [Ca2+]i imaging (see Figs. 4, 5). Apart from exhibiting an increase Ca2+ permeability, neurons with reduced or absent GluR2 [(+/−) and (−/−), respectively] exhibited increased kainate potency and larger ionic currents (see Fig.2E, Table 1). The dependence of excitotoxic vulnerability on GluR2 level paralleled the magnitude of the anticipated kainate-activated ionic current, not the predicted Ca2+ permeability. Thus insults that used equi-effective kainate concentrations were equally neurotoxic (see Fig.3Ci,Cii) despite eliciting higher [Ca2+]i elevations in GluR2-deficient neurons (see Fig.4Ai,Aii). However, vulnerability to equivalent concentrations of kainate was higher in GluR2(−/−)neurons (see Fig. 3Ciii), also as predicted by the larger ionic currents (Fig. 2E). Removing Na+, the major charge carrier of AMPAR-mediated currents, significantly protected neurons against kainate-evoked excitotoxicity (see Fig. 6). GluR2 mutant neurons did not exhibit detectable alterations in AMPAR distribution (see Fig.7A,B) or expression (see Fig. 7B) or in Ca2+ buffering as gauged by [Ca2+]i imaging (see Fig. 4), by responses to NMDAR-mediated toxicity (see Fig.3Cv), or by staining for Ca2+-binding proteins (see Fig. 9). Also, despite exhibiting various phenotypic changes, animals lacking GluR2 did not exhibit increased vulnerability to kainate toxicity (see Fig.8).
Despite our demonstration of a strong relationship of toxicity to the magnitude of the kainate-evoke ionic current and a poor correlation with AMPAR Ca2+ permeability, our data do not necessarily exclude an excitotoxic role for Ca2+ ions in AMPAR-mediated excitotoxicity. Indeed, early experiments that used ion substitution already have suggested that removal of extracellular Ca2+ appears to protect neurons against kainate-evoked AMPAR-mediated neuronal damage (Brorson et al., 1994). Ca2+ entry in our neurons could occur directly, because GluR2 containing AMPARs have a low but nonzero Ca2+ permeability (Brorson et al., 1999), or could occur indirectly via secondary pathways. For example, increased Na+ loading incurred by GluR2(−/−)as compared with GluR2(+/+) neurons could activate secondary means of toxic Ca2+entry such as the reverse operation of Na+/Ca2+exchange (Kiedrowski et al., 1994; Itoh et al., 1998) or other as yet uncharacterized means of Ca2+ entry. Notably, increased Ca2+ loading in response to Na+ influx would have been reflected in our [Ca2+]imeasurements, which did not predict excitotoxicity. Thus toxicity attributable to secondary means of Ca2+entry is less likely. Another possibility is that excessive Na+ entry is directly toxic or that toxicity is the result of other ions that were not addressed in our study, such as Cl− ions. It is also possible that the GluR2 subunit somehow governs the interactions between the receptor and intracellular Ca2+ stores such as mitochondria and that, even in the absence of extracellular Ca2+entry, increased Ca2+ release from internal stores could mediate the increased neurotoxic consequences of GluR2(−/−)AMPAR activation. Such possibilities have yet to be determined.
Although we have not resolved completely the degree to which Ca2+ ions are essential to AMPAR-mediated excitotoxicity, we have suggested that removing extracellular Ca2+ ions had some protective effect irrespective of GluR2 level (see Fig. 6D). This observation raises the possibility that some threshold Ca2+ level is permissive and necessary for triggering events leading to AMPAR-mediated damage. For example, a baseline cytosolic or mitochondrial Ca2+load may be required to facilitate the initiation or propagation of damaging downstream second messenger cascades. If so, then our data suggest that, once such a baseline level is attained, additional Ca2+ loading during kainate exposure would have no additional effect. Under this hypothesis, although Ca2+ ions would play a role in the excitotoxicity process, increased Ca2+entry because of GluR2 deficiency would not have an additional neurotoxic consequence. This explanation would account for the apparent protective effect of removing Ca2+ ions from the extracellular medium while also accounting for the independence of kainate toxicity on the level of free Ca that is reached with kainate exposure (see Figs. 3, 4).
Regardless of the magnitude of the role of Ca2+ ions in AMPAR-mediated toxicity, our data suggest strongly that Ca2+permeability, as governed by GluR2 level, is not the chief determinant of excitotoxic vulnerability. In a recent study Vandenberghe and colleagues (2000) combined whole-cell patch-clamp electrophysiology and single-cell RT-PCR to examine the relationship among AMPA receptor-mediated excitotoxicity, the relative Ca2+ permeability of AMPA receptors, and the fractional expression of GluR2 in spinal motor neurons. They found that, although anterior horn motor neurons were more vulnerable to AMPA agonist toxicity, their GluR2 expression and Ca2+ permeability characteristics were not significantly different from dorsal horn cells, which were more resilient. Thus they concluded that the selective vulnerability of motor neurons to AMPA receptor agonists is not determined solely by whole-cell relative Ca2+ permeability of AMPA receptors. Our present findings also support this view and raise the possibility that GluR2 also governs other aspects of AMPAR function that may cause neurotoxicity.
Current knowledge about the molecular interactions of GluR2 subunits with other cellular elements is consistent with the idea that GluR2 mutations may have an impact on neuronal function in a manner that is not necessarily governed by Ca2+ ions. For example, GluR2 subunits bind with high specificity to diverse submembrane proteins. These include glutamate receptor-interacting protein (GRIP), which participates in the synaptic localization and clustering of AMPARs (Dong et al., 1997), andN-ethylmaleimide-sensitive factor (NSF), a protein involved in membrane fusion events (Osten et al., 1998; Song et al., 1998). GluR2 deficiency therefore may interfere with the molecular organization, function, and plasticity of glutamatergic synapses. Consistent with this are observations in recently generated mice with targeted GluR2 subunit alleles producing AMPARs with increased Ca2+ permeability. The numbers of neurons in the hippocampus and cortex of such mice were unchanged (Feldmeyer et al., 1999). However, they exhibited reduced dendritic length and arborization in CA3 pyramidal cells, correlating with developmental delays, neurological dysfunction, and early mortality (Feldmeyer et al., 1999). The GluR2 subunit therefore may have a role in neuronal function and development that exceeds its role in governing Ca2+ permeability; thus GluR2 deficiency could have adverse consequences because of a combination of mechanisms, some related to its effects on synaptic organization in the intact brain rather than solely because of altered Ca2+ permeability.
We have reported previously that the extent of Ca2+ influx into neurons does not necessarily predict excitotoxic vulnerability. In cultured spinal (Tymianski et al., 1993) and cortical (Sattler et al., 1998) neurons, a chief determinant of Ca2+-triggered excitotoxicity is the route, not the quantity, of Ca2+ influx; Ca2+ influx via NMDA receptors is lethal, whereas equivalent Ca2+ entry through VSCCs is innocuous (Tymianski et al., 1993; Sattler et al., 1998). One mechanism that governs the toxicity of NMDAR-mediated Ca2+ entry is the coupling of NMDARs to neurotoxic intracellular second messengers by submembrane scaffolding proteins such as postsynaptic density-95 (PSD-95) protein (Sattler et al., 1999). This precedent opens the possibility that submembrane scaffolding proteins that interact specifically with AMPARs (Dong et al., 1997) also could couple AMPAR-mediated ionic fluxes to neurotoxic second messengers. If so, then AMPAR-mediated and NMDAR-mediated excitotoxic signaling is fundamentally different, because the initiation of AMPAR-mediated toxic cascades may be less dependent on a high permeability to Ca2+.
Our findings may have implications beyond the understanding of excitotoxic mechanisms. Excitotoxicity likely reflects a dysfunction of processes that also govern physiological neuronal functioning. AMPARs mediate rapid synaptic transmission and play key roles in long-term potentiation, learning, memory, and behavior. Many recent efforts to understand the mechanisms by which AMPARs modulate these processes have focused on their Ca2+ permeability characteristics (Brorson et al., 1994; Turetsky et al., 1994; Brusa et al., 1995; Jia et al., 1996; Lu et al., 1996; Feldmeyer et al., 1999). AMPAR subunit composition, particularly the GluR2 subunit, clearly has important functional consequences. However, our findings raise the possibility that such consequences depend on GluR2-associated features other than, or in addition to, Ca2+permeability.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS 39060 (M.T.) and Ontario Heart and Stroke Foundation Grant NA-3694 (B.A.O. and M.T.). We thank Drs. J. F. MacDonald and M. Salter for a critical review of this manuscript and E. Czerwinska and L. Teves for technical assistance. K.I. is a fellow and R.S. is a student of the Ontario Heart and Stroke Foundation; D.J. is a Medical Research Council fellow; M.T. is a Medical Research Council Clinician–Scientist.
Correspondence should be addressed to Dr. Michael Tymianski, Lab 11-416, MC-PAV, Toronto Western Hospital, 399 Bathurst Street, Toronto, Ontario M5T-2S8, Canada. E-mail: mike_t@uhnres.utoronto.ca.
REFERENCES
- 1.Allison DW, Gelfand VI, Spector I, Craig AM. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18:2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ben-Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- 3.Bennett MV, Pellegrini-Giampietro DE, Gorter JA, Aronica E, Connor JA, Zukin RS. The GluR2 hypothesis: Ca2+-permeable AMPA receptors in delayed neurodegeneration. Cold Spring Harb Symp Quant Biol. 1996;61:373–384. [PubMed] [Google Scholar]
- 4.Bindonkas VP, Miller RJ. Excitotoxic degeneration is initiated at nonrandom sites in cultured rat cerebellar neurons. J Neurosci. 1995;15:6999–7011. doi: 10.1523/JNEUROSCI.15-11-06999.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaustein MP. Calcium transport and buffering in neurons. Trends Neurosci. 1988;11:438–443. doi: 10.1016/0166-2236(88)90195-6. [DOI] [PubMed] [Google Scholar]
- 6.Bleakman D, Ballyk BA, Schoepp DD, Palmer AJ, Bath CP, Sharpe EF, Woolley ML, Bufton HR, Kamboj RK, Tarnawa I, Lodge D. Activity of 2,3-benzodiazepines at native rat and recombinant human glutamate receptors in vitro: stereo specificity and selectivity profiles. Neuropharmacology. 1996;35:1689–1702. doi: 10.1016/s0028-3908(96)00156-6. [DOI] [PubMed] [Google Scholar]
- 7.Brorson JR, Manzolillo PA, Miller RJ. Ca2+ entry via AMPA/KA receptors and excitotoxicity in cultured cerebellar Purkinje cells. J Neurosci. 1994;14:187–197. doi: 10.1523/JNEUROSCI.14-01-00187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brorson JR, Zhang Z, Vandenberghe W. Ca2+ permeation of AMPA receptors in cerebellar neurons expressing Glu receptor 2. J Neurosci. 1999;19:9149–9159. doi: 10.1523/JNEUROSCI.19-21-09149.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruno VMG, Goldberg MP, Dugan LL, Giffard RG, Choi DW. Neuroprotective effect of hypothermia in cortical cultures exposed to oxygen–glucose deprivation or excitatory amino acids. J Neurochem. 1994;63:1398–1406. doi: 10.1046/j.1471-4159.1994.63041398.x. [DOI] [PubMed] [Google Scholar]
- 10.Brusa R, Zimmermann F, Koh DS, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270:1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- 11.Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- 12.Burnashev N, Zhou Z, Neher E, Sakmann B. Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA, and kainate receptor subtypes. J Physiol (Lond) 1995;485:403–418. doi: 10.1113/jphysiol.1995.sp020738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carriedo SG, Yin HZ, Sensi SL, Weiss JH. Rapid Ca2+ entry through Ca2+-permeable AMPA/kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. J Neurosci. 1998;18:7727–7738. doi: 10.1523/JNEUROSCI.18-19-07727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988;11:465–467. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- 15.Cotton JL, Partin KM. The contributions of GluR2 to allosteric modulation of AMPA receptors. Neuropharmacology. 2000;39:21–31. doi: 10.1016/s0028-3908(99)00105-7. [DOI] [PubMed] [Google Scholar]
- 16.David JC, Yamada KA, Bagwe MR, Goldberg MP. AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J Neurosci. 1996;16:200–209. doi: 10.1523/JNEUROSCI.16-01-00200.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong H, O'Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386:279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- 18.Feldmeyer D, Kask K, Brusa R, Kornau HC, Kolhekar R, Rozov A, Burnashev N, Jensen V, Hvalby O, Sprengel R, Seeburg PH. Neurological dysfunction in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci. 1999;2:57–64. doi: 10.1038/4561. [DOI] [PubMed] [Google Scholar]
- 19.Friedman LK. Selective reduction of GluR2 protein in adult hippocampal CA3 neurons following status epilepticus but prior to cell loss. Hippocampus. 1998;8:511–525. doi: 10.1002/(SICI)1098-1063(1998)8:5<511::AID-HIPO9>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 20.Friedman LK, Veliskova J. GluR2 hippocampal knockdown reveals developmental regulation of epileptogenicity and neurodegeneration. Brain Res Mol Brain Res. 1998;61:224–231. doi: 10.1016/s0169-328x(98)00213-7. [DOI] [PubMed] [Google Scholar]
- 21.Gallo V, Kingsbury A, Balazs R, Jorgensen OS. The role of depolarization in the survival and differentiation of cerebellar granule cells in culture. J Neurosci. 1987;7:2203–2213. doi: 10.1523/JNEUROSCI.07-07-02203.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 23.Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- 24.Gerlai R, Henderson JT, Roder JC, Jia Z. Multiple behavioral anomalies in GluR2 mutant mice exhibiting enhanced LTP. Behav Brain Res. 1998;95:37–45. doi: 10.1016/s0166-4328(98)00002-3. [DOI] [PubMed] [Google Scholar]
- 25.Golovina VA, Blaustein MP. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science. 1997;275:1643–1648. doi: 10.1126/science.275.5306.1643. [DOI] [PubMed] [Google Scholar]
- 26.Gorter JA, Petrozzino JJ, Aronica EM, Rosenbaum DM, Opitz T, Bennett MV, Connor JA, Zukin RS. Global ischemia induces downregulation of GluR2 mRNA and increases AMPA receptor-mediated Ca2+ influx in hippocampal CA1 neurons of gerbil. J Neurosci. 1997;17:6179–6188. doi: 10.1523/JNEUROSCI.17-16-06179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 28.Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA–AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 29.Hume RI, Dingledine R, Heinemann SF. Identification of a site in glutamate receptor subunits that controls calcium permeability. Science. 1991;253:1028–1031. doi: 10.1126/science.1653450. [DOI] [PubMed] [Google Scholar]
- 30.Itoh T, Itoh A, Horiuchi K, Pleasure D. AMPA receptor-mediated excitotoxicity in human NT2-N neurons results from loss of intracellular Ca2+ homeostasis following marked elevation of intracellular Na+. J Neurochem. 1998;71:112–124. doi: 10.1046/j.1471-4159.1998.71010112.x. [DOI] [PubMed] [Google Scholar]
- 31.Jensen AG, Schousboe A, Pickering DS. Role of desensitization and subunit expression for kainate receptor-mediated neurotoxicity in murine neocortical cultures. J Neurosci Res. 1999;55:208–217. doi: 10.1002/(SICI)1097-4547(19990115)55:2<208::AID-JNR8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 32.Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996;17:945–956. doi: 10.1016/s0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- 33.Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H. Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron. 1994;12:1281–1289. doi: 10.1016/0896-6273(94)90444-8. [DOI] [PubMed] [Google Scholar]
- 34.Joo DT, Xiong Z, MacDonald JF, Jia Z, Roder J, Sonner J, Orser BA. Blockade of glutamate receptors and barbiturate anesthesia: increased sensitivity to pentobarbital-induced anesthesia despite reduced inhibition of AMPA receptors in GluR2 null mutant mice. Anesthesiology. 1999;91:1329–1341. doi: 10.1097/00000542-199911000-00025. [DOI] [PubMed] [Google Scholar]
- 35.Kask K, Zamanillo D, Rozov A, Burnashev N, Sprengel R, Seeburg PH. The AMPA receptor subunit GluR-B in its Q/R site-unedited form is not essential for brain development and function. Proc Natl Acad Sci USA. 1998;95:13777–13782. doi: 10.1073/pnas.95.23.13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato K, Puttfarcken PS, Lyons WE, Coyle JT. Developmental time course and ionic dependence of kainate-mediated toxicity in rat cerebellar granule cell cultures. J Pharmacol Exp Ther. 1991;256:402–411. [PubMed] [Google Scholar]
- 37.Kiedrowski L, Brooker G, Costa E, Wroblewski JT. Glutamate impairs neuronal calcium extrusion while reducing sodium gradient. Neuron. 1994;12:295–300. doi: 10.1016/0896-6273(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 38.Kondo M, Sumino R, Okado H. Combinations of AMPA receptor subunit expression in individual cortical neurons correlate with expression of specific calcium-binding proteins. J Neurosci. 1997;17:1570–1581. doi: 10.1523/JNEUROSCI.17-05-01570.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lewis CA. Ion concentration dependence of the reversal potential and the single channel conductance of ion channels at the frog neuromuscular junction. J Physiol (Lond) 1979;286:417–445. doi: 10.1113/jphysiol.1979.sp012629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin JW, Sheng M. NSF and AMPA receptors get physical. Neuron. 1998;21:267–270. doi: 10.1016/s0896-6273(00)80534-6. [DOI] [PubMed] [Google Scholar]
- 41.Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- 42.Lu YM, Yin HZ, Chiang J, Weiss JH. Ca2+-permeable AMPA/kainate and NMDA channels: high rate of Ca2+ influx underlies potent induction of injury. J Neurosci. 1996;16:5457–5465. doi: 10.1523/JNEUROSCI.16-17-05457.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luthi A, Chittajallu R, Duprat F, Palmer MJ, Benke TA, Kidd FL, Henley JM, Isaac JT, Collingridge GL. Hippocampal LTD expression involves a pool of AMPARs regulated by the NSF–GluR2 interaction. Neuron. 1999;24:389–399. doi: 10.1016/s0896-6273(00)80852-1. [DOI] [PubMed] [Google Scholar]
- 44.Mainen ZF, Jia Z, Roder J, Malinow R. Use-dependent AMPA receptor block in mice lacking GluR2 suggests postsynaptic site for LTP expression. Nat Neurosci. 1998;1:579–586. doi: 10.1038/2812. [DOI] [PubMed] [Google Scholar]
- 45.Man YH, Lin JW, Ju WH, Ahmadian G, Liu L, Becker LE, Sheng M, Wang YT. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25:649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 46.Matsuda S, Launey T, Mikawa S, Hirai H. Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J. 2000;19:2765–2774. doi: 10.1093/emboj/19.12.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller TM, Johnson EM., Jr Metabolic and genetic analyses of apoptosis in potassium/serum-deprived rat cerebellar granule cells. J Neurosci. 1996;16:7487–7495. doi: 10.1523/JNEUROSCI.16-23-07487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nadler JV, Perry BW, Cotman CW. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. 1978;271:676–677. doi: 10.1038/271676a0. [DOI] [PubMed] [Google Scholar]
- 49.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nishimune A, Isaac JT, Molnar E, Noel J, Nash SR, Tagaya M, Collingridge GL, Nakanishi S, Henley JM. NSF binding to GluR2 regulates synaptic transmission. Neuron. 1998;21:87–97. doi: 10.1016/s0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
- 51.Ohno K, Okada M, Tsutsumi R, Kohara A, Yamaguchi T. Kainate excitotoxicity is mediated by AMPA- but not kainate-preferring receptors in embryonic rat hippocampal cultures. Neurochem Int. 1997;31:715–722. doi: 10.1016/s0197-0186(97)00011-9. [DOI] [PubMed] [Google Scholar]
- 52.Okazaki MM, Nadler JV. Protective effects of mossy fiber lesions against kainic acid-induced seizures and neuronal degeneration. Neuroscience. 1988;26:763–781. doi: 10.1016/0306-4522(88)90097-8. [DOI] [PubMed] [Google Scholar]
- 53.Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, States BA, Einheber S, Milner TA, Hanson PI, Ziff EB. The AMPA receptor GluR2 C terminus can mediate a reversible, ATP-dependent interaction with NSF and alpha- and beta-SNAPs. Neuron. 1998;21:99–110. doi: 10.1016/s0896-6273(00)80518-8. [DOI] [PubMed] [Google Scholar]
- 54.Pellegrini-Giampietro DE, Zukin RS, Bennett MVL, Cho S, Pulsinelli WA. Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc Natl Acad Sci USA. 1992;89:10499–10503. doi: 10.1073/pnas.89.21.10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pellegrini-Giampietro DE, Gorter JA, Bennett MV, Zukin RS. The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997;20:464–470. doi: 10.1016/s0166-2236(97)01100-4. [DOI] [PubMed] [Google Scholar]
- 56.Pozzan T, Rizzuto R, Volpe P, Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev. 1994;74:595–635. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- 57.Pruss RM, Akeson RL, Racke MM, Wilburn JL. Agonist-activated cobalt uptake identifies divalent cation-permeable kainate receptors on neurons and glial cells. Neuron. 1991;7:509–518. doi: 10.1016/0896-6273(91)90302-g. [DOI] [PubMed] [Google Scholar]
- 58.Sattler R, Charlton MP, Hafner M, Tymianski M. Determination of the time-course and extent of neurotoxicity at defined temperatures in cultured neurons using a modified multi-well plate fluorescence scanner. J Cereb Blood Flow Metab. 1997;17:455–463. doi: 10.1097/00004647-199704000-00011. [DOI] [PubMed] [Google Scholar]
- 59.Sattler R, Charlton MP, Hafner M, Tymianski M. Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J Neurochem. 1998;71:2349–2364. doi: 10.1046/j.1471-4159.1998.71062349.x. [DOI] [PubMed] [Google Scholar]
- 60.Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- 61.Sattler R, Xiong Z, Lu WY, MacDonald JF, Tymianski M. Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J Neurosci. 2000;20:22–33. doi: 10.1523/JNEUROSCI.20-01-00022.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21:393–400. doi: 10.1016/s0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- 63.Strain SM, Tasker RA. Hippocampal damage produced by systemic injections of domoic acid in mice. Neuroscience. 1991;44:343–352. doi: 10.1016/0306-4522(91)90059-w. [DOI] [PubMed] [Google Scholar]
- 64.Stryker MP, Tonegawa S, Wang Y, Wolfer DP. Mutant mice and neuroscience: recommendations concerning genetic background. Banbury conference on genetic background in mice. Neuron. 1997;19:755–759. doi: 10.1016/s0896-6273(00)80958-7. [DOI] [PubMed] [Google Scholar]
- 65.Swanson GT, Kamboj SK, Cull-Candy SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Turetsky DM, Canzoniero LMT, Sensi S, Weiss JH, Goldberg MP, Choi DW. Cortical neurons exhibiting kainate-activated Co2+ uptake are selectively vulnerable to AMPA/kainate receptor-mediated toxicity. Neurobiol Dis. 1994;1:101–110. doi: 10.1006/nbdi.1994.0013. [DOI] [PubMed] [Google Scholar]
- 67.Tymianski M. Cytosolic calcium concentrations and cell death in vitro. In: Siesjo BK, Wieloch T, editors. Advances in neurology: cellular and molecular mechanisms of ischemic brain damage. Lippincott-Raven; Philadelphia: 1996. pp. 85–105. [PubMed] [Google Scholar]
- 68.Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tymianski M, Wang LY, MacDonald JF. Enhancement of neuroprotection by chronic depolarization in cultured spinal neurons. Brain Res. 1994;648:291–295. doi: 10.1016/0006-8993(94)91129-0. [DOI] [PubMed] [Google Scholar]
- 70.Vandenberghe W, Robberecht W, Brorson JR. AMPA receptor calcium permeability, GluR2 expression, and selective motoneuron vulnerability. J Neurosci. 2000;20:123–132. doi: 10.1523/JNEUROSCI.20-01-00123.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Van Eldik LJ, Zendegui JG, Marshak DR, Watterson DM. Calcium-binding proteins and the molecular basis of calcium action. Int Rev Cytol. 1982;77:1–61. doi: 10.1016/s0074-7696(08)62463-8. [DOI] [PubMed] [Google Scholar]
- 72.Villalba M, Bockaert J, Journot L. Concomitant induction of apoptosis and necrosis in cerebellar granule cells following serum and potassium withdrawal. NeuroReport. 1997;8:981–985. doi: 10.1097/00001756-199703030-00032. [DOI] [PubMed] [Google Scholar]
- 73.Wood SA, Allen ND, Rossant J, Auerbach A, Nagy A. Noninjection methods for the production of embryonic stem cell–embryo chimeras. Nature. 1993;365:87–89. doi: 10.1038/365087a0. [DOI] [PubMed] [Google Scholar]
- 74.Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22:179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- 75.Yin HZ, Sensi SL, Carriedo SG, Weiss JH. Dendritic localization of Ca2+-permeable AMPA/kainate channels in hippocampal pyramidal neurons. J Comp Neurol. 1999;409:250–260. [PubMed] [Google Scholar]
- 76.Ying HS, Weishaupt JH, Grabb M, Canzoniero LMT, Sensi SL, Sheline CT, Monyer H, Choi DW. Sublethal oxygen–glucose deprivation alters hippocampal neuronal AMPA receptor expression and vulnerability to kainate-induced death. J Neurosci. 1997;17:9536–9544. doi: 10.1523/JNEUROSCI.17-24-09536.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu SP, Choi DW. Na+–Ca2+ exchange currents in cortical neurons: concomitant forward and reverse operation and effect of glutamate. Eur J Neurosci. 1997;9:1273–1281. doi: 10.1111/j.1460-9568.1997.tb01482.x. [DOI] [PubMed] [Google Scholar]
- 78.Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]