

Abstract
Leptin is involved in the hypothalamic control of food intake and body weight. Fos immunohistochemistry has been used to functionally map leptin target neurons involved in these regulatory processes. However, only a subset of hypothalamic neurons expressing the long form of the leptin receptor (Ob-Rb) also coexpress the neuronal activation marker Fos after leptin stimulation. To functionally map all leptin target neurons, regardless of whether leptin-mediated neuronal activation or inhibition occurs, we immunohistochemically investigated the leptin-induced nuclear translocation of the signal transducer and activator of transcription molecule STAT3, which represents a crucial step in the regulation of leptin-dependent gene expression. As proven by colocalization studies with the nuclear 4′,6-diamidino-2-phenylindole dilactate stain, intracerebroventricular leptin treatment, but not intracerebroventricular application of pyrogen-free saline, induced a time-dependent nuclear translocation of STAT3 immunoreactivity in hypothalamic nuclei, with strong nuclear STAT3 signals detectable in the arcuate nucleus, the lateral hypothalamus, and the ventromedial and dorsomedial hypothalamic nuclei. This leptin-induced STAT3 translocation pattern proved to be distinct from that induced by interleukin-6, another cytokine using STAT3 in its signaling pathway. Combined immunohistochemical STAT3 and Fos detection after leptin treatment revealed a higher number of STAT3-positive than Fos-positive cell nuclei in the aforementioned hypothalamic structures and showed that Fos immunoreactivity colocalized only in a subset of all leptin-responsive STAT3 nuclei. These results suggest that the detection of nuclear STAT3 immunoreactivity represents a new neuroanatomical tool to functionally map central leptin actions. They further support the importance of ventrally located caudal hypothalamic structures representing the main leptin targets involved in body weight regulation.
Keywords: hypothalamus, food intake, appetite control, leptin, interleukin-6, cytokines, transcription factors, signal transducers and activator of transcription, STAT3, c-Fos, immunohistochemistry, confocal microscopy
Leptin is a cytokine that acts in the hypothalamus as a hormone to regulate food intake and body weight (BW) (Friedman, 1998; Elmquist et al., 1999; Meister, 2000). Its discovery has raised questions on the mechanisms and locations of its receptive systems involved in these regulatory processes. Five receptor subtypes (Ob-Ra–Ob-Re) have been identified (Lee et al., 1996; Fei et al., 1997; Leibel et al., 1997; Friedman, 1998) with the second isoform, Ob-Rb, representing the only splice variant capable of full activation of the leptin receptor-specific signal transduction pathway, the Janus kinase signal transducer and activator of transcription (Jak–STAT) signaling cascade (Chen et al., 1996; Lee et al., 1996). Leptin acts through Jak2 and STAT3 isoforms of this signal transduction pathway (Bjorbaek et al., 1997; Ghilardi and Skoda, 1997). Leptin receptor activation in vivo resulted in a time-dependent stimulation of STAT3 tyrosine phosphorylation in the rodent hypothalamus (Vaisse et al., 1996; McCowen et al., 1998), suggesting the presence of functionally active Ob-Rb receptors there. Indeed,in situ hybridization analysis demonstrated the presence of Ob-Rb mRNA in various brain structures, with high levels in hypothalamic nuclei described previously to control body weight regulation, such as the arcuate nucleus (ARC), the paraventricular nucleus (PVN), the dorsomedial hypothalamic nucleus (DMH), the ventromedial hypothalamic nucleus (VMH), and the lateral hypothalamic area (LHA) (Hakansson et al., 1996; Mercer et al., 1996; Cheung et al., 1997; Fei et al., 1997; Guan et al., 1997; Elmquist et al., 1998a).
To functionally map the actions of leptin on the brain, Fos immunohistochemical studies were performed, revealing Fos induction in all of the aforementioned hypothalamic structures, but with a distinct activation pattern in some of their subnuclei (van Dijk et al., 1996;Woods and Stock, 1996; Elmquist et al., 1997, 1998b, 1999; Yokosuka et al., 1998; Niimi et al., 1999; Elias et al., 2000). Whereas ventrolateral parts of the ARC showed Fos activation, there was an almost complete lack of Fos immunoreactivity in ventromedial areas, an ARC subnucleus with intense Ob-Rb expression. This unexpected observation was explained by an inhibitory leptin action on these neurons not detectable by the neuronal activation marker c-Fos (Elmquist et al., 1998b). By studying the mRNA expression of SOCS-3, a member of the suppressors of cytokine signaling proteins and a leptin-induced inhibitor of the leptin signaling cascade, a neuroanatomical method that was useful as a marker of direct leptin actions regardless of whether leptin-mediated neuronal excitation or inhibition has occurred, was consequently established (Bjorbaek et al., 1998; Elias et al., 1999). This method proved to be helpful for the characterization of leptin effects in distinct ARC subnuclei; however, a comprehensive hypothalamic map of direct leptin actions is still missing.
We therefore hypothesized that the combination of central leptin treatment with the subsequent immunohistochemical detection of STAT3 should be a good tool to reveal direct leptin actions on the brain. Various studies examined the distribution of STAT3 immunoreactivity within the hypothalamus (Hakansson and Meister, 1998; Hakansson et al., 1999; Wilkinson et al., 2000) and the whole CNS (Strömberg et al., 2000). However, a functional analysis studying the leptin-induced nuclear STAT3 translocation within hypothalamic nuclei has not yet been performed. This study describes a new mapping procedure that is useful for the neuroanatomical characterization of the centrally mediated effects of leptin.
MATERIALS AND METHODS
Animals. Adult male Wistar rats (Tierzucht Schönwalde GmbH, Schönwalde, Germany), with an initial BW ranging from 180 to 210 gm, were housed in individual cages in the animal house (Max Rubner Laboratory, Bergholz-Rehbrücke, Germany) and used in accordance with ethics authorities and local regulations (ethics approval number 48-3560-1/2). Lights were on from 6:00 A.M. to 6:00 P.M., and room temperature (RT) was adjusted to 22 ± 1°C. Animals had constant access to water and were fed standard lab chow available ad libitum. BW was monitored daily, and thereby the rats were handled at least once a day.
Intracerebroventricular cannulation of the lateral ventricle. Animals were anesthetized with intraperitoneal injections of a ketamine–xylazine solution [100 mg/kg BW Ketamin Gräub (Albrecht, Aulendorf, Germany); 10 mg/kg BW Rompun (2%; Bayer Vital, Leverkusen, Germany)]. During surgery, body temperature was kept constant at ∼37°C using a self-regulating heating pad (Fine Science Tools, Heidelberg, Germany). A modified 23 gauge cannula (Braun, Melsungen, Germany) attached to a PP30 tubing (Portex, Hythe, UK) and a 1 ml syringe (Braun) was filled with pyrogen-free saline (Sigma, Deisenhofen, Germany) and inserted stereotaxically (Ultra Precise Small Animal Stereotaxic Instrument, model 963; David Kopf Instruments, Tujunga, CA) into the left lateral ventricle of the brain. With a general +0.5 mm tooth bar adjustment, the coordinates used from bregma point were 0.4 mm posterior, 1.5 mm lateral, and 4 mm ventral (brain surface). Three small screws with a diameter of 1.4 mm (M1,4 V4A; Föhr Medical Instruments, Seeheim, Germany) were secured on the skull before the screws and the inserted cannula were embedded into dental cement. Finally, the cannula was sealed at the remaining opening with a plastic blocker, and the probe was checked daily during BW measurements to ensure that it remained sealed.
Accuracy of the cannula placement was tested by performing a drink test with a 50 ng intracerebroventricular bolus application of angiotensin II (Bachem Biochemica GmbH, Heidelberg, Germany) dissolved in 5 μl of 0.9% pyrogen-free saline (Sigma). The cumulative water intake was monitored for the following 60 min. Only those animals that showed an adequate drinking response of 9.8 ± 0.6 ml (n = 35; mean ± SEM) were used for additional studies. These drink tests were conducted 7–8 d after intracerebroventricular cannulation and 3–4 d before experimental stimulation.
Intracerebroventricular cytokine stimulation. On the day of experimental stimulation, BW of the rats reached 222.5 ± 3.4 gm compared with 198.5 ± 3.6 gm on the day of surgery (n = 35; mean ± SEM); hence, the rats had recovered from the surgical procedure and were gaining weight. In vivo cytokine stimulation was conducted in conscious animals with a 25 μl Hamilton syringe attached to the intracerebroventricular cannula via PP30 tubing. The central treatments consisted of an intracerebroventricular bolus injection of either 3.5 μg of recombinant murine leptin (kindly provided by Aventis Pharma, Frankfurt, Germany) or 200 ng of species homologous rat interleukin-6 (IL-6) (kindly provided by Dr. S. Poole, National Institute for Biological Standard and Control, Potters Bar, UK) (for details, seeRees et al., 1999), both diluted in 5 μl of pyrogen-free saline. The leptin and IL-6 doses used in this study were based on previous intracerebroventricular application experiments in which leptin proved to inhibit food intake by 40% at a dose of 4 μg per rat (Luheshi et al., 1999) and in which IL-6 caused a maximum febrile response at a dose of 200 ng per rat (Lenczowski et al., 1999; our unpublished observations). In control experiments, 5 μl of pyrogen-free saline alone was applied. Animals were then left 15–180 min before being deeply anesthetized with sodium pentobarbital (60 mg/kg BW Narcoren; Merial GmbH, Hallbergmoos, Germany) and transcardially perfused with 0.9% saline kept at RT, followed by ice-cold 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2. The animal's brains were removed and post-fixed in the same fixative for 1 hr at RT, and then the tissue was cryoprotected in 20% sucrose in phosphate buffer overnight at 4°C. Tissue was cut the following day.
Catalyzed reporter deposition amplification protocol for STAT3 detection. Initially, standard immunohistochemical procedures were used for STAT3 detection (rabbit anti-STAT3 antibody, sc-482; Santa Cruz Biotechnology, Heidelberg, Germany), resulting in a very weak and nonsatisfying STAT3 signal. As shown in a recent publication investigating STAT3 distribution in the rat CNS, amplification procedures are a helpful tool to investigate STAT3 immunoreactivity (Strömberg et al., 2000). Therefore, to enhance basal as well as cytokine-induced STAT3 signals in brain sections, we used a commercial tyramide amplification kit (NEL700; NEN Life Science Products GmbH, Köln, Germany) based on the catalyzed reporter deposition method. The specificity of the Santa Cruz Biotechnology STAT3 antibody used in the present study was clearly proven in a recent publication (Strömberg et al., 2000). Our own preabsorption control experiments with the control STAT3 peptide (data not shown) confirmed these findings.
Coronal 20–40 μm free-floating hypothalamic sections (bregma levels, 0.00 to −4.80 mm) were cut on a freezing microtome (model 1205; Jung, Heidelberg, Germany) for double- and triple-labeling experiments, and 40 μm free-floating sections of the forebrain, midbrain, and hindbrain were used for STAT3 detection alone. Sections were placed into 10% fetal calf serum and 0.3% Triton X-100 in 0.1 mphosphate buffer, pH 7.2, for 1 hr at RT. For tyramide signal amplification, sections were then transferred to the blocking reagent provided in the kit for 30 min at RT. This was followed by the primary STAT3 antibody incubation (1:12000 diluted in 0.1 mphosphate buffer, 2% fetal calf serum, and 0.1% Triton X-100) for 24–48 hr at 4°C. The tyramide amplification protocol was continued according to the kit description but using a phosphate buffer system and not the suggested borate buffer. Primary STAT3 antibody was detected with a secondary biotinylated anti-rabbit antibody (Vector BA-1000; Linaris Biologische Produkte, Wertheim-Bettingen, Germany) for 1 hr at RT (diluted 1:200 in 0.1 m phosphate buffer and 2% fetal calf serum). The additional immunohistochemical processing was performed with an avidin–biotin–horseradish peroxidase complex (Vector Elite Kit; Linaris Biologische Produkte), which was visualized by either diaminobenzidine hydrochloride (Sigma) reaction in the presence of hydrogen peroxide or fluorescein (FITC)-conjugated avidin D (Vector A-200; Linaris Biologische Produkte).
Immunohistochemical colocalization studies and nuclear 4′,6-diamidino-2-phenylindole dilactate stain. In double-labeling experiments, coronal 20–40 μm free-floating hypothalamic sections already stained for STAT3 were coanalyzed using additional antibodies. A coincubation was performed with a 1:2000 mouse anti-glial fibrillary acidic protein (GFAP) antibody (MAB3402; Chemicon, Hofheim, Germany) and with a 1:200 mouse anti-adenomatous polyposis coli (APC) protein (OP80; Oncogene Research Products, Calbiochem, Bad Soden, Germany). Both antibodies are used as cytoskeletal and cytoplasmatic markers of glia cells in the CNS for either astrocytes (GFAP) or mature oligodendrocytes (APC).
STAT3 detection was also combined with the immunohistochemical analysis of the immediate early gene Fos. Fos-like immunoreactivity was detected with an anti-c-fos antibody (sc-52; Santa Cruz Biotechnology) at a 1:500–1:1000 dilution. In this experiment, both primary antisera were raised in rabbits, and therefore a consecutive detection of first STAT3 and then Fos was performed according to the method described previously by Shindler and Roth (1996). In detail, STAT3 was visualized using the catalyzed reporter deposition amplification protocol with FITC-conjugated avidin D as described above. With a STAT3 antibody dilution of 1:12000 that proved to be below the detection limit of a fluorescent-labeled secondary antibody, yet being still sufficient for detection with the catalyzed reporter deposition amplification system, the STAT3 primary antibody was effectively neglected during the visualization steps necessary to detect the second primary Fos antibody. Fos-like immunoreactivity was then localized using a Cy3-conjugated anti-rabbit (1:200–1:400) antibody (Sigma). All primary antibodies were diluted in 0.1m phosphate buffer with 2% fetal calf serum and 0.1% Triton X-100, and the sections were incubated 12–48 hr at 4°C. The visualization for the monoclonal primary mouse antibodies were performed with a secondary Cy3-conjugated anti-mouse (1:2000) antibody (Sigma).
In a final step, all sections were stained for 5 min at RT with the nuclear stain 4′,6-diamidino-2-phenylindole dilactate (DAPI) (Molecular Probes Europe BV, Leiden, Netherlands), which was diluted 1:400 in phosphate buffer.
Microscopical analysis. The free-floating sections were mounted onto gelatin-coated slides and for the enzyme-histochemical detection coverslipped with Entellan (Merck, Darmstadt, Germany), whereas for fluorescent detection, slides were coverslipped with Crystal/Mount (Biomedia, Foster City, CA). The sections were analyzed using a conventional Zeiss (Jena, Germany) Axioplan light microscope and a Zeiss confocal microscope. Confocal images were taken with an inverted Zeiss Laser Scanning Microscope (model LSM 410) attached to an internal helium–neon ion laser and two external lasers, one argon ion and one argon UV laser with individual excitation outputs of 543, 488, and 364 nm, respectively. Images were individually processed for the color channel red, green, and blue (RGB) using a 570 long-pass emission filter for Cy3 detection (R), a 510–525 bandpass emission filter for FITC detection (G), and a 397 long-pass emission filter for DAPI detection (B). The images shown are the result of one optical section; the color images show individual color channels or the overlay of two of such individually taken RGB images. Image editing software (Adobe Photoshop; Adobe Systems, San Jose, CA) was used to change the graphic mode from RGB to CMYK (cyan, magenta, yellow, and black) and to combine the images into plates.
RESULTS
Intracerebroventricular leptin treatment induces a time-dependent nuclear translocation of STAT3 immunoreactivity within the hypothalamus
To investigate the basal and the leptin-induced hypothalamic STAT3 expression at the cellular level, immunohistochemical procedures were used. Basal STAT3 expression levels in the rat forebrain were evaluated in controls via intracerebroventricular pyrogen-free saline application and found to be highest in the caudal hypothalamus (Fig.1A). In particular, ventrally located hypothalamic structures, such as the ARC (Figs.1A,2A), its adjacent periarcuate area (PAA), and areas ventral to the fornix in the LHA (Figs. 1A, 2G), were labeled. Weaker STAT3 immunoreactivity was observed in several other hypothalamic nuclei, such as the VMH, the DMH, the retrochiasmatic area (RCH), the PVN, the supraoptic nucleus (SON), and the medial preoptic area (MPO). By confocal microscopy and the nuclear DAPI stain, the subcellular distribution of the basal STAT3 expression was investigated. Under control conditions, STAT3 immunoreactivity was predominantly found in the cell cytoplasm and in nerve fibers (Fig. 2A–C,G–I, insets in B/C,H/I), with nuclear STAT3 labeling being rarely detectable in the aforementioned hypothalamic structures.
Fig. 1.
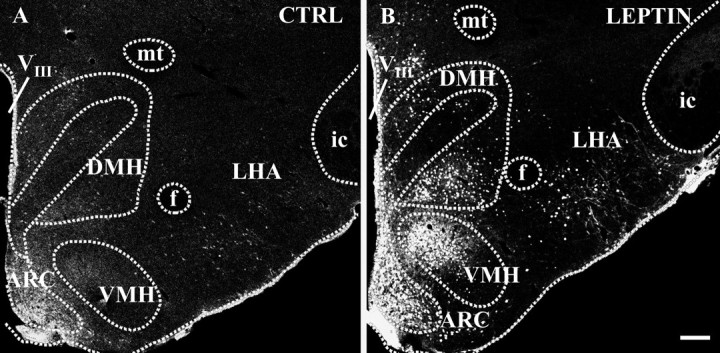
Hypothalamic leptin targets mapped by the detection of STAT3 immunoreactivity under control conditions (A) and after intracerebroventricular leptin application (B). Immunofluorescence photomicrographs show an overview at caudal hypothalamic levels approximately −3.3 mm posterior to bregma point under control conditions (CTRL; 30 min after 5 μl of pyrogen-free saline) and 30 min after central leptin treatment (3.5 μg of leptin in 5 μl of pyrogen-free saline). Note the massive increase of STAT3 immunoreactivity in various hypothalamic structures, such as within the ARC, the ventral parts of the DMH, the dorsal parts of the VMH, and the LHA, in the leptin-treated animal (B) compared with the basal STAT3 expression detected in the control (A). VIII, Third ventricle; f, fornix; mt, mamillothalamic tract; ic, internal capsule. Scale bar, 200 μm.
Fig. 2.
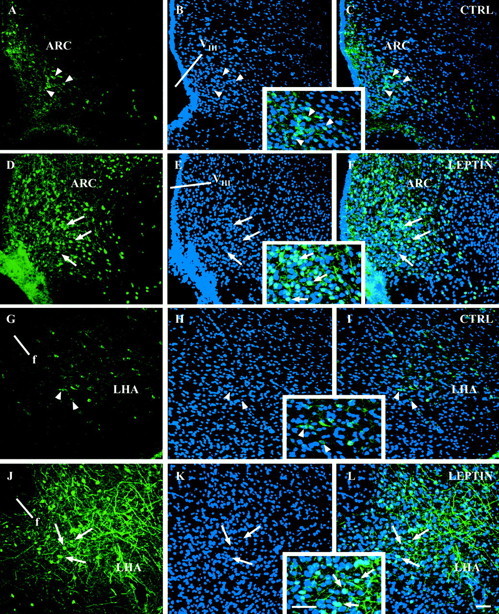
Leptin induces a nuclear translocation of STAT3 immunoreactivity in hypothalamic nuclei. Immunofluorescence photomicrographs of hypothalamic sections of the ARC (A–F) and ventral aspects of the LHA (G–L) are shown under control conditions (CTRL; A–C, G–I; 30 min after pyrogen-free saline) and leptin-stimulated conditions (D–F, J–L; 30 min after leptin). STAT3 immunoreactivity in green (A,D, G, J) is compared with the blue nuclear DAPI stain (B, E, H,K). The overlays of these individual confocal images are shown in C, F,I, and L. Note that, under basal control conditions, STAT3 immunoreactivity is predominantly located within the cytoplasm and also nerve fibers of ARC and LHA neurons, whereas most of the cell nuclei seem to be devoid of STAT3 labeling (see arrowheads in A–Cand G–I and also the insets inB/C and H/I showing this at higher magnification). Leptin treatment (30 min) not only led to a nuclear translocation of STAT3 immunoreactivity in both hypothalamic structures the ARC and the LHA (see arrows showing the light blue double-labeled cell nuclei inF and L and also theinsets in E/F and K/Lshowing this at higher magnification) but also to a general increase of STAT3 labeling (compare A with D andG with J).VIII, Third ventricle;f, fornix. Scale bars, 50 μm.
The subcellular distribution and the intensity of STAT3 signals within some of these hypothalamic structures dramatically changed after the intracerebroventricular leptin administration. As shown in overview in Figure 1B, leptin treatment led to a marked increase of STAT3 immunoreactivity in the caudal hypothalamus. Although all parts of the ARC seemed to be heavily labeled, a distinct subdistribution of the STAT3 signal became obvious for the VMH or the DMH, with dorsal (VMH) and ventral (DMH) parts being the most leptin-responsive hypothalamic targets besides the ARC. Within the PAA, the RCH, and the LHA, intense but more scattered labeling of individual medium- to large-sized neurons occurred. In addition, cells within the ependymal lining of the third ventricle, the adjacent periventricular nucleus, and in ventrally located meninges showed strong STAT3 immunoreactivity. A subcellular analysis of the leptin-induced STAT3 signals revealed that a massive shift of STAT3 immunoreactivity from the cytoplasm into the cell nucleus occurred 30 min after the intracerebroventricular leptin application, as shown for example within the ARC (Fig. 2D–F, inset inE/F) or the LHA (Fig. 2J–L,inset in K/L). Such a nuclear translocation of STAT3 was observed in various hypothalamic but also extrahypothalamic forebrain structures (Table 1). Besides the induction of this intense nuclear STAT3 signal, intracerebroventricular leptin treatment additionally induced a massive increase of the STAT3 signal in nerve fibers and axons, being most prominent within the ARC (Fig.2D,F, inset inE/F) and the ventral LHA (Fig.2J,L, inset inK/L).
Table 1.
Forebrain1-a distribution of leptin- and interleukin-6 (IL-6)-induced nuclear STAT3 immunoreactivity1-b compared with basal nuclear STAT3 expression in the control
| Forebrain1-astructures | Cytokine-induced nuclear STAT3 labeling1-b | ||
|---|---|---|---|
| i.c.v. Leptin | i.c.v. IL-6 | Control | |
| Hypothalamus | |||
| Anterior hypothalamic area (AHA) | − | ++ | − |
| Arcuate nucleus (ARC) | +++ | + | + |
| Dorsal hypothalamic area (DA) | + | ++ | + |
| Dorsomedial nucleus (DMH) | +++ | + | − |
| Lateral hypothalamic area (LHA) | ++ | ++ | − |
| Mammillary body | |||
| medial parts | + | + | − |
| lateral parts | + | ++ | − |
| Medial preoptic area (MPO) | + | ++ | − |
| Ventromedial preoptic nucleus (VMPO) | + | +++ | − |
| Median preoptic nucleus (MnPO) | + | + | − |
| Paraventricular nucleus (PVN) | + | + | − |
| Periarcuate area (PAA) | ++ | +++ | − |
| Perifornical area (PFA) | ++ | − | − |
| Periventricular nucleus (PE) | ++ | + | − |
| Posterior hypothalamic area (PA) | ++ | ++ | − |
| Retrochiasmatic area (RCH) | ++ | ++ | − |
| Subfornical organ (SFO) | − | − | |
| near to the big lateral vessels | + | ||
| Suprachiasmatic nucleus (SCN) | + | + | − |
| Supraoptic nucleus (SON) | − | +++ | − |
| Vascular organ of the lamina terminalis (OVLT) | − | − | |
| lateral parts | ++ | ||
| Ventromedial nucleus (VMH) | +++ | + | − |
| Septal and basal telencephalic regions | |||
| Bed nucleus of the stria terminalis (BNST) | − | − | − |
| Lateral preoptic area (LPO) | − | − | |
| only ventral parts | ++ | ||
| Lateral septum (LS) | − | ++ | − |
| Substantia innominata (SI) | − | − | − |
| Others | |||
| Ependymal lining of all ventricles | +++ | +++ | + |
| Meninges, in particular at ventral sites | ++ | +++ | − |
| Periventricular | |||
| lateral ventricles (cannula side) | +(+) | ++(+++) | −(−) |
| third ventricle | + | +++ | − |
| aqueduct | + | + | − |
| fourth ventricle | − | ++ | − |
| Piriform cortex, layer I+II | +++ | + | − |
| Probe site | +++ | +++ | +++ |
| Thalamic nuclei | − | − | |
| medial and lateral habenular nucleus | ++ | ||
| mediodorsal thalamic nuclei | +++ | ||
| Cerebellum | − | − | − |
The nomenclature used is modified from that used by Paxinos and Watson (1997) and Swanson (1992).
Relative values are given as qualitative estimates of the densities of nuclear STAT3 labeling. A four-point scale was used to rate the data: +++, high density; ++, moderate density; +, low density; −, no nuclear signal. The data are based on the light microscopical analysis of representative animals treated with 30 min intracerebroventicular (i.c.v.) application of leptin, IL-6, or pyrogen-free saline (Control).
The time dependency of this nuclear STAT3 translocation is demonstrated for the ARC using a post-application period ranging from 15 to 180 min (Fig. 3). Compared with the basal STAT3 expression in the control (Fig. 3A), intracerebroventricular leptin application rapidly induced a nuclear translocation of STAT3 immunoreactivity already present at 15 min (Fig. 3B), which became even more pronounced at 30 min (Fig.3C). When using post-application periods longer than 30 min, the high intensity of the STAT3 signal present at 30 min slowly decreased (Fig. 3D–F), with only a small number of STAT3-positive cell nuclei being left 180 min after leptin treatment (Fig. 3F).
Fig. 3.
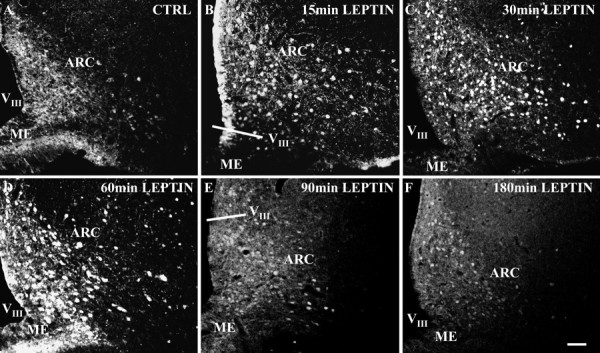
Leptin induces a time-dependent nuclear translocation of STAT3 immunoreactivity in the ARC. Immunofluorescence photomicrographs of STAT3 immunoreactivity in ARC sections are shown under control conditions (A, CTRL; 30 min pyrogen-free saline) and under leptin-stimulated conditions (B–F). Under basal control conditions, STAT3 immunoreactivity is detectable in all subnuclei of the ARC and is predominantly located in the cytoplasm and nerve fibers of ARC neurons (A). As soon as 15 min after leptin application, a massive increase in nuclear STAT3 labeling is detectable in all parts of the ARC (B), which becomes even more pronounced after 30 min (C). However, 60–180 min after leptin application (D–F), the nuclear STAT3 signal slowly starts to decrease again, which is indicated by the shift from a sharp-edged round-shaped STAT3 signal in the nucleus (B, C) to a more blurred and less defined nuclear labeling and a decrease in the intensity of STAT3 immunoreactivity (D–F).VIII, Third ventricle;ME, median eminence. Scale bar, 50 μm.
Intracerebroventricular leptin treatment shows a nuclear STAT3 translocation pattern distinct from that induced by intracerebroventricular interleukin-6 application
The specificity of the leptin-induced nuclear STAT3 translocation in distinct forebrain structures was additionally checked using IL-6, another cytokine also capable of activating the Jak–STAT3 signal transduction pathway. Indeed, for both cytokines, a specific pattern of nuclear STAT3 translocation became obvious within the rat forebrain (Fig. 4, Table 1). Whereas in controls most (extra)hypothalamic structures proved to be devoid of nuclear STAT3 labeling, both intracerebroventricular leptin and intracerebroventricular IL-6 induced a shift of the STAT3 signal into the cell nucleus (Table 1). However, the densities of nuclear STAT3 labeling and the brain sites affected were different for the two cytokines. Whereas leptin seemed to predominantly target ventral parts of the caudal hypothalamus, IL-6 additionally induced a massive STAT3 translocation in the rostral hypothalamus, in particular within the MPO and its ventromedial part, the VMPO (Fig. 4B,inset in A/B), which was not detectable in the leptin-treated animals (Fig. 4A). When looking at other forebrain structures, the two cytokine stimuli expressed a differing pattern of nuclear STAT3 translocations. This included the anterior piriform cortex (PIR), which showed a high density of nuclear STAT3-labeled layer II neurons in the intracerebroventricular leptin but not in the intracerebroventricular IL-6-treated animals (Fig.4C,D), and also the SON (Fig.4E,F) and some thalamic nuclei (Table 1) in which IL-6 but not leptin induced a nuclear translocation of STAT3 immunoreactivity. In the caudal hypothalamus, the overall pattern of STAT3 immunoreactivity induced by leptin or IL-6 again proved to be different (Fig. 4G,H). Whereas leptin induced nuclear STAT3 translocation in medial and lateral parts of the ARC as well as in dorsal parts of the VMH, IL-6 did not lead to STAT3 translocation in the medial ARC and only to a minor nuclear labeling in the dorsal VMH.
Fig. 4.
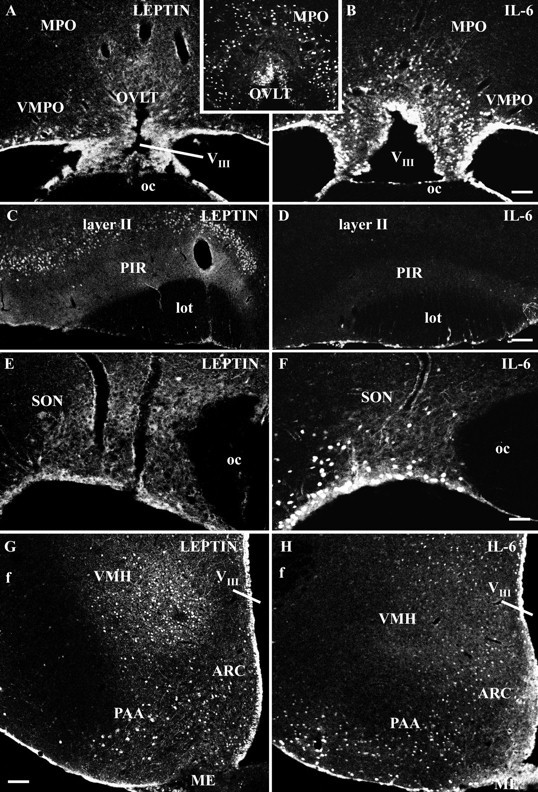
Distinct cytokine-induced nuclear STAT3 translocation patterns in forebrain structures induced by intracerebroventricular leptin or by intracerebroventricular IL-6 treatment. Immunofluorescence photomicrographs of STAT3 immunoreactivity are shown at rostral (A–D), medial (E, F), and caudal (G, H) hypothalamic levels under leptin-stimulated (A, C,E, G) or IL-6-stimulated (B, D, F,H, inset in A/B) conditions. Only basal, predominantly non-nuclear STAT3 signals are expressed in the 30 min leptin-treated animal within lateral parts of the organum vasculosum of the lamina terminalis (OVLT) and the adjacent ventromedial preoptic area (VMPO) (A). IL-6 treatment on the other hand induced an increase and shift of STAT3 immunoreactivity into cell nuclei, particularly in the MPO, including the ventromedial preoptic area, as well as in the lateral and dorsal aspects of the organum vasculosum of the lamina terminalis (B, inset A/B). Intracerebroventricular leptin (C) but not IL-6 application (D) induced nuclear STAT3 labeling in neurons of layer II of the PIR. For orientation, note thatC and D have been rotated. Within the hypothalamic SON, IL-6 (F) but not leptin treatment (E) led to nuclear STAT3 labeling. Note that the basal expression of STAT3 immunoreactivity depicted from the leptin-treated animal (E) is almost completely located within the cytoplasm of magnocellular SON neurons. In caudal hypothalamic aspects shown in overview for leptin (G) and IL-6 treatment (H), a distinct cytokine-induced pattern of nuclear STAT3 immunoreactivity could be detected in the ARC, the PAA, and the VMH. VIII, Third ventricle; oc, optic chiasm; lot, lateral olfactory tract; f, fornix; ME, median eminence. Scale bars: A–D, G,H, 100 μm; E, F,inset in A/B, 50 μm.
Leptin-induced nuclear STAT3 immunoreactivity within the piriform cortex and the hypothalamus does not colocalize with glial cell markers
For the light microscopic analysis, the enzyme immunohistochemical STAT3 detection was combined with cresyl violet counterstaining, indicating a predominant neuronal location of the STAT3 immunoreactivity under both basal and leptin-stimulated conditions within the hypothalamus (data not shown). However, non-neuronal, nuclear STAT3 labeling was also detected, e.g., in ependymal and meningeal cells, and also near to the probe site in small, noncircular-shaped cell nuclei, which presumably belonged to glial cells (Table 1). Former electron microscopic investigations have shown that, within the brain tissue, leptin receptors are located on both neurons and glia. It was therefore of importance to analyze the cell types in which intracerebroventricular administration of leptin induced nuclear translocation of STAT3. To demonstrate the cellular origin of the STAT3 signals, double-labeling experiments combining the STAT3 detection with the immunohistochemical localization of the cytoskeletal and cytoplasmatic glial cell markers GFAP (Fig.5A–E) and APC (Fig.5F) were performed after intracerebroventricular leptin application. The size and shape of the STAT3-stained nuclei within most cells of the PIR (Fig. 5A) and the hypothalamus (Fig. 5B–F) again indicated a predominant neuronal cell origin. Indeed, no cellular colocalization of the nuclear STAT3 signal was found within the astrocytes in layer II of the anterior PIR (Fig. 5A) or within the astrocytes in the main hypothalamic leptin targets, such as the ARC (Fig. 5B), the LHA (Fig.5C), and the DMH and VMH (Fig.5D,E). In addition, leptin-induced nuclear STAT3 signals within the VMH did not colocalize with the cytoplasmatic labeling of oligodendrocytes (Fig. 5F). Within the most intense nuclear STAT3-labeled hypothalamic regions, in no single case did a colocalization occur, suggesting that glia cells in these areas may express a leptin receptor isoform that is not capable to activate the full cytokine specific Jak–STAT pathway.
Fig. 5.

Leptin-translocated nuclear STAT3 immunoreactivity does not colocalize with the glial cell markers GFAP (A–E) and APC (F) in forebrain structures. Immunofluorescence photomicrographs of leptin responsive-forebrain structures, such as the PIR (layer II, A), the ARC (B), ventral aspects of the LHA (C), the DMH (D), and the VMH (E,F) are shown 30 min after intracerebroventricular leptin stimulation. Nuclear STAT3 immunoreactivity (green) is compared with the cellular location of glial cells (red). Note that the STAT3-labeled nuclei do not colocalize with the cytoplasm or cytoskeletal structures of labeled astrocytes or oligodendrocytes, thereby indicating that within these forebrain structures the leptin-induced nuclear STAT3-translocation has occurred in neurons. f, Fornix. Scale bars: A, C, D, 25 μm; B, E, F, 10 μm.
Nuclear STAT3 and Fos immunoreactivity colocalizes only in a subset of leptin-responsive forebrain neurons
In double-labeling experiments, the leptin-induced nuclear STAT3 immunoreactivity within the forebrain was compared with the Fos response 60 min after intracerebroventricular leptin application. The time point used in these experiments was chosen as a compromise between the time necessary for nuclear translocation of STAT3 (15–30 min) (Fig. 3) and the time known to show the first peak of the nuclear Fos signal (90 min) in neurons. A clear difference between the number of STAT3-stained (green nuclei) versus Fos-stained (red nuclei) nuclei could be detected, with more STAT3-stained than Fos-stained nuclei after central leptin treatment (Fig.6A–G,I,J). Individual detection of STAT3 and Fos immunoreactivity (Fig.6A–D) showed a similar distribution of nuclear signals in layer II of the PIR (Fig.6A,B) but distribution differences within the caudal hypothalamus. STAT3 was predominantly induced in medial parts of the ARC and dorsal aspects of the VMH (Fig.6C), whereas nuclear Fos was mainly detectable in the ventrolateral ARC and the adjacent periarcuate area (Fig.6D). At higher magnification, nuclear STAT3 and Fos immunoreactivity proved to be colocalized (yellownuclei) only in a subset of neurons of the anterior PIR (Fig.6E), the ventrolateral ARC (Fig.6F), and ventral parts of the DMH (Fig.6I). Minor colocalization of the two nuclear signals was detected within the LHA (Fig. 6G) and the dorsal part of the VMH (Fig. 6J).
Fig. 6.
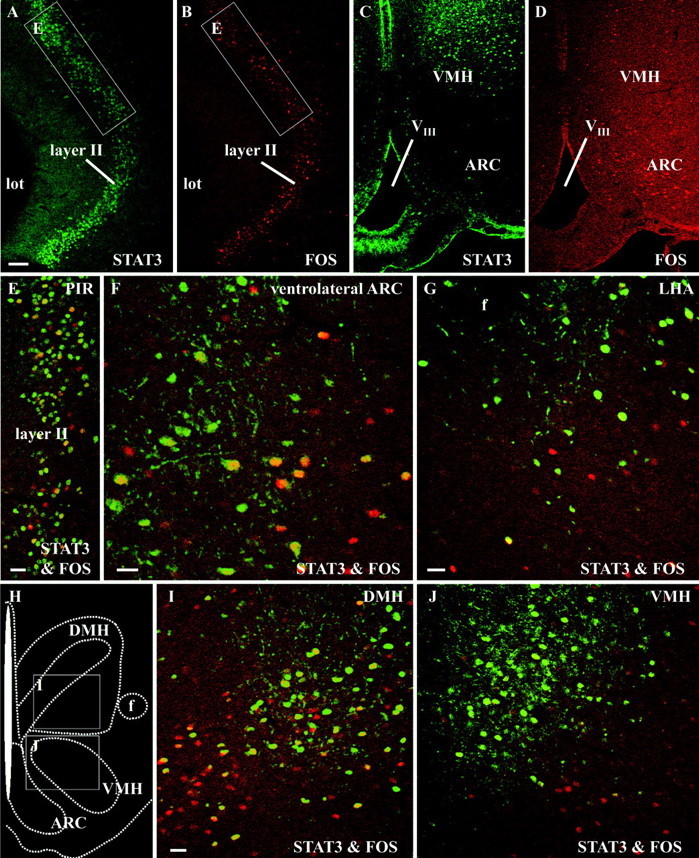
Leptin-translocated nuclear STAT3 and Fos immunoreactivity colocalizes only in a subset of leptin-responsive forebrain neurons. Immunofluorescence photomicrographs of leptin-responsive forebrain structures, such as the PIR (A, B, E), the ARC (C, D, F), ventral aspects of the LHA (G), the DMH (I), and the VMH (C,D, J), are shown 60 min after intracerebroventricular leptin stimulation. The brain map (H) indicates the position of the photomicrographs shown in I and J. Nuclear STAT3 immunoreactivity (green) is compared with the nuclear localization of the protein product of the immediate early gene c-fos (red). Although within layer II of the PIR many STAT3-labeled (A) and Fos-labeled (B) nuclei could be detected, the overlay (E) at higher magnification (the exact location indicated by the white rectangles in A and B) clearly showed that only a small subset of layer II neurons coexpressed both transcription factors (yellow nuclei), whereas the majority responded with a nuclear translocation of STAT3 only (green nuclei). Red nuclei belong to those layer II neurons that responded with a nuclear translocation of Fos but not STAT3. Similar results, but with varying degrees of STAT3 and Fos colocalization, were also obtained in the hypothalamic leptin target structures, such as the ventrolateral ARC (C, D, F), the LHA (G), the ventral DMH (I), and the dorsal VMH (J). VIII, Third ventricle; lot, lateral olfactory tract;f, fornix. Scale bars: A–D, 100 μm;E–G, I, J, 25 μm.
DISCUSSION
The ventrobasal hypothalamus has been described as the primary central leptin target involved in the control of food intake and energy balance. Many physiological responses regulated by leptin, such as the inhibition of appetite via intracerebroventricular leptin treatment in rats, characteristically start with some delay and show long-lasting effects (Cusin et al., 1996; Woods et al., 1998). This is in agreement with the long-duration intracellular genomic mode of the Jak–STAT signaling pathway downstream to Ob-Rb receptor activation (Schindler and Darnell, 1995; Darnell, 1997). Thus, it is likely that the weight-reducing effects of leptin are mediated via the Jak–STAT signal transduction of hypothalamic Ob-Rb receptors.
The leptin-induced nuclear STAT3 pattern matches with Ob-Rb receptor expression in the ventrobasal hypothalamus
One open question in the past was whether the extensive distribution of the Ob-Rb receptor within the rodent hypothalamus, detected via in situ hybridization techniques (Mercer et al., 1996; Fei et al., 1997; Guan et al., 1997; Elmquist et al., 1998a) or immunohistochemistry (Hakansson et al., 1998; Baskin et al., 1999), really represents the active leptin receptor isoform. With the Ob-Rb receptor being the major brain-intrinsic leptin receptor isoform capable of full activation of the Jak–STAT signaling cascade, (Chen et al., 1996; Lee et al., 1996) the characterization of the leptin-induced nuclear STAT3 translocation could be a functional proof of active Ob-Rb expression. Indeed, our results demonstrate a leptin-induced nuclear STAT3 translocation in the rat hypothalamus. The hypothalamic location of strong Ob-Rb receptor expression on both mRNA and protein level described in the literature (Mercer et al., 1996; Elmquist et al., 1998a; Hakansson et al., 1998; Baskin et al., 1999) matches with the most intense nuclear STAT3 signals detected in the present study, and this overlap becomes even more evident in distinct subnuclei of the ventrobasal hypothalamus. In detail, all ARC subnuclei, a caudal DMH subnucleus located ventrolaterally to its compact formation and the dorsomedial part of the VMH, can be regarded as functionally active major hypothalamic leptin targets. Hypothalamic structures described to express less intense Ob-Rb mRNA levels, such as the LHA, the perifornical area, the periventricular nucleus, the parvocellular part of the paraventricular nucleus, the posterior hypothalamic nucleus, and the medial mammillary body (Elmquist et al., 1998a), showed an individual spread and/or a weaker nuclear STAT3 response in our study. Although for the above mentioned structures a relative clear-cut match of nuclear STAT3 signals and Ob-Rb expression was demonstrated, discrepant cases also existed. Strong leptin receptor immunoreactivity was found in the SON (Hakansson et al., 1998); however, leptin treatment failed to induce a nuclear STAT3 translocation there. This suggests that these immunohistochemically identified SON leptin receptors are not able to induce leptin-dependent signaling because they do not represent the active full-length receptor variant.
The present data also neuroanatomically confirm the finding of Vaisse et al. (1996) and McCowen et al. (1998) that leptin induces STAT3 phosphorylation in the rodent hypothalamus in vivo. Furthermore, the time course evaluated in the present study with nuclear STAT3 translocation already present at 15 min and being maximal at 30 min is identical to that reported with biochemical and molecular–biological approaches (Vaisse et al., 1996; McCowen et al., 1998). So in summary, our experiments further extend these studies by mapping the functionally active hypothalamic leptin targets, with their exact neuroanatomical localization being essential for the understanding of leptin-induced physiological responses.
The leptin-induced nuclear STAT3 pattern is distinct from that induced by interleukin-6
The specificity of the leptin-induced STAT3 translocation was verified with intracerebroventricular treatment of the endogenous pyrogen IL-6. Central IL-6 administration elicits a variety of (patho)physiological functions, such as the reduction of food intake and locomotor activity, the activation of the hypothalamic–pituitary–adrenocortical axis, and the mediation of fever responses (Lenczowski et al., 1999). The IL-6 receptor has been described to act through similar signaling pathways as the Ob-Rb receptor, that is by altering gene expression via STAT3 binding to gene promoter regions (Darnell, 1997; Takeda and Akira, 2000). Indeed, intracerebroventricular IL-6 application also induced nuclear STAT3 translocation but with a pattern clearly different from that induced by leptin treatment. Whereas leptin proved to be almost ineffective in rostral hypothalamic regions such as the preoptic area, IL-6 induced a strong nuclear STAT3 translocation in the MPO, especially in its ventromedial part. Not surprisingly, these brain regions have been reported previously to participate in the central control of body temperature and fever regulation (Saper, 1998). Within the MPO, the observed IL-6-induced nuclear STAT3 staining pattern 15–30 min after intracerebroventricular application was similar to that seen with Fos analysis 2 hr after endotoxin (lipopolysaccharide) treatment (Elmquist et al., 1996), suggesting a STAT3 action upstream of Fos induction in those neurons.
STAT3 immunohistochemistry: a novel tool to map neurons responding to leptin
Leptin binding to the Ob-Rb receptor leads to nuclear translocation of the activated STAT3 isoform and finally induces transcriptional activation of genes (Ihle, 1996; Darnell, 1997; Takeda and Akira, 2000), among them the immediate early gene c-fos(Elmquist et al., 1997, 1998b; Banks et al., 2000) and the SOCS-3 gene (Bjorbaek et al., 1998; Elias et al., 1999). Fos immunohistochemistry has been thoroughly used to investigate leptin actions on the brain. Systemic (Elmquist et al., 1997, 1998b; Wang et al., 1998; Elias et al., 2000; Wilkinson et al., 2000) but also central leptin treatment (van Dijk et al., 1996; Niimi et al., 1999) induced Fos expression in the ventrobasal hypothalamus, with a similar pattern also detected in the present study. However, using Fos analysis, certain hypothalamic Ob-Rb receptor populations seemed to be inaccessible to leptin treatment. One hypothalamic structure in which a discrepancy of high Ob-Rb expression level (Elmquist et al., 1998a; Baskin et al., 1999) with the lack of Fos activation became most evident is the ventromedial ARC (Elmquist et al., 1997, 1998b; Elias et al., 1998). Because inhibitory responses in neurons may not be associated with Fos expression (Chan et al., 1993; Elmquist et al., 1998b), SOCS-3 mRNA expression was consequently used as the first neuroanatomical approach to identify direct leptin-responsive neurons (Bjorbaek et al., 1998;Elias et al., 1999). Within the rat ventrobasal hypothalamus, leptin treatment induced a SOCS-3 expression in areas with high densities of Ob-Rb receptors (Bjorbaek et al., 1998). Both hypothalamic distribution patterns, the leptin-dependent SOCS-3 expression and the constitutive Ob-Rb receptor expression, again resemble the leptin-induced nuclear STAT3 pattern found in the present study. In particular, the SOCS-3 expression and the nuclear STAT3 signals proved to be almost identical in the caudal DMH ventral to its compact formation, as well as in all parts of the ARC, including medial subnuclei, which have been demonstrated to lack a Fos response after leptin treatment. The data collected with both methods, the detection of SOCS-3 mRNA expression and the STAT3 immunohistochemistry, strongly suggest that, unlike Fos immunohistochemistry, these two methods are functional mapping procedures for all leptin targets, whether or not cells are activated or inhibited by leptin.
Recent electrophysiological data have clearly demonstrated an inhibitory leptin action on the neuronal discharge rate of orexin-sensitive ARC neurons, suggesting that neuronal inhibition and not activation is the predominant mode of leptin action in this nucleus. Although in many cases a leptin-induced direct inhibition of neuronal activity in the ARC occurred, some direct activation of neuronal activity also existed (Rauch et al., 2000). In any case, the neuronal discharge rate was rapidly changed in these experiments and therefore, at least initially, these changes occurred independently from genomic effects, e.g., the leptin-induced Fos expression or gene regulation via binding of phosphorylated STAT3 dimers to distinct promoter regions. The putative participation of the STAT3 molecule and its mode of action in such nongenomic, fast neuronal leptin effects remains to be elucidated; however, the intense nerve fiber and axonal STAT3 labeling occurring after leptin application favors the hypothesis of an involvement of axodendritic neuronal processes in fast responses to leptin. In line with electrophysiological data, less Fos-stained than STAT3-labeled cell nuclei may indicate that leptin predominantly induces neuronal inhibition rather than activation in those structures (Spanswick et al., 1997; Rauch et al., 2000). It should be mentioned, however, that these results could also be a reflection of the 60 min time point used in the present study, it being a compromise between the peak of leptin-induced nuclear STAT3 (15–30 min) and nuclear Fos expression (90 min). Those neurons that showed both a nuclear Fos and a STAT3 signal proved to be a minor subpopulation of all STAT3-responsive cells and should be classified as directly activated neurons. Such double-labeled neurons seemed to be prominent in the ventrolateral part of the ARC and in the caudal DMH in a subnucleus ventral to its compact formation. In contrast, within the dorsomedial VMH and in particular within the ventral LHA, only minor colocalization was found, possibly reflecting independent leptin target entities and pathways in the very same nuclei.
Footnotes
This work was supported in part by European Commission Grant QLG3-CT-1999–00908 to W.M. We thank Dr. E. Simon for critical reading of this manuscript.
Correspondence should be addressed to Dr. Thomas Hübschle, Institute of Veterinary Physiology, Justus-Liebig-University, Frankfurter Straße 100, D-35392 Giessen, Germany. E-mail:thomas.huebschle@vetmed.uni-giessen.de.
REFERENCES
- 1.Banks AS, Davis SM, Bates SH, Myers MG., Jr Activation of downstream signals by the long form of the leptin receptor. J Biol Chem. 2000;275:14563–14572. doi: 10.1074/jbc.275.19.14563. [DOI] [PubMed] [Google Scholar]
- 2.Baskin DG, Schwartz MW, Seeley RJ, Woods SC, Porte D, Jr, Breininger JF, Jonak Z, Schaefer J, Krouse M, Burghardt C, Campfield LA, Burn P, Kochan JP. Leptin receptor long-form splice-variant protein expression in neuron cell bodies of the brain and co-localization with neuropeptide Y mRNA in the arcuate nucleus. J Histochem Cytochem. 1999;47:353–362. doi: 10.1177/002215549904700309. [DOI] [PubMed] [Google Scholar]
- 3.Bjorbaek C, Uotani S, da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem. 1997;272:32686–32695. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- 4.Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- 5.Chan RK, Brown ER, Ericsson A, Kovacs KJ, Sawchenko PE. A comparison of two immediate-early genes, c-fos and NGFI-B, as markers for functional activation in stress-related neuroendocrine circuitry. J Neurosci. 1993;13:5126–5138. doi: 10.1523/JNEUROSCI.13-12-05126.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, Duyk GM, Tepper RI, Morgenstern JP. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 7.Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology. 1997;138:4489–4492. doi: 10.1210/endo.138.10.5570. [DOI] [PubMed] [Google Scholar]
- 8.Cusin I, Rohner-Jeanrenaud F, Stricker-Krongrad A, Jeanrenaud B. The weight-reducing effect of an intracerebroventricular bolus injection of leptin in genetically obese fa/fa rats. Reduced sensitivity compared with lean animals. Diabetes. 1996;45:1446–1450. doi: 10.2337/diab.45.10.1446. [DOI] [PubMed] [Google Scholar]
- 9.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 10.Elias CF, Lee C, Kelly J, Aschkenasi C, Ahima RS, Couceyro PR, Kuhar MJ, Saper CB, Elmquist JK. Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron. 1998;21:1375–1385. doi: 10.1016/s0896-6273(00)80656-x. [DOI] [PubMed] [Google Scholar]
- 11.Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- 12.Elias CF, Kelly JF, Lee CE, Ahima RS, Drucker DJ, Saper CB, Elmquist JK. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol. 2000;423:261–281. [PubMed] [Google Scholar]
- 13.Elmquist JK, Scammell TE, Jacobson CD, Saper CB. Distribution of Fos-like immunoreactivity in the rat brain following intravenous lipopolysaccharide administration. J Comp Neurol. 1996;371:85–103. doi: 10.1002/(SICI)1096-9861(19960715)371:1<85::AID-CNE5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 14.Elmquist JK, Ahima RS, Maratos-Flier E, Flier JS, Saper CB. Leptin activates neurons in ventrobasal hypothalamus and brainstem. Endocrinology. 1997;138:839–842. doi: 10.1210/endo.138.2.5033. [DOI] [PubMed] [Google Scholar]
- 15.Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998a;395:535–547. [PubMed] [Google Scholar]
- 16.Elmquist JK, Ahima RS, Elias CF, Flier JS, Saper CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci USA. 1998b;95:741–746. doi: 10.1073/pnas.95.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22:221–232. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 18.Fei H, Okano HJ, Li C, Lee GH, Zhao C, Darnell R, Friedman JM. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci USA. 1997;94:7001–7005. doi: 10.1073/pnas.94.13.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friedman JM. Leptin, leptin receptors, and the control of body weight. Nutr Rev. 1998;56:s38–s46. doi: 10.1111/j.1753-4887.1998.tb01685.x. [DOI] [PubMed] [Google Scholar]
- 20.Ghilardi N, Skoda RC. The leptin receptor activates janus kinase 2 and signals for proliferation in a factor-dependent cell line. Mol Endocrinol. 1997;11:393–399. doi: 10.1210/mend.11.4.9907. [DOI] [PubMed] [Google Scholar]
- 21.Guan XM, Hess JF, Yu H, Hey PJ, van der Ploeg LH. Differential expression of mRNA for leptin receptor isoforms in the rat brain. Mol Cell Endocrinol. 1997;133:1–7. doi: 10.1016/s0303-7207(97)00138-x. [DOI] [PubMed] [Google Scholar]
- 22.Hakansson M, de Lecea L, Sutcliffe JG, Yanagisawa M, Meister B. Leptin receptor- and STAT3-immunoreactivities in hypocretin/orexin neurones of the lateral hypothalamus. J Neuroendocrinol. 1999;11:653–663. doi: 10.1046/j.1365-2826.1999.00378.x. [DOI] [PubMed] [Google Scholar]
- 23.Hakansson ML, Meister B. Transcription factor STAT3 in leptin target neurons of the rat hypothalamus. Neuroendocrinology. 1998;68:420–427. doi: 10.1159/000054392. [DOI] [PubMed] [Google Scholar]
- 24.Hakansson ML, Hulting AL, Meister B. Expression of leptin receptor mRNA in the hypothalamic arcuate nucleus: relationship with NPY neurones. NeuroReport. 1996;7:3087–3092. doi: 10.1097/00001756-199611250-00059. [DOI] [PubMed] [Google Scholar]
- 25.Hakansson ML, Brown H, Ghilardi N, Skoda RC, Meister B. Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J Neurosci. 1998;18:559–572. doi: 10.1523/JNEUROSCI.18-01-00559.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ihle JN. STATs: signal transducers and activators of transcription. Cell. 1996;84:331–334. doi: 10.1016/s0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- 27.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 28.Leibel RL, Chung WK, Chua SC., Jr The molecular genetics of rodent single gene obesities. J Biol Chem. 1997;272:31937–31940. doi: 10.1074/jbc.272.51.31937. [DOI] [PubMed] [Google Scholar]
- 29.Lenczowski MJ, Bluthe RM, Roth J, Rees GS, Rushforth DA, van Dam AM, Tilders FJ, Dantzer R, Rothwell NJ, Luheshi GN. Central administration of rat IL-6 induces HPA activation and fever but not sickness behavior in rats. Am J Physiol. 1999;276:R652–R658. doi: 10.1152/ajpregu.1999.276.3.R652. [DOI] [PubMed] [Google Scholar]
- 30.Luheshi GN, Gardner JD, Rushforth DA, Loudon AS, Rothwell NJ. Leptin actions on food intake and body temperature are mediated by IL-1. Proc Natl Acad Sci USA. 1999;96:7047–7052. doi: 10.1073/pnas.96.12.7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCowen KC, Chow JC, Smith RJ. Leptin signaling in the hypothalamus of normal rats in vivo. Endocrinology. 1998;139:4442–4447. doi: 10.1210/endo.139.11.6301. [DOI] [PubMed] [Google Scholar]
- 32.Meister B. Control of food intake via leptin receptors in the hypothalamus. Vitam Horm. 2000;59:265–304. doi: 10.1016/s0083-6729(00)59010-4. [DOI] [PubMed] [Google Scholar]
- 33.Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996;387:113–116. doi: 10.1016/0014-5793(96)00473-5. [DOI] [PubMed] [Google Scholar]
- 34.Niimi M, Sato M, Yokote R, Tada S, Takahara J. Effects of central and peripheral injection of leptin on food intake and on brain Fos expression in the Otsuka Long–Evans Tokushima Fatty rat with hyperleptinaemia. J Neuroendocrinol. 1999;11:605–611. doi: 10.1046/j.1365-2826.1999.00368.x. [DOI] [PubMed] [Google Scholar]
- 35.Rauch M, Riediger T, Schmid HA, Simon E. Orexin A activates leptin-responsive neurons in the arcuate nucleus. Pflügers Arch. 2000;440:699–703. doi: 10.1007/s004240000342. [DOI] [PubMed] [Google Scholar]
- 36.Rees GS, Ball C, Ward HL, Gee CK, Tarrant G, Mistry Y, Poole S, Bristow AF. Rat interleukin 6: expression in recombinant Escherichia coli, purification and development of a novel ELISA. Cytokine. 1999;11:95–103. doi: 10.1006/cyto.1998.0408. [DOI] [PubMed] [Google Scholar]
- 37.Saper CB. Neurobiological basis of fever. Ann NY Acad Sci. 1998;856:90–94. doi: 10.1111/j.1749-6632.1998.tb08317.x. [DOI] [PubMed] [Google Scholar]
- 38.Schindler C, Darnell JE., Jr Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu Rev Biochem. 1995;64:621–651. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 39.Shindler KS, Roth KA. Double immunofluorescent staining using two unconjugated primary antisera raised in the same species. J Histochem Cytochem. 1996;44:1331–1335. doi: 10.1177/44.11.8918908. [DOI] [PubMed] [Google Scholar]
- 40.Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford ML. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature. 1997;390:521–525. doi: 10.1038/37379. [DOI] [PubMed] [Google Scholar]
- 41.Strömberg H, Svensson SP, Hermanson O. Distribution of the transcription factor signal transducer and activator of transcription 3 in the rat central nervous system and dorsal root ganglia. Brain Res. 2000;853:105–114. doi: 10.1016/s0006-8993(99)02260-x. [DOI] [PubMed] [Google Scholar]
- 42.Takeda K, Akira S. STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev. 2000;11:199–207. doi: 10.1016/s1359-6101(00)00005-8. [DOI] [PubMed] [Google Scholar]
- 43.Vaisse C, Halaas JL, Horvath CM, Darnell JE, Jr, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet. 1996;14:95–97. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 44.van Dijk G, Thiele TE, Donahey JC, Campfield LA, Smith FJ, Burn P, Bernstein IL, Woods SC, Seeley RJ. Central infusions of leptin and GLP-1-(7–36) amide differentially stimulate c-FLI in the rat brain. Am J Physiol. 1996;271:R1096–R1100. doi: 10.1152/ajpregu.1996.271.4.R1096. [DOI] [PubMed] [Google Scholar]
- 45.Wang L, Martinez V, Barrachina MD, Tache Y. Fos expression in the brain induced by peripheral injection of CCK or leptin plus CCK in fasted lean mice. Brain Res. 1998;791:157–166. doi: 10.1016/s0006-8993(98)00091-2. [DOI] [PubMed] [Google Scholar]
- 46.Wilkinson M, Morash B, Ur E. The brain is a source of leptin. Front Horm Res. 2000;26:106–125. doi: 10.1159/000061018. [DOI] [PubMed] [Google Scholar]
- 47.Woods AJ, Stock MJ. Leptin activation in hypothalamus. Nature. 1996;381:745. doi: 10.1038/381745a0. [DOI] [PubMed] [Google Scholar]
- 48.Woods SC, Seeley RJ, Porte D, Jr, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science. 1998;280:1378–1383. doi: 10.1126/science.280.5368.1378. [DOI] [PubMed] [Google Scholar]
- 49.Yokosuka M, Xu B, Pu S, Kalra PS, Kalra SP. Neural substrates for leptin and neuropeptide Y (NPY) interaction: hypothalamic sites associated with inhibition of NPY-induced food intake. Physiol Behav. 1998;64:331–338. doi: 10.1016/s0031-9384(98)00065-1. [DOI] [PubMed] [Google Scholar]