

Abstract
Axonal neurofilament (NF) accumulations occur before development of symptoms and before other pathological changes among idiopathic neurodegenerative diseases and toxic neuropathies, suggesting a cause–effect relationship. The dependence of symptoms and axonal degeneration on neurofilament accumulation has been tested here in a transgenic mouse model (Eyer and Peterson, 1994) lacking axonal NFs and using two prototypic toxicant models. Chronic acrylamide (ACR) or 2,5-hexanedione exposure resulted in progressive and cumulative increases in sensorimotor deficits. Neurobehavioral tests demonstrated similar expression of neurotoxicity in transgenic (T) mice and their nontransgenic (NT) littermates (containing normal numbers of axonal NFs). Axonal lesions were frequently observed after exposure to either toxicant. Quantitation of ACR-induced lesions demonstrated the distal location of pathology and equal susceptibility of T and NT axons. We conclude that axonal NFs have no effect on neurotoxicity and the pattern of pathology in these mammalian toxic neuropathies. These results also suggest that the role of neurofilament accumulation in the pathogenesis of neurodegenerative diseases requires careful evaluation.
Keywords: neurofilaments; acrylamide; 2,5-hexanedione; γ-diketones; neuropathies; transgenic mice
The accumulation of axonal neurofilaments (NF) and the subsequent onset of axonal dysfunction and/or pathology are common in neurodegenerative disorders and toxicant-induced neuropathies. The accumulations occur contemporaneous with, or before, development of pathologies. This spatiotemporal pattern of neuropathology has led to the widespread assumption of a cause–effect relationship. However, overexpression of NF or axonal NF accumulation by β,β′-iminodipropionitrile is best correlated with axonal atrophy (reduced diameter) rather than axonal degeneration (Griffin et al., 1978; Cote et al., 1993; Xu et al., 1993), indicating that NF accumulation may not be causally related to axonal loss. A direct test of the relationship between axonal NF accumulation and the development of symptoms and axonal degeneration is lacking.
Neurotoxicants serve as tools for identifying the function(s) of cellular components and the consequences of their alteration, as well as models for elucidating general pathogenic mechanisms. Acrylamide (ACR) and γ-diketones are prototypic chemicals causing a neurofilamentous axonopathy characterized by accumulation of NF along with tubulovesicular profiles, mitochondria, and dense bodies (Prineas, 1969). The accumulations and degeneration are correlated with symptoms (Spencer and Schaumburg, 1974). Direct covalent modification of NFs (Graham et al., 1982a,b; DeCaprio and O'Neill, 1985; Sayre et al., 1985; Lapadula et al., 1989) and/or NF cross-linking (Anthony et al., 1983a,b; Sayre et al., 1985; Genter-St Clair et al., 1988;St Clair et al., 1989; Graham et al., 1990) has been hypothesized to block fast axonal transport of vital nutrients to the distal axon. However, there is no direct proof that accumulation of NF by these toxicants, or under any condition, produces a compromise of fast transport or axonal degeneration. Simultaneous modification of other molecules can equally explain the neuropathology; the accumulation of NF could be epiphenomenal.
Support for the latter, alternative, interpretation exists. Other proteins are chemically modified by γ-diketones in the same manner as NFs (DeCaprio et al., 1982). The temporal onset of neurotoxicity by 2,5-hexanedione (2,5-HD) and ACR does not correlate with the onset or the magnitude of NF accumulation (Anthony et al., 1983c; Sickles and Goldstein, 1985; Spencer and Schaumburg, 1991). Progressive NF accumulation does not correlate with transient and repeated block of fast anterograde axonal transport by 2,5-HD and ACR (Sickles, 1989a,b,1991, 1992). Toxicological (Griffin et al., 1978; Papasozemenos et al., 1982) and transgenic (Cote et al., 1993; Xu et al., 1993) models demonstrate axonal NF accumulation without degeneration.
To test directly the relationship of axonal NF accumulation to the pathogenesis of ACR and γ-diketone neuropathy, we have compared the development of symptoms and pathology in transgenic mice lacking axonal NFs (Eyer and Peterson, 1994) with those of their normal littermates. We demonstrate, qualitatively and quantitatively, no difference in symptoms, pathology, and fast axonal transport effects (Stone et al., 1999, 2001) between mice possessing or lacking axonal NFs. We conclude that NF accumulation is epiphenomenal to ACR and γ-diketone neurotoxicity. Although NF accumulation may play a role in other conditions in which NF might be more uniquely targeted, the relevance of axonal NF accumulation to pathogenesis should be interpreted cautiously.
MATERIALS AND METHODS
Animal model. The transgenic (T; strain 44A) and nontransgenic (NT) mice used in the present experiments were offspring from a colony maintained at McGill University (Alan P. Peterson); details of the production and phenotype of the T mice are available in the previous publication (Eyer and Peterson, 1994). Briefly, the T mouse was produced by initial ligation of the heavy neurofilament subunit (NFH) gene in-frame to the lacZ gene in the pGNA vector. This transgene was then isolated, gel purified, and microinjected into male pronuclei of B6C3F2 zygotes. The embryos were transferred to the oviducts of B6C3F1 females. At birth, tail biopsies were performed, and DNA was extracted and analyzed for the presence of transgene sequences using PCR and Southern blotting. The fusion protein encoded by this transgene includes the entire N-terminal and central α-helical rod domains and 45 KSP repeats from the C-terminal domain of NFH followed by the complete amino acid sequence ofEscherichia coli β-galactosidase. The absence of NFs from the axons of T mice resulted in axonal diameters approximately half those of nonafflicted littermates. However, axons of T mice formed normal associations with their final targets, and most important, the mice were physically and reproductively normal through 14 months of age (Eyer and Peterson, 1994). Positive offspring are referred to throughout this report as T mice, whereas animals testing negative are referred to as NT littermates. All animal classifications were verified (blindly) at the end of the experiment by the presence or absence of axonal NFs in electron micrographs of sciatic nerves. Two-month-old (ACR study) to 3-month-old (HD study) animals (20–30 gm) were air-freighted to the Medical College of Georgia animal care facility where they were maintained on a 12 hr light/dark schedule and provided food and water ad libitum. Animals were adapted to the new surroundings for at least 3 d before experimentation.
Toxicant exposure. All chemicals used were purchased from Sigma (St. Louis, MO) except as follows. Acrylamide (electrophoretic grade) was purchased from Bio-Rad (Hercules, CA), whereas propionamide, 2,5-hexanedione, and 3,4-hexanedione were purchased from Aldrich (Milwaukee, WI). T mice and their NF-containing littermates were given intraperitoneal injections of either ACR (50 mg · kg−1 · d−1;n = 11 T and 11 NT), equimolar doses of the non-neurotoxic analog propionamide (n = 8 T and 8 NT), or physiological saline (controls; n = 8 T and 8 NT). These injections were given daily over a consecutive 18 d treatment period. Previous studies suggested that mice were unsusceptible to γ-diketones (Graham and Gottfried, 1984). Thus, preliminary experiments were conducted to identify whether a neuropathy could be produced with a different dosing regimen. Neurobehavioral testing was conducted in this preliminary study; no microscopy was performed on these groups. Three different dosing regimens of 2,5-HD or 3,4-HD (non-neurotoxic analog of 2,5-HD) were used as follows: (1) 4 mmol · kg−1 · d−1for 14 d, followed by 6 mmol · kg−1 · d−1for 14 d and 8 mmol · kg−1 · d−1for 8 d, (2) 6 mmol · kg−1 · d−1for 14 d and then 8 mmol · kg−1 · d−1for 10 d, or (3) 8 mmol · kg−1 · d−1for 19 d. The latter dosing regimen consistently resulted in the development of symptoms and rotarod failure. Therefore, all subsequent experiments were conducted using the 8 mmol · kg−1 · d−1dose (2,5-HD, n = 8 T and 8 NT; 3,4-HD,n = 6 T and 6 NT).
Neurobehavioral testing. Before each daily injection, all animals were, without the investigator having any knowledge of experimental group, examined for signs of ataxia, hindlimb paralysis, and foot drop, as well as any other gross abnormalities in gait. Numerous functional observational batteries have been developed to identify sensorimotor deficits. The rotarod test has proven value in both ACR and γ-diketone neuropathy and was chosen for the present experiments. Body weights were obtained for each animal and followed with testing of each animal's ability to remain on a rotating cylinder 1 inch in diameter and rotating at the constant speed of 10 rpm for 30 sec. Two opportunities were provided to each animal during each testing session. The animal was designated as failing the rotarod test only after falling off the rotarod on both trials. Because of an observed trend (nonsignificant), potential differences in rotarod performance between transgenic and nontransgenic animals were further considered with a more discriminating accelerated rotarod test. Immediately after the standard rotarod test at 10 rpm, the speed of the rotarod was incrementally increased by 2 rpm every 10 sec, and the speed, in revolutions per minute, at which the animal could no longer remain on the rotarod was recorded. If the animal fell off the rotarod immediately after being placed on the rod, a speed of “0” was recorded.
Morphological studies. One day after rotarod failure, all ACR- and 2,5-HD-injected animals were anesthetized with sodium pentobarbital and perfused via aortic cannula with saline followed by freshly prepared fixative (0.5% paraformaldehyde and 2.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, at room temperature; Fisher Scientific, Pittsburgh, PA). All saline-, propionamide-, and 3,4-HD-injected T and NT animals were killed on the same day as their toxicant-exposed counterparts. The following nerves (midthigh sciatic, midleg sural, peroneal distal to the knee, and midleg tibial) were gently dissected free and placed in room temperature fixative for 24 hr. Samples were post-fixed in 4% osmium tetroxide (Electron Microscopy Sciences, Fort Washington, PA) and embedded in Epon (Electron Microscopy Sciences). Ultra-thin sections were stained with lead citrate and viewed with a Philips 400 electron microscope. All mice in each group were processed for histological analysis. For all ACR, saline, and propionamide T and NT treatment groups, the number of axons with two or more pathological lesions (including accumulations of mitochondria, dense bodies, multilaminar bodies, and/or tubulovesicular profiles) was quantitated within randomly selected areas of sciatic and tibial nerves at 21,500× magnification. The regions were selected without the investigator having knowledge of the experimental groups and included equal samples from superficial and deep regions of the nerves. All axons whose axoplasm was completely within the section were included.
Statistical analysis. Significant differences in the ability to remain on the constant-speed rotarod between T and NT mice of ACR-, propionamide-, 2,5-HD-, 3,4-hexanedione-, and saline-injected groups were determined using the Kaplan and Meier survival analysis with Breslow's statistic. Differences in accelerated rotarod performance among each of the T and NT treatment groups were determined using a two-way ANOVA for repeated measures. In addition, statistically significant differences in average animal weights in each of the experimental groups were determined using a two-way ANOVA for repeated measures. Significant differences in the number of axons with pathological lesions in T and NT animals injected with saline (controls) or ACR were determined using a two-way ANOVA for repeated measures followed by Tukey's highly significant differencespost hoc test at a preset significance level of p<0.05.
RESULTS
Both T and NT animals receiving either saline or propionamide either maintained their starting weight or gained weight over the treatment period (Fig.1A,B), whereas those receiving ACR stopped gaining weight after day 12 (Fig.1A,B). During the time frame of the experiment no significant weight differences were observed between any of the groups. Regardless of the dosing regimen, all 2,5-HD mice showed statistically significant reductions in weight (Fig. 1C,D, 8 mmol/kg dosing regimen; others not shown) in comparison with saline- and 3,4-hexanedione-exposed mice (Fig. 1C,D). No significant weight differences were observed between T and NT mice in any group, including the ACR and 2,5-HD treatments. Data are expressed as a percentage of starting weight; therefore, SEs are extremely small and cannot be appropriately represented on the graph (Fig. 1).
Fig. 1.

A, B, Animal weights (shown as % of starting weight) for saline-treated (n = 8 T and 8 NT), ACR-treated (n = 11 T and 11 NT), and propionamide-treated (n = 8 T and 8 NT) groups of T (A) and NT (B) mice over the 18 d treatment period are shown. ACR-injected T and NT mice failed to gain weight after experimental day 12; however, no significant weight differences were observed between any of the groups.C, D, T (C) and NT (D) mice exposed to 2,5-HD (8 mmol · kg−1 · d−1;n = 8 T and 8 NT) over a 19 d treatment period exhibited a statistically significant reduction in body weight (p < 0.05) compared with either saline-exposed (n = 6 T and 6 NT) or 3,4-HD-exposed (n = 6 T and 6 NT) mice. No difference was found between saline- and 3,4-HD-treated mice. Error bars are not included for appearance and because most were within the font size of the datapoints. Statistical differences were determined using a two-way ANOVA for repeated measures. Differences in weight gain of the control animals of the ACR study versus those of the HD study were attributed to differences in the starting age of the mice (ACR, 2 months; HD, 3 months). PROP, Propionamide.
Development of the characteristic symptoms of ACR and γ-diketone neurotoxicity in T and NT mice was comparable with those reported previously for other mammalian models. Neurobehavioral changes caused by either neurotoxicant included ataxia, foot drop, and mild hindlimb paralysis. These symptoms were consistently observed in both T and NT mice. The development of these neurobehavioral changes was progressive; the frequency and severity of these changes increased with continued neurotoxicant exposure. There were no observable differences in the temporal onset, progression, or severity of the symptoms between T and NT mice for ACR or 2,5-HD. In contrast, no behavioral changes were observed in any saline-, propionamide-, or 3,4-hexanedione-injected mice.
Failures in the rotarod test confirmed the development of neurotoxicity in ACR- and 2,5-HD-exposed T and NT mice. All saline- (data not shown), propionamide-, and 3,4-hexanedione-injected groups successfully completed the rotarod test every day of the treatment period (Fig. 2). Some ACR-T animals failed the 30 sec rotarod test starting on day 10, more failed each subsequent day, and on day 18 all animals failed (Fig.2A). NT mice began to fail on day 12; all of the animals failed by day 18 (Fig. 2A). Statistical analysis revealed that the ACR-injected animals were different from those injected with either saline or propionamide. Although there appeared to be some difference in onset and time course of rotarod failure between T and NT mice, no statistical difference was demonstrated. With the observed variability, an n of 50 animals would be required to demonstrate significance. Both T and NT mice exposed to 2,5-HD (8 mmol · kg−1 · d−1) began to fail the rotarod test on day 17 with daily increasing frequency until total failure of all animals at day 18 (Fig.2B). Using the other two dosing regimens, rotarod failure occurred consistently only after the higher 8 mmol · kg−1 · d−1dose was administered for several days. Regardless of the dosing regimen, T and NT mice failed the rotarod test on comparable time courses (data not shown). For all 2,5-HD-dosing regimens, statistically significant differences were observed between 2,5-HD- and saline- or 3,4-HD-treated groups. However, no differences were observed between 2,5-HD-exposed T and NT animals. Graphically, rotarod data are expressed as a percentile; thus SE is minimal. Therefore, error bars cannot be appropriately expressed on the graphs. Accelerated rotarod performance remained consistent for T and NT mice exposed to either physiological saline (Fig. 3), propionamide (Fig. 3A,B), or 3,4-hexanedione (Fig.3C,D). In contrast, rotarod speeds for ACR-treated (Fig.3A,B) and 2,5-HD-treated (Fig. 3C,D; 8 mmol/kg) T (Fig. 3A,C) and NT (Fig. 3B,D) animals began to decline after several injections and continued to decrease over the treatment periods until these toxicant-exposed animals could no longer perform this test. Failures in the accelerated rotarod test produced the precipitous drop in average revolutions per minute that was achieved. Complete failure of the test after ACR (50 mg · kg−1 · d−1) occurred at day 18 for NT mice (Fig. 3B) and day 15 for T mice (Fig. 3A); with 8 mmol · kg−1 · d−12,5-HD, both T (Fig. 3C) and NT (Fig. 3D) mice failed on day 17. Statistically significant differences were observed between 2,5-HD- and either saline- or 3,4-HD-treated groups and between ACR- and either saline- or propionamide-treated groups for both T and NT mice. No difference was found between T and NT mice exposed to either ACR or 2,5-HD.
Fig. 2.

Effects of ACR and 2,5-HD on rotarod performance. Animals were tested daily for their ability to remain on a rotating, 1-inch-diameter cylinder rotating at a speed of 10 rpm for 30 sec. An animal was designated as failing if it fell off the rod on two trials. Data are illustrated as the percentage of animals tested that failed the test on each treatment day. Daily injections of 50 mg/kg ACR (A), equimolar propionamide (A), 8 mmol/kg 2,5-HD (B), or equimolar 3,4-HD (B) produced a similar rotarod failure in T and NT mice. T and NT ACR-exposed animals (n = 11 T and 11 NT) were significantly different from corresponding propionamide-injected (n = 8 T and 8 NT) and saline-injected (n = 8 T and 8 NT; data not shown) mice. Similarly, both T and NT 2,5-HD-exposed animals (n = 8 T and 8 NT) were significantly different from corresponding 3,4-HD-exposed (n = 6 T and 6 NT) and saline-exposed (n = 6 T and 6 NT; data not shown) animals. No statistically significant differences were found between any T and NT animals. For illustration purposes only, data from T and NT mice exposed to non-neurotoxic propionamide (A) or 3,4-HD (B) were combined. Survival analysis data do not allow the presentation of error bars. Statistical differences were determined using the Kaplan and Meier survival analysis with Breslow's statistic.
Fig. 3.

A, B, Accelerated rotarod performance of T (A) and NT (B) mice exposed to ACR (50 mg · kg−1 · d−1;n = 3 T and 3 NT), propionamide (Prop; 50 mg · kg−1 · d−1;n = 3 T and 3 NT), or saline (n= 1 T and 1 NT) is shown. ACR-treated animals were statistically different from saline- or propionamide-exposed animals (p < 0.05). C, D, T (C) and NT (D) mice receiving 2,5-HD (8 mmol · kg−1 · d−1;n = 3 T and 3 NT) were statistically different from 3,4-HD-treated (equimolar dose; n = 3 T and 3 NT) or saline-treated (n = 1 T and 1 NT) groups in the performance of the accelerated rotarod test at the preset significance (p < 0.05). No differences were found between T and NT mice for any experimental group. Statistical differences were determined using a two-way ANOVA for repeated measures.
As indicated above, PCR and Southern blotting of tail biopsies verified the presence of the transgene in T mice. Electron micrographs verified the absence of NFs in axons of T mice (Fig.4). NT littermates showed a normal neurofilament appearance, number, and distribution in control as well as in propionamide- and 3,4-HD-exposed mice (data not shown). Morphological examination of axons from both ACR-injected T mice (Fig.4) and ACR-injected NT littermates (Fig.5) as well as nerves from 2,5-HD-injected T (Fig. 6) and 2,5-HD-injected NT (Fig.7) mice revealed similar pathological axonal lesions. Common to all four groups of mice were the accumulation of mitochondria and other membrane-bound organelles, dense bodies, multilaminar bodies and tubulovesicular profiles (Figs. 4-7). Frank Wallerian degeneration was infrequent. One important difference was the presence of neurofilament accumulations in the NT ACR- and 2,5-HD-exposed groups (Figs. 5, 7) and the absence of this hallmark in all T animals (Figs. 4, 6). The spatial distribution of pathology was objectively determined by comparison of the number of axons with lesions in the midthigh sciatic nerve with the number of lesions in the midleg tibial nerve. This quantitation was limited in ACR experiments because of the difficulty in obtaining successful perfusions in ACR-exposed mice. Furthermore, quantitation was precluded in the γ-diketone group by even greater difficulty in obtaining an acceptable sample size in the 2,5-HD-exposed group. Numerous modifications of the perfusion protocol were attempted, including using a peristaltic pump to deliver the fixative at a constant pressure, administering the fixative via a syringe directly into the heart, as well as injecting the animal with heparin before perfusion of the fixative. None of these modifications produced any improvement in the preservation of structure. Quantitation of the number of axons containing pathological lesions is illustrated in Figure8. ACR-injected animals demonstrated significantly higher numbers of axons with lesions in both sciatic and tibial nerves compared with the same nerves from saline controls (Fig.8). This was true for both T and NT animals. A statistically significant difference was observed between sciatic and tibial nerves, the more distal tibial nerve demonstrating a greater frequency of lesions. No differences between T and NT mice, for either nerve, were observed.
Fig. 4.

A–C, Axons from ACR-injected (50 mg · kg−1 · d−1 for 18 d) T mice tibial nerves. Typical pathological lesions induced by ACR include accumulations of mitochondria, dense bodies, and other membrane-bound vesicles (arrows), vacuole formation (asterisks), and multilaminar bodies (arrowheads) of both myelinated and unmyelinated axons. Abnormal axoplasm is also found within glial compartments (C). Neurofilament accumulations were not observed. A–C, 21,500×. Scale bars, 1 μm.
Fig. 5.

A–C, Tibial nerve axons from NT mice injected daily with 50 mg/kg ACR for 18 d. Morphological lesions similar to those observed in other mammalian species were observed. These include the accumulation of mitochondria, dense bodies, and other membrane-bound vesicles (arrows), vacuole formation (asterisks), and multilaminar bodies (arrowhead). Numerous examples of neurofilament accumulations (B) were observed, along with abnormal organelles (B; arrow). A, C, 21,500×; B, 43,000×. Scale bars, 1 μm.
Fig. 6.
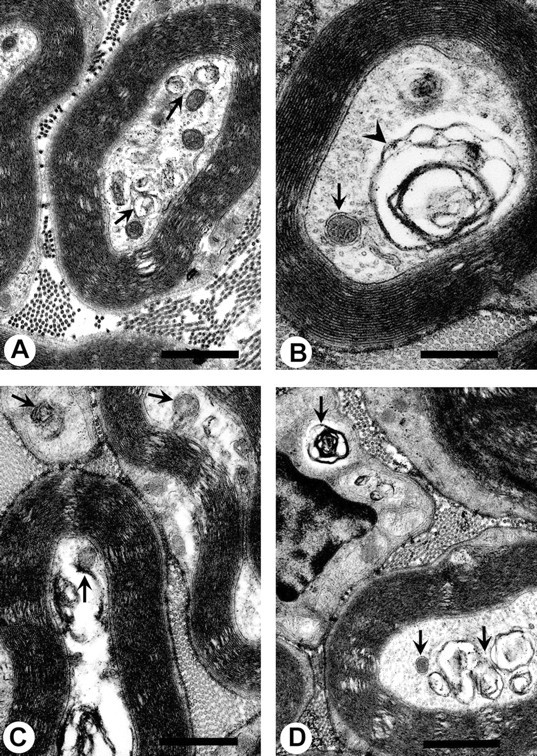
A–D, Pathological lesions in tibial nerves of T mice chronically injected with 2,5-HD (8 mmol · kg−1 · d−1) for 19 d. Typical lesions include accumulations of mitochondria, dense bodies, and other membrane-bound vesicles (arrows), as well as the presence of multilaminar bodies (arrowheads). These lesions are very similar to those observed in NT animals, except neurofilaments are absent from these nerve sections (see Fig. 7 to compare). These lesions are also similar to those observed with chronic ACR exposure (see Figs. 4, 5 to compare). A–D, 21,500×. Scale bars, 1 μm.
Fig. 7.
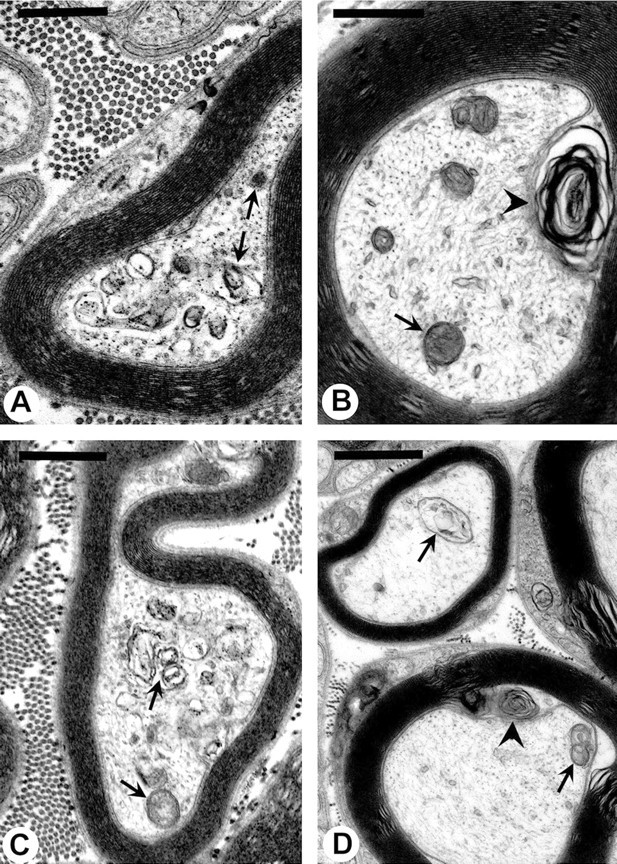
A–D, Pathological lesions in tibial nerves of NT mice chronically injected with 2,5-HD (8 mmol · kg−1 · d−1) for 19 d. Typical lesions include accumulations of mitochondria, dense bodies, and other membrane-bound vesicles (arrows), as well as the presence of multilaminar bodies (arrowheads) in axons and surrounding glia. Neurofilaments are present in all of these tibial nerve sections. A–D, 21,500×. Scale bars, 1 μm.
Fig. 8.

Quantitative comparison of frequency of pathological lesions within axons of sciatic and tibial nerves of T and NT mice injected with 50 mg · kg−1 · d−1 ACR or saline for 18 d. The number of axons counted in each group is provided above each bar. ACR significantly increased the frequency of lesions over controls. A significant difference was also observed between sciatic (proximal) and tibial (distal) nerves in both T and NT mice. No differences were found between T and NT mice under any experimental condition. Comparable data for 2,5-HD were unavailable because of an unresolvable difficulty in the perfusion of 2,5-HD-exposed mice. Statistical differences were determined using a two-way ANOVA for repeated measures followed by Tukey's highly significant differences post hoc test. *p < 0.05, significantly different from the corresponding saline control; #p < 0.05, significantly different from the corresponding sciatic nerve.
DISCUSSION
The NFH-lacZ transgenic mouse provides a useful model for testing the relevance of axonal NF modification and/or accumulation to the development of symptoms and pathology of neurodegeneration. The present study demonstrates that ACR or 2,5-HD produce symptoms, behavioral indices, and morphological expression of neurotoxicity in a transgenic mouse model lacking axonal NFs (Eyer and Peterson, 1994) similar to those in nontransgenic littermates possessing normal NF content. Changes in gait, ataxia, and hindlimb paralysis, as well as the objective measurement of deficits in the ability to remain on a rotating rod (a sensitive test for sensorimotor function in rodents), were equally expressed in T and NT mice. Furthermore, the time course of rotarod deficits in the T and NT mice was similar for both ACR and 2,5-HD. NFs were observed in all NT mice, and NF accumulations were observed in NT axons after exposure to either ACR or 2,5-HD. Neither 10 nm filaments, nor any other filaments, accumulated in any T mice. Other pathological lesions characteristic of ACR- and 2,5-HD-induced neuropathy, including tubulovesicular profile accumulation, mitochondria accumulation, vacuole formation, and dense and multilaminar body formation (Prineas, 1969; Schaumburg et al., 1974), were consistently observed in both T and NT mice. The dissociation of symptoms, behavioral indices, and pathological lesions from the presence of axonal NFs in a mammalian model clearly demonstrates that modification of another target is sufficient to produce neurotoxicity by these agents. This observation for two different classes of prototypic toxicants for neurofilamentous axonopathies suggests that great caution must be exercised when assuming that NF accumulation precipitates pathologies in other degenerative diseases. Other toxicants or etiologies may produce a different type of neurofilament accumulation that can have pathogenic relevance. For example, altered NF expression has been observed to trap organelles and potentially reduce fast axonal transport (Collard et al., 1995). Direct quantitation of fast transport in this model does need to be experimentally determined. However, the mere presence of NF accumulations does not necessitate pathogenic involvement. Studies with β,β′-iminodipropionitrile and transgenic mice overexpressing neurofilaments similarly indicate dissociation of NF from the pathogenesis of distal axonal degeneration (Griffin et al., 1978;Papasozemenos et al., 1982; Cote et al., 1993; Xu et al., 1993). Axonal degeneration was not determined in the current study because animals were killed at the initial rotarod failure.
The current results are consistent with observations in nonmammalian models. Crayfish, a crustacean lacking NFs (Miller et al., 1987), and a mutant light neurofilament subunit (NFL)-deficient quail with a greatly reduced axonal NF content (Takahashi et al., 1994) developed axonal degeneration with ACR (Sickles et al., 1994; Takahashi et al., 1994, 1995). 2,5-HD produced neurotoxicity and axonal pathology in crayfish (Sickles et al., 1994); the mutant quail was extremely sensitive to 2,5-HD, and premature death of the animals, likely because of asphyxia, precluded determination of neurotoxicological outcomes in this model (Hirai et al., 1999). Unfortunately, crayfish possess unique axonal cytoskeletal proteins, potentially substituting for NFs (Hirokawa, 1986; Weaver and Viancour, 1991), that could react with neurotoxicants to produce mechanistic similarities. However, no filamentous accumulations in crayfish were observed with either ACR or 2,5-HD. The quail model is limited by the presence of a minor amount of other NF subunits (Takahashi et al., 1994, 1995) and the questionable relevance of avian data to mammalian systems. The current model eliminates these potential limitations and permits direct comparison of the effects of these toxicants on mammalian axons with and without axonal NFs. Compensation for the axonal loss of NF in the transgenic model appears unlikely because extensive mRNA and protein analysis of nerves indicates no unique protein expression or compensatory adjustment in cytoskeletal proteins, except a higher than normal microtubule density (Eyer and Peterson, 1994). This increased microtubule density may be considered a compensation for neurofilament absence. However, the increased density is caused by the smaller axonal diameters present in T mice and is not an actual increase in the number of microtubules per cell. This number remains unchanged; therefore, we do not consider the increase in microtubule density a compensation.
The presence of NFs in the soma of T mice does not appear to be problematic to the interpretation of the present data. The comparable performance of control T and non-T mice on both the regular and accelerated rotarod tests indicates that the accumulation of NFs within the cell body does not significantly alter neuronal function. The absence of an identifiable phenotype and the longevity of NFH-lacZ mice and their neurons support the conclusion that the neuron is capable of accommodating the somal NF load. Furthermore, the outflow of fast anterogradely transported proteins in control T mice is identical to the outflow observed in NT animals, demonstrating that somal NF accumulations do not interrupt de novo synthesis and loading of proteins for fast axonal transport (Stone et al., 2001). It is possible that toxicant modification of somal NFs contributes to neurotoxicity. However, this is inconsistent with the comparable outflow of radiolabeled fast anterogradely transported proteins in both T and NT mice after ACR and 2,5-HD exposure (Stone et al., 2001) and comparable reductions in membrane-bound organelle flow within isolated axons from T and NT mice (Stone et al., 1999), which are independent of the neuron soma.
NFs could be an alternative target resulting in neurotoxicity or at least accentuate the response of axons to these toxicants. Takahashi et al. (1995) reported that Wallerian degeneration was less prominent in ACR-exposed NF-deficient quails compared with normal quails, suggesting that the presence of NFs influences the extent of ACR pathology. The current results from this mammalian model indicate that there is no difference in response to either toxicant related to NF content. In fact, the rotarod test demonstrated a trend toward ACR-exposed T mice failing the rotarod test 2 d earlier than ACR-exposed NT animals; using 8–11 animals per group resulted in no statistical difference (p = 0.09) in the rotarod performance of T and NT mice. If a larger sample size resulted in a statistical difference, this would indicate a protective action of NFs rather than an alternative or supplemental action. However, statistical analyses have determined that a sample size of 50 animals would be required to produce a statistically significant difference; this would seem to be an excessive use of animals in that it would not significantly alter the conclusions of the present study. 2,5-HD produced an identical time to onset and level of rotarod failure (both standard and accelerated) in T and NT mice. This is surprising because most of the data supporting the NF hypothesis have been generated with γ-diketones.
ACR- and 2,5-HD-induced neuropathies are characterized by a very distinct and reproducible spatial distribution of pathology (Spencer and Schaumburg, 1974, 1976) that has been useful in proposing hypothetical mechanisms of action. The distal pattern of initial pathology is reproduced here in both toxicant-exposed T and NT mice. The predominance of pathology in the more distal locations suggests the same pattern of “dying-back” neuropathy (Cavanagh, 1964) in T and NT mice as well as other mammalian models (Kuperman, 1958; Fullerton and Barnes, 1966; Barnes, 1970; Hopkins, 1970; Kaplan and Murphy, 1972;Kaplan et al., 1973; Post, 1978). Another characteristic similar in T mice and their NF-containing littermates was a comparable specificity of action as demonstrated by the lack of effect of propionamide, a non-neurotoxic analog of ACR (Lin et al., 1993), or 3,4-HD, a non-neurotoxic analog of 2,5-HD (O'Donoghue et al., 1984), on symptoms or morphology.
Several studies have reported previously ACR neurotoxicity in mice (Edwards and Parker, 1977; Von Burg et al., 1981; Gilbert and Maurissen, 1982; Teal and Evans, 1982; Miller et al., 1984; Bradley and Asbury, 1991). However, mice have been considered resistant to γ-diketone neurotoxicity (Graham and Gottfried, 1984). Axon length was presumed to be insufficient to permit a threshold of toxicant attacks on NF proteins to produce neurotoxicity. The current report indicates that doubling the dose from the typical 4 to 8 mmol · kg−1 · d−1is required, but comparable symptoms and pathology were observed. The similarity of the response in mice to that in other mammalian species, including humans, supports extrapolation of T mice data across species.
The current results indicate that covalent modification of NFs and their accumulation contribute little or nothing to the pathogenesis of symptoms and pathology in ACR- and γ-diketone-induced neurotoxicity. Other studies using this model have identified the contributions of NFs to normal axonal transport as well as the contribution of axonal NFs and their accumulation to changes in fast axonal transport by these toxicants. Fast axonal transport was reduced comparably in both transgenic and nontransgenic mice by these toxicants (Stone et al., 1999, 2001). Collectively, these studies indicate that future research should examine axonal proteins other than NFs. For example, kinesin, the motor protein for fast anterograde axonal transport, is inhibited by ACR (Sickles et al., 1996) and 2,5-HD (Sickles and Tsai, 1996); it remains to be determined whether this inhibition is epiphenomenal or whether it represents a critical site of action. Although NF may not be a critical site of action, the formation of pyrrole adducts and/or cross-linking of γ-diketones and the covalent modification of sulfhydryl groups by ACR (Hashimoto and Aldridge, 1970; DeCaprio and O'Neill, 1985; Sayre et al., 1985;Lapadula et al., 1989; Graham et al., 1990) should be considered as critical to neurotoxicity. These chemical reactions, identified on NF, should be considered in future studies of other axonal proteins.
Footnotes
This work was supported by National Institute of Environmental Health Sciences Grant ES 06150 to D.W.S.
Correspondence should be addressed to Dr. Dale W. Sickles, Department of Cellular Biology and Anatomy, Medical College of Georgia, Augusta, GA 30912. E-mail: dsickles@mail.mcg.edu.
REFERENCES
- 1.Anthony DC, Boekelheide K, Graham DG. The effect of 3,4-dimethyl substitution on the neurotoxicity of 2,5-hexanedione. I. Accelerated clinical neuropathy is accompanied by more proximal axonal swellings. Toxicol Appl Pharmacol. 1983a;71:362–371. doi: 10.1016/0041-008x(83)90023-6. [DOI] [PubMed] [Google Scholar]
- 2.Anthony DC, Boekelheide K, Anderson CW, Graham DG. The effect of 3,4-dimethyl substitution on neurotoxicity of 2,5-hexanedione. II. Dimethyl substitution accelerates pyrrole formation and protein crosslinking. Toxicol Appl Pharmacol. 1983b;71:372–382. doi: 10.1016/0041-008x(83)90024-8. [DOI] [PubMed] [Google Scholar]
- 3.Anthony DC, Giangaspero F, Graham DG. The spatio-temporal pattern of the axonopathy associated with the neurotoxicity of 3,4-dimethyl-2,5-hexanedione in the rat. J Neuropathol Exp Neurol. 1983c;42:548–560. doi: 10.1097/00005072-198309000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Barnes JM. Observations on the effects on rats of compounds related to acrylamide. Br J Ind Med. 1970;27:147–149. doi: 10.1136/oem.27.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradley WG, Asbury AK. Radioautographic studies of Schwann cell behavior. I. Acrylamide neuropathy in the mouse. J Neuropathol Exp Neurol. 1991;29:500–506. doi: 10.1097/00005072-197007000-00011. [DOI] [PubMed] [Google Scholar]
- 6.Cavanagh JB. The significance of the “dying-back” process in experimental and human neurological disease. Int Rev Exp Pathol. 1964;3:219–267. [PubMed] [Google Scholar]
- 7.Collard JF, Cote F, Julien JP. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- 8.Cote F, Collard JF, Julien JP. Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Cell. 1993;73:35–46. doi: 10.1016/0092-8674(93)90158-m. [DOI] [PubMed] [Google Scholar]
- 9.DeCaprio AP, O'Neill EA. Alterations in rat axonal cytoskeletal proteins induced by in vitro and in vivo 2,5-hexanedione exposure. Toxicol Appl Pharmacol. 1985;78:235–247. doi: 10.1016/0041-008x(85)90287-x. [DOI] [PubMed] [Google Scholar]
- 10.DeCaprio AP, Olajos EJ, Weber P. Covalent binding of a neurotoxic n-hexane metabolite: conversion of primary amines to substituted pyrrole adducts by 2,5-hexanedione. Toxicol Appl Pharmacol. 1982;65:440–450. doi: 10.1016/0041-008x(82)90389-1. [DOI] [PubMed] [Google Scholar]
- 11.Edwards PM, Parker VH. A simple, sensitive, and objective method for early assessment of acrylamide neuropathy in rats. Toxicol Appl Pharmacol. 1977;40:589–591. doi: 10.1016/0041-008x(77)90083-7. [DOI] [PubMed] [Google Scholar]
- 12.Eyer J, Peterson A. Neurofilament-deficient axons and perikaryal aggregates in viable transgenic mice expressing a neurofilament-B-galactosidase fusion protein. Neuron. 1994;12:398–405. doi: 10.1016/0896-6273(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 13.Fullerton PM, Barnes JM. Peripheral neuropathy in rats produced by acrylamide. Br J Ind Med. 1966;23:210–221. doi: 10.1136/oem.23.3.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Genter-St Clair MB, Amarnath V, Moody MA, Anthony DC, Anderson CW, Graham DG. Pyrrole oxidation and protein cross-linking as necessary steps in the development of γ-diketone neuropathy. Chem Res Toxicol. 1988;1:179–185. doi: 10.1021/tx00003a009. [DOI] [PubMed] [Google Scholar]
- 15.Gilbert SG, Maurissen PJ. Assessment of the effects of acrylamide, methylmercury, and 2,5-hexanedione on motor functions in mice. J Toxicol Environ Health. 1982;10:31–41. doi: 10.1080/15287398209530228. [DOI] [PubMed] [Google Scholar]
- 16.Graham DG, Gottfried MR. Cross-species extrapolation in hydrocarbon neuropathy. Neurobehav Toxicol Teratol. 1984;6:433–435. [PubMed] [Google Scholar]
- 17.Graham DG, Anthony DC, Boekelheide K. In vitro and in vivo studies of the molecular pathogenesis of n-hexane neuropathy. Neurobehav Toxicol Teratol. 1982a;4:629–634. [PubMed] [Google Scholar]
- 18.Graham DG, Anthony DC, Boekelheide K, Maschmann NA, Richards RG, Wolfram JW, Shaw BR. Studies of the molecular pathogenesis of hexane neuropathy. Toxicol Appl Pharmacol. 1982b;64:415–422. doi: 10.1016/0041-008x(82)90237-x. [DOI] [PubMed] [Google Scholar]
- 19.Graham DG, Genter-St Clair MB, Amarnath V, Anthony DC. Molecular mechanisms of γ-diketone neuropathy. Biol React Intermed. 1990;4:427–431. doi: 10.1007/978-1-4684-5877-0_58. [DOI] [PubMed] [Google Scholar]
- 20.Griffin JW, Hoffman PN, Clark AW, Carroll PT, Price DL. Slow axonal transport of neurofilament proteins: impairment by B, B′-iminodipropionitrile administration. Science. 1978;202:633–635. doi: 10.1126/science.81524. [DOI] [PubMed] [Google Scholar]
- 21.Hashimoto K, Aldridge WN. Biochemical studies on acrylamide, a neurotoxic agent. Biochem Pharmacol. 1970;19:2591–2604. doi: 10.1016/0006-2952(70)90009-2. [DOI] [PubMed] [Google Scholar]
- 22.Hirai T, Mizutani M, Kimura T, Ochiai K, Umemura T, Itakura C. Neurotoxic effects of 2,5-hexanedione on normal and neurofilament-deficient quail. Toxicol Pathol. 1999;27:348–353. doi: 10.1177/019262339902700311. [DOI] [PubMed] [Google Scholar]
- 23.Hirokawa N. 270K microtubule-associated protein cross-reacting with anti-MAP2 IgG in the crayfish peripheral nerve axon. J Cell Biol. 1986;103:33–39. doi: 10.1083/jcb.103.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopkins A. The effect of acrylamide on the peripheral nervous system of the baboon. J Neurol Neurosurg Psychiatry. 1970;33:805–816. doi: 10.1136/jnnp.33.6.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaplan ML, Murphy SD. Effect of acrylamide on rotarod performance and sciatic nerve B-glucuronidase activity of rats. Toxicol Appl Pharmacol. 1972;22:259–268. doi: 10.1016/0041-008x(72)90176-7. [DOI] [PubMed] [Google Scholar]
- 26.Kaplan ML, Murphy SD, Gilles FH. Modification of acrylamide neuropathy in rats by selected factors. Toxicol Appl Pharmacol. 1973;24:564–579. doi: 10.1016/0041-008x(73)90218-4. [DOI] [PubMed] [Google Scholar]
- 27.Kuperman AS. Effects of acrylamide on the central nervous system of the cat. J Pharmacol Exp Ther. 1958;123:180–192. [PubMed] [Google Scholar]
- 28.Lapadula DM, Bowe M, Carrington CD, Dulak L, Friedman MA, Abou-Donia MB. In vitro binding of [14C]acrylamide to neurofilament and microtubule proteins of rats. Brain Res. 1989;481:157–161. doi: 10.1016/0006-8993(89)90496-4. [DOI] [PubMed] [Google Scholar]
- 29.Lin W, Corcoran JJ, Abou-Donia MB, Friedman MA. Differential neurotoxicity of acrylamide, glycidamide, and propionamide on PC-12 cells. Toxicologist. 1993;13:127. [Google Scholar]
- 30.Miller MJ, Miller MS, Burks TF, Sipes IG. A simple, sensitive method for detecting early peripheral nerve dysfunction in the rat following acrylamide treatment. Neurotoxicology. 1984;5:15–24. [PubMed] [Google Scholar]
- 31.Miller RH, Lasek RJ, Katz MJ. Preferred microtubules for vesicle transport in lobster axons. Science. 1987;235:220–222. doi: 10.1126/science.2432661. [DOI] [PubMed] [Google Scholar]
- 32.O'Donoghue JL, Krasavage WJ, Divincenzo GD, Katz GV. Further studies on ketone neurotoxicity and interactions. Toxicol Appl Pharmacol. 1984;72:201–209. doi: 10.1016/0041-008x(84)90304-1. [DOI] [PubMed] [Google Scholar]
- 33.Papasozemenos SCH, Yoon M, Crane R, Autilio-Gambetti L. Redistribution of proteins of fast axonal transport following administration of B,B′-iminodipropionitrile: a quantitative autoradiographic study. J Cell Biol. 1982;95:672–675. doi: 10.1083/jcb.95.2.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Post EJ. Unmyelinated nerve fibres in feline acrylamide neuropathy. Acta Neuropathol (Berl) 1978;42:19–24. doi: 10.1007/BF01273262. [DOI] [PubMed] [Google Scholar]
- 35.Prineas J. The pathogenesis of dying-back polyneuropathies. Part 2. An ultrastructural study of experimental acrylamide intoxication in the cat. J Neuropathol Exp Neurol. 1969;28:598–621. doi: 10.1097/00005072-196910000-00004. [DOI] [PubMed] [Google Scholar]
- 36.Sayre LM, Autilio-Gambetti L, Gambetti P. Pathogenesis of experimental giant neurofilamentous axonopathies: a unified hypothesis based on chemical modification of neurofilaments. Brain Res Rev. 1985;10:69–83. doi: 10.1016/0165-0173(85)90008-6. [DOI] [PubMed] [Google Scholar]
- 37.Schaumburg HH, Wisniewski HM, Spencer PS. Ultrastructural studies of the dying-back process. I. Peripheral nerve terminal and axon degeneration in systemic acrylamide intoxication. J Neuropathol Exp Neurol. 1974;33:260–284. doi: 10.1097/00005072-197404000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Sickles DW. Toxic neurofilamentous axonopathies and fast anterograde axonal transport. I. The effects of single doses of acrylamide on the rate and capacity of transport. Neurotoxicology. 1989a;10:91–102. [PubMed] [Google Scholar]
- 39.Sickles DW. Toxic neurofilamentous axonopathies and fast anterograde axonal transport. II. The effects of single doses of neurotoxic and non-neurotoxic diketones and B,B′-iminodipropionitrile (IDPN) on the rate and capacity of transport. Neurotoxicology. 1989b;10:103–112. [PubMed] [Google Scholar]
- 40.Sickles DW. Toxic neurofilamentous axonopathies and fast anterograde axonal transport. III. Recovery from single injections and multiple dosing effects of acrylamide and 2,5-hexanedione. Toxicol Appl Pharmacol. 1991;108:390–396. doi: 10.1016/0041-008x(91)90085-s. [DOI] [PubMed] [Google Scholar]
- 41.Sickles DW. Toxic neurofilamentous axonopathies and fast anterograde axonal transport. IV. In vitro analysis of transport following acrylamide and 2,5-hexanedione. Toxicol Lett. 1992;61:199–204. doi: 10.1016/0378-4274(92)90146-b. [DOI] [PubMed] [Google Scholar]
- 42.Sickles DW, Goldstein BD. Acrylamide alters oxidative enzyme activity in rat motoneurons. Toxicol Lett. 1985;26:111–118. doi: 10.1016/0378-4274(85)90154-7. [DOI] [PubMed] [Google Scholar]
- 43.Sickles DW, Tsai J. 2,5-Hexanedione directly affects kinesin-based microtubule motility. Fundam Appl Toxicol [Suppl] 1996;30:302. [Google Scholar]
- 44.Sickles DW, Pearson JK, Beall A, Testino A. Toxic axonal degeneration occurs independent of neurofilament proteins. J Neurosci Res. 1994;39:347–354. doi: 10.1002/jnr.490390312. [DOI] [PubMed] [Google Scholar]
- 45.Sickles DW, Brady ST, Testino A, Friedman MA, Wrenn RW. Direct effect of the neurotoxicant acrylamide on kinesin-based microtubule motility. J Neurosci Res. 1996;46:7–17. doi: 10.1002/(SICI)1097-4547(19961001)46:1<7::AID-JNR2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 46.Spencer PS, Schaumburg HH. A review of acrylamide neurotoxicity. Part l. Properties, uses and human exposure. Can J Neurol Sci. 1974;1:143–150. doi: 10.1017/s0317167100019739. [DOI] [PubMed] [Google Scholar]
- 47.Spencer PS, Schaumburg HH. Central-peripheral distal axonopathy—the pathology of dying-back polyneuropathies. Prog Neuropathol. 1976;3:253–295. [Google Scholar]
- 48.Spencer PS, Schaumburg HH. A review of acrylamide neurotoxicity. Part 2. Experimental animal neurotoxicity and pathologic mechanisms. Can J Neurol Sci. 1991;152:60–69. doi: 10.1017/s0317167100119201. [DOI] [PubMed] [Google Scholar]
- 49.St Clair MBG, Anthony DC, Wikstrand CJ, Graham DG. Neurofilament protein crosslinking in gamma-diketone neuropathy: in vitro and in vivo studies using the seaworm Myxicola infundibulum. Neurotoxicology. 1989;10:743–756. [PubMed] [Google Scholar]
- 50.Stone JD, Peterson AP, Eyer J, Oblak TG, Sickles DW. Axonal neurofilaments are nonessential elements of toxicant-induced reductions in fast axonal transport: video-enhanced differential interference microscopy in peripheral nervous system axons. Toxicol Appl Pharmacol. 1999;161:50–58. doi: 10.1006/taap.1999.8780. [DOI] [PubMed] [Google Scholar]
- 51.Stone JD, Peterson AP, Eyer J, Sickles DW. Neurofilaments are non-essential elements of toxicant-induced reductions in fast axonal transport. Pulse labeling in CNS neurons. Neurotoxicology. 2000;21:447–458. [PubMed] [Google Scholar]
- 52.Takahashi A, Mizutani M, Agr B, Itakura C. Acrylamide-induced neurotoxicity in the central nervous system of Japanese quails. Comparative studies of normal and neurofilament-deficient quails. J Neuropathol Exp Neurol. 1994;53:276–283. doi: 10.1097/00005072-199405000-00009. [DOI] [PubMed] [Google Scholar]
- 53.Takahashi A, Mizuatani M, Itakura C. Acrylamide-induced peripheral neuropathy in normal and neurofilament-deficient Japanese quails. Acta Neuropathol (Berl) 1995;89:17–22. doi: 10.1007/BF00294254. [DOI] [PubMed] [Google Scholar]
- 54.Teal JJ, Evans HL. Behavioral effects of acrylamide in the mouse. Toxicol Appl Pharmacol. 1982;63:470–480. doi: 10.1016/0041-008x(82)90275-7. [DOI] [PubMed] [Google Scholar]
- 55.Von Burg R, Penny DP, Conroy PJ. Acrylamide neurotoxicity in the mouse: a behavioral, electrophysiological and morphological study. J Appl Toxicol. 1981;1:227–233. doi: 10.1002/jat.2550010409. [DOI] [PubMed] [Google Scholar]
- 56.Weaver DJ, Viancour TA. The crayfish neuronal cytoskeleton: an investigation of proteins having neurofilament-like immunoreactivity. Brain Res. 1991;544:49–58. doi: 10.1016/0006-8993(91)90884-x. [DOI] [PubMed] [Google Scholar]
- 57.Xu S, Cork LC, Griffin JW, Cleveland DW. Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell. 1993;73:23–33. doi: 10.1016/0092-8674(93)90157-l. [DOI] [PubMed] [Google Scholar]