

Abstract
Previous research has suggested that cGMP-dependent protein kinases (cGKs) may play a role in long-term potentiation in hippocampus, but their site of action has been unknown. We examined this question at synapses between pairs of hippocampal neurons in dissociated cell culture. Injection of a specific peptide inhibitor of cGK into the presynaptic but not the postsynaptic neuron blocked long-lasting potentiation induced by tetanic stimulation of the presynaptic neuron. As controls, injection of a scrambled peptide or a peptide inhibitor of cAMP-dependent protein kinase into either neuron did not block potentiation. Conversely, injection of the α isozyme of cGK type I into the presynaptic but not the postsynaptic neuron produced activity-dependent potentiation that did not require NMDA receptor activation. Evidence from Western blots, reverse transcription-PCR, activity assays, and immunocytochemistry indicates that endogenous cGK type I is present in the neurons, including presynaptic terminals. These results support the idea that cGK plays an important presynaptic role during the induction of long-lasting potentiation in hippocampal neurons.
Keywords: presynaptic, cGMP, protein kinase, potentiation, hippocampus, culture
The mechanisms of induction and expression of long-lasting potentiation at hippocampal synapses have been the subject of long-standing debate (Bliss and Collingridge, 1993;Kullmann and Siegelbaum, 1995). A large body of evidence now supports the idea that potentiation involves postsynaptic mechanisms (Luscher et al., 2000), but other evidence suggests that potentiation may also involve retrograde signaling and a presynaptic increase in transmitter release (Hawkins et al., 1993; Choi et al., 2000). More specifically, previous research has suggested that cGMP-dependent protein kinase (cGK) may function as a presynaptic target of nitric oxide (NO) during the induction of long-term potentiation (LTP) in hippocampus (Hawkins et al., 1998). Bath application of inhibitors of cGK such as Rp-8-Br-cGMPS or KT5823 can block LTP in hippocampal slices (Zhuo et al., 1994b; Blitzer et al., 1995; Son et al., 1998), and relatively selective activators of cGK such as 8-pCPT-cGMP and PET-cGMP produce activity-dependent long-lasting potentiation that does not require NMDA receptor activation (Zhuo et al., 1994b; Son et al., 1998). These results are consistent with the idea that cGK may play a presynaptic role in LTP, but they do not provide direct evidence for that hypothesis.
The hippocampal cell culture system provides a good model for studying presynaptic mechanisms (Bekkers and Stevens, 1990; Malgaroli and Tsien, 1992) because substances injected into the cell body have access to the presynaptic terminals as well as the postsynaptic dendrites within minutes (Alder et al., 1992; Popov and Poo, 1992; Arancio et al., 1996). Furthermore, hippocampal neurons in culture can undergo long-lasting potentiation with many of the key features of LTP in the CA1 region of hippocampus in slices or in vivo, including a requirement for Ca2+ influx through postsynaptic NMDA receptor channels (Bekkers and Stevens, 1990;Malgaroli and Tsien, 1992; Arancio et al., 1995, 1996; Diesseroth et al., 1996; Bi and Poo, 1998; Tao et al., 2000). Arancio et al. (1995)found that long-lasting potentiation can be induced reliably at synapses between pairs of hippocampal neurons in culture by high-frequency stimulation of the presynaptic neuron during brief removal of Mg2+ from the extracellular solution, which unblocks the NMDA receptor channels. This procedure is effective even when both cells are held under ruptured whole-cell voltage clamp, permitting injection of substances into either neuron. Further experiments using this method have suggested that, during induction of the potentiation, a retrograde messenger, NO, is synthesized in the postsynaptic neuron by a Ca2+-dependent enzyme, NO-synthase, and diffuses to the presynaptic terminals, where it produces a long-lasting increase in transmitter release (O'Dell et al., 1991; Arancio et al., 1996). NO appears to act by stimulating soluble guanylyl cyclase leading to the production of cGMP, and injection of cGMP into the presynaptic (but not the postsynaptic) neuron produces activity-dependent long-lasting potentiation (Arancio et al., 1995). However, cGMP might act through several pathways besides cGK, including cyclic nucleotide-gated channels and phosphodiesterases (Lincoln and Cornwell, 1993), both of which are also present in hippocampal neurons (Repasko et al., 1993; Kingston et al., 1996; Bradley et al., 1997). We have therefore now investigated the possible presynaptic role of cGK during the induction of long-lasting potentiation in hippocampal cell culture.
MATERIALS AND METHODS
Electrophysiology. Sprague Dawley rats (Hilltop Laboratory, Scottdale, PA) were housed and cared for in accordance with the guidelines of Columbia University. Dissociated cell cultures of hippocampal neurons from postnatal day 1 animals were prepared as described previously (O'Dell et al., 1991). The bath and electrode solutions were also as described previously (Arancio et al., 1995). We examined monosynaptic EPSCs between pairs of pyramidal-shaped neurons 7–21 d after plating. Both the presynaptic and postsynaptic neurons were maintained under ruptured whole-cell voltage clamp throughout the experiments, and the holding currents and input resistances of both cells were checked for constancy. EPSCs were evoked once every 10 sec by applying a 10 msec positive voltage step to the presynaptic neuron of sufficient amplitude to elicit an inward current in that neuron. Currents were recorded with an Axopatch 1A and Axopatch 200B (Axon Instruments, Foster City, CA) and filtered at 1 kHz. EPSC amplitudes were measured automatically between the peak and the mean during the interval just before the start of the EPSC using the pClamp program from Axon Instruments. A peptide inhibitor of cGK (synthesized at Albert Einstein College of Medicine), a peptide containing the same amino acids with their sequence scrambled (synthesized at Columbia University), or a peptide inhibitor of cAMP-dependent protein kinase (Upstate Biotechnology, Lake Placid, NY) were included in the presynaptic or postsynaptic electrode solution throughout the experiments. The α isozyme of cGK type I (Promega, Madison, WI) was delivered to the tip of the electrode by a fast internal perfusion method that has been described previously (Arancio et al., 1995). All other chemicals were from Sigma (St. Louis, MO) unless otherwise indicated. Data are shown as mean ± SEM percent of the baseline EPSC amplitude. The data were analyzed by a two-way ANOVA (treatment and time) followed by planned pairwise comparisons of the treatments overall and at each time point.
Western blotting. Hippocampi microdissected from male Sprague Dawley rats (150–200 gm, anesthetized with Nembutal) were homogenized in PBS containing protease inhibitors (5 μg/ml leupeptin, 1.5 mm benzamidine, 200 U/ml aprotinin, 2 μg/ml pepstatin A, 10 μg/ml PMSF, and 1 mm EDTA), and then the homogenates were mixed with SDS-PAGE stop solution. Cultured hippocampal cells were scraped together directly in stop solution. Samples were analyzed by 8% SDS-PAGE and Western blotting with ECL detection (Amersham Pharmacia Biotech, Piscataway, NJ) using cGK I antiserum diluted 1:3000 or affinity-purified cGK II antibody (Markert et al., 1995) diluted 1:1000. In separate experiments, the immunolabeling specificity of the cGK antibodies was demonstrated by antibody preabsorption with recombinant cGK I or cGK II purified from Sf9 cells.
Reverse transcription-PCR. Total RNA from cultured hippocampal neurons was isolated using Trizol Reagent from Life Technologies (Grand Island, NY), and reverse transcription (RT) was performed using a kit from Life Technologies with oligo-dT priming. For cDNA amplification, the primers used for human cGK I were 5′-GGAGACGTGGGGTCACT-GGTG-3′ (sense, position 503–523 nt) and 5′-AAAGAAGGTGTCCCC-TCTTGC-3′ (antisense, position 860–880 nt), and for rat cGK II were 5′-CGAGGGTAGACTGGAGGTGTT-3′ (sense, position 674–694 nt) and 5′-AATGGGGAGGTTGAGGAGAAT-3′ (antisense, position 1376–1396 nt). The cDNA was submitted to 35 PCR cycles (94°C, 40 sec; 56°C, 40 sec; and 72°C, 1 min), followed by final elongation at 72°C for 10 min. Samples of PCR products were analyzed by electrophoresis on 1.5% agarose gels and staining with ethidium bromide.
Kinase activity. The cultured cells were washed with PBS and incubated in serum-free medium with or without 250 μm 8-pCPT-cGMP for 30 min at 37°C. The reaction was terminated by addition of SDS-PAGE stop solution, and samples were analyzed by Western blotting as described above. The nitrocellulose blot was divided into two pieces, and the high molecular weight protein range was incubated with cGK I antibody, whereas the lower range was incubated with monoclonal 16C2 antibody (Smolenski et al., 1998), which recognizes the endogenous cGK substrate vasodilator-stimulated phosphoprotein (VASP) phosphorylated on Ser-239 as a 46 kDa protein (VASP-P). VASP-P was identified by comparison with a standard prepared from a lysate of human platelets that had been incubated with sodium nitroprusside to cause phosphorylation of VASP.
Immunocytochemistry. Hippocampal cultures on glass coverslips were fixed in 4% paraformaldehyde in PBS, pH 7.4, at 4°C for 1 hr, washed three times with PBS, permeabilized in 0.1% Triton X-100 for 10 min at room temperature, and washed three more times in PBS. The free aldehydes were then quenched with 50 mm ammonium chloride in PBS for 15 min at room temperature, and nonspecific antibody binding was blocked with 10% normal goat serum in PBS for 30 min at room temperature. The cultures were then incubated with the two primary antibodies, rabbit anti-cGK I (Markert et al., 1995) or preimmune serum diluted 1:300 and either mouse anti-microtubule-associated protein 2 (MAP2) (Sigma, 5 μg/ml) or mouse monoclonal anti-synaptophysin (5 μg/ml; Boehringer Mannheim, Indianapolis, IN) at 4°C overnight. The cultures were washed three times in PBS and then incubated with the secondary antibodies, goat anti-rabbit antibody conjugated with Cy3 (Jackson ImmunoResearch, West Grove, PA) diluted 1:100 and goat anti-mouse antibody conjugated with Cy5 (Jackson ImmunoResearch) diluted 1:100 in 10% goat serum in PBS at room temperature for 1 hr. The cultures were then washed four to five times with PBS, mounted in Fluoromount-G, and examined with a Bio-Rad (Hercules, CA) MRC 1000 laser confocal scanning system coupled to a Zeiss (Oberkochen, Germany) Axiovert 100 inverted microscope with a 100×, 1.3 numerical aperture oil immersion objective. The cultures were excited using the 568 and 647 lines of a krypton–argon laser to image Cy3 and Cy5. Kalman averages of four scans were collected for each image.
RESULTS
We first replicated the previous finding (Arancio et al., 1995,1996) that three trains of tetanic stimulation (50 Hz, 2 sec) of the presynaptic neuron during brief perfusion with Mg2+-free saline reliably produced rapid and long-lasting potentiation of the EPSC at synapses between individual hippocampal neurons in culture (Fig.1). In control experiments with no tetanic stimulation, there was a slight rundown of the baseline EPSC attributable to the whole-cell ruptured patch recording. To test the role of cGK in long-lasting potentiation, we injected a peptide inhibitor of cGK (cGKi) into either the presynaptic or postsynaptic neuron. This novel peptide Gly-Arg-Thr-Gly-Arg-Arg-Asn-(D-Ala)lle-NH2 blocks the phospho-acceptor binding site of the kinase and is the most specific cGK inhibitory pseudosubstrate currently available (Lev-Ram et al., 1997). After injection of cGKi (100 μm;Ki of 14.8 μm,Lev-Ram et al., 1997) into the postsynaptic neuron tetanic stimulation still induced long-lasting potentiation, although the potentiation was somewhat reduced compared with normal. In contrast, injection of cGKi into the presynaptic neuron completely blocked potentiation. In control experiments with no tetanic stimulation, injection of cGKi into either the presynaptic or postsynaptic neuron did not affect the stability of the baseline EPSC. A two-way ANOVA with one repeated measure (test time) revealed that the six training procedures in Figure 1Cproduced significantly different amounts of potentiation (F(5,88) = 8.72; p < 0.01). Subsequent pairwise comparisons showed that tetanic stimulation with postsynaptic cGKi produced significantly less potentiation than tetanic stimulation alone (p < 0.05), but that either produced significantly greater potentiation than the other four training procedures (including test alone control and tetanic stimulation with presynaptic cGKi; p < 0.01 in each case), which were not significantly different from each other. These results suggest that presynaptic cGK is necessary for the potentiation and that postsynaptic cGK may also play a role.
Fig. 1.
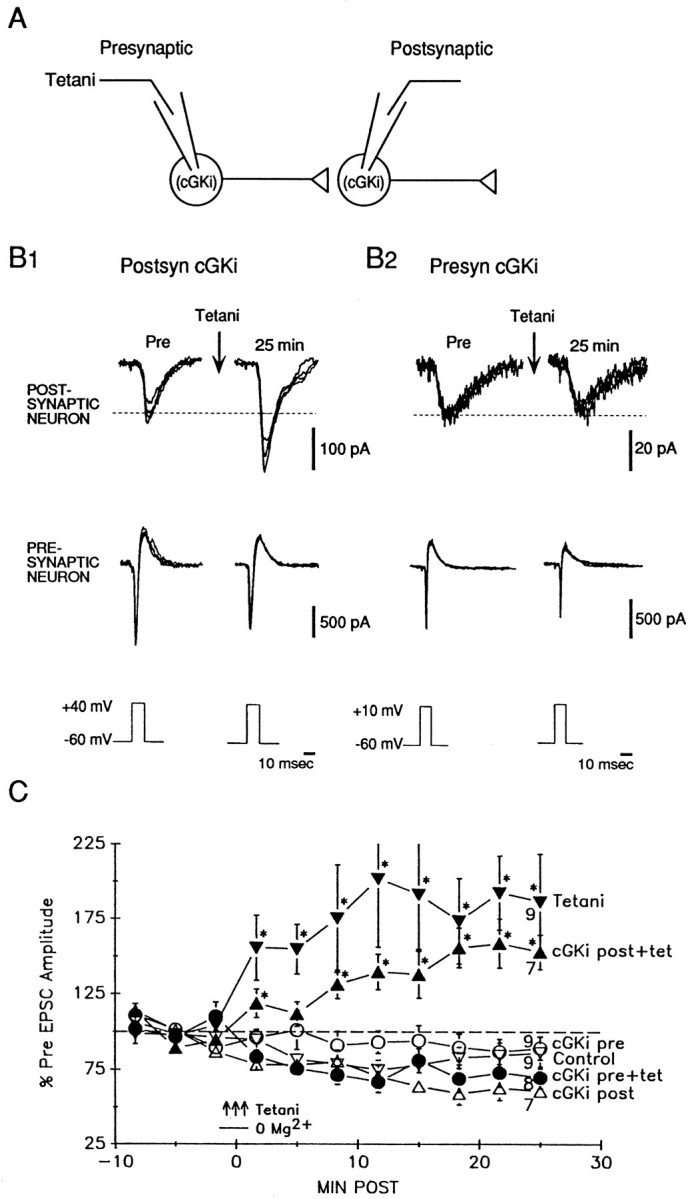
Presynaptic injection of cGKi blocks potentiation by tetanic stimulation. A, Experimental arrangement. B1, Example of potentiation by tetanic stimulation of the presynaptic neuron after injection of cGKi into the postsynaptic neuron. EPSCs were produced in the postsynaptic neuron by step depolarization sufficient to elicit an inward current in the presynaptic neuron once every 10 sec. The current in the presynaptic neuron has had leakage subtracted. Both recordings are a.c.-coupled. Sample traces are shown before (Pre) and 25 min after tetanic stimulation of the presynaptic neuron (three 50 Hz, 2 sec trains of depolarizations at 20 sec intervals) during brief perfusion with Mg2+-free solution. Four successive traces are superimposed at each time period. The dashed line shows the average Pre value.B2, Example of block of potentiation by injection of cGKi into the presynaptic neuron. C, Average potentiation produced by tetanic stimulation (a; filled inverted triangles) and slight rundown of the EPSC in test-alone controls (b; open inverted triangles). Injection of cGKi into the postsynaptic neuron reduced but did not block potentiation by tetanic stimulation (c; filled triangles), whereas injection of cGKi into the presynaptic neuron completely blocked the potentiation (d; filled circles). Injection of cGKi alone into either the postsynaptic neuron (e; open triangles) or the presynaptic neuron (f;open circles) had no effect on the baseline EPSC. EPSC amplitude has been normalized to the average value during the 10 min before training (% of Pre) in each experiment. Average Pre values in picoamperes were as follows: (a) 137 ± 33,n = 21; (b) 157 ± 86, n = 15; (c) 123 ± 39, n = 22; (d) 110 ± 43,n = 11; (e) 325 ± 191, n= 12; and (f) 178 ± 108, n = 13; not significantly different by a one-way ANOVA. Tetanic stimulation (3 arrows) occurred at time 0. The horizontal bar shows the time during which the bath solution was briefly changed to one with 0 Mg2+. The inhibitor was present throughout the experiment. Each point represents the average of 20 successive trials, and the numbers at 25 min indicate the n at that point. Some experiments were terminated earlier if the electrode seal was lost or the input resistance changed in either the presynaptic or postsynaptic cell. Thepoints indicate the means, the error bars indicate SEM, and asterisks indicate a significant difference from both the Pre level (dashed line) and the nontetanized controls.
To test whether the presynaptic injection procedure itself might have interfered with the ability of the cell to undergo potentiation, we injected a peptide containing the same amino acids as cGKi with their sequence scrambled (ScGKi). Unlike cGKi, injection of ScGKi (100 μm) into the presynaptic neuron did not prevent long-lasting potentiation by tetanic stimulation (Fig.2). Injection of cGKi might also have blocked potentiation by inhibiting cAK, although cGKi is 70 times more potent an inhibitor of cGK than of cAK (Lev-Ram et al., 1997). Therefore, as an additional control, we injected a specific peptide inhibitor of cAK [cAKi or PKI-(6–22)-amide, 20 μm;Ki of 1.7 nm] (Glass et al., 1989) into either the presynaptic or postsynaptic neuron and found that it also did not block long-lasting potentiation. Presynaptic cAKi may have produced some reduction of the potentiation but it was less effective than cGKi, although cAKi was injected at a much higher concentration relative to its Ki. The ScGKi and cAKi injections also did not have effects on the baseline EPSC. A two-way ANOVA revealed that the six training procedures in Figure 2Cproduced significantly different amounts of potentiation (F(5,69) = 9.76; p < 0.01). Subsequent pairwise comparisons showed that the three groups with tetanic stimulation were not significantly different from each other and that each produced significantly greater potentiation than the three groups without tetanic stimulation (p < 0.05 in each case), which were also not significantly different from each other. These results suggest that the block of long-lasting potentiation by injection of cGKi is not attributable to nonspecific effects of the injection and also that cGKi did not block the potentiation by inhibiting cAK.
Fig. 2.
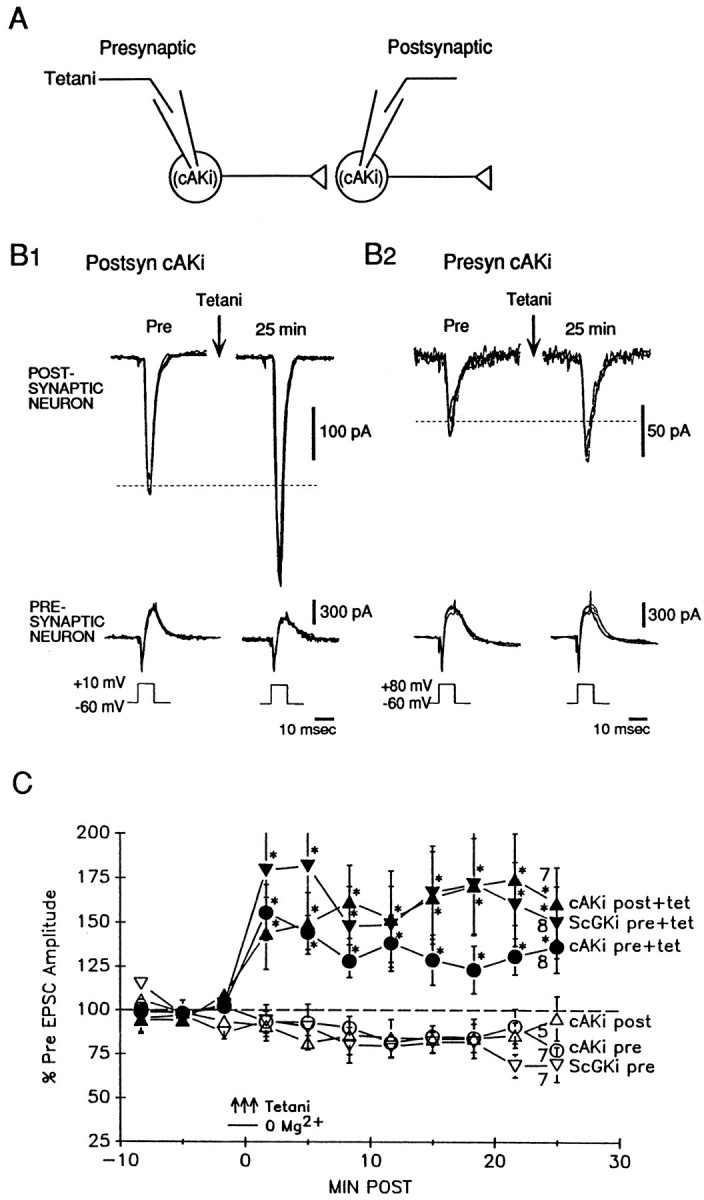
Injection of ScGKi or cAKi does not block potentiation. A, Experimental arrangement.B1, Example of potentiation by tetanic stimulation after injection of cAKi into the postsynaptic neuron.B2, Example of potentiation by tetanic stimulation after injection of cAKi into the presynaptic neuron.C, Average potentiation by tetanic stimulation after injection of ScGKi into the presynaptic neuron (a; filled inverted triangles). Tetanic stimulation also still produced potentiation after injection of cAKi into either the postsynaptic neuron (b; filled triangles) or the presynaptic neuron (c; filled circles). Injection of ScGKi alone into the presynaptic neuron (d; open inverted triangles) or injection of cAKi alone into either the postsynaptic neuron (e;open triangles) or the presynaptic neuron (f;open circles) did not have an effect on EPSC amplitude. Average Pre values in picoamperes were as follows: (a) 116 ± 46,n = 8; (b) 60 ± 24, n = 13; (c) 90 ± 24, n = 19; (d) 83 ± 30,n = 13; (e) 130 ± 44, n = 7; and (f) 136 ± 49, n = 15; not significantly different by a one-way ANOVA.
The results of the inhibitor experiments suggest that presynaptic cGK plays an important role in long-lasting potentiation and that postsynaptic cGK may also be involved. However, they do not say which type of cGK is involved. Two genes have been identified in vertebrates that encode for cGK type I (which is soluble) and cGK type II (which is membrane-associated) (Wernet et al., 1989; Uhler, 1993; Jarchau et al., 1994; Orstavik et al., 1997). cGK type I mRNA has been detected in several types of neurons, including pyramidal neurons in the CA1 and CA3 regions of hippocampus (Kingston et al., 1996; Kleppisch et al., 1999), and cGK type II is also present in a number of brain areas, including hippocampus (El Husseini et al., 1995; Kleppisch et al., 1999). We tested for the presence of the two types of cGK protein by Western blotting. Both cultured hippocampal cells and microdissected hippocampus contained an endogenous protein that migrated like cGK type I (Fig.3A1), and this signal was blocked by preabsorption of the antibody with recombinant cGK type I, demonstrating its specificity. In contrast, cGK type II was detected in microdissected hippocampus but not (or only very faintly) in cultured hippocampal cells (Fig.3A2). Similarly, cGK type I but not cGK type II mRNA was detected in cultured hippocampal cells by RT-PCR (Fig.3B). As additional evidence for the presence of cGK as well as its functional activity, treatment of intact cultured hippocampal cells with 8-pCPT-cGMP caused an increase in phosphorylation of an endogenous substrate, VASP, at a residue (Ser-239) that is preferentially phosphorylated by cGKs (Smolenski et al., 1998) (Fig.3C). cGK type I immunolabeling in the same samples was constant, indicating that loading of the − and + 8-pCPT-cGMP lanes was equal and that the increased VASP-P signal in the + lanes was attributable to increased kinase activity.
Fig. 3.
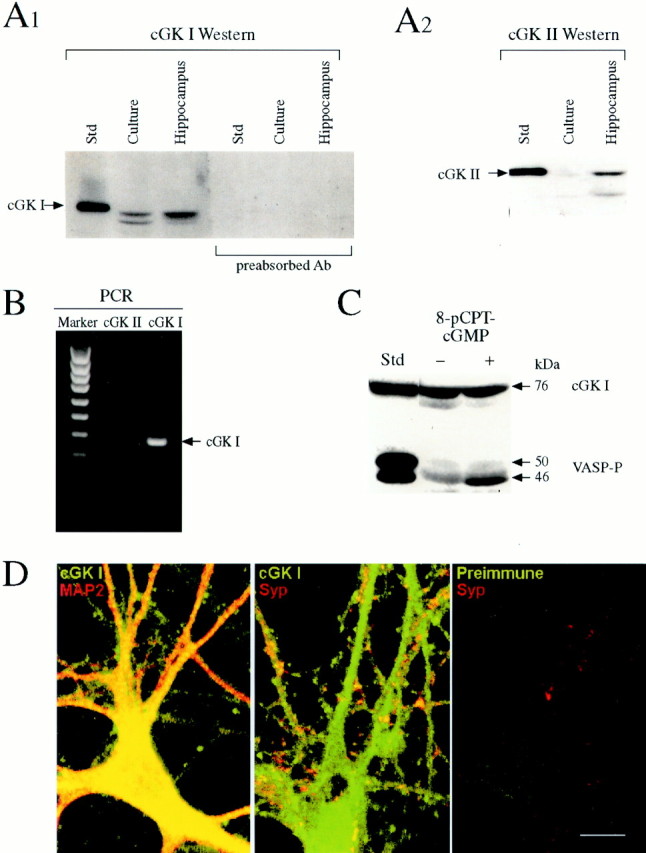
Cultured hippocampal neurons contain cGK type I.A, Western blot demonstration of endogenous cGK type I in cultured hippocampal cells. Standards of recombinant cGK I and cGK II (76 and 86 kDa, respectively) purified from Sf9 cells were used for identifying cGKs in samples (25 μg of protein) from hippocampal cultures or microdissected hippocampus. Both cultured hippocampal cells and microdissected hippocampus contained a protein that migrated like cGK I (A1). The cGK I signals were blocked by antibody (Ab) preabsorption, indicating that they are specific. Microdissected hippocampus also contained a protein that migrated like cGK II, but cultured hippocampal cells did not (A2). B, In agreement with the Western blot analysis, the RT-PCR technique detected cGK I but not cGK II mRNA in samples from cultured hippocampal cells. RT-PCR products obtained using oligonucleotide primers specific for cGK I and cGK II were analyzed using 1.5% agarose gels and ethidium bromide staining. DNA molecular weight standards (Marker) and the PCR product for cGK I (378 kb; arrow) are indicated. A cGK II product of expected size, 725 kb, was not detected.C, cGMP-dependent phosphorylation of an endogenous substrate protein, VASP. Hippocampal cultures were incubated in serum-free media with or without 8-pCPT-cGMP, and samples (30 μg of protein) were analyzed by Western blotting using a monoclonal antibody that recognized VASP phosphorylated on Ser-239 (which is preferentially phosphorylated by cGKs) as a 46 kDa protein. VASP-P was identified by comparison with a standard prepared from human platelets (left). cGK I was also assayed in the same samples, as described for A1. D, Immunocytochemical localization of cGK type I in cultured hippocampal neurons. Cultured hippocampal cells showed positive immunoreactivity for cGK type I (green) compared with control cells treated with preimmune serum. The same cells that displayed positive labeling for cGK type I also showed positive labeling for MAP2 (red), a specific neuronal marker located in cell bodies and dendrites, confirming that the cGK I-labeled cells were neurons (left). They also showed positive labeling for synaptophysin (red), a presynaptic vesicle-associated protein, indicating that the localization of cGK type I includes presynaptic terminals (middle). Double-labeled structures appear yellow or orange. Scale bar, 10 μm.
These results demonstrate that cGK type I is present and functional in the cell cultures. However, they do not show that cGK type I is present in neurons (as opposed to glia) and more specifically in presynaptic terminals, as would be expected for a presynaptic function. To test these possibilities, we used immunocytochemical techniques with a primary antibody against cGK type I. As illustrated in Figure3D, cultured hippocampal neurons showed positive immunoreactivity compared with control neurons treated with the preimmune serum. The immunoreactivity appeared to be fairly evenly distributed throughout the neuron except for the nucleus, as would be expected from the cytoplasmic localization of cGK type I. The same cells that showed positive immunoreactivity for cGK type I also double-labeled with an antibody against MAP2, a specific neuronal marker located in cell bodies and dendrites, confirming that the cells were neurons. To test whether cGK type I is also present in presynaptic terminals, we double-labeled with an antibody against synaptophysin, a vesicle-associated protein that is commonly used as a presynaptic marker. Most of the structures that labeled for synaptophysin also double-labeled for cGK type I (Fig. 3D, yellow ororange), indicating that the localization of endogenous cGK type I includes presynaptic terminals.
We next examined whether injection of cGK type I is able to produce long-lasting potentiation. The protein was introduced into either the presynaptic or postsynaptic neuron by means of a fast internal perfusion method (Arancio et al., 1995). Because we have found previously that presynaptic injection of cGMP or presynaptic release of caged NO produce potentiation only if they are paired with weak tetanic stimulation of the presynaptic neuron (Arancio et al., 1995, 1996), we injected cGK type I either by itself or paired with a weak tetanus (50 Hz, 0.5 sec). The cultures were bathed in saline containing normal Mg2+ and the NMDA antagonist APV (50 μm) throughout the experiments to ensure that the weak tetanus by itself did not produce potentiation (Fig.4). Injection of the α isozyme of cGK type I (0.5 μm) into the presynaptic neuron, paired with weak tetanic stimulation of that neuron, produced rapid and long-lasting potentiation of the EPSC. It was not necessary to coinject cGMP with the cGK type I, suggesting either that the kinase has some cGMP-independent activity or that basal levels of endogenous cGMP can produce sufficient stimulation of the kinase if it is present in excess. In contrast, injection of cGK type I into the postsynaptic neuron paired with weak tetanus produced no potentiation. Injection of cGK type I into either neuron also did not affect the baseline EPSC. A two-way ANOVA revealed that the five training procedures in Figure4C produced significantly different amounts of potentiation (F(4,48) = 4.67; p < 0.01). Subsequent pairwise comparisons showed that presynaptic injection of cGK type I paired with weak tetanus produced significantly greater potentiation than each of the other training procedures (p < 0.01 in each case), which were not significantly different from each other. These results indicate that presynaptic cGK, in conjunction with weak tetanic stimulation, is sufficient as well as necessary for long-lasting potentiation in cultured hippocampal neurons.
Fig. 4.
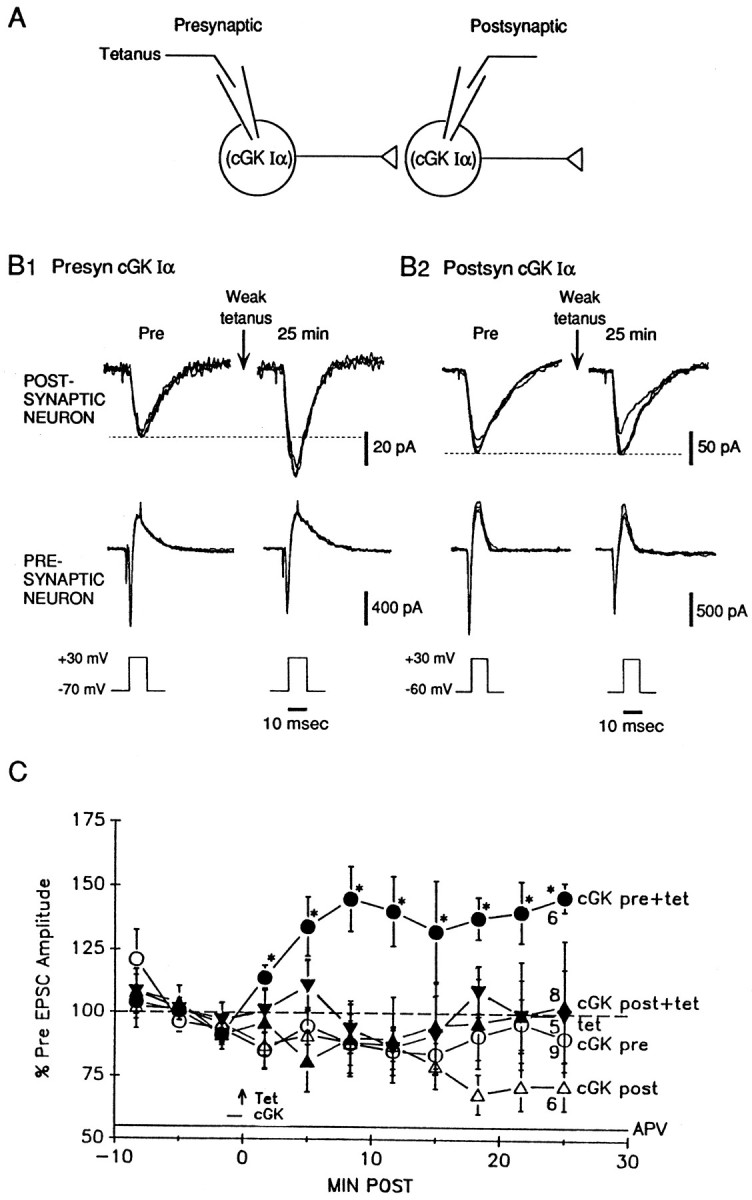
Presynaptic injection of cGK type I produces activity-dependent long-lasting potentiation. A, Experimental arrangement. B1, Example of long-lasting potentiation produced by injection of the α isozyme of cGK type I into the presynaptic neuron paired with weak tetanic stimulation (50 Hz, 0.5 sec) of that neuron in the presence of 50 μm APV. B2, Example of lack of potentiation by injection of cGK type I into the postsynaptic neuron paired with weak tetanic stimulation.C, Average potentiation by presynaptic injection of cGK type I paired with weak tetanic stimulation (a; filled circles). The weak tetanus (arrow) occurred at time 0. The horizontal bar shows the time during which cGK type I was injected into the neuron. The cultures were perfused with APV throughout the experiment. There was no potentiation after either postsynaptic injection of cGK type I paired with weak tetanic stimulation (b; filled triangles) or weak tetanic stimulation alone (c; filled inverted triangles). Injection of cGK type I alone into the presynaptic neuron (d;open circles) or the postsynaptic neuron (e; open triangles) did not have an affect on EPSC amplitude. Average Pre values in picoamperes were as follows: (a) 43 ± 9,n = 15; (b) 173 ± 44, n = 11; (c) 22 ± 4, n = 8; (d) 153 ± 53,n = 11; and (e) 178 ± 92,n = 8.
DISCUSSION
cGKs are cGMP-binding proteins that function as mediators of intracellular signaling in many cell types (Lohmann et al., 1997), but their function in the nervous system is generally poorly understood. Previous research has suggested that cGMP and cGK play a role in LTP in the CA1 region of hippocampal slices, although the results have been somewhat mixed. A number of studies have found that LTP is blocked by inhibitors of either guanylyl cyclase or cGK (Zhuo et al., 1994b; Blitzer et al., 1995; Boulton et al., 1995; Son et al., 1998; Lu et al., 1999), but other studies have found no effect of those inhibitors (Schuman et al., 1994; Gage et al., 1997). Similarly, several studies have found that cGMP analogs can produce activity-dependent long-lasting potentiation (Haley et al., 1992; Zhuo et al., 1994b; Son et al., 1998; Lu et al., 1999), whereas other studies have failed to replicate that effect (Schuman et al., 1994;Selig et al., 1996). One possible explanation for the differing results in these different studies is that LTP in hippocampal slices has multiple components involving several different mechanisms (only some of which involve NO, cGMP, and cGK) and that the different mechanisms make larger or smaller contributions depending on the experimental procedures (Son et al., 1998; Zhuo et al., 1998; Lu et al., 1999). For example, Son et al. (1998) found that activity-dependent potentiation by cGMP analogs is optimal when a relatively large, rapid rise in cGMP is combined with relatively brief, high-frequency activity. A smaller or slower rise in cGMP or presynaptic activity can actually lead to long-lasting depression (Zhuo et al., 1994a; Gage et al., 1997) and, at intermediate levels, potentiation and depression may cancel out. These findings might help to explain the negative results from studies in which cGMP analogs were applied for relatively long times (Schuman et al., 1994; Selig et al., 1996).
Recently, Kleppisch et al. (1999) took a different approach to examining the role of cGK in LTP in hippocampal slices by testing mice with either single or double knock-outs of cGK type I and cGK type II and found that LTP was normal in those mice. They therefore concluded that cGK is not involved in LTP, although it is possible that there was compensation by other pathways in the knock-out animals. Furthermore,Kleppisch et al. (1999) also found no effect of a guanylyl cyclase inhibitor or of cGMP analogs on LTP in wild-type animals. Thus, it appears likely that they performed their experiments under conditions in which the cGMP–cGK pathway makes a minimal contribution, so that knock-out of cGK would be expected to have little effect.
In light of the more complicated results in hippocampal slices, it is somewhat surprising that the NO–cGMP–cGK pathway appears to make a very large, if not dominant, contribution to long-lasting potentiation in culture. The potentiation in culture can be almost completely blocked by cGKi (Fig. 1) or pharmacological inhibitors of NO synthase, guanylyl cyclase, or cGK (Arancio et al., 1995, 1996). Conversely, exogenous cGK (Fig. 4), NO, or cGMP (Arancio et al., 1995, 1996) can produce activity-dependent potentiation comparable with that produced by tetanic stimulation. In these studies, the exogenous substances were applied as rapidly as possible just before the presynaptic activity, which may have optimized their effects. In addition, the cells in culture probably are not fully mature so that the potentiation may engage end-stage developmental processes that involve NO, cGMP, and cGK (Hindley et al., 1997; Leamey and Sur, 1999; Van Wagenen and Rehder, 1999) in addition to mature plasticity mechanisms.
One question that has been difficult to address in hippocampal slices is whether various molecules such as NO, cGMP, and cGK exert their effects in the presynaptic or postsynaptic neuron. Blitzer et al. (1995) found that the cGK inhibitor Rp-8-Br-cGMPS blocked LTP in slices when the inhibitor was applied to the bath but not when it was injected into the postsynaptic neuron, suggesting that cGK may act in the presynaptic neuron. However, testing this idea by injecting substances presynaptically has not been possible in hippocampal slices. This limitation, among others, has motivated research on hippocampal neurons in dissociated cell culture, in which both sides of the synapse are accessible to substances injected into the presynaptic or postsynaptic cell body (Alder et al., 1992; Popov and Poo, 1992; Arancio et al., 1996). Our results with injections of cGKi or cGK type I into the presynaptic neuron are consistent with previous results showing that presynaptic NO or cGMP can produce activity-dependent long-lasting potentiation in culture (Arancio et al., 1995, 1996) and suggest that they act predominantly by stimulating presynaptic cGK during the induction of the potentiation.
In addition to these presynaptic effects, we found that intracellular injection of cGKi into the postsynaptic neuron reduced but did not completely block potentiation, suggesting a possible role for postsynaptic cGK as well. Lu et al. (1999) have also provided evidence for a postsynaptic role of cGK in late-phase LTP in hippocampal slices. However, a postsynaptic effect of cGKi would appear to be inconsistent with negative results from postsynaptic injections of cGK type I (Fig.4), NO, or cGMP (Arancio et al., 1995, 1996). Because in those experiments the intracellular effects of cGK type I, NO, or cGMP were all tested with the NMDA antagonist APV in the bath, one possible explanation is that the postsynaptic but not the presynaptic actions of cGK may require coincident NMDA receptor activation (Son et al., 1998).
Presynaptic cGK type I, like cGMP or NO (Arancio et al., 1995, 1996) produced potentiation only when it was paired with weak tetanic stimulation. Because this potentiation was not blocked by APV, the cGK did not simply act to make the weak tetanus stronger and thereby enhance Ca2+ current through postsynaptic NMDA receptor channels. Previous studies have also shown that cGMP analogs do not alter the postsynaptic current during the tetanus either in culture (Arancio et al., 1995) or in slices (Son et al., 1998). One other possibility is that cGK may act synergistically with Ca2+ that enters the presynaptic terminal during the weak tetanus, perhaps by converging on a common molecular target. Consistent with that possibility, Antonova et al. (2000)recently found that the NO–cGMP–cGK pathway contributes to an increase in the number of synaptophysin- and synapsin-immunoreactive presynaptic terminals during long-lasting potentiation in hippocampal cell culture. Those results provide independent evidence that cGK acts presynaptically and indicate that it in some way affects vesicle-associated proteins. It will now be interesting to examine how activation of cGK contributes to changes in presynaptic immunoreactivity and synaptic transmission during long-lasting potentiation.
Footnotes
The research was supported by National Institutes of Health Grants MH50733 and GM45989 and grants from the Whitehall Foundation, the Deutsche Forschungsgemeinschaft, and the Howard Hughes Medical Institute. We thank A. MacDermott, E. R. Kandel, and S. Siegelbaum for their comments, H. Ayers, A. Krawetz, and M. Pellan for typing this manuscript, and C. Lam for help with the figures.
Correspondence should be addressed to Dr. Robert D. Hawkins, Center for Neurobiology and Behavior, Columbia University, 1051 Riverside Drive, New York, NY 10032. E-mail: rhawkins@pi.cpmc.columbia.edu.
REFERENCES
- 1.Alder J, Xie ZP, Valtorta F, Greengard P, Poo M. Antibodies to synaptophysin interfere with transmitter secretion at neuromuscular synapses. Neuron. 1992;9:759–768. doi: 10.1016/0896-6273(92)90038-f. [DOI] [PubMed] [Google Scholar]
- 2.Antonova I, Trillat A-C, Arancio O, Zablow L, Kandel ER, Hawkins RD. Rapid increase in immunoreactivity for presynaptic proteins during long-lasting potentiation. Soc Neurosci Abstr. 2000;26:363. [Google Scholar]
- 3.Arancio O, Kandel ER, Hawkins RD. Activity-dependent long-term enhancement of transmitter release by presynaptic 3′,5′-cyclic GMP in cultured hippocampal neurons. Nature. 1995;376:74–80. doi: 10.1038/376074a0. [DOI] [PubMed] [Google Scholar]
- 4.Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell. 1996;87:1025–1035. doi: 10.1016/s0092-8674(00)81797-3. [DOI] [PubMed] [Google Scholar]
- 5.Bekkers JM, Stevens CF. Presynaptic mechanisms for long-term potentiation in the hippocampus. Nature. 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- 6.Bi Q-Q, Poo M-M. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10469–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 8.Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 9.Boulton CL, Southam E, Garthwaite J. Nitric oxide dependent long-term potentiation is blocked by a specific inhibitor of soluble guanylyl cyclase. Neuroscience. 1995;69:699–703. doi: 10.1016/0306-4522(95)00349-n. [DOI] [PubMed] [Google Scholar]
- 10.Bradley T, Zhang Y, Bakin R, Lester HA, Ronnett GV, Zin K. Functional expression of the heteromeric “olfactory” cyclic nucleotide-gated channel in the hippocampus: a potential effector of synaptic plasticity in brain neurons. J Neurosci. 1997;17:1993–2005. doi: 10.1523/JNEUROSCI.17-06-01993.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi S, Klingauf J, Tsien RW. Postfusional regulation of cleft glutamate concentration during LTP at “silent synapses.”. Nat Neurosci. 2000;3:330–336. doi: 10.1038/73895. [DOI] [PubMed] [Google Scholar]
- 12.Diesseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 13.El Husseini AE-D, Bladen C, Vincent SR. Molecular characterization of type II cyclic GMP-dependent protein kinase expressed in the rat brain. J Neurochem. 1995;64:2814–2817. doi: 10.1046/j.1471-4159.1995.64062814.x. [DOI] [PubMed] [Google Scholar]
- 14.Gage AT, Reyes M, Stanton PK. Nitric-oxide-guanylyl-cyclase-dependent and independent components of multiple forms of long-term synaptic depression. Hippocampus. 1997;7:286–295. doi: 10.1002/(SICI)1098-1063(1997)7:3<286::AID-HIPO4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 15.Glass DB, Cheng HC, Mende-Mueller L, Reed J, Walsh DA. Primary structural determinants essential for potent inhibition of cAMP-dependent protein kinase by inhibitory peptides corresponding to the active portion of the heat-stable inhibitor protein. J Biol Chem. 1989;264:8802–8810. [PubMed] [Google Scholar]
- 16.Haley JE, Wilcox GL, Chapman PF. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- 17.Hawkins RD, Kandel ER, Siegelbaum SA. Learning to modulate transmitter release: themes and variations in synaptic plasticity. Annu Rev Neurosci. 1993;16:625–665. doi: 10.1146/annurev.ne.16.030193.003205. [DOI] [PubMed] [Google Scholar]
- 18.Hawkins RD, Son H, Arancio O. Nitric oxide as a retrograde messenger during long-term potentiation in hippocampus. Prog Brain Res. 1998;118:155–172. doi: 10.1016/s0079-6123(08)63206-9. [DOI] [PubMed] [Google Scholar]
- 19.Hindley S, Juurlink BH, Gysberg JW, Middlemiss PJ, Herman MA, Rathbone MP. Nitric oxide donors enhance neurotrophin-induced neurite outgrowth through a cGMP-dependent mechanism. J Neurosci Res. 1997;47:427–439. [PubMed] [Google Scholar]
- 20.Jarchau T, Hausler C, Markert T, Pohler D, Vanderkerckhove J, DeJonge HR, Lohmann SM, Walter U. Cloning, expression, and in situ localization of rat intestinal cGMP-dependent protein kinase II. Proc Natl Acad Sci USA. 1994;91:9426–9430. doi: 10.1073/pnas.91.20.9426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kingston PA, Zufall F, Barnstable CT. Rat hippocampal neurons express genes for both rod retinal and olfactory cyclic nucleotide-gated channels: novel targets for cAMP/cGMP function. Proc Natl Acad Sci USA. 1996;93:10440–10445. doi: 10.1073/pnas.93.19.10440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleppisch T, Pfeifer A, Klatt P, Rutyh P, Montkowski A, Fassler R, Hofmann F. Long-term potentiation in the hippocampal CA1 region of mice lacking cGMP-dependent kinases is normal and susceptible to inhibition of nitric oxide synthesis. J Neurosci. 1999;19:48–55. doi: 10.1523/JNEUROSCI.19-01-00048.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kullmann DM, Siegelbaum SA. The site of expression of NMDA receptor-dependent LTP: new fuel for an old fire. Neuron. 1995;15:997–1002. doi: 10.1016/0896-6273(95)90089-6. [DOI] [PubMed] [Google Scholar]
- 24.Leamey CA, Sur M. The soluble guanylyl cyclase-protein kinase G pathway is a target of nitric oxide during on/off sublimination in the retinogeniculate projection. Soc Neurosci Abstr. 1999;25:1006. [Google Scholar]
- 25.Lev-Ram V, Jiang T, Wood J, Lawrence DS, Tsien RY. Synergies and coincidence requirements between NO, cGMP, and Ca2+ in the induction of cerebellar long-term depression. Neuron. 1997;18:1025–1038. doi: 10.1016/s0896-6273(00)80340-2. [DOI] [PubMed] [Google Scholar]
- 26.Lincoln TM, Cornwell TL. Intracellular cyclic GMP receptor proteins. FEBS J. 1993;7:328–338. doi: 10.1096/fasebj.7.2.7680013. [DOI] [PubMed] [Google Scholar]
- 27.Lohmann SM, Vaandrager AB, Smolenski A, Walter U, De Jonge HR. Distinct and specific functions of cGMP-dependent protein kinases. Trends Biochem Sci. 1997;22:307–312. doi: 10.1016/s0968-0004(97)01086-4. [DOI] [PubMed] [Google Scholar]
- 28.Lu Y-F, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luscher C, Nicoll RA, Malenka RC, Muller D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci. 2000;3:545–550. doi: 10.1038/75714. [DOI] [PubMed] [Google Scholar]
- 30.Malgaroli A, Tsien RW. Glutamate-induced long-term potentiation of the frequency of miniature synaptic currents in cultured hippocampal neurons. Nature. 1992;357:134–139. doi: 10.1038/357134a0. [DOI] [PubMed] [Google Scholar]
- 31.Markert T, Vaandrager AB, Gambaryan S, Pohler D, Hausler C, Walter U, De Jonge HR, Jarchau T, Lohmann SM. Endogenous expression of type II cGMP-dependent protein kinase mRNA and protein in rat intestine. Implications for cystic fibrosis transmembrane conductance regulator. J Clin Invest. 1995;96:822–830. doi: 10.1172/JCI118128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation. Proc Natl Acad Sci USA. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orstavik S, Natarajan V, Tasken K, Jahnsen T, Sandberg M. Characterization of the human gene encoding the type I alpha and type I beta cGMP-dependent protein kinase (PRK G1). Genomics. 1997;42:311–318. doi: 10.1006/geno.1997.4743. [DOI] [PubMed] [Google Scholar]
- 34.Popov S, Poo M-M. Diffusional transport of macromolecules in developing nerve processes. J Neurosci. 1992;12:77–85. doi: 10.1523/JNEUROSCI.12-01-00077.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Repasko DR, Corbin JG, Conti M, Goy MF. A cyclic GMP-stimulated cyclic nucleotide phosphodiesterase gene is highly expressed in the limbic system of the rat brain. Neuroscience. 1993;56:673–686. doi: 10.1016/0306-4522(93)90364-l. [DOI] [PubMed] [Google Scholar]
- 36.Schuman EM, Meffert MK, Schulman H, Madison DV. An ADP-ribosyl transferase as a potential target for nitric oxide action in hippocampal long-term potentiation. Proc Natl Acad Sci USA. 1994;91:11958–11962. doi: 10.1073/pnas.91.25.11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Selig DK, Segal MR, Liao D, Malenka RC, Malinow R, Nicoll RA, Lisman JE. Examination of the role of cGMP in long-term potentiation in the CA1 region of the hippocampus. Learn Mem. 1996;3:42–48. doi: 10.1101/lm.3.1.42. [DOI] [PubMed] [Google Scholar]
- 38.Smolenski A, Bachmann C, Reinhard K, Honig-Liedl P, Jarchau T, Hoschuetzky H, Walter U. Analysis and regulation of vasodilator-stimulated phosphoprotein serine 239 phosphorylation in vitro and in intact cells using a phosphospecific monoclonal antibody. J Biol Chem. 1998;273:20029–20035. doi: 10.1074/jbc.273.32.20029. [DOI] [PubMed] [Google Scholar]
- 39.Son H, Lu Y-F, Zhuo M, Arancio O, Kandel ER, Hawkins RD. The specific role of cGMP in hippocampal LTP. Learn Mem. 1998;5:231–245. [PMC free article] [PubMed] [Google Scholar]
- 40.Tao H-ZW, Zhang LI, Bi G-Q, Poo M-M. Selective presynaptic propagation of long-term potentiation in defined neural networks. J Neurosci. 2000;20:3233–3243. doi: 10.1523/JNEUROSCI.20-09-03233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uhler MD. Cloning and expression of a novel cyclic GMP-dependent protein kinase from mouse brain. J Biol Chem. 1993;268:13586–13591. [PubMed] [Google Scholar]
- 42.Van Wagenen S, Rehder V. Regulation of neuronal growth cone filopdia by nitric oxide. J Neurobiol. 1999;39:168–185. doi: 10.1002/(sici)1097-4695(199905)39:2<168::aid-neu2>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 43.Wernet W, Flockerzi V, Hutmann F. The cDNA of the two isoforms of bovine cGMP-dependent protein kinase. FEBS Lett. 1989;251:191–196. doi: 10.1016/0014-5793(89)81453-x. [DOI] [PubMed] [Google Scholar]
- 44.Zhuo M, Kandel ER, Hawkins RD. Nitric oxide and cGMP can produce either synaptic depression or potentiation depending on the frequency of presynaptic stimulation in hippocampus. NeuroReport. 1994a;5:1033–1036. doi: 10.1097/00001756-199405000-00004. [DOI] [PubMed] [Google Scholar]
- 45.Zhuo M, Hu Y, Schultz C, Kandel ER, Hawkins RD. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994b;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]
- 46.Zhuo M, Laitinen JT, Li X-C, Hawkins RD. On the respective roles of nitric oxide and carbon monoxide in long-term potentiation in the hippocampus. Learn Mem. 1998;5:467–480. [PMC free article] [PubMed] [Google Scholar]