

Abstract
T-cell autoimmunity to myelin basic protein was recently shown to be neuroprotective in injured rat optic nerves. In the present study, using the mouse optic nerve, we examined whether active immunization rather than passive transfer of T-cells can be beneficial in protecting retinal ganglion cells (RGCs) from post-traumatic death. Before severe crush injury of the optic nerve, SJL/J and C3H.SW mice were actively immunized with encephalitogenic or nonencephalitogenic peptides of proteolipid protein (PLP) or myelin oligodendrocyte glycoprotein (MOG), respectively. At different times after the injury, the numbers of surviving RGCs in both strains immunized with the nonencephalitogenic peptides pPLP 190–209 or pMOG 1–22 were significantly higher than in injured controls treated with the non-self-antigen ovalbumin or with a peptide derived from β-amyloid, a non-myelin-associated protein. Immunization with the encephalitogenic myelin peptide pPLP 139–151 was beneficial only when the disease it induced, experimental autoimmune encephalomyelitis, was mild. The results of this study show that survival of RGCs after axonal injury can be enhanced by vaccination with an appropriate self-antigen. Furthermore, the use of nonencephalitogenic myelin peptides for immunization apparently allows neuroprotection without incurring the risk of an autoimmune disease. Application of these findings might lead to a promising new approach for treating optic neuropathies such as glaucoma.
Keywords: vaccination, neuroprotection, myelin, T-cells, CNS injury, autoimmunity, optic nerve
Injury to the CNS of adult mammals results in primary damage to the directly affected neurons and is often compounded by a gradual secondary loss of undamaged neurons in the vicinity (Faden and Salzman, 1992; Faden, 1993; McIntosh, 1993;Yoles and Schwartz, 1998). The primary lesion causes changes in extracellular ion concentrations, increased circulation of free radicals, release of neurotransmitters, depletion of growth factors, and local inflammation (Liu et al., 1994). These changes trigger a cascade of intracellular destructive events leading to delayed neuronal death (Villegas-Perez et al., 1993; Berkelaar et al., 1994;Garcia-Valenzuela et al., 1994).
In the case of the optic nerve, primary injury of the nerve fibers leads to death of the corresponding cell bodies, the retinal ganglion cells (RGCs) (Villegas-Perez et al., 1993). In the adult rat, for example, >90% of RGCs die within 2 weeks of optic nerve axotomy performed close to the eye (Villegas-Perez et al., 1988). In mice, RGCs undergo apoptosis after trauma to the axon (Li et al., 1999), decreasing to 50–80% of their original number by 30 d after intraorbital optic nerve transection (Allcutt et al., 1984).
It was recently shown in our laboratory that optic nerve injury in the rat is followed by a transient accumulation of activated T-cells at the lesion site, irrespective of their antigenic specificity. Although the accumulation is nonselective (Hirschberg et al., 1998; Moalem et al., 1999a), only T-cells that are specific to myelin basic protein (MBP) were found to reduce secondary degeneration (i.e., the spread of damage to neurons that had escaped the primary lesion) (Moalem et al., 1999b). These findings indicated that specific autoimmunity in the CNS may not always be detrimental and, under certain circumstances, might have a physiological role in protecting the damaged CNS.
The aim of the present study was to determine whether immune neuroprotection can promote post-traumatic survival of RGCs with damaged axons, and whether this can be achieved by active immunization. The model chosen for the study was the mouse optic nerve, which offers the possibility of examining various knock-out mice and thus gaining insight into the genetic control of immune neuroprotection.
The myelin sheath of the nerve is composed mainly of proteolipid protein (PLP) and MBP (Wucherpfennig, 1994). Some glycoproteins are also present in the myelin sheath but constitute only a minor fraction of the total protein. Among these, myelin oligodendrocyte glycoprotein (MOG) comprises 0.01–0.05% of the total myelin protein (Amiguet et al., 1992). A large body of evidence indicates that MBP, PLP, and MOG are all encephalitogenic proteins capable of causing experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis (Zamvil and Steinman, 1990; Linington et al., 1993).
The MOG peptide encompassing amino acids 35–55 (pMOG 35–55) of the mouse MOG sequence is encephalitogenic in H-2b (C3H.SW, C56BL/6J) mice (Kerlero de Rosbo et al., 1995; Mendel et al., 1995) but not in H-2k mice, and the disease in H-2b mice is similar to the chronic EAE induced by MBP or PLP. Histologically, the perivascular inflammatory foci seen in the CNS of C3H.SW mice with EAE induced by pMOG 35–55 are equivalent in number and intensity to those seen in mice with EAE induced by MBP or PLP (Kerlero de Rosbo et al., 1995). Whereas pPLP 139–151 and pMOG 35–55 are known to induce EAE in SJL/J (Bebo et al., 1998) and C3H.SW (Mendel et al., 1995) mice, respectively, the peptides pPLP 190–209 and pMOG 1–22 are not encephalitogenic in these strains (Mendel et al., 1995; Nicholson et al., 1995).
In this study, using the injured mouse optic nerve as a model, we demonstrate that active immunization with the encephalitogenic PLP peptide leads to neuroprotection only in cases in which the induced EAE is mild; the immunization has no beneficial effect in mice that develop severe EAE. Immunization with nonencephalitogenic myelin-associated peptides derived from PLP or MOG leads to neuroprotection. The unexpected finding that the post-traumatic death of RGCs might be slowed down by immunization, and especially by immunization with an autoimmune antigen, compels us to reassess the relationship between the immune system (in particular autoimmunity) and the injured CNS.
MATERIALS AND METHODS
Animals. Adult female C3H.SW and SJL/J mice (8–12 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME). The mice were housed in a light- and temperature-controlled room and matched for age in each experiment.
Antigens. MOG peptides 1–22 (GQFRVIGPGHPIRALVGDEAEL) and 35–55 (MEVGWYRSPFSRVVHLYRNGK) of rat origin, PLP peptides 190–209 (SKTSASIGSLCADARMYGVL) and 139–151 (HSLGKWLGHPDKF) of mouse origin, and βAP peptides 1–20 (DAEFGHDSGFEVRHQKLVFF) and 10–30 (EVRHQKLVFFAEDVGSNKGA) of rat and mouse origin were synthesized in the laboratory of Prof. M. Fridkin, at the Department of Chemistry of the Weizmann Institute of Science, using the fluorenylmethoxycarbonyl amino acids by automatic multiple peptide synthesizer (AMS422; Abimed, Langenfeld, Germany). Ovalbumin (OVA), fraction V, was purchased from Sigma Israel (Rehovot, Israel).
Active immunization. For EAE induction, mice were injected subcutaneously in the flank with 200 μl of an emulsion containing either 300 μg of rat MOG peptide (35–55) or 200 μg of rat PLP peptide (139–151) in complete Freund's adjuvant (CFA) supplemented with 500 μg of Mycobacterium tuberculosis H37RA (Difco, Detroit, MI). In the mice injected with pMOG 35–55, an identical injection was given in the other flank as a booster 1 week later. Immunization with nonencephalitogenic myelin peptides was done as follows. Mice were injected subcutaneously at one site in the flank with 200 μl of emulsion consisting of MOG 1–22 or OVA (C3H.SW mice, 300 μg/mouse) or PLP 190–209, βAP 1–30, or OVA (SJL/J mice, 200 μg/mouse), emulsified in CFA supplemented with 500 μg of M. tuberculosis. In the mice injected with pMOG 1–22, an identical injection was given in the other flank as a booster 1 week later. Control mice were injected only with PBS and CFA.
After encephalitogenic challenge, the mice were observed daily, and clinical manifestations of EAE were scored on a scale of 0–5 (0, no clinical signs; 1, flaccid tail; 2, hind-leg paralysis; 3, hind-leg paralysis with lower-body paresis; 4, hind-leg and foreleg paralysis; 5, death).
Preinjury application of stereotactic dye. For baseline labeling of RGCs, a stereotactic dye was applied before the crush injury. Eleven days after the first active immunization, mice were deeply anesthetized by intraperitoneal injection of xylazine (14 mg/kg; Vitamed, Bat Yam, Israel) and ketamine (60 mg/kg; Fort Dodge Laboratories, Fort Dodge, IA) and placed in a small stereotactic instrument. The skull was exposed and kept dry and clean using 3% hydrogen peroxide. The bregma was identified and marked. A hole was drilled above the superior colliculus of each hemisphere (0.292 mm behind and 0.05 mm lateral to the midline). Using a stereotactic measuring device and a Hamilton injector, the mice were injected with FluoroGold (3% in saline; ; 1 μl; Fluorochrome, Denver, CO) at one point in the superior colliculus of each hemisphere, at a depth of 0.16 mm (in C3H.SW mice) or 0.175 mm (in SJL/J mice) from the bony surface of the brain. After completion of the injection, the wound was sutured. Retrograde uptake of the dye provides a marker of the living cells.
Three days after dye application, the right optic nerve of each mouse was subjected to a crush injury severe enough to cause primary damage to most of the axons. After such an injury, the number of labeled cell bodies at a given time provides an indication of the rate of primary degeneration and cell body death.
RGC survival was assessed 1, 2, and 4 weeks after the crush injury. Mice were killed, eyes showing signs of ischemia or infection were discarded, and only eyes that looked healthy were used. Each retina was detached from the eye, prepared as a flattened whole mount in 4% paraformaldehyde solution, and examined for labeled RGCs by fluorescence microscopy. Labeled RGCs from five to six fields of identical size (0.196 mm2), located 1 mm from the optic disk, were counted under the fluorescence microscope and averaged. Calculation of the number of labeled RGCs per square millimeter in the right retina provided a quantitative measure of the total degeneration.
Crush injury of the mouse optic nerve. Three days after stereotactic dye application (14 d after the first active immunization), the mice were deeply anesthetized by intraperitoneal injection of xylazine (14 mg/kg) and ketamine (60 mg/kg). Using a binocular operating microscope, the conjuctiva of the right eye was incised, and the optic nerve was exposed. With the aid of cross-action forceps, the optic nerve was subjected to a severe crush injury 1–2 mm from the eyeball. The uninjured contralateral nerve was left undisturbed. Anterograde labeling of RGCs revealed no continuity of fibers beyond the site of injury, indicating that the crush was severe enough to cause primary damage to most of the axons (E. Yoles and M. Schwartz, unpublished observations). This model has yielded a high degree of reproducibility with minimal variation among animals in the same group. Thus, significant results can be obtained even when the number of mice used in each group is small.
Immunocytochemistry of T-cells. Longitudinal cryosections of the excised nerves (10 μm thick) were placed on gelatin-coated glass slides and frozen until preparation for fluorescence staining. Sections were fixed in ethanol for 10 min at room temperature, washed twice in double-distilled water, and incubated for 3 min in PBS containing 0.05% polyoxyethylene-sorbitan monolaurate (Tween 20). Sections were then incubated for 1 hr at room temperature with rat antibody to human CD3 (Serotec, Oxford, UK) or mouse antibody to mouse glial fibrillary acid protein (GFAP) (PharMingen, San Diego, CA), diluted (1:50) in PBS containing 3% fetal calf serum and 2% bovine serum albumin. The sections were washed three times with PBS containing 0.05% Tween 20 and incubated with either fluorescein isothiocyanate-conjugated goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA) or Cy3-conjugated goat anti-rat IgG (Jackson ImmunoResearch) for 1 hr at room temperature. The sections were then washed with PBS containing Tween 20 and treated with glycerol containing 1,4-diazobicyclo-(2,2,2) octane to inhibit quenching of the fluorescence. Sections were viewed with either a confocal or light microscope, and cells were observed. Staining in the absence of the first antibody was negative.
Statistical analysis. The number of RGCs per square millimeter was calculated for each experiment. Statistical analysis was performed by one-way ANOVA or Student's t test.
RESULTS
Establishment of the mouse optic nerve model for the study of RGC death
Injury of optic nerve axons leads to delayed death of their corresponding RGCs, mostly by apoptosis (Berkelaar et al., 1994). The model usually used to study this degenerative process is the rat optic nerve. However, the availability of a mouse model would confer certain unique advantages. Unlike the rat model, a model of optic nerve injury in the mouse will enable us to study post-traumatic death of RGCs in various transgenic and knock-out mice. It would also allow us to study autoimmune neuroprotection in mice with different susceptibilities to EAE. To establish such a model, we developed a method for inflicting and quantifying well-controlled injuries of the mouse optic nerve, using a relatively simple technique in which all RGCs are stereotactically labeled before the injury. This approach yields reproducible labeling of viable RGCs, with almost no variation among individuals or among strains. Testing of several mouse strains yielded similar numbers of RCGs (Table 1). The efficiency of labeling [i.e., the percentage of RGCs stained after a single injection of FluoroGold, calculated as the average number of RGCs observed per square millimeter (Table 1) multiplied by the approximate size of a mouse retina (24 mm2) divided by the average number of RGCs per retina (previously shown to be 100,000)] was found to be 80–90%.
Table 1.
No significant variation among labeled RGCs in different mouse strains
| Mouse strain | SJL/J | C3H.SW | C57BL/6 |
|---|---|---|---|
| RGCs per mm2 | 3386 ± 43.06 | 3694 ± 50.76 | 3529 ± 47 |
| (n = 10) | (n = 10) | (n = 5) |
Possible link between EAE severity and neuroprotection
Having established a technique for reproducible labeling of RGCs in the mouse, our objective was to use this method to determine whether active immunization with myelin-associated peptides in mice would result in neuroprotection. To ensure that the T-cell response would reach its peak at the time of injury (which presumably is when it is needed), we had to suit the timing of the injury to the course of EAE induced by the encephalitogenic MOG and PLP peptides (pMOG 35–55 and pPLP 139–151).
EAE was induced in C3H.SW mice (n = 4) by two inoculations (1 week apart) of pMOG 35–55 in CFA. Clinical signs of EAE began to develop 12–14 d after the initial challenge, with a chronic course of ascending paralysis that affected the tail and the hind legs (Fig. 1a). EAE was induced in SJL/J mice (n = 5) by a single inoculation of pPLP 139–151 in CFA. Starting 12–14 d after the initial challenge, clinical signs of EAE developed in a relapsing–remitting manner (Fig.1b).
Fig. 1.

Clinical course of EAE, induced by either pMOG 35–55 in C3H.SW mice (n = 4) (a) or pPLP 139–151 in SJL/J mice (n = 5) (b). For induction of disease, mice were injected with pMOG 35–55 or pPLP 190–209 in CFA supplemented with M. tuberculosis, as described in Materials and Methods. EAE was evaluated according to a neurological paralysis scale. The mean ± SEM daily clinical score is shown for each mouse strain. Note that, in the C3H.SW mice, the induced EAE had a chronic course, whereas in the SJL/J mice, it was relapsing–remitting in nature.
To determine whether the active immunization leads to neuroprotection, we immunized mice with the encephalitogenic peptide PLP 139–151 and determined the effect on RGC survival after optic nerve injury. The EAE score was assessed daily, starting on day 10 after the immunization and ending on the day of retinal excision. Only those mice (two of five) in which no further signs of EAE were detectable by day 14 (peak EAE score of 1) showed significantly larger numbers of surviving RGCs than the numbers measured in the OVA-injected controls (n = 4). RGC survival in mice that still had EAE symptoms at the time of retinal excision (three of five) was no better than in the OVA controls. The persistence of EAE symptoms 4 weeks after injury is an indication that the disease had been severe at its peak. RGC survival measured 4 weeks after immunization was indeed found to be negatively correlated with disease severity 2 weeks after optic nerve injury (Fig.2). Our interpretation of these findings is that autoimmunity, although beneficial in terms of neuroprotection, has a threshold beyond which it is detrimental.
Fig. 2.

Negative correlation between the number of surviving RGCs of injured nerves from mice immunized with PLP 139–151 and EAE disease severity. Each point indicates the mean number of labeled RGCs per square millimeter 2 weeks after optic nerve injury. Two weeks before injury, mice were injected with pPLP 139–151 or OVA. The neurotracer dye FluoroGold was applied stereotactically 3 d before injury. Two weeks after the injury, the retinas were excised and flat-mounted, and the EAE score was determined. Labeled RGCs from three to five randomly selected fields in each retina (all located 1 mm from the optic disk) were counted by fluorescence microscopy. In mice immunized with PLP 139–151, in which no protection was observed, the average number of RGCs (563 RGCs/mm2; n = 3) was similar to that found in OVA-immunized mice (613 RGCs/mm2;n = 4). The correlation coefficient (r) was −0.96 according to linear regression; the p value was 0.009 according to one-way ANOVA.
Active immunization with nonencephalitogenic myelin-associated peptides slows down RGC degeneration
In view of the above findings, together with previous findings in our laboratory that T-cells against nonencephalitogenic myelin-associated peptides have a neuroprotective effect, we examined whether preinjury immunization with the nonencephalitogenic myelin-associated peptide pPLP 190–209 or pMOG 1–22 would lead to neuroprotection in SJL/J and C3H.SW mice, respectively. βAP was used as a non-myelin-associated self-peptide control, and OVA was used as a foreign antigen control. The numbers of labeled RGCs per square millimeter (reflecting cell bodies that were still viable 2 weeks after the injury) are shown in Figure 3. The mean number of surviving RGCs in the retinas of mice immunized with pPLP 190–209 (1725 RGCs/mm2;n = 10) was significantly higher than that in the retinas of OVA-immunized control mice (965 RGCs/mm2; n = 10;p < 0.001; one-way ANOVA). The mean number of surviving RGCs in the retinas of βAP-injected mice (1240 RGCs/mm2; n = 8) was not significantly higher than that found in the OVA controls (p > 0.05; one-way ANOVA).
Fig. 3.

Immunization with pPLP 190–209 slows down death of RGCs. The histograms record the mean number of labeled RGCs 2 weeks after optic nerve injury. Two weeks before injury, SJL/J mice were injected with pPLP 190–209, OVA, or βAP. The neurotracer dye FluoroGold was applied stereotactically 3 d before injury. Two weeks after the injury, the retinas were excised and flat-mounted. Labeled RGCs from three to five randomly selected fields in each retina (all located 1 mm from the optic disk) were counted by fluorescence microscopy. Survival in each group of injured nerves was expressed as the mean ± SEM number of labeled RGCs per square millimeter. The neuroprotective effect of pPLP 190–209 was significant compared with that of OVA (p < 0.001; one-way ANOVA). βAP did not differ significantly from OVA in its protective effect on neurons that had escaped the primary injury (p > 0.05; one-way ANOVA). The results are a summary of three experiments.
A similar survival pattern relative to the controls (n= 14) was observed in mice immunized with pMOG 1–22 (n= 12), in which ∼1017 RGCs/mm2 were found to be labeled 2 weeks after injury compared with only 728 in the retinas of the OVA-immunized controls (n = 14;p < 0.05; Student's t test).
Because active immunization with pMOG 1–22 or pPLP 190–209 performed 2 weeks before the injury reduced the post-traumatic loss of RGCs, we were interested in examining the effect of active immunization performed immediately after the injury. As shown in Figure4a, in the retinas of mice immunized immediately after the injury with pMOG 1–22 (n = 5), ∼1049 RGCs/mm2were still viable 2 weeks after the injury, whereas the mean number of surviving RGCs in the retinas of OVA-injected controls (n = 4) was significantly lower (∼777 RGCs/mm2) (p< 0.02; Student's t test).
Fig. 4.
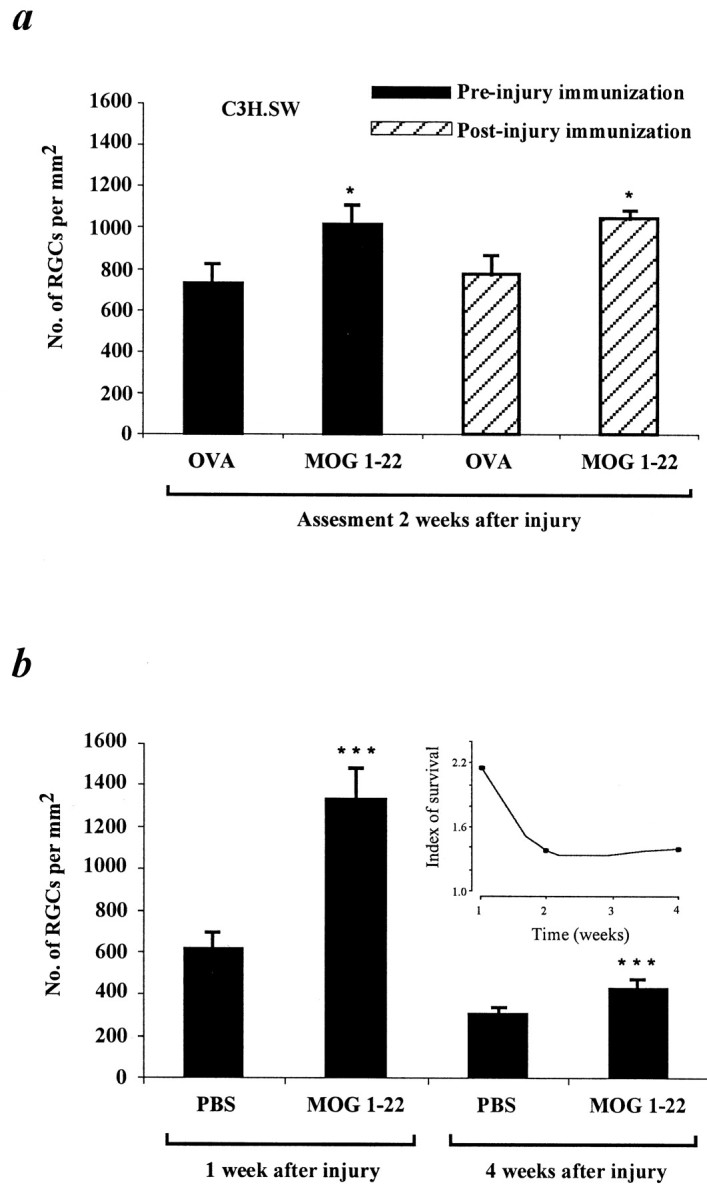
Immunization with pMOG 1–22 slows down RGC degeneration. The histograms record the mean ± SEM number of labeled RGCs per square millimeter. a, The number of labeled RGCs assessed 2 weeks after injury in retinas of mice before or after injury immunized with pMOG 1–22. For preinjury immunization, C3H.SW mice were injected with pMOG 1–22 or OVA, 14 d before optic nerve injury. Dye application, preparation, and counting of RGCs, as well as calculation of RGC survival, were as described for Figure 3. The number of labeled RGCs in the retinas of mice pretreated with pMOG 1–22 was significantly higher (p< 0.05; Student's t test) than in mice pretreated with OVA. The results are a summary of three experiments. For postinjury immunization, FluoroGold was applied stereotactically 3 d before optic nerve injury, and the mice were injected with either pMOG 1–22 or OVA immediately after the injury. Two weeks later, the retinas were prepared and counted as before. The number of labeled RGCs in the retinas of mice inoculated with pMOG 1–22 after the injury was significantly higher (p < 0.02; Student'st test) than in the mice inoculated with OVA before injury. The results shown are from one experiment only. Each group contained four to five mice. b, The number of labeled RGCs in preinjury immunized mice assessed 1 and 4 weeks after the injury. The histograms record the mean ± SD number of labeled RGCs per square millimeter. The RGC number in pMOG 1–22-immunized mice was significantly higher than that of PBS control mice (p < 0.0001; Student's ttest) either 1 or 4 weeks after the injury. The insetshows the ratio between the number of surviving RGCs in the retinas of MOG-immunized mice and the control mice, at each of the time points tested. The results shown are from one experiment only. Each group contained five to nine mice.
To verify that immunization with MOG had indeed slowed down the rate of RGC death caused by the crush injury, we repeated the experiment and analyzed the retinas of C3H.SW mice at two additional times, 1 and 4 weeks after the injury. As shown in Figure 4b, in the retinas of mice preimmunized with pMOG 1–22 (n = 9), ∼1323 RGCs/mm2 were still viable 1 week after the injury, whereas in the retinas of PBS-injected controls (n = 5), the mean number of viable RGCs was significantly lower (∼610 RGCs/mm2;p < 0.0001; Student's t test). In contrast, 4 weeks after the injury, only 422 RGCs/mm2 were still viable in the preimmunized mice, whereas the mean number of surviving RGCs in the controls (n = 7) was 296 RGCs/mm2 (p > 0.0001; Student's t test). It thus appeared that at any time point tested, the number of surviving RGCs in the MOG-immunized mice was higher than in the control. These results thus confirm that the MOG immunization affects the rate of RGC loss and that it has a long-lasting effect.
T-cell accumulation at the injury site
The finding that adoptively transferred activated T-cells accumulate at the site of optic nerve injury in rats (Hirschberg et al., 1998) prompted us to analyze crush-injured optic nerves of mice for the presence of T-cells after active immunization with pMOG 1–22 or OVA or after injection of PBS with CFA. Staining with GFAP and CD3 antibodies showed that T-cells were present from day 3 after injury with a peak at day 7 and were restricted to the site of injury. T-cell accumulation at the site of optic nerve injury in C3H.SW mice after active immunization with pMOG 1–22 is indicated in Figure5. Similar patterns were observed in control mice immunized with either OVA or PBS, in line with a previous finding of no antigenic selectivity in the post-traumatic accumulation of T-cells in the rat optic nerve.
Fig. 5.

Confocal microscopy pictures showing T-cell accumulation at the optic nerve injury site. One week after the injury, the optic nerves of C3H.SW mice immunized with pMOG 1–22 before injury were excised and labeled immunocytochemically. Serial optic nerve sections immunolabeled for GFAP (a) delineate the site of injury (indicated by the arrows). Immunolabeling for CD3 (b) indicates the presence of T-cells at the injury site.
DISCUSSION
This work shows that active immunization with myelin-associated peptides leads to neuroprotection. The neuroprotective effect could be achieved with either immunodominant encephalitogenic epitopes or with nonencephalitogenic epitopes. The former were found to be beneficial only when the disease they caused was mild. These findings support our contention that beneficial autoimmunity is functionally distinct from autoimmune disease (Schwartz et al., 1999) and that autoimmunity might be a benign physiological response as long as it is well controlled (Cohen and Schwartz, 1999; Schwartz et al., 1999). Thus, autoimmunity, although beneficial in terms of neuroprotection, has a threshold beyond which it is detrimental. Accordingly, the autoimmune response induced here by active immunization with an immunodominant encephalitogenic epitope failed to lead to neuroprotection when it exceeded the upper limit of benign autoimmunity, becoming what we would define as “out of control.” Such a response would involve substantially more T-cells and macrophages than the response induced by adoptive transfer of T-cells (Moalem et al., 1999a). In fact, our studies of active immunization and passive immunization in the injured spinal cord have yielded similar results (Hauben et al., 2000a).
Immunization with nonencephalitogenic myelin peptides was shown here to trigger an immune response that slows down the degeneration of RGCs after optic nerve injury in mice. This neuroprotective effect is not restricted to peptides associated with MBP, because nonencephalitogenic peptides derived from PLP and MOG were similarly neuroprotective. After passive immunization using the rat optic nerve model, the percentage of labeled RGCs was about twofold higher in the retinas of rats inoculated with anti-MBP T-cells than in controls (Moalem et al., 1999b). In the present study, the retinas of mice actively immunized with pPLP 190–209 or pMOG 1–22 showed ∼80 or 40% more surviving RGCs, respectively, than those of the controls. The differences in degree of protection between the rat and the mouse might be attributable to the type of immunization (passive vs active), species differences, or differences between the parameters examined in the two models. In the mouse, we examined the effects of different autoimmune responses on the death rate of RGCs of damaged axons, with such death being an inevitable consequence of the primary insult (Berkelaar et al., 1994;Garcia-Valenzuela et al., 1994). Indeed, although the beneficial effect of the immunization was long-lasting, it decreased with time; 1 week after the injury, the neuroprotective effect was higher than the effect 2 and 4 weeks after the injury. In contrast, the parameter measured in the rat optic nerve model was the effect of passive transfer of anti-MBP T-cells after partial injury (Moalem et al., 1999b) on the secondary degeneration of neurons that had escaped the primary lesion (Yoles and Schwartz, 1998). It should be noted that, unlike passive immunization, which in our experiments involved only T-cells, active immunization involves additional immune elements, such as macrophages and antibodies. Macrophages might affect RGC survival in a way that influences the potency of active but not of passive immunization (Hirschberg and Schwartz, 1995). The slight difference in the degree of protection conferred by immunization with PLP and with MOG in the mouse might be attributable to the relative abundance of these proteins in the myelin sheath (Wucherpfennig, 1994); PLP and MBP are the major constituents of the myelin sheath, whereas MOG comprises only ∼0.05% of the total myelin protein (Amiguet et al., 1992).
The neuroprotective effect induced by immunization with myelin peptides can, in principle, be mediated by either T-cells or antibodies that were secreted from B-cells and managed to pass through the disrupted blood–brain barrier as a result of the injury. The previous finding in the rat model that anti-MBP T-cells accumulate at the injury site (Konno et al., 1990; Hirschberg et al., 1998; Moalem et al., 1999a) and are involved in neuroprotection (Moalem et al., 1999b), together with the present finding that some T-cells “home” to the site of injury, suggests that the neuroprotective effect seen here is mediated by T-cells. Moreover, the period between the time of immunization (and hence the onset of the immune response) and the time when this response is needed by the damaged nerve is not long enough for the antibodies to be produced. In a recent study by Huang et al. (1999), it was suggested that active immunization with a myelin homogenate emulsified in incomplete Freund's adjuvant promotes nerve regrowth via antibody activity. Studies in our laboratory have shown that vaccination with MBP is effective only in neuroprotection, because no recovery could be demonstrated in the completely transected spinal cord (Hauben et al., 2000a). Moreover, passive transfer of autoimmune anti-MBP T-cells (Moalem et al., 1999b; Hauben et al., 2000b), but not of antibodies against MBP, led to neuroprotection of the partially injured spinal cord (Hauben et al., 2000a). It is therefore possible that the observed neuroprotection obtained with autoimmune T-cells represents a physiological mode of recovery that is spontaneously evoked by the injury but is apparently insufficient and needs boosting to be effectively manifested. In contrast, the regrowth obtained by Huang et al. (1999) represents a pharmacological intervention achieved by anti-myelin antibodies, similar to that reported previously bySchnell et al. (1997).
The results of our recent studies, and of the present work, argue in favor of autoimmunity as a benign physiological response, which, unless it gets out of control, reduces postinjury degeneration in the CNS. Other studies have demonstrated an accumulation of T-cells in the injured CNS (Schnell et al., 1997; Raivich et al., 1998), as well as a systemic T-cell immune response against MBP in animals with CNS injuries (Popovich et al., 1996). It was not clear, however, whether the effect of these T-cells was beneficial or harmful.
The molecular mechanisms by which autoimmune T-cells protect the injured nerve from degeneration are not yet fully understood. Previous electrophysiological findings indicated that the anti-MBP T-cells exert neuroprotection by causing a transient reduction in the electrophysiological activity of the nerve (Moalem et al., 1999b). It was suggested recently that the neuroprotection mediated by autoimmune T-cells might be attributable, at least in part, to antigen-dependent secretion of neurotrophins by the autoimmune T-cells reactivated at the lesion site when encountering their specific antigen (Moalem et al., 2000). The production of neurotrophins by T-cells was reported recently (Ehrhard et al., 1993; Santambrogio et al., 1994; Besser and Wank 1999;Kerschensteiner et al., 1999). There is evidence showing that various neurotrophins exert neuroprotective effects after axotomy (Sawai et al., 1996). If, however, the effect is indeed exerted via trophic factors, the therapeutic administration of T-cells would have an advantage over the administration of any single factor, because the T-cells are capable of producing a repertoire of factors whose production can be regulated by signals emanating from the damaged tissue.
In summary, vaccination with CNS myelin-associated self-antigens was shown here to be capable of leading to post-traumatic neuroprotection in mice and hence to delayed death of RGCs. The therapeutic window of the vaccination-induced neuroprotection will be determined by the choice of peptides and the route of administration. On the basis of these results, it seems reasonable to suggest that, if an autoimmune response to specific antigens exposed by CNS injury could be selectively augmented and well regulated, it might be possible to achieve neuroprotection without the threat of an autoimmune disease (Schwartz et al., 1999). Immune activity in general, and autoimmune activity in particular, have been shown to have conflicting effects depending on the type of the immune cells, the timing, and the tissue context. Thus, for example, autoimmunity may be destructive for myelin in cases in which the axons are intact (Sriram et al., 1989), whereas in traumatized axons, well-controlled autoimmunity, as shown here and in previous studies from our laboratory (Moalem et al., 1999b; Hauben et al., 2000b), is beneficial. Vaccination with myelin-associated peptides around the time of CNS axonal damage may thus be viewed as a way of boosting a beneficial physiological response, which is awakened after injury. These findings have promising clinical implications for the development of a vaccination against optic nerve neuropathies such as glaucoma, a disease of the optic nerve in which degeneration starts at the nerve fibers and ends with the death of the RGCs (Jonas and Budde, 2000). In many cases of optic neuropathy, degeneration continues to progress even after removal of the primary risk factor(s), and the end result is RGC death (Yoles and Schwartz, 1998). Neuroprotection might therefore serve as a complementary treatment for optic nerve neuropathies in general and for glaucoma in particular (Schwartz et al., 1996).
Footnotes
This work was supported in part by a Glaucoma Research Foundation grant awarded to M.S. A.B.-N. is the incumbent of the Eugene and Marcia Appelbaum Professorial Chair. M.S. holds the Maurice and Ilse Katz Professorial Chair in Neuroimmunology. We thank S. Smith for editorial assistance and A. Shapira for animal assistance.
Correspondence should be addressed to Dr. Michal Schwartz, Department of Neurobiology, The Weizmann Institute of Science, 76100 Rehovot, Israel. E-mail: bnschwartz@wiccmail.weizmann.ac.il.
REFERENCES
- 1.Allcutt D, Berry M, Sievers J. A qualitative comparison of the reactions of retinal ganglion cell axons to optic nerve crush in neonatal and adult mice. Brain Res. 1984;318:231–240. doi: 10.1016/0165-3806(84)90028-2. [DOI] [PubMed] [Google Scholar]
- 2.Amiguet P, Gardinier MV, Zanetta JP, Matthieu JM. Purification and partial structural and functional characterization of mouse myelin/oligodendrocyte glycoprotein. J Neurochem. 1992;58:1676–1682. doi: 10.1111/j.1471-4159.1992.tb10040.x. [DOI] [PubMed] [Google Scholar]
- 3.Bebo BF, Jr, Zelinka-Vincent E, Adamus G, Amundson D, Vandenbark AA, Offner H. Gonadal hormones influence the immune response to PLP 139–151 and the clinical course of relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. 1998;84:122–130. doi: 10.1016/s0165-5728(97)00214-2. [DOI] [PubMed] [Google Scholar]
- 4.Berkelaar M, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J Neurosci. 1994;14:4368–4374. doi: 10.1523/JNEUROSCI.14-07-04368.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Besser M, Wank R. Clonally restricted production of the neurotrophin brain-derived neurotrophic factor and neurotrophin-3 mRNA by human immune cells and Th1/Th2-polarized expression of their receptors. J Immunol. 1999;162:6303–6306. [PubMed] [Google Scholar]
- 6.Cohen IR, Schwartz M. Autoimmune maintenance and neuroprotection of the central nervous system. J Neuroimmunol. 1999;100:111–114. doi: 10.1016/s0165-5728(99)00190-3. [DOI] [PubMed] [Google Scholar]
- 7.Ehrhard PB, Erb P, Graumann U, Otten U. Expression of nerve growth factor and nerve growth factor receptor tyrosin kinase Trk in activated CD4-positive T-cell clones. Proc Natl Acad Sci USA. 1993;90:10984–10988. doi: 10.1073/pnas.90.23.10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faden AI. Experimental neurobiology of central nervous system trauma. Crit Rev Neurobiol. 1993;7:175–186. [PubMed] [Google Scholar]
- 9.Faden AI, Salzman S. Pharmacological strategies in CNS trauma. Trends Pharmacol Sci. 1992;13:29–35. doi: 10.1016/0165-6147(92)90013-v. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Valenzuela E, Gorczyca W, Darzynkiewicz Z, Sharma SC. Apoptosis in adult retinal ganglion cells after axotomy. J Neurobiol. 1994;25:431–438. doi: 10.1002/neu.480250408. [DOI] [PubMed] [Google Scholar]
- 11.Hauben E, Butovsky O, Nevo U, Yoles E, Moalem G, Agranov E, Mor F, Leibowitz-Amit R, Pevsner E, Akselrod S, Neeman M, Cohen IR, Schwartz M. Passive or active immunization with myelin basic protein promotes recovery from spinal cord contusion. J Neurosci. 2000a;20:6421–6430. doi: 10.1523/JNEUROSCI.20-17-06421.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hauben E, Nevo U, Yoles E, Moalem G, Agranov E, Mor F, Akselrod S, Neeman M, Cohen IR, Schwartz M. Autoimmune T cells as potential neuroprotective therapy for spinal cord injury. Lancet. 2000b;354:286–287. doi: 10.1016/s0140-6736(99)05140-5. [DOI] [PubMed] [Google Scholar]
- 13.Hirschberg DL, Schwartz M. Macrophage recruitment to acutely injured central nervous system is inhibited by a resident factor: a basis for an immune-brain barrier. J Neuroimmunol. 1995;61:89–96. doi: 10.1016/0165-5728(95)00087-i. [DOI] [PubMed] [Google Scholar]
- 14.Hirschberg DL, Moalem G, He J, Mor F, Cohen IR, Schwartz M. Accumulation of passively transferred primed T cells independently of their antigen specificity following central nervous system trauma. J Neuroimmunol. 1998;89:88–96. doi: 10.1016/s0165-5728(98)00118-0. [DOI] [PubMed] [Google Scholar]
- 15.Huang DW, McKerracher L, Braun PE, David S. A therapeutic vaccine approach to stimulate axon regeneration in the adult mammalian spinal cord. Neuron. 1999;24:639–647. doi: 10.1016/s0896-6273(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 16.Jonas JB, Budde WM. Diagnosis and pathogenesis of glaucomatous optic neuropathy: morphological aspects. Prog Retin Eye Res. 2000;19:1–40. doi: 10.1016/s1350-9462(99)00002-6. [DOI] [PubMed] [Google Scholar]
- 17.Kerlero de Rosbo N, Mendel I, Ben-Nun A. Chronic relapsing experimental autoimmune encephalomyelitis with a delayed onset and an atypical clinical course, induced in PL/J mice by myelin oligodendrocyte glycoprotein (MOG)-derived peptide: preliminary analysis of MOG T cell epitopes. Eur J Immunol. 1995;25:985–993. doi: 10.1002/eji.1830250419. [DOI] [PubMed] [Google Scholar]
- 18.Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Misgeld T, Klinkert WE, Kolbeck R, Hoppe E, Oropeza-Wekerle RL, Bartke I, Stadelmann C, Lassmann H, Wekerle H, Hohlfeld R. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med. 1999;189:865–870. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Konno H, Yamamoto T, Suzuki H, Yamamoto H, Iwasaki Y, Ohara Y, Terunuma H, Harata N. Targeting of adoptively transferred experimental allergic encephalitis lesion at the sites of wallerian degeneration. Acta Neuropathol (Berl) 1990;80:521–526. doi: 10.1007/BF00294613. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Schlamp CL, Nickells RW. Experimental induction of retinal ganglion cell death in adult mice. Invest Ophthalmol Vis Sci. 1999;40:1004–1008. [PubMed] [Google Scholar]
- 21.Linington C, Berger T, Perry L, Weerth S, Hinze-Selch D, Zhang Y, Lu HC, Lassmann H, Wekerle H. T cells specific for the myelin oligodendrocyte glycoprotein mediate an unusual autoimmune inflammatory response in the central nervous system. Eur J Immunol. 1993;23:1364–1372. doi: 10.1002/eji.1830230627. [DOI] [PubMed] [Google Scholar]
- 22.Liu D, Yang R, Yan X, McAdoo DJ. Hydroxyl radicals generated in vivo kill neurons in the rat spinal cord: electrophysiological, histological, and neurochemical results. J Neurochem. 1994;62:37–44. doi: 10.1046/j.1471-4159.1994.62010037.x. [DOI] [PubMed] [Google Scholar]
- 23.McIntosh TK. Novel pharmacologic therapies in the treatment of experimental traumatic brain injury: a review. J Neurotrauma. 1993;10:215–261. doi: 10.1089/neu.1993.10.215. [DOI] [PubMed] [Google Scholar]
- 24.Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. 1995;25:1951–1959. doi: 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- 25.Moalem G, Monsonego A, Shani Y, Cohen IR, Schwartz M. Differential T cell response in central and peripheral nerve injury: connection with immune privilege. FASEB J. 1999a;13:1207–1217. doi: 10.1096/fasebj.13.10.1207. [DOI] [PubMed] [Google Scholar]
- 26.Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med. 1999b;5:49–55. doi: 10.1038/4734. [DOI] [PubMed] [Google Scholar]
- 27.Moalem G, Gdalyahu A, Leibowitz-Amit R, Yoles E, Muller-Gilor S, Shani Y, Mor F, Otten U, Cohen IR, Schwartz M. Production of neurotrophins by activated T cells: implications for neuroprotective autoimmunity. J Autoimmun. 2000;15:331–345. doi: 10.1006/jaut.2000.0441. [DOI] [PubMed] [Google Scholar]
- 28.Nicholson LB, Greer JM, Sobel RA, Lees MB, Kuchroo VK. An altered peptide ligand mediates immune deviation and prevents autoimmune encephalomyelitis. Immunity. 1995;3:397–405. doi: 10.1016/1074-7613(95)90169-8. [DOI] [PubMed] [Google Scholar]
- 29.Popovich PG, Stokes BT, Whitacre CC. Concept of autoimmunity following spinal cord injury: possible roles for T lymphocytes in the traumatized central nervous system. J Neurosci Res. 1996;45:349–363. doi: 10.1002/(SICI)1097-4547(19960815)45:4<349::AID-JNR4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 30.Raivich G, Jones LL, Kloss CUA, Werner A, Neumann H, Kreutzberg GW. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J Neurosci. 1998;18:5804–5816. doi: 10.1523/JNEUROSCI.18-15-05804.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santambrogio L, Benedetti M, Chao MV, Muzaffar R, Kulig K, Gabellini N, Hochwald G. Nerve growth factor production by lymphocytes. J Immunol. 1994;153:4488–4495. [PubMed] [Google Scholar]
- 32.Sawai H, Clarke DB, Kittlerova P, Bray GM, Aguayo AJ. Brain-derived neurotrophic factor and neurotrophin-4/5 stimulate growth of axonal branches from regenerating retinal ganglion cells. J Neurosci. 1996;16:3887–3894. doi: 10.1523/JNEUROSCI.16-12-03887.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnell L, Schneider R, Berman MA, Perry VH, Schwab ME. Lymphocyte recruitment following spinal cord injury in mice is altered by prior viral exposure. Eur J Neurosci. 1997;9:1000–1007. doi: 10.1111/j.1460-9568.1997.tb01450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwartz M, Belkin M, Yoles E, Solomon A. Potential treatment modalities for gaucomatous neuropathy: neuroprotection and neurodegeneration. J Glaucoma. 1996;5:427–432. [PubMed] [Google Scholar]
- 35.Schwartz M, Moalem G, Leibowitz-Amit R, Cohen IR. Innate and adaptive immune responses can be beneficial for CNS repair. Trends Neurosci. 1999;22:295–299. doi: 10.1016/s0166-2236(99)01405-8. [DOI] [PubMed] [Google Scholar]
- 36.Sriram S, Topham DJ, Huang SK, Rodriguez M. Treatment of encephalomyocarditis virus-induced central nervous system demyelination with monoclonal anti T cell antibodies. J Virol. 1989;63:4242–4248. doi: 10.1128/jvi.63.10.4242-4248.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Villegas-Perez MP, Vidal-Sanz M, Bray GM, Aguayo AJ. Influences of peripheral nerve grafts on the survival and regrowth of axotomized retinal ganglion cells in adult rats. J Neurosci. 1988;8:265–280. doi: 10.1523/JNEUROSCI.08-01-00265.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villegas-Perez MP, Vidal-Sanz M, Rasminsky M, Bray GM, Aguayo AJ. Rapid and protracted phases of retinal ganglion cell loss follow axotomy in the optic nerve of adult rats. J Neurobiol. 1993;24:23–36. doi: 10.1002/neu.480240103. [DOI] [PubMed] [Google Scholar]
- 39.Wucherpfennig KW. Autoimmunity in the central nervous system: mechanisms of antigen presentation and recognition. Clin Immunol Immunopathol. 1994;72:293–306. doi: 10.1006/clin.1994.1145. [DOI] [PubMed] [Google Scholar]
- 40.Yoles E, Schwartz M. Potential neuroprotective therapy for glaucomatous optic neuropathy. Surv Ophthalmol. 1998;42:367–372. doi: 10.1016/s0039-6257(97)00123-9. [DOI] [PubMed] [Google Scholar]
- 41.Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]