

Abstract
Excessive light can cause retinal degeneration and may be an environmental cofactor accelerating retinal dystrophies and age-related diseases. In rodent models, the light damage susceptibility (LDS) of the retina is determined genetically. In two mouse strains, with different degrees of LDS, a Leu450Met variation in the pigment epithelial protein RPE65 was shown recently to cosegregate with low LDS. Because light damage is rhodopsin-mediated, and RPE65 is essential for the regeneration of rhodopsin in the visual cycle, we analyzed this variation regarding rhodopsin metabolism and LDS in four mouse strains. We found that, in contrast to previous assertions, LDS does not correlate with the maximal retinal content of rhodopsin present after dark adaptation. Instead, LDS correlated positively with the kinetics of rhodopsin regeneration, which determine rhodopsin availability during light exposure. Light damage occurred after absorption of a threshold dose of photons and thus fast regeneration, as observed in those two strains having Leu at position 450 of RPE65, was correlated with the occurrence of photoreceptor apoptosis after short exposure. In contrast, mice with the Leu450Met variation of Rpe65regenerated rhodopsin with slow kinetics and showed an increased resistance to light-induced retinal degeneration. In these mice, RPE65 protein levels were reduced by a post-transcriptional mechanism. F1 hybrid mice, carrying one normal and one variantRpe65 gene, had intermediate levels of the corresponding protein and showed intermediate rhodopsin regeneration kinetics and an intermediate LDS. Thus, none of the two variants of Rpe65had a dominant effect.
Keywords: retinal degeneration, light damage, photoreceptor, pigment epithelium, rhodopsin, RPE65
Retinal degeneration induced by exposure to excessive doses of light is a model to study photoreceptor apoptosis, the common final pathway of cell loss in human age-related macular degeneration and some forms of inherited retinal dystrophies (Remé et al., 1998a). In patients with these retinal diseases, permanent exposure to high levels of light may also be an environmental factor accelerating loss of vision (Taylor et al., 1990; Cruickshanks et al., 1993, Simons, 1993; Cideciyan et al., 1998; Mata et al., 2000). This view is supported by the observation that several animal models for human retinal degeneration show a higher light damage susceptibility (LDS) than do controls (Sanyal and Hawkins, 1986; Wang et al., 1997; C. K. Chen et al., 1999; J. Chen et al., 1999; LaVail et al., 1999; Organisciak et al., 1999).
Light damage to photoreceptors is triggered by the excess absorption of photons by the visual pigment rhodopsin (Grimm et al., 2000). Once bleached, rhodopsin is regenerated metabolically in a multistep process called the visual cycle, involving photoreceptors and the adjacent retinal pigment epithelium (RPE) (Saari, 2000). One essential protein for this cycle is RPE65, which is specifically expressed in the RPE (Redmond et al., 1998). In mice, the sensitivity for retinal damage induced by light depends on the genetic background (LaVail et al., 1987). The hereditary factors determining LDS, however, are largely unknown. Recently, a sequence variation in Rpe65, causing a Leu450Met amino acid substitution, was shown to cosegregate with an increased resistance against acute light damage and reduced photoreceptor loss with age in C57BL/6J mice (Danciger et al., 2000). Furthermore, mice lacking RPE65 are completely resistant to light damage (Grimm et al., 2000). How the genetic variation in Rpe65 confers a low LDS and a reduced age-related dropout of visual cells is unknown at present.
Here, we analyzed four strains of mice for the Rpe65Leu450Met variation and correlated its presence or absence with the amount of RPE65 protein, the kinetics of rhodopsin regeneration, and finally the LDS of these strains. Based on these correlations, we delineate mechanistically how the genetic variation phenotypically alters the resistance of photoreceptors against light-induced apoptosis and suggest how age-related loss of photoreceptors may be modulated genetically.
MATERIALS AND METHODS
Mice. All procedures concerning animals were in accord with the regulations of the Veterinary Authority of Zurich and with the statement of The Association for Research in Vision and Ophthalmology for the use of animals in research. We obtained 21-d-old mice from the following suppliers: BALB/c (Wiga, Sulzfeld, Germany), C57BL/6J (RCC, Itigen, Switzerland), and 129/Ola (Dr. I. Tobler, Institute of Pharmacology, University of Zurich, Switzerland). Pigmented B6;129S(N2) and albinotic littermates (B6;129S(N2)-a) were offspring from mice purchased from The Jackson Laboratory (Bar Harbor, ME). B6;129S(N2)-a mice were seldom born and were used only for morphological studies. C57BL/6 × BALB/c F1(B6CF1) mice were obtained from Biological Research Laboratories (Basel, Switzerland). All animals were reared in 12 hr (6:00 A.M.–6:00 P.M.) light/dark cycles with 60–100 lux within the cages. Experiments were performed at the age of 5–12 weeks.
Induction and analysis of light damage. Light damage was induced in dark-adapted mice with dilated pupils by exposure to 5 klux of diffuse white fluorescent light for 10, 20, 30, 40, and 50 min and 1, 2, 4, 6, 8, and 10 hr (lights on at 10:00 A.M.) (Wenzel et al., 2000). Animals were then kept in darkness for 24 hr, their retinas were examined histologically, and the minimal time required to induce photoreceptor apoptosis was determined.
Rhodopsin content and regeneration. The rhodopsin content was determined as described recently (Kueng-Hitz et al., 2000). R0 was measured after 16 hr of dark adaptation. The existence of a negative correlation between light damage susceptibility and R0 was tested in the four strains homozygous for the individual Rpe65 variant using an ordered logit model (Powers and Xie, 2000). The continuous variable was R0 and the ordinal response variables were the time intervals to induce light damage. The analysis was performed using “R” software version 1.1 (http://www. r-project.org) on Windows NT 4 (Ihaka and Gentleman, 1996). P values of <0.05 were considered significant.
To assess the rhodopsin regeneration kinetics, dark-adapted mice, with dilated pupils, were exposed to white light (10 min, 5 klux). Rhodopsin was measured at various times in darkness (Table1). Its regeneration rate constants were calculated. These constants, multiplied by the rhodopsin at steady state, give the number of photons absorbed per minute during exposure. The existence of a steady state was verified in B6;129S(N2) mice by showing that rhodopsin levels during 10–60 min of light exposure did not change (data not shown).
Table 1.
Rhodopsin regeneration after bleaching
| BALB/c | 129/Ola | B6;129S(N2) | C57BL/6J | B6CF1 | |
|---|---|---|---|---|---|
| R01-a | 0.43 ± 0.04, 31-b | 0.45 ± 0.04, 4 | 0.48 ± 0.08, 10 | 0.52 ± 0.04, 2 | 0.53 ± 0.05, 7 |
| 0 min after bleaching | 0.04 ± 0.00, 2 | 0.03 ± 0.01, 3 | 0.04 ± 0.01, 4 | 0.03 ± 0.01, 2 | 0.06 ± 0.02, 3 |
| 5 min after bleaching | 0.11 ± 0.02, 2 | 0.08 ± 0.01, 2 | 0.05, 1 | 0.08 ± 0.03, 2 | 0.07 ± 0.02, 3 |
| 15 min after bleaching | 0.22 ± 0.07, 2 | 0.18 ± 0.02, 3 | 0.07 ± 0.04, 3 | 0.12 ± 0.00, 2 | 0.13 ± 0.03, 3 |
| 30 min after bleaching | 0.32 ± 0.01, 2 | 0.22 ± 0.03, 2 | 0.13, 1 | 0.11 ± 0.01, 2 | 0.22 ± 0.01, 3 |
| 45 min after bleaching | 0.35, 1 | 0.34 ± 0.05, 3 | 0.19 ± 0.02, 2 | 0.20 ± 0.01, 2 | 0.29 ± 0.01, 4 |
| 60 min after bleaching | 0.42 ± 0.05, 2 | 0.48 ± 0.00, 2 | 0.26 ± 0.09, 7 | 0.18 ± 0.01, 2 | 0.34 ± 0.05, 3 |
| 120 min after bleaching | 0.44 ± 0.05, 2 | 0.52 ± 0.05, 2 | 0.31 ± 0.07, 5 | 0.36 ± 0.06, 2 | 0.49 ± 0.04, 5 |
| 180 min after bleaching | n.d.1-c | n.d. | 0.47 ± 0.12, 2 | 0.43 ± 0.02, 2 | n.d. |
| 240 min after bleaching | n.d. | n.d. | 0.47 ± 0.06, 6 | 0.51 ± 0.01, 2 | n.d. |
All values for rhodopsin are in nanomoles per retina.
Rhodopsin content per retina after 16 hr of dark adaptation.
Numbers in italics represent numbers of mice.
n.d., not determined.
Western blotting. Eyecups (including the retina) from dark-adapted mice were homogenized in 100 mm Tris/HCl, pH 7.4, and analyzed for protein content. Standard SDS-PAGE (7.5 or 10%) and Western blotting were performed. For immunodetection, polyclonal rabbit antisera directed against RPE65 (raised against amino acids 150–164 of human/bovine RPE65) (Redmond and Hamel, 2000), interphotoreceptor retinoid binding protein (IRBP) (Smith et al., 1997), cellular retinaldehyde-binding protein (CRALBP) (Crabb et al., 1991), transducin (Hamm et al., 1987), rhodopsin-kinase (Zhao et al., 1998), arrestin (Kueng-Hitz et al., 2000), and actin (Lessard, 1988) were applied. HRP-conjugated secondary antibodies were applied (catalog #sc 2004 and 2031; Santa Cruz Biotechnology, Santa Cruz, CA), and immunoreactivity was visualized using the Renaissance–Western blot detection kit (PerkinElmer Life Sciences, Emeryville, CA).
RT-PCR and Rpe65 sequence analysis. Total RNA was prepared from eyecups using the RNeasy kit (Qiagen, Hilden, Germany). Remaining genomic DNA was digested with deoxyribonuclease RQ (Promega, Madison, WI), and 600 ng of total RNA was reverse-transcribed with oligo(dT) and M-MLV (Promega). A 367-bp fragment of the Rpe65 cDNA was amplified using exon 4 forward primer and exon 6 reverse primer (Redmond et al., 1998). The amplified fragment was cut with MboI and the 222-bp and the 20-bp fragments were ligated (T4 DNA ligase; Roche, Basel, Switzerland). The resulting 242-bp product was amplified using the above primers, gel-purified, and used as mimic DNA. Levels of β-actin mRNA in 10 ng of total RNA were determined by exponential RT-PCR (primers: 5′-CAA CGG CTC CGG CAT GTGC-3′; 5′-CTC TTG CTC TGG GCC TCG-3′; annealing: 62°C; cycles: 24) and amounts of cDNA for Rpe65 mimic PCR were adjusted accordingly. Semiquantitative determination ofRpe65 mRNA levels was done by mimic PCR using the above primers with end-labeled 32P reverse primer; cDNA representing ∼20 ng of total RNA and increasing amounts of mimic DNA. For amplification (30 cycles), the annealing temperature was 56°C. Products were separated by native PAGE (6%), stained with ethidium bromide, and quantified on a PhosphorImager (Molecular Dynamics, Amersham Pharmacia Biotech Europe, Freiburg, Germany).
To determine the sequence of Rpe65 by PCR, tail DNA was isolated and amplified using the following primer pair: up, 5′-CTG ACA AGC TCT GTA AG-3′; down, 5′-CAT TAC CAT CAT CTT CTT CCA-3′. Amplification was done during 35 cycles with a 30 sec denaturing step at 94°C, a 1 min annealing step at 52°C, and a 1 min extension step at 72°C. The amplified PCR product from C57BL/6 mice had a length of 126-bp and contained one AluI site. Restriction digestion with AluI resulted in two fragments of 118 and 8 bp, respectively. The BALB/c sequence contains one additionalAluI site resulting in three products of 109, 9, and 8 bp, after digestion. Undigested and digested PCR products were analyzed on an 8% polyacrylamide gel.
In some cases, results from genotyping were verified as follows: total RNA was reverse-transcribed, and Rpe65 mRNA was amplified using the following primer pair: up, 5′-TTA CGT GAG AAT TGG GAA GAA GTT-3′; down, 5′-AGA AGG GCA GTC AGT GTG GTA CTG-3′ (Danciger et al., 2000). PCR products were purified (QIAquick; Qiagen) and sequenced (Microsynth, Balgach, Switzerland).
RESULTS
Light exposure duration required to induce light damage depends on the genetic background
To judge LDS, the minimal time of light exposure that induced condensation of nuclear chromatin and of rod inner segments in the majority of photoreceptors of the lower central retina was determined. In BALB/c and 129/Ola mice, these distinct morphological signs of apoptosis were observed after 20 min of light exposure (Fig.1). After 10 min of exposure, such damage was absent in 129/Ola mice and rare in BALB/c mice, indicating that the threshold for apoptosis was reached between 10 and 20 min of light exposure in both strains (apparently closer to 10 min in BALB/c mice). The slightly increased LDS of BALB/c mice compared with 129/Ola mice may have resulted from slightly elevated RPE65 levels (Fig.2A), minimally faster rhodopsin regeneration (Table2), and/or the expression of slightly more transducin (Fig. 2B) protein, which may increase signal gain (Fung et al., 1981) during exposure. In B6;129S(N2) mice, the threshold for apoptosis was passed between 40 and 50 min of light exposure (Fig. 1); in C57BL/6J mice, no apoptosis was detected after 6 h of exposure. In the latter mice, apoptosis was restricted to a few scattered nuclei (Fig. 1) even after 8–10 hr of exposure. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling-positive nuclei and internucleosomal DNA fragmentation 24 hr after light exposure confirmed the apoptotic nature of cell death in photoreceptors (data not shown). Thus, different degrees of LDS were found: BALB/c ≥ 129/Ola ≫ B6;129S(N2) >≫ C57BL/6J.
Fig. 1.
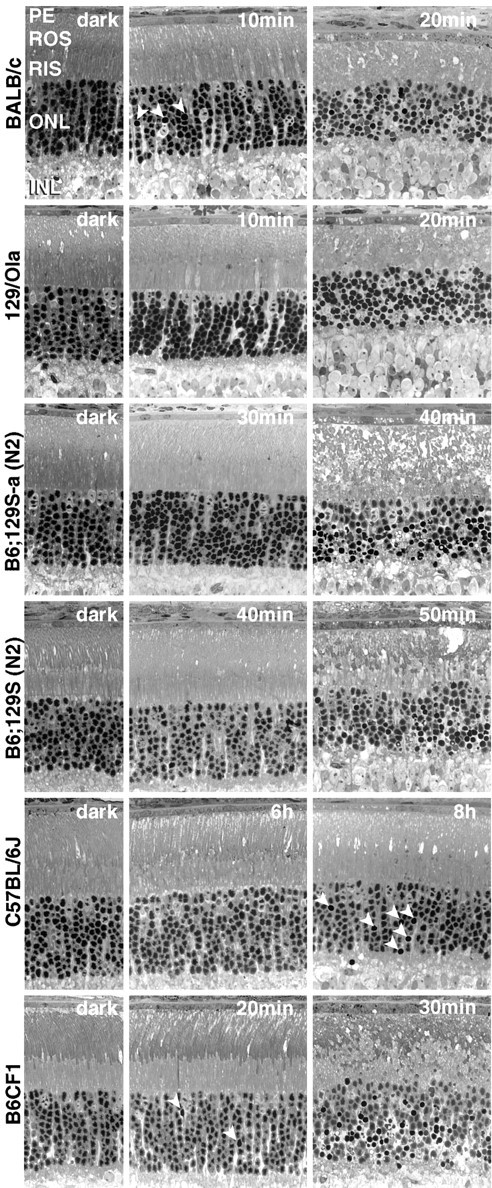
Light damage susceptibilities in different mouse strains. Light micrographs of control mice (dark, left column) or mice exposed to light (5 klux) for the times indicated. Different exposure times were required to induce photoreceptor apoptosis (right column). Transition from reversible damage (middle column), characterized by vesiculation of ROS, to irreversible (apoptotic) damage occurred in the interval between the times indicated. Arrowheads indicate initial apoptotic lesions involving only few photoreceptor nuclei in the outer nuclear layer (ONL). INL, Inner nuclear layer;PE, pigment epithelium. Four to six eyes were examined for each condition 24 hr after light exposure.
Fig. 2.
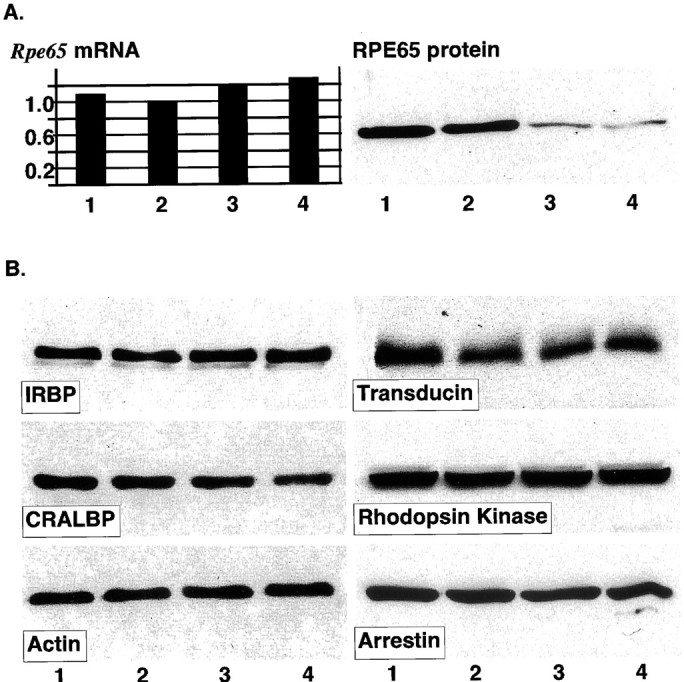
A, Post-transcriptional control of RPE65 protein levels. Comparison of Rpe65 mRNA levels (arbitrary units) revealed only minor differences between the four mouse strains. In contrast, significantly more RPE65 protein was found in eyecups of BALB/c (lane 1) and 129/Ola (lane 2) mice as compared with B6;129S(N2) (lane 3) and C57BL/6J (lane 4) mice, suggesting a post-transcriptional control of RPE65 protein levels (25 μg of protein was loaded). These results are representative of three independent experiments. B, Proteins of visual cycle and signal transduction. Eyecup homogenates of BALB/c (lane 1), 129/Ola (lane 2), B6;129S(N2) (lane 3), and C57BL/6J mice (lane 4) were analyzed for immunoreactivity of proteins of the visual cycle: IRBP, CRALBP, or the visual transduction cascade (transducin, rhodopsin kinase, arrestin). Actin immunoreactivity served as control for equal load. Protein load was 12.5 μg for IRBP, arrestin, actin, and CRALBP; 25 μg for transducin; and 50 μg for rhodopsin kinase. These results are representative of three independent experiments.
Table 2.
RPE65, rhodopsin regeneration, photon catch, and light damage
| BALB/c | 129/Ola | B6;129S(N2) | C57BL/6J | B6CF1 | |
|---|---|---|---|---|---|
| Rpe65 sequence at codon 450 | CTG = Leu | CTG = Leu | ATG = Met | ATG = Met | CTG + ATG |
| Rpe65mRNA2-a | 1.1 | 1.0 | 1.2 | 1.3 | n.d. |
| RPE65 protein2-b | ++++ | ++++ | + | + | ++(+) |
| R0(nmol/retina) | 0.43 | 0.45 | 0.48 | 0.52 | 0.53 |
| Regeneration rate constant2-c | 0.036/min | 0.028/min | 0.008/min | 0.009/min | 0.017/min |
| Photons/retina/min2-c | 8.4 × 1012 | 7.1 × 1012 | 2.2 × 1012 | 2.5 × 1012 | 4.8 × 1012 |
| Damage occurs after | 10–20 min | 20 min | 50 min | 8 hr | 30 min |
| Photons absorbed until damage | 8–17 × 1013 | 14 × 1013 | 11 × 1013 | 120 × 1013 | 14 × 1013 |
B6CF1 (C57BL/6 × BALB/c F1) mice showed distinct signs of photoreceptor apoptosis after 30 min of light exposure. Twenty minutes of exposure induced apoptosis in very few scattered photoreceptors of the most affected area (Fig. 1). Thus, the threshold for distinct light damage was passed close to 30 min.
Albinotic B6;129S(N2) mice, showing light damage after exposure for 30–40 min, were only slightly more vulnerable to light damage than pigmented littermates and distinctly less vulnerable than albinotic BALB/c and 129/Ola mice.
Correlation of rhodopsin regeneration kinetics with LDS
Light-induced apoptosis of rods is triggered by the excessive absorption of photons by rhodopsin (Grimm et al., 2000). Use of different mouse strains allowed us to separately analyze the effect of the maximal rhodopsin content after dark adaptation (R0) and the rate of rhodopsin regeneration on LDS. The highest R0 values were found in the most resistant strain (C57BL/6J) followed by B6;129S(N2), 129/Ola, and BALB/c (Table1). This ranking reversed the order found for LDS (see above). Thus, in mice homozygous for one or the other variation of Rpe65, R0 was negatively correlated with LDS (p < 0.041). Compared with these strains, B6CF1 hybrid mice, carrying one copy of each of the variants, had higher R0 values. In this particular case, the negative correlation was invalid, because these animals were more sensitive than C57BL/6J or B6;129S(N2) mice, which showed lower R0 values.
Because rhodopsin regeneration during light exposure provides the chromophore to absorb light, fast rhodopsin regeneration might be predictive of a high LDS. Indeed, the rhodopsin regeneration rate constants correlated directly with the LDS rank for all mice, including B6CF1 mice (Table 2).
Leu450Met variation in Rpe65 is associated with reduced protein levels and slowed rhodopsin regeneration
Analysis of the Rpe65 sequences showed that not only BALB/c (Danciger et al., 2000), but also 129/Ola mice were homozygous for the normal Rpe65 sequence in exon 13 at codon 450 (CTG = Leu). In contrast, B6;129S(N2) and C57BL/6J mice were homozygous for the variant Rpe65 gene (ATG = Met). Although this variation apparently did not affect the expression ofRpe65 mRNA (Fig.2A), it reduced the steady-state levels of the RPE65 protein in B6;129S(N2) and C57BL/6J mice (Fig. 2A), suggesting a post-transcriptional regulation of RPE65 in vivo. With regard to their RPE65 levels, the strains were ranked as follows: BALB/c ≥ 129/Ola >≫ B6;129S(N2) = C57BL/6J (judged by visual impression; summarized in Table 2). The different regeneration rates for rhodopsin (Table 2) appear to reflect these differences in the amount of RPE65; other ancillary proteins of the visual cycle, such as CRALBP and IRBP, did not show major differences among the four strains (Fig.2B). Furthermore, no differences in the amount of arrestin, which might affect the kinetics of the visual cycle (Palczewski et al., 1999), were found (Fig.2B).
Mice heterozygous for the L450M variation in Rpe65 are intermediates regarding amount of RPE65, rhodopsin regeneration kinetics, and LDS
B6CF1 mice, carrying one normal copy and one variant copy of Rpe65, expressed intermediate amounts of RPE65 (Fig.3) when compared with pure BALB/c and C57BL/6 mice. Likewise, the rate of rhodopsin regeneration in those F1 mice (0.017/min) was slower than in BALB/c mice (0.036/min) but faster than in C57BL/6 mice (0.009/min). Consequently, the photon catch capacity of F1 mice was intermediate compared with that of animals from their parental strains (Table 2), and their LDS was decreased compared with BALB/c mice but increased compared with C57BL/6J mice. Thus, phenotypically, none of the two copies of Rpe65 displayed a dominant effect.
Fig. 3.

RPE65 immunoreactivity in eyecup homogenates from age-matched pure BALB/c, B6CF1, and pure C57BL/6 mice. F1 mice, heterozygous for normal and variantRpe65, show intermediate levels of the RPE65 protein, suggesting that a single copy of normal Rpe65 is insufficient to restore normal levels of the protein. A total of 25 μg of protein was loaded. Results are representative of three independent experiments.
Light damage occurs after absorption of threshold amount of photons
From the number of photons absorbed per retina per minute in different strains (Table 2), we calculated the number of photons absorbed during the minimal time of exposure needed to induce light damage: BALB/c = 16.8 × 1013, 129/Ola = 14.2 × 1013, B6;129S(N2) = 11 × 1013, B6CF1 = 14.4 × 1013, and C57BL/6J = 120 × 1013.
The number calculated for BALB/c mice may be overestimated because it is based on a minimal time of 20 min. However, slight damage was already observed after 10 min of exposure, during which 8.4 × 1013 photons would have been absorbed (see above). Thus, the threshold dose of photons to be absorbed to induce irreversible retinal light damage was 8–14 × 1013 photons, except for C57BL/6J mice (see Discussion).
DISCUSSION
Genetic factors reducing LDS in C57BL/6J mice have been postulated already in 1987 (LaVail et al., 1987), but the underlying molecular mechanisms have not yet been identified. Recently, by comparing BALB/c with C57BL/6J mice, a sequence variation in theRpe65 gene was shown to cosegregate with low LDS in C57BL/6J mice (Danciger et al., 2000). Here we show that thisRpe65 Leu450Met variation leads to reduced levels of the corresponding protein, a decreased capacity of the pigment epithelium to regenerate rhodopsin, a decreased number of photons (pro-apoptotic stimulus) that can be absorbed by a retina per time, and finally a decreased LDS. The presence of one normal and one variant allele ofRpe65 in B6CF1 heterozygous mice resulted in intermediate levels of RPE65, an intermediate rate of rhodopsin regeneration, and an intermediated LDS, suggesting that neither copy had a dominant effect.
Dark-adapted levels of rhodopsin do not determine LDS
Light-induced apoptosis of rods is mediated by rhodopsin (Grimm et al., 2000), and it was previously asserted that R0 positively correlates with LDS (Noell and Albrecht, 1971; Noell et al., 1966;Organisciak et al., 1987; Remé et al., 1998b). However, BALB/c mice, showing the highest LDS, had the lowest R0 but regenerated rhodopsin with the fastest kinetics. Thus, the critical determinant for LDS, at least in mice homozygous for their respective Rpe65 variant, seems to be the regeneration rate and thus photon absorption over time. Pigmentation, at least when pupils were dilated, had very little influence on LDS, because albinotic B6;129S(N2) mice were only slightly more vulnerable to light damage than pigmented littermates and distinctly less vulnerable than albinotic BALB/c and 129/Ola mice.
Rpe65 genotype determines LDS phenotype
RPE65, expressed in the pigment epithelium, is essential for the isomerization of all-trans into 11-cis-retinoids, required for the generation/regeneration of rhodopsin in the visual cycle. The lack of RPE65 prevents rhodopsin generation/regeneration and consequently leads to the absence of the visual pigment from photoreceptors (Redmond et al., 1998). Here, the two strains with slow rhodopsin regeneration kinetics and low LDS (B6;129S(N2) and C57BL/6J) carried theRpe65 Leu450Met variation, whereas both strains with fast kinetics and high LDS had the normal sequence found in most species, including humans (Danciger et al., 2000). In strains with variant Rpe65 gene, only low protein levels of RPE65 were detectable, whereas high amounts of the protein were found in BALB/c and 129/Ola mice with the normal sequence. B6CF1mice that were heterozygous for normal and variant RPE65 showed intermediate levels of the protein and an intermediate LDS. Thus, presence of one normal allele was insufficient to restore normal levels of RPE65; this finding is in line with the observation of 50% reduced RPE65 levels in Rpe65+/− mice (data not shown; Van Hooser et al., 2000). Despite different amounts of the protein, no differences were found in Rpe65mRNA levels among the four strains tested. This suggests that either the translation rate of the C57BL/6J variant of Rpe65 was decreased and/or that the turnover of the variant protein was accelerated due to its different amino acid sequence. A post-transcriptional regulation of Rpe65 was also observed in pigment epithelial cells in vitro (Hamel et al., 1993).
Two other ancillary proteins of the visual cycle, IRBP and CRALBP, which likewise may have an impact on rhodopsin regeneration (Smith et al., 1997; Ripps et al., 2000;Saari, 2000), were not differentially expressed in the four strains of mice. Thus, in line with findings fromRpe65+/− mice (50% reduced levels of RPE65) that show a slower regeneration rate thanRpe65+/+ mice (data not shown; Van Hooser et al., 2000), the amount of RPE65 protein appeared to be the critical determinant for the kinetics of rhodopsin regeneration.
Proteins involved in signal transduction [e.g., rhodopsin kinase or arrestin (Baylor and Burns, 1998), which terminate signaling of activated rhodopsin, or transducin, the G-protein activated by rhodopsin (Fung et al., 1981)], may influence LDS (C. K. Chen et al., 1999; J. Chen et al., 1999). However, no major differences were found in levels of rhodopsin kinase, arrestin, or transducin among the four strains of mice.
Thus, it appears very likely that the RPE65 amino acid substitution found in mice with a C57BL/6J genetic background is the primary genetic factor reducing LDS. Indeed, it accounts for ∼47% of the light damage protective effect in these mice (Danciger et al., 2000).
Additional factors in C57BL/6J mice
Our findings point to factors apart from L450M in RPE65 that increase resistance against light damage in pure C57BL/6J mice. Despite being indistinguishable from B6;129S(N2) mice in terms of RPE65 variant, ancillary protein levels, and rhodopsin regeneration (see above), it took >10 times more photons (120 × 1013) to induce light damage in C57BL/6J mice compared with B6;129S(N2) mice (11 × 1013photons). Likewise, the genetic study by Danciger et al. (2000) suggests several (unidentified) genetic loci apart fromRpe65 conferring low LDS in C57BL/6J mice. Biochemically, several candidates such as glutathione-dependent enzymes, fatty acid composition, or cytoplasmic amino acids were excluded in previous work (Naash et al., 1989). In contrast to the L450M variation, these unknown genetic factors may exert their protective effect only at homozygocity. In B6CF1 mice, the threshold dose of photons required to induce light damage was similar to that observed in all mice but C57BL/6J mice. Thus, although one copy each of these putative protective genes of C57BL/6J was present in the F1 animals, their LDS appeared to be solely determined by their RPE65 status.
Relevance for age-related loss of photoreceptors?
Is kinetics of rhodopsin regeneration a factor relevant for age-related human retinal diseases that are suspect to be accelerated by light (Taylor et al., 1990; Cruickshanks et al., 1993; Simons, 1993; Cideciyan et al., 1998; Mata et al., 2000)? This may indeed be the case: mice carrying the normal RPE65 protein (such as BALB/c mice), and thus having fast kinetics of rhodopsin regeneration, show significant loss of photoreceptors with age, whereas mice carrying the variant Rpe65 sequence (such as C57BL/6J mice) do not (Danciger et al., 2000). Interestingly, preliminary data from a clinical study on patients with a slowed dark adaptation (rhodopsin regeneration) indicate that these patients show a lower prevalence for neovascular age-related macular degeneration (F. Hafezi, C. Grimm, A. Wenzel, C. E. Remé, and A. Thölen, unpublished observations). Thus, by modulating the daily dose of photons absorbed by the retina, differences in rhodopsin regeneration kinetics might indeed modulate retinal susceptibility to the environmental risk-factor light.
Footnotes
This work was supported by the Swiss National Science Foundation (Zürich, Switzerland) and the E&B Grimmke Foundation (Düsseldorf, Germany) (C.G., F.H., C.R., and A.W.). T.P.W. was an Alexander von Humboldt Fellow. We thank G. Hoegger, C. Imsand, and D. Greuter for skilled technical assistance and Drs. B. N. Wiggert, H. E. Hamm, J. W. Crabb, K. Palczewski, T. M. Redmond, T. vanVeen, and U. Wolfrum for providing antisera. We also thank Fabio Valeri from Hypothesis Scientific Data Evaluation (Zürich, Switzerland) for the statistical analysis.
Correspondence should be addressed to A. Wenzel, University Hospital Zürich, Frauenklinikstrasse 24, CH-8091 Zurich, Switzerland. E-mail: awenzel@opht.unizh.ch.
REFERENCES
- 1.Baylor DA, Burns ME. Control of rhodopsin activity in vision. Eye. 1998;12:521–525. doi: 10.1038/eye.1998.140. [DOI] [PubMed] [Google Scholar]
- 2.Chen CK, Burns ME, Spencer M, Niemi GA, Chen J, Hurley JB, Baylor DA, Simon MI. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci USA. 1999;96:3718–3722. doi: 10.1073/pnas.96.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J, Simon MI, Matthes MT, Yasumura D, LaVail MM. Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness). Invest Ophthalmol Vis Sci. 1999;40:2978–2982. [PubMed] [Google Scholar]
- 4.Cideciyan AV, Hood DC, Huang Y, Banin E, Li ZY, Stone EM, Milam AH, Jacobson SG. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci USA. 1998;95:7103–7108. doi: 10.1073/pnas.95.12.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crabb JW, Gaur VP, Garwin GG, Marx SV, Chapline C, Johnson CM, Saari JC. Topological and epitope mapping of the cellular retinaldehyde-binding protein from retina. J Biol Chem. 1991;266:16674–16683. [PubMed] [Google Scholar]
- 6.Cruickshanks KJ, Klein R, Klein BE. Sunlight and age-related macular degeneration: the Beaver Dam Eye Study. Arch Ophthalmol. 1993;111:514–518. doi: 10.1001/archopht.1993.01090040106042. [DOI] [PubMed] [Google Scholar]
- 7.Danciger M, Matthes MT, Yasumura D, Akhmedov NB, Rickabaugh T, Gentleman S, Redmond TM, LaVail MM, Farber DB. A QTL on distal Chr 3 that influences the severity of light-induced damage to mouse photoreceptors. Mamm Genome. 2000;11:422–427. doi: 10.1007/s003350010081. [DOI] [PubMed] [Google Scholar]
- 8.Fung BK, Hurley JB, Stryer L. Flow of information in the light-triggered cyclic nucleotide cascade of vision. Proc Natl Acad Sci USA. 1981;78:152–156. doi: 10.1073/pnas.78.1.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grimm C, Wenzel A, Hafezi F, Yu S, Redmond TM, Reme CE. Protection of Rpe65-deficient mice identifies rhodopsin as a mediator of light-induced retinal degeneration. Nat Genet. 2000;25:63–66. doi: 10.1038/75614. [DOI] [PubMed] [Google Scholar]
- 10.Hamel CP, Tsilou E, Pfeffer BA, Hooks JJ, Detrick B, Redmond TM. Molecular cloning and expression of RPE65, a novel retinal pigment epithelium-specific microsomal protein that is post-transcriptionally regulated in vitro. J Biol Chem. 1993;268:15751–15757. [PubMed] [Google Scholar]
- 11.Hamm HE, Deretic D, Hofmann KP, Schleicher A, Kohl B. Mechanism of action of monoclonal antibodies that block the light activation of the guanyl nucleotide-binding protein, transducin. J Biol Chem. 1987;262:10831–10838. [PubMed] [Google Scholar]
- 12.Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Computational Graphical Statistics. 1996;5:299–314. [Google Scholar]
- 13.Kueng-Hitz N, Grimm C, Lansel N, Hafezi F, He L, Fox D, Remé CE, Niemeyer G, Wenzel A. The retina of c-fos+/+ and c-fos−/− mice: electrophysiological, morphological, and biochemical aspects. Invest Ophthalmol Vis Sci. 2000;41:909–916. [PubMed] [Google Scholar]
- 14.LaVail MM, Gorrin GM, Repaci MA, Thomas LA, Ginsberg HM. Genetic regulation of light damage to photoreceptors. Invest Ophthalmol Vis Sci. 1987;28:1043–1048. [PubMed] [Google Scholar]
- 15.LaVail MM, Gorrin GM, Yasumura D, Matthes MT. Increased susceptibility to constant light in nr and pcd mice with inherited retinal degeneration. Invest Ophthalmol Vis Sci. 1999;40:1020–1024. [PubMed] [Google Scholar]
- 16.Lessard JL. Two monoclonal antibodies to actin: one muscle selective and one generally reactive. Cell Motil Cytoskeleton. 1988;10:349–362. doi: 10.1002/cm.970100302. [DOI] [PubMed] [Google Scholar]
- 17.Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci USA. 2000;97:7154–7159. doi: 10.1073/pnas.130110497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naash MI, LaVail MM, Anderson RE. Factors affecting the susceptibility of the retina to light damage. Prog Clin Biol Res. 1989;314:513–522. [PubMed] [Google Scholar]
- 19.Noell WK, Albrecht A. Irreversible effects of visible light on the retina: role of vitamin A. Science. 1971;172:76–79. doi: 10.1126/science.172.3978.76. [DOI] [PubMed] [Google Scholar]
- 20.Noell WK, Walker VS, Kang BS, Berman S. Retinal damage by light in rats. Invest Ophthalmol. 1966;5:450–473. [PubMed] [Google Scholar]
- 21.Organisciak DT, Wang HM, Noell WK. Aspects of the ascorbate protective mechanism in retinal light damage of rats with normal and reduced ROS docosahexaenoic acid. Prog Clin Biol Res. 1987;247:455–468. [PubMed] [Google Scholar]
- 22.Organisciak DT, Li M, Darrow RM, Farber DB. Photoreceptor cell damage by light in young Royal College of Surgeons rats. Curr Eye Res. 1999;19:188–196. doi: 10.1076/ceyr.19.2.188.5333. [DOI] [PubMed] [Google Scholar]
- 23.Palczewski K, Van Hooser JP, Garwin GG, Chen J, Liou GI, Saari JC. Kinetics of visual pigment regeneration in excised mouse eyes and in mice with a targeted disruption of the gene encoding interphotoreceptor retinoid-binding protein or arrestin. Biochemistry. 1999;38:12012–12019. doi: 10.1021/bi990504d. [DOI] [PubMed] [Google Scholar]
- 24.Powers DA, Xie Y. Statistical methods for categorical data analysis. Academic; San Diego: 2000. [Google Scholar]
- 25.Redmond TM, Hamel CP. Genetic analysis of RPE65: from human disease to mouse model. Methods Enzymol. 2000;316:705–724. doi: 10.1016/s0076-6879(00)16758-8. [DOI] [PubMed] [Google Scholar]
- 26.Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, Goletz P, Ma JX, Crouch RK, Pfeifer K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet. 1998;20:344–351. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- 27.Remé CE, Grimm C, Hafezi F, Marti A, Wenzel A. Apoptotic cell death in retinal degenerations. Prog Retin Eye Res. 1998a;17:443–464. doi: 10.1016/s1350-9462(98)00009-3. [DOI] [PubMed] [Google Scholar]
- 28.Remé CE, Hafezi F, Munz K, Reinboth JJ. Light damage to the retina and pigment epithelium. In: Marmor MF, Wolfensberger TJ, editors. The pigment epithelium, current aspects of function and disease. Oxford UP; New York: 1998b. pp. 563–586. [Google Scholar]
- 29.Ripps H, Peachey NS, Xu X, Nozell SE, Smith SB, Liou GI. The rhodopsin cycle is preserved in IRBP “knockout” mice despite abnormalities in retinal structure and function. Vis Neurosci. 2000;17:97–105. doi: 10.1017/s095252380017110x. [DOI] [PubMed] [Google Scholar]
- 30.Saari JC. Biochemistry of visual pigment regeneration. Invest Ophthalmol Vis Sci. 2000;41:337–348. [PubMed] [Google Scholar]
- 31.Sanyal S, Hawkins RK. Development and degeneration of retina in rds mutant mice: effects of light on the rate of degeneration in albino and pigmented homozygous and heterozygous mutant and normal mice. Vision Res. 1986;26:1177–1785. doi: 10.1016/0042-6989(86)90099-4. [DOI] [PubMed] [Google Scholar]
- 32.Simons K. Artificial light and early-life exposure in age-related macular degeneration and in cataractogenic phototoxicity. Arch Ophthalmol. 1993;111:297–298. doi: 10.1001/archopht.1993.01090030015002. [DOI] [PubMed] [Google Scholar]
- 33.Smith SB, McClung J, Wiggert BN, Nir I. Delayed rhodopsin regeneration and altered distribution of interphotoreceptor retinoid binding protein (IRBP) in the mi(vit)/mi(vit) (vitiligo) mouse. J Neurocytol. 1997;26:605–613. doi: 10.1023/a:1018589609622. [DOI] [PubMed] [Google Scholar]
- 34.Taylor HR, Munoz B, West S, Bressler NM, Bressler SB, Rosenthal FS. Visible light and risk of age-related macular degeneration. Trans Am Ophthalmol Soc. 1990;88:163–173. [PMC free article] [PubMed] [Google Scholar]
- 35.Van Hooser JP, Aleman TS, He YG, Cideciyan AV, Kuksa V, Pittler SJ, Stone EM, Jacobson SG, Palczewski K. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc Natl Acad Sci USA. 2000;97:8623–8628. doi: 10.1073/pnas.150236297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang M, Lam TT, Tso MO, Naash MI. Expression of a mutant opsin gene increases the susceptibility of the retina to light damage. Vis Neurosci. 1997;14:55–62. doi: 10.1017/s0952523800008750. [DOI] [PubMed] [Google Scholar]
- 37.Wenzel A, Grimm C, Marti A, Kueng-Hitz N, Hafezi F, Niemeyer G, Remé CE. c-Fos controls the “private pathway” of light-induced apoptosis of retinal photoreceptors. J Neurosci. 2000;20:80–88. doi: 10.1523/JNEUROSCI.20-01-00081.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao X, Huang J, Khani SC, Palczewski K. Molecular forms of human rhodopsin kinase (GRK1). J Biol Chem 273 1998. 5124 5131 damage by light in young Royal College of Surgeons rats. Curr Eye Res 19:188–196. [DOI] [PubMed] [Google Scholar]