

Abstract
Changes in BDNF expression after different types of brain insults are related to neuroprotection, stimulation of sprouting, and synaptic reorganization. In the cerebral cortex, an autocrine–paracrine mechanism for BDNF has been proposed because the distribution patterns of BDNF and TrkB expression are almost identical. Moreover, cortical BDNF is anterogradely transported to the striatum, suggesting a role of BDNF in the functional interaction between the two brain regions. Here we have examined the expression of this neurotrophin in the cerebral cortex after various striatal lesions. Intrastriatal injection of quinolinate, kainate, 3-nitropropionic acid, or colchicine increased BDNF mRNA levels in cerebral cortex. In contrast, stimulation of neuronal activity in the striatum did not change cortical BDNF expression. Both excitatory amino acids increased BDNF expression in neurons of cortical layers II/III, V, and VI that project to the striatum. Moreover, grafting a BDNF-secreting cell line prevented both the loss of striatal neurons and the cortical upregulation of BDNF induced by excitotoxins. Because retrograde transport in the corticostriatal pathway was intact after striatal lesions, our results suggest that striatal damage upregulates endogenous BDNF in corticostriatal neurons by a transneuronal mechanism, which may constitute a protective mechanism for striatal and/or cortical cells.
Keywords: corticostriatal pathway, anterograde transport, excitatory amino acids, 3-nitropropionic acid, neurotrophins, synaptic activity, Huntington's disease
Neurotrophins have been implicated in the regulation of development, neuronal survival, and adult synaptic plasticity (Thoenen, 1995; Reichardt and Fariñas, 1997;McAllister et al., 1999). Although the survival of specific neurons in the PNS is frequently promoted by individual neurotrophins (Snider, 1994; Lindsay, 1996), the situation in the CNS is further complicated by the fact that trophic support may be derived from different sources (Korsching, 1993). Moreover, trophic dependencies in the adult CNS may be altered by several insults, and the lack of target support could be compensated by the enhancement of autocrine–paracrine trophic factors (Giehl et al., 1998; Venero et al., 2000).
Afferents from all areas of the cortex regulate neuronal activity in the striatal neurons via glutamate release. Excessive activation of glutamate receptors in the striatum produces selective degeneration of striatal projection neurons that resembles the pathological findings observed in Huntington's disease (Beal et al., 1986, 1991; DiFiglia, 1990). This neurological disorder is also characterized by a decrease in the number of neurons in the layer V of the cerebral cortex (Cudkowicz and Kowall, 1990). Pyramidal neurons in that layer provide the main neocortical and mesocortical projection to the striatum, but additional inputs come from layers II/III and VI (Gerfen, 1992). It has been described recently that the corticostriatal projection transports BDNF from cerebral cortex to the striatum (Altar et al., 1997; Kokaia et al., 1998), showing a trophic relationship between both brain regions. Increased levels of BDNF mRNA have been observed in cortical and striatal neurons after brain insults, including epileptic seizures, cerebral ischemia, excitotoxicity, and traumatic injury (Rocamora et al., 1996; Canals et al., 1998; Kokaia et al., 1998; McAllister et al., 1999). Because BDNF exerts neuroprotective effects against excitotoxicity in the striatum (Martínez-Serrano and Björklund, 1996; Pérez-Navarro et al., 1999, 2000b), it has been postulated that enhancement of this neurotrophin in the cortex may be involved in protection of striatal neurons against damage via anterograde transport (Altar et al., 1997; Kokaia et al., 1998).
Increase of cortical BDNF can also constitute an autocrine–paracrine trophic response for cortical neurons because BDNF protects cortical neurons from various types of insults (Shimohama et al., 1993; Kume et al., 1997). The importance of BDNF in the cerebral cortex has also been demonstrated by a decrease in the number of neurons that express neuropeptides or calcium-binding proteins in BDNF knock-out mice (Jones et al., 1994). Furthermore, an increase in cortical cell death has been described in the null mutant mouse of the BDNF receptor, TrkB, during development (Alcántara et al., 1997) and in adulthood (Xu et al., 2000).
Our previous results showing that striatal excitotoxic lesions differentially regulated neurotrophin expression in various brain regions (Canals et al., 1998; Checa et al., 2000) suggest that changes in the levels of neurotrophins, including BDNF, could underlie the differential vulnerability of striatal neurons observed in Huntington's disease. The main goal of the present study is to characterize the regulation of BDNF in the cortex of rat models of Huntington's disease. Here we show that cortical BDNF was upregulated after different types of striatal lesions and after blocking retrograde transport with colchicine. Moreover, protection of striatal cells with exogenous administration of BDNF prevented this enhancement. Therefore, the present results suggest that the upregulation of cortical BDNF may be an endogenous trophic response to counteract cortical and/or striatal cell death.
MATERIALS AND METHODS
Animal injury. Male Sprague Dawley rats (140–210 gm) were anesthetized with pentobarbital (50 mg/kg, i.p.) and placed in a David Kopf Instruments (Tujunga, CA) stereotaxic apparatus (DK 900) with the incisor bar 5 mm above the interaural line. We used four to seven animals for each condition and time point studied. Quinolinate (QUIN) (34 or 68 nmol; Sigma, St. Louis, MO), kainate (KA) (2 or 6 nmol; Sigma), 3-nitropropionic acid (3-NPA) (500 nmol; Sigma), KCl (100 mm; Merck, Darmstadt, Germany), and PBS, pH 7.5, were injected in a volume of 1 μl into the left striatum at two coordinates [anteroposterior (AP), +2.3; lateral (L), −2.4 from bregma; −5.0 from dura; and AP, +1.3; L, −3.0 from bregma; −5.0 from dura] as described by Canals et al. (1998). The effectiveness of the lesion induced by QUIN, KA, and 3-NPA was assessed 7 d after intrastriatal injection by the number of turns induced after apomorphine administration (0.5 mg/kg, s.c.; 1.33–2.5 rpm; Sigma).
Some injections of QUIN, KA, or PBS to the striatum were performed from the contralateral hemisphere to avoid the effects of the needle-track injury in the ipsilateral cortex (AP, −1; L, −3.6 from bregma; −8.5 from dura; and AP, −2; L, −3.2 from bregma; −8.9 from dura; incisor bar, −11.6; angle of injection, 62°; n = 4 for each condition).
In another set of experiments, glutamate antagonists were coinjected with 3-NPA at the following doses: kynurenic acid (Sigma), 60 nmol; dizocilpine maleate (MK801) (Tocris Cookson, Bristol, UK), 30 nmol; 6-cyano-7-nitroquinoxaline-2,3-dione disodium (CNQX) (Tocris Cookson), 1 nmol (n = 3 per group). Control rats were injected with the same doses of glutamate antagonists alone (n = 3 per group).
After lesioning, animals were housed separately with access to food and water ad libitum in a colony room maintained at a constant temperature (19–22°C) and humidity (40–50%) on a 12 hr light/dark cycle. All animal-related procedures were in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the local animal care committee of the Universitat de Barcelona (99/01) and by the Generalitat de Catalunya (99/1094).
Morphological analysis of the lesion size. Brains intrastriatally injected with QUIN, KA, 3-NPA, 3-NPA plus kynurenic acid, 3-NPA plus MK801, or 3-NPA plus CNQX were morphologically analyzed 3 d after lesion. The animals were deeply anesthetized in a CO2 chamber and transcardially perfused with 4% paraformaldehyde solution in 0.1m sodium phosphate, pH 7.2. The brains were post-fixed 2 hr in the same solution, cryoprotected in 10% sucrose–PBS for 15 hr, and frozen in dry ice-cooled isopentane. Serial horizontal cryostat sections (14 μm) were processed for cresyl violet staining. Lesions were measured using Scion NIH Image on a computer attached to an Olympus Optical (Tokyo, Japan) microscope. Consecutive sections (26–29 sections per animal) were visualized, and the border of the lesion was outlined. The volume of the lesion was calculated by multiplying the sum of all the sectional areas (in square millimeters) by the distance between successive sections (0.2 mm) as described previously (Pérez-Navarro et al., 2000a).
RNase protection assay. Rats were killed by decapitation at 2, 4, 6, 10, 16, or 24 hr after injury, brains were removed, and cerebral cortex was quickly dissected out, frozen on dry ice, and stored at −80°C. Total RNA was isolated by the guanidine isothiocyanate method (Chomczynski and Sacchi, 1987). BDNF mRNA levels were estimated by RNase protection assay using the RPAII Kit (Ambion Inc., Austin, TX) as described previously by Canals et al. (1998). In each reaction, total RNA (20 μg) was hybridized with the BDNF cRNA probe (Lindefors et al., 1995) together with a glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cRNA probe (Fort et al., 1985). Protected fragments were separated on 4% polyacrylamide gels in denaturing conditions, and the gels were exposed to BIOMAX (Eastman Kodak, Rochester, NY) films at −80°C with an intensifying screen. The optical density of the autoradiograms was quantified with the Phoretix 1D gel analysis (Phoretix International Ltd., Newcastle, UK). The amount of BDNF mRNA in each lane was standardized to GAPDH.
In situ hybridization studies. Animals were killed 6 hr after intrastriatal injection of PBS or excitatory amino acids (EAAs). Brains were dissected out and frozen in dry ice-cooled isopentane. Serial horizontal cryostat sections (14 μm) were processed for hybridization with radioactive BDNF riboprobe (Lindefors et al., 1995) as follows. After rinsing in PBS, tissue was deproteinated in 0.2m HCl for 10 min, acetylated with 0.25% acetic anhydride in 0.1 m ethanolamine for 10 min, post-fixed in 4% paraformaldehyde, and dehydrated with increasing concentrations of ethanol. Slices were incubated for 16 hr in a humidified chamber at 53°C with 3 × 105 cpm of antisense BDNF probe in 150 μl of hybridization cocktail (50% formamide, 20 mm Tris-HCl, pH 7.6, 1 mmEDTA, pH 8.0, 0.3 m NaCl, 0.1m dithiothreitol, 0.5 mg/ml yeast tRNA, 0.1 mg/ml poly(A+) RNA, 1× Denhardt's solution, and 10% dextran sulfate). Antisense cRNA probe to detect the BDNF transcript was prepared by in vitro transcription using T7 RNA polymerase (Promega, Madison, WI) and [35S]UTP or [32P]UTP (Amersham Pharmacia Biotech, Uppsala, Sweden). For control experiments, sense cRNA probe was obtained by in vitro transcription using T3 RNA polymerase (Promega). After hybridization, slices were first washed at room temperature in 1× SSC, followed by two washes in 1× SSC at 37°C for 15 min each. Single-stranded RNA was digested by RNase treatment (40 μg/ml) for 30 min at 37°C in 0.5 m NaCl, 20 mm Tris-HCl, pH 7.6, and 2 mm EDTA. Tissue was washed twice in 1× SSC in 65°C for 10 min, dehydrated in ethanol, and air-dried. Slices were exposed to β-max x-ray film (Amersham Pharmacia Biotech) for 20 d and dipped in LM-1 photoemulsion (Amersham Pharmacia Biotech), exposed at 4°C for 2 months, developed with D19 (Eastman Kodak), fixed, and counterstained with cresyl violet staining. Some sections were processed for immunohistochemistry after the in situhybridization. For this purpose, immediately after the last washes with SSC, the slides were coincubated with the primary antibodies GFAP (1:500; Dako A/S, Glostrup, Germany) and neuron-specific nuclear protein (NeuN) (1:100; Chemicon, Temecula, CA) overnight at 4°C. After three washes in PBS, the sections were coincubated with both secondary antibodies (anti-rabbit-FITC conjugated; 1:100; Vector Laboratories, Burlingame, CA; and anti-mouse-Texas Red conjugated; 1:100; Jackson ImmunoResearch, West Grove, PA), washed overnight in PBS, and dipped as mentioned above. Triple-labeling analysis was performed using a confocal microscope.
BDNF immunohistochemistry. Sections obtained with a cryostat (40 μm) were collected in PBS as free-floating sections and preincubated for 1 hr with PBS containing 10% methanol and 3% H2O2. Sections were then washed three times in PBS, permeabilized with 0.5% Triton X-100 (Sigma), and preincubated for 2 hr with 1.5% goat serum in PBS. After three washes in 0.1% Triton X-100 in PBS, tissue was incubated with the primary anti-BDNF antibody (1:200; Santa Cruz Biotechnology, Santa Cruz, CA) in PBS containing 0.1% Triton X-100 and 1.5% goat serum for 16 hr at 4°C. Sections were washed three times as above and incubated with a biotinylated goat anti-rabbit antibody (1:200; Vector Laboratories) for 1–2 hr at room temperature in the same buffer as the primary antibody. The immunohistochemical reaction was developed using the Vectastain ABC kit (Vector Laboratories) and intensified by incubation with 0.01% NiCl2.
Cell grafting of a BDNF-secreting cell line. Fischer 344 rat 3T3 fibroblasts transfected with BDNF (F3N-BDNF) (Neveu and Arenas, 1996) were used for the cell-grafting experiments. Mock-transfected fibroblasts (F3A-MT) were used as control (Arenas and Persson, 1994). Cells in active growth phase were washed and collected in serum-free media at a concentration of 2.5 × 105 cells/μl as described byPérez-Navarro et al. (1999). A microinjection cannula was implanted into the left striatum, and 7.5 × 105 cells in 3 μl were injected (1 μl/min) at the following coordinates: AP, +1.8; L, −2.7 from bregma; and −5.0 from dura. Four days later, two injections of glutamate receptor agonists were placed anterior and posterior to the grafting site at the coordinates described above. The cerebral cortex was quickly dissected out and processed for the BDNF mRNA analysis at 6 hr of the striatal injury.
Studies of the corticostriatal transport. Studies of corticostriatal transport were performed by injecting a retrograde transport tracer (0.2 μl of Fluorogold; 2 mg/ml in PBS; Fluorochrome, Denver, CO) into the striatum in both hemispheres 3 d after unilateral QUIN or KA lesion (n = 3). The coordinates used for the tracer were the same as for the EAAs. Rats were transcardially perfused with 4% paraformaldehyde solution 48 hr after Fluorogold injection, and the brains were post-fixed and cryoprotected in 10% sucrose. Cryostat serial horizontal sections (25 μm) were mounted with immunofluore mounting medium (ICN, Costa Mesa, CA) and visualized under fluorescent microscope. All retrogradely labeled cells were counted in three separate regions (250 μm2/field): frontal, motor, and somatosensorial cortex. Retrograde signal was analyzed in 10–12 sections per animal separated by 175 μm. The number of positive cells on the side ipsilateral to the lesion was normalized to the number of cells on the contralateral side (nonlesioned).
To study the effect of blockage of axonal transport, a double injection of colchicine (2 × 1 μl at 20 μg/μl; Sigma) was performed 30 min before striatal excitotoxic injury. The coordinates used were the same as those described above for EAAs.
Statistical analysis. Results were normalized to the mean of sham-injected animals and expressed as a percentage of these data. Statistical analysis was performed using one-way ANOVA, followed by LSD (least significant difference t test) post hoctest.
RESULTS
Striatal excitotoxic injury increases BDNF mRNA levels in the ipsilateral cortex
Morphological analysis showed that intrastriatal injection of QUIN (68 nmol), KA (6 nmol), or 3-NPA (500 nmol) produced lesion sizes with the following volume rank order: KA (3.60 ± 0.10 mm3) > 3-NPA (3.23 ± 0.09 mm3) > QUIN (2.98 ± 0.08 mm3) (Fig.1; see Fig. 6).
Fig. 1.
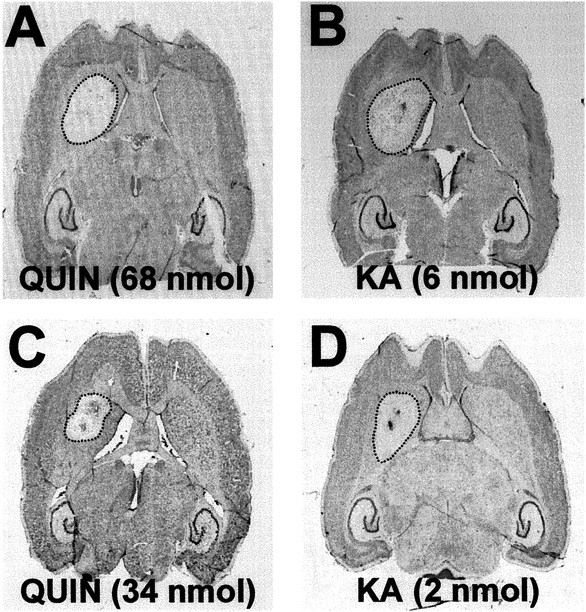
Nissl-stained sections showing the striatal lesion (area inside dotted lines) induced by intrastriatal injection: A, C, QUIN (68 and 34 nmol, respectively); B, D, KA (6 and 2 nmol, respectively).
Fig. 6.
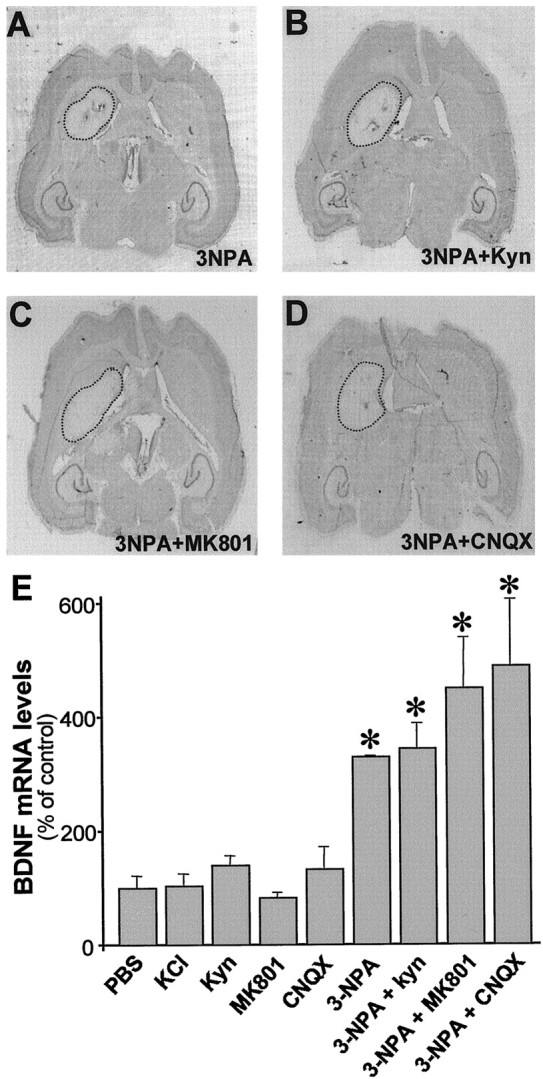
BDNF mRNA is transneuronally upregulated in the cortex by striatal cell death and not by synaptic activity per se.A–D, Nissl-stained sections in which unilateral lesions were performed. Note the areas of reduced staining (area insidedotted lines) in striata injected with the following:A, 3-NPA (500 nmol); B, 3-NPA (500 nmol) and kynurenic acid (Kyn; 60 nmol); C, 3-NPA (500 nmol) and an NMDA receptor antagonist (MK801; 30 nmol); andD, 3-NPA (500 nmol) and a non-NMDA receptor antagonist (CNQX; 1 nmol). E, Histogram represents BDNF mRNA levels from cerebral cortex 6 hr after intrastriatal injection of PBS, KCl, or 3-NPA alone or in combination with different glutamate receptor antagonists [kynurenic acid (Kyn), MK801, and CNQX]. Values, from three to four animals per condition, were normalized to those obtained in PBS-injected animals and are represented as mean ± SEM (*p < 0.05).
To examine BDNF expression in the cerebral cortex, after intrastriatal injection of QUIN or KA, we performed a sensitive RNase protection assay. Both glutamate receptor agonists increased BDNF mRNA in the cerebral cortex with a different profile (Fig.2A). QUIN (68 nmol) induced a peak between 4 and 6 hr after lesion, reaching a maximum at 6 hr (306 ± 73%). The increase induced by KA occurred between 4 and 16 hr after injury, showing the highest levels of BDNF expression at 6 hr after injection (524 ± 115%).
Fig. 2.

A, BDNF mRNA is upregulated in the cortex ipsilateral to excitotoxic lesion in the striatum. Intrastriatal QUIN injection (circles) induced a peak of expression between 4 and 6 hr. BDNF mRNA levels were also increased between 4 and 16 hr after KA-induced striatal injury (squares).Triangles represent sham-injected striata. Values are the mean ± SEM (n = 4–7; *p < 0.05; **p < 0.005). Autoradiograms show two representative experiments. B, Injection of QUIN or KA in the striatum through the contralateral cortex upregulated BDNF mRNA in the ipsilateral cortex. Values are mean ± SEM (n = 4; *p < 0.05).
To determine whether the regulation of cortical BDNF could be attributable to a local mechanical damage by the cannula, we performed intrastriatal injections from the contralateral hemisphere. In these conditions, BDNF mRNA was upregulated at 6 hr in the cortex ipsilateral to the intrastriatal QUIN or KA injection (270 ± 73 and 378 ± 87%, respectively) (Fig. 2B). No significant increase of BDNF expression was detected in the contralateral cortex.
In situ hybridization studies showed high levels of BDNF expression in the cortex ipsilateral to the lesioned striata (Fig.3). At the two tested doses of QUIN (34 or 68 nmol) or KA (2 or 6 nmol), the upregulation of BDNF depended on the intensity of striatal damage (compare Figs. 1, 3). However, both NMDA and non-NMDA glutamate receptor agonists produced a similar pattern of expression in cortical layers II/III, V, and VI (Fig.3D,E). This increase in BDNF mRNA levels was observed along the cerebral cortex, including the frontal, motor, and somatosensorial areas (Fig. 3B–E). No regulation of BDNF mRNA was observed in the hippocampus, indicating that the effects of QUIN or KA were specific to striatal neurons (Fig.3B,C). Triple-labeling showed that BDNF mRNA was upregulated in cortical neurons (NeuN-positive cells) but not in glial cells (GFAP-positive cells) (Fig.4A,B).
Fig. 3.
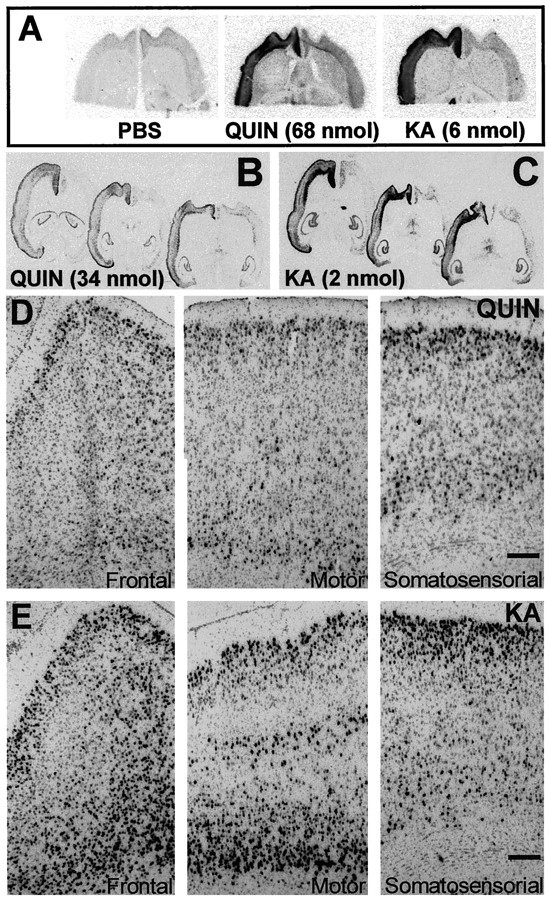
In situ hybridization shows specific BDNF mRNA increased levels in the ipsilateral cortex after striatal EAA lesions. A, Autoradiograms showing a 20 d exposure of horizontal sections through brains receiving intrastriatal injection of PBS or high doses of EAAs (QUIN, 68 nmol; or KA, 6 nmol). B, C, Autoradiograms showing a 20 d exposure of intrastriatal injected brains with lower QUIN or KA doses (34 and 2 nmol, respectively). D, Bright-field photomicrographs of frontal, motor, and sensory cortical areas of QUIN-lesioned brains. E, Photomicrographs showing BDNF upregulation in the same cortical areas of KA intrastriatally injected animals. Scale bars, 150 μm.
Fig. 4.

A, Photomicrographs showing triple-labeling of cortical layer II/III. NeuN-positive neurons are labeled in red, GFAP-positive astrocytes are labeled ingreen, and blue corresponds to the BDNF hybridization signal as assessed in radioactive in situhybridization. B, High magnification ofA. Note that GFAP-positive cells are negative for BDNF hybridization (open arrows), whereas NeuN-positive neurons are positive for BDNF mRNA signal (filled arrows). Some NeuN-positive neurons are negative for BDNF (arrowheads). Scale bars: A, 30 μm;B, 15 μm.
Immunohistochemical studies showed that the increase in BDNF protein coincided with the location of BDNF mRNA upregulation (Fig.5). BDNF-positive cells were mainly located in layers II/III, V, and VI after intrastriatal QUIN or KA injury (Fig. 5B,C). The strongest BDNF immunolabeling was observed on cell bodies and apical dendrites of the pyramidal projecting neurons of layer V in the prefrontal cortex after QUIN or KA striatal injection (Fig.5E,F).
Fig. 5.
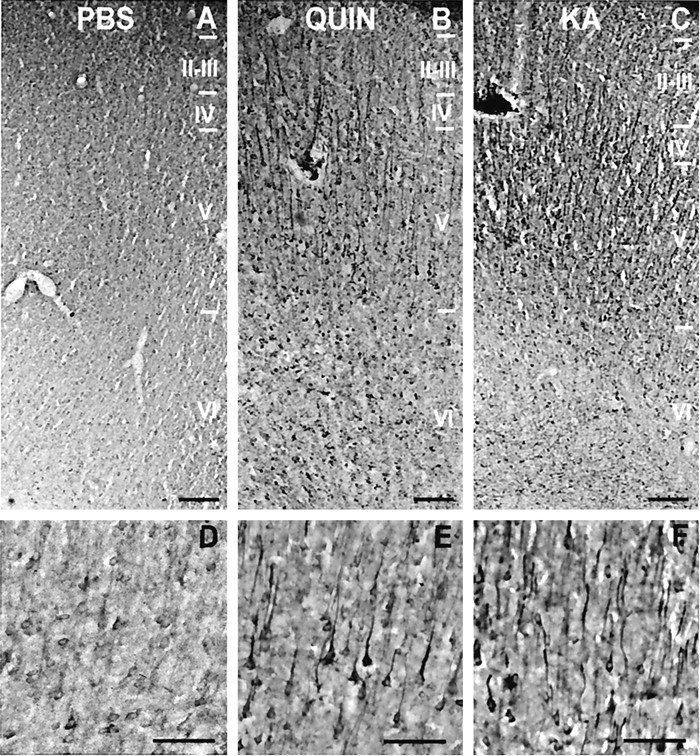
BDNF immunohistochemistry in the cerebral cortex reveals a pattern of expression similar to that observed for BDNF mRNA after striatal excitotoxic lesions. A, B,C, Immunolocalization of BDNF protein in the prefrontal cortex ipsilateral to PBS-, QUIN-, or KA-injected striata, respectively. D, E, F, Detailed photomicrographs of prefrontal layer V after injection of PBS, QUIN, or KA in the striatum, respectively. Scale bars:A–C, 200 μm; D–F, 100 μm.
Thus, our findings suggested that cortical BDNF is upregulated in response to either the loss of striatal cells or the increased neuronal activity by EAA. We therefore tested whether a striatal neurotoxin working on an energy-dependent mechanism could modify the cortical BDNF mRNA.
Cortical BDNF mRNA upregulation is specifically induced by striatal damage but not by increased neuronal activity
It has been shown that 3-NPA, an irreversible inhibitor of succinate dehydrogenase, produces selective loss of striatal projection neurons (Beal et al., 1993; Miller and Zaborszky, 1997). Intrastriatal injection of 3-NPA increased BDNF mRNA levels in the cortex ipsilateral to the lesioned striata at 6 hr (329 ± 5%) (Fig.6A,E). This neurotoxin induced glutamate-independent lesions because simultaneous coinjection of glutamate receptor antagonists with 3-NPA did not prevent the striatal damage (Fig. 6B–D). Intrastriatal coinjection of 3-NPA with a generic glutamate receptor antagonist, kynurenic acid, increased BDNF mRNA in the ipsilateral cortex similarly to 3-NPA alone (344 ± 44%) (Fig.6E). Moreover, coinjection of 3-NPA with either a specific NMDA (MK-801) or non-NMDA (CNQX) receptor antagonists produced similar changes in cortical BDNF expression (450 ± 89 and 488 ± 118%, respectively) (Fig. 6E).
We also examined whether increased neuronal activity induced by KCl administration into the striatum modified BDNF mRNA expression in the cortex. Intrastriatal injection of 100 mm KCl did not affect cortical BDNF mRNA levels at 6 hr (104 ± 21%) (Fig. 6). Thus, combined, our observations suggested that cortical BDNF is upregulated after striatal damage. We therefore tested whether administration of neuroprotective factors for striatal neurons, such as BDNF, could prevent the cortical upregulation of BDNF expression.
The upregulation of BDNF mRNA in the cerebral cortex after striatal injury is prevented by intrastriatal administration of exogenous BDNF
We next examined whether administration of exogenous BDNF to the striatum modified the increase of cortical BDNF mRNA levels induced by intrastriatal EAA injections (Fig. 7). A BDNF-secreting cell line that protects striatal neurons against excitotoxicity (Pérez-Navarro et al., 1999, 2000b) was grafted into the striatum before QUIN or KA injection. Intrastriatal grafting of F3N-BDNF but not the F3A-MT cell line prevented the upregulation of BDNF mRNA in the cerebral cortex at 6 hr after intrastriatal EAA injections (Fig. 7). BDNF mRNA levels in the cortex were not modified by intrastriatal grafting of F3A-MT or F3N-BDNF cells in animals injected with PBS (Fig. 7A). These results suggest that a striatal-derived signal might be retrogradely transported to the cortex, in which it regulates BDNF expression. To test this possibility, we studied the retrograde transport in corticostriatal neurons.
Fig. 7.

Intrastriatal grafting of a BDNF-secreting cell line (F3N-BDNF) prevents the upregulation of BDNF mRNA in the cerebral cortex of EAA-lesioned animals. A, Values obtained from intrastriatal PBS injection in the F3A-MT-grafted striata were taken as 100%, and all other values were normalized to these data. Values are the mean ± SEM of four animals (***p < 0.001). B, Autoradiograms showing a 10 d exposure of brains hybridized with a BDNF 32P riboprobe in situ. Horizontal sections were obtained from animals intrastriatally grafted with the F3N-BDNF cell line and injected with PBS, QUIN, or KA. Although BDNF mRNA was detected in the cell line, no upregulation of cortical BDNF mRNA was observed.
Blockade of axonal transport does not prevent the BDNF upregulation in the cortex induced by striatal excitotoxic lesions
Intrastriatal injection of Fluorogold, a retrograde transport tracer, showed that the corticostriatal pathway was not affected by excitotoxic lesions in the target area (Fig.8). The retrograde transport was studied in frontal, motor, and somatosensorial cortical areas (Fig. 8A,B,C, respectively). No significant differences in cortical Fluorogold-labeled cells were observed after QUIN or KA intrastriatal lesion compared with the contralateral (nonlesioned) hemisphere.
Fig. 8.
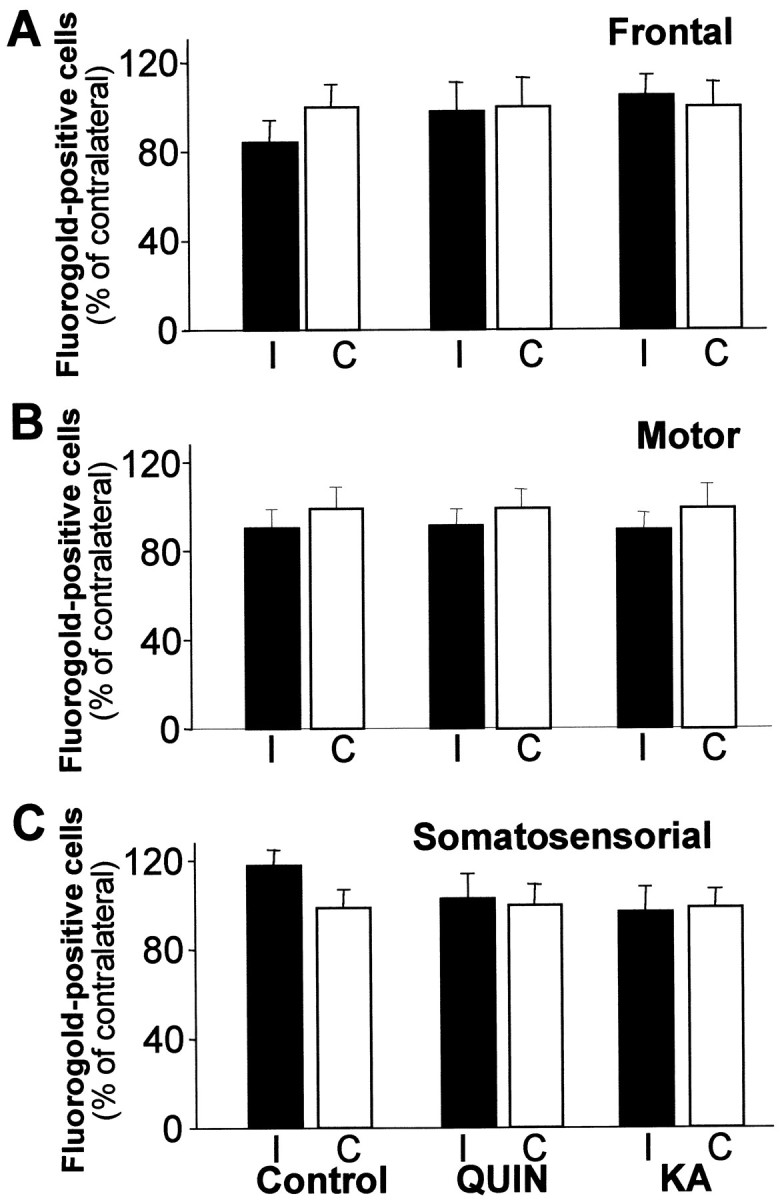
Excitotoxic striatal lesions do not affect the corticostriatal transport. Fluorogold-labeled cortical cells were counted in animals receiving bilateral injection of this retrograde tracer and unilateral injection of PBS, QUIN, or KA in the striatum. No differences between the three groups were observed. Number of labeled cells in the ipsilateral cortex to the lesioned striatum (I) were normalized to the number of Fluorogold-positive neurons from the contralateral hemisphere (nonlesioned; C). The values are represented as mean ± SEM of three animals per group.
To determine whether axonal transport is involved in the induction of BDNF mRNA expression in the cerebral cortex, colchicine was injected into the striatum. No retrograde transport of Fluorogold was observed in the cerebral cortex after intrastriatal colchicine administration, showing that retrograde transport was completely blocked (Fig.9A, B). However, intrastriatal injection of colchicine alone or in combination with QUIN or KA upregulated BDNF expression similarly to EAA injection alone at 6 hr in the cortex (Fig. 9C). Thus, our results show that cortical neurons upregulate BDNF expression in the absence of either retrograde transport or in the absence of striatal neurons.
Fig. 9.
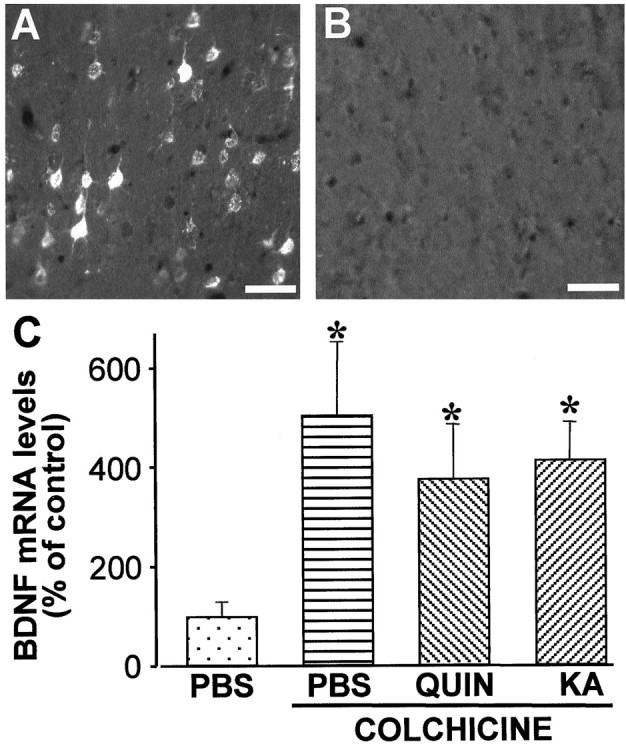
BDNF mRNA is upregulated in the cerebral cortex 6 hr after disruption of corticostriatal transport. A,B, Photomicrographs comparing the retrograde transport of Fluorogold to the cortical layer V after injection of KA or colchicine, respectively. No labeling in the cerebral cortex was observed in colchicine-injected animals. Scale bars, 100 μm.C, Histogram represents cortical BDNF mRNA levels after intrastriatal injection of colchicine alone or in combination with QUIN or KA. Values are the mean ± SEM of four different animals for each condition, normalized to those obtained from PBS-injected animals (*p < 0.05).
DISCUSSION
Here we demonstrate that striatal damage or blockade of retrograde transport in corticostriatal neurons results in an upregulation of BDNF mRNA and protein in the cerebral cortex. Moreover, protection of the neostriatal neurons with exogenous BDNF prevented the upregulation of cortical BDNF induced by EAA. These results suggest that cortical BDNF upregulation may be an endogenous protective response to striatal damage.
Although QUIN and KA act through two different types of glutamate receptors, NMDA and non-NMDA, respectively, intrastriatal injection of either agonist at excitotoxic doses induced the same morphological pattern of cortical BDNF upregulation. The expression of BDNF was also increased in the cerebral cortex after intrastriatal injection of another neurotoxin, the 3-NPA (Beal et al., 1993; Miller and Zaborszky, 1997). Although the involvement of glutamate in the mechanism of neuronal death induced by 3-NPA remains controversial (Behrens et al., 1995; Pang and Geddes, 1997; Lee et al., 2000), this neurotoxin has no effect on the extracellular glutamate levels (Erecinska and Nelson, 1994). Moreover, here we observed that injection of NMDA or non-NMDA antagonists simultaneously with 3-NPA neither prevented striatal cell death nor blocked the upregulation of cortical BDNF mRNA. Furthermore, intrastriatal injection of KCl, at doses that increases synaptic activity in the striatum (Samuel et al., 1996; Kantor et al., 1999), did not modify BDNF mRNA levels in the cerebral cortex. Although it has been widely demonstrated that synaptic activity regulates neurotrophin expression (Thoenen, 1995; McAllister et al., 1999), it has been suggested that neuronal activity alone is not a sufficient stimulus for the induction of BDNF mRNA after seizures (Dugich-Djordjevic et al., 1992). In previous studies, we have shown that intrastriatal injection of glutamate receptor agonists induced a differential regulation of the expression of neurotrophins and their receptors (Canals et al., 1998,1999; Checa et al., 2000). These results could be related to a direct activation of specific glutamate receptors, although injury-induced changes in neurotrophin expression cannot be ruled out. However, the present results demonstrate that both QUIN and KA induce an increase in cortical BDNF depending on the severity of striatal excitotoxic injury. These findings together with the induction of cortical BDNF by 3-NPA and the lack of effect of KCl after intrastriatal injection indicate that BDNF expression in the cerebral cortex is upregulated by striatal damage.
We next examined whether axonal communication is involved in the regulation of endogenous cortical BDNF mRNA levels. Intrastriatal injection of colchicine alone or in combination with QUIN or KA produced a large increase in cortical BDNF mRNA levels, reinforcing the idea that transneuronal BDNF expression is stimulated by the lack of the target area. These results suggest the existence of a constitutive factor released by striatal neurons that is retrogradely transported and regulates the cortical BDNF expression. In agreement with this hypothesis, it has been reported recently the existence of a neuron-restrictive silencer element that constitutively inhibits specific gene expression within the nervous system (Pathak et al., 1994; Bessis et al., 1997; Kallunki et al., 1998), including thebdnf transgene expression (Timmusk et al., 1999). Thus, the mechanism by which BDNF upregulation could take place may involve the blockade or downregulation of the BDNF silencer element after striatal damage or blockade of retrograde transport.
It has been shown previously that, after colchicine treatment, BDNF immunoreactivity is increased in cell bodies of cortical neurons, which send afferents to the striatum (Altar et al., 1997), suggesting that this neurotrophin is anterogradely transported. Thus, it has been proposed that cortical neurons provide an afferent supply of BDNF to target neurons in the striatum (Altar et al., 1997; Kokaia et al., 1998). Our findings are consistent with this hypothesis, because enhancement of BDNF mRNA was observed in neurons of cortical layers II/III, V, and VI, which project to the striatum (Gerfen, 1992). In addition, the results observed after administration of exogenous BDNF to the striatum provide further evidence for an anterograde protective role of cortical BDNF. We have reported previously that grafting of a BDNF-secreting cell line protects striatal neurons against excitotoxicity (Pérez-Navarro et al., 1999, 2000b), and we now show that intrastriatal grafting of this cell line prevented the increase in cortical BDNF mRNA by striatal lesion. Thus, induction of cortical BDNF after damage of the striatum could be a compensatory response to the loss of target neurons, which, in turn, may provide trophic support to striatal neurons. In keeping with this view, changes in BDNF protein after focal ischemia have been postulated to play a role in promoting the survival and plasticity of cortical and striatal neurons (Kokaia et al., 1998).
Although our results are consistent with a long-distance effect of BDNF, we cannot rule out the possibility of a local action of BDNF in the cortex. Cortical BDNF upregulation could also constitute part of an autocrine–paracrine trophic response to protect cortical neurons from the loss of its target, the striatum. The study with Fluorogold also showed that retrograde transport is not affected in corticostriatal neurons by the lack of target neurons, raising the possibility of a potential role of BDNF in the maintenance of the cortical neurons. In fact, Giehl et al. (1998) demonstrated that endogenous cortical BDNF is involved in providing a trophic support to corticospinal neurons after axotomy. Furthermore, a BDNF receptor, TrkB, has been shown recently to be essential for the survival of pyramidal neurons of layers II/III and V in the adult brain (Xu et al., 2000), confirming the requirement of TrkB ligands for the cerebral cortex.
Analyses of neurotrophin expression in animal models of neurodegenerative diseases may help advance future understanding of the mechanism responsible for brain disease and development of neuroprotective treatments. We have shown previously that activation of glutamate receptors located on striatal neurons results in a specific pattern of neurotrophin expression during the development (Checa et al., 2000) and in adult excitotoxic models of Huntington's disease (Canals et al., 1998). Combined, all these results indicate that changes in neurotrophin expression may be involved in the selective vulnerability of striatal populations observed in Huntington's disease (Ferrante et al., 1985; Reiner et al., 1988; Richfield et al., 1995). Moreover, the present new data suggest that the regulation of BDNF in the cerebral cortex by striatal neurons is required to provide trophic support to striatal and/or cortical neurons in physiological and pathological conditions.
In conclusion, our results show that striatal damage or blockade of retrograde transport in the corticostriatal pathway increases the levels of cortical BDNF, suggesting that this upregulation may constitute an adaptive mechanism against the progression of striatal and/or cortical neurodegeneration. Thus, stimulation of endogenous production of BDNF or administration of exogenous trophic factor may be a good therapeutic approach to prevent the progression of Huntington's disease.
Footnotes
This study was supported by Comisión Interministerial de Ciencia y Tecnología Grant SAF-99–0017 (Ministerio de Educación y Ciencia, Spain), Marató TV-3 Grant 97-TV1009, a grant from the Fundación Ramón Areces, a grant from the Swedish Medical Research Council (MFR), and BIOMED2 program of the European Commission Grant BMH4-CT96–0273. J.M.C. was supported by a short-term Human Frontiers Science Program fellowship. N.C. was supported by IDIBAPS, and S.M. was a fellow of CIRIT (Generalitat de Catalunya, Spain). We thank Dr. B. A. Sieber for critical reading of this manuscript, M. T. Muñoz and A. Nonell for technical assistance, and Anna Bosch and the Serveis Científico-Tècnics (Universitat de Barcelona) for support and advice in the use of confocal microscopy. We are also very grateful to Robin Rycroft for English language revision. This article is dedicated to the memory of Oriol Riba Canals, nephew of J.M.C.
Correspondence should be addressed to Dr. Jordi Alberch, Departament de Biologia Cel·lular i Anatomia Patològica, Facultat de Medicina, Universitat de Barcelona, Casanova 143, 08036 Barcelona, Spain. E-mail:alberch@medicina.ub.es.
REFERENCES
- 1.Alcántara S, Frisén J, del Rio JA, Soriano E, Barbacid M, Silos-Santiago I. TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. J Neurosci. 1997;17:3623–3633. doi: 10.1523/JNEUROSCI.17-10-03623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- 3.Arenas E, Persson H. NT-3 prevents the death of adult central noradrenergic neurons in vivo. Nature. 1994;367:368–371. doi: 10.1038/367368a0. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- 5.Beal MF, Ferrante RJ, Swartz KJ, Kowall NW. Chronic quinolinic acid lesions in rats closely resemble Huntington's disease. J Neurosci. 1991;11:1649–1659. doi: 10.1523/JNEUROSCI.11-06-01649.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Behrens MI, Koh J, Canzoniero LMT, Sensi SL, Csernansky CA, Choi DW. 3-Nitropropionic acid induces apoptosis in cultured striatal and cortical neurons. NeuroReport. 1995;6:545–548. doi: 10.1097/00001756-199502000-00034. [DOI] [PubMed] [Google Scholar]
- 8.Bessis A, Champtiaux N, Chatelin L, Changeux JP. The neuron-restricter silencer element: a dual enhancer/silencer crucial for patterned expression of a nicotinic receptor gene in the brain. Proc Natl Acad Sci USA. 1997;94:5906–6011. doi: 10.1073/pnas.94.11.5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canals JM, Marco S, Checa N, Michels A, Pérez-Navarro E, Arenas E, Alberch J. Differential regulation of the expression of NGF, BDNF and NT-3 after excitotoxicity in a rat model of Huntington's disease. Neurobiol Dis. 1998;5:357–364. doi: 10.1006/nbdi.1998.0211. [DOI] [PubMed] [Google Scholar]
- 10.Canals JM, Checa N, Marco S, Michels A, Pérez-Navarro E, Alberch J. The neurotrophin receptors trkA, trkB and trkC are differentially regulated after excitotoxic lesions in rat striatum. Mol Brain Res. 1999;69:242–248. doi: 10.1016/s0169-328x(99)00130-8. [DOI] [PubMed] [Google Scholar]
- 11.Checa N, Canals JM, Alberch J. Developmental regulation of BDNF and NT-3 expression by quinolinic acid in the striatum and its main connections. Exp Neurol. 2000;165:118–124. doi: 10.1006/exnr.2000.7451. [DOI] [PubMed] [Google Scholar]
- 12.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 13.Cudkowicz M, Kowall NW. Degeneration of pyramidal projection neurons in Huntington's disease cortex. Ann Neurol. 1990;27:200–204. doi: 10.1002/ana.410270217. [DOI] [PubMed] [Google Scholar]
- 14.DiFiglia M. Excitotoxic injury of the neostriatum: a model for Huntington's disease. Trends Neurosci. 1990;13:286–289. doi: 10.1016/0166-2236(90)90111-m. [DOI] [PubMed] [Google Scholar]
- 15.Dugich-Djordjevic MM, Tocco G, Willoughby DA, Najm I, Pasinetti G, Thompson RF, Baudry M, Lapchak PA, Hefti F. BDNF mRNA expression in the developing rat brain following kainic acid-induced seizure activity. Neuron. 1992;8:1127–1138. doi: 10.1016/0896-6273(92)90133-x. [DOI] [PubMed] [Google Scholar]
- 16.Erecinska M, Nelson D. Effects of 3-nitropropionic acid on synaptosomal energy and transmitter metabolism: relevance to neurodegenerative brain diseases. J Neurochem. 1994;63:1033–1041. doi: 10.1046/j.1471-4159.1994.63031033.x. [DOI] [PubMed] [Google Scholar]
- 17.Ferrante RJ, Kowall NW, Beal MF, Richardson EP, Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington's disease. Science. 1985;230:561–563. doi: 10.1126/science.2931802. [DOI] [PubMed] [Google Scholar]
- 18.Fort P, Marty L, Piechaczyk M, Sabrouty SE, Dani C, Jeanteur P, Blanchard JM. Various rat adult tissues express only one major mRNA species from the glyceraldehyde-3-phosphate-dehydrogenase multigenic family. Nucleic Acids Res. 1985;13:1431–1441. doi: 10.1093/nar/13.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320. doi: 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- 20.Giehl KM, Schütte A, Mestres P, Yan Q. The survival-promoting effect of glial cell line-derived neurotrophic factor on axotomized corticospinal neurons in vivo is mediated by an endogenous brain derived-derived neurotrophic factor mechanism. J Neurosci. 1998;18:7351–7360. doi: 10.1523/JNEUROSCI.18-18-07351.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones KR, Fariñas I, Backus C, Reichardt LF. Target disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kallunki P, Edelman GM, Jones FS. The neuronal restrictive silencer element can act as both repressor and enhancer of L1 cell adhesion molecule gene expression during postnatal development. Proc Natl Acad Sci USA. 1998;95:3233–3238. doi: 10.1073/pnas.95.6.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kantor L, Keikilani Hewlett GH, Gnegy ME. Enhanced amphetamine- and K+-mediated dopamine release in rat striatum after repeated amphetamine: differential requirements for Ca2+- and calmodulin-dependent phosphorylation and synaptic vesicles. J Neurosci. 1999;19:3801–3808. doi: 10.1523/JNEUROSCI.19-10-03801.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kokaia Z, Andsberg G, Yan Q, Lindvall O. Rapid alterations of BDNF protein levels in the rat brain after focal ischemia: evidence for increased synthesis and anterograde axonal transport. Exp Neurol. 1998;154:289–301. doi: 10.1006/exnr.1998.6888. [DOI] [PubMed] [Google Scholar]
- 25.Korsching S. The neurotrophic factor concept: a reexamination. J Neurosci. 1993;13:2739–2748. doi: 10.1523/JNEUROSCI.13-07-02739.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kume T, Kouchiyama H, Kaneko S, Maeda T, Akaike A, Shimohama S, Kihara T, Kimura J, Wada K, Koizumi S. BDNF prevents NO mediated glutamate cytotoxicity in cultured cortical neurons. Brain Res. 1997;756:200–204. doi: 10.1016/s0006-8993(97)00195-9. [DOI] [PubMed] [Google Scholar]
- 27.Lee WT, Shen YZ, Chang C. Neuroprotection effect of Lamotrigine and MK-801 on a rat brain lesioned by 3-nitropropionic acid: evaluation by magnetic resonance spectroscopy. Neuroscience. 2000;95:89–95. doi: 10.1016/s0306-4522(99)00410-8. [DOI] [PubMed] [Google Scholar]
- 28.Lindefors N, Brondin E, Metsis M. Spatiotemporal selective effects on brain-derived neurotrophic factor and trkB messenger RNA in rat hippocampus by electroconvulsive shock. Neuroscience. 1995;65:661–670. doi: 10.1016/0306-4522(94)00550-o. [DOI] [PubMed] [Google Scholar]
- 29.Lindsay RM. Role of neurotrophins and trk receptors in the development and maintenance of sensory neurons: an overview. Philos Trans R Soc Lond B Biol Sci. 1996;351:365–373. doi: 10.1098/rstb.1996.0030. [DOI] [PubMed] [Google Scholar]
- 30.Martínez-Serrano A, Björklund A. Protection of the neostriatum against excitotoxic damage by neurotrophin-producing, genetically modified neural stem cells. J Neurosci. 1996;16:4604–4616. doi: 10.1523/JNEUROSCI.16-15-04604.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 32.Miller PJ, Zaborszky L. 3-nitropropionic acid neurotoxicity: visualization by silver staining and implications for use as an animal model of Huntington's disease. Exp Neurol. 1997;146:212–229. doi: 10.1006/exnr.1997.6522. [DOI] [PubMed] [Google Scholar]
- 33.Neveu I, Arenas E. Neurotrophins promote the survival and development of neurons in the cerebellum of hypothyroid rats in vivo. J Cell Biol. 1996;133:631–646. doi: 10.1083/jcb.133.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pang Z, Geddes JW. Mechanism of cell death induced by the mitochondrial toxin 3-nitropropionic acid: acute excitotoxic necrosis and delayed apoptosis. J Neurosci. 1997;17:3064–3073. doi: 10.1523/JNEUROSCI.17-09-03064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pathak BG, Neumann JC, Croyle M, Lingrel JB. The presence of both negative and positive elements in the 5′-flanking sequence of the rat Na,K-ATPase alpha 3 subunit gene are required for brain expression in transgenic mice. Nucleic Acids Res. 1994;22:4748–4755. doi: 10.1093/nar/22.22.4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pérez-Navarro E, Alberch J, Neveu I, Arenas E. BDNF, NT-3 and NT-4/5 differentially regulate the phenotype and prevent degenerative changes of striatal projection neurons after excitotoxicity in vivo. Neuroscience. 1999;91:1257–1264. doi: 10.1016/s0306-4522(98)00723-4. [DOI] [PubMed] [Google Scholar]
- 37.Pérez-Navarro E, Akerud P, Marco S, Canals JM, Tolosa E, Arenas E, Alberch J. Neurturin protects striatal projection neurons but not interneurons in a rat model of Huntington's disease. Neuroscience. 2000a;98:89–96. doi: 10.1016/s0306-4522(00)00074-9. [DOI] [PubMed] [Google Scholar]
- 38.Pérez-Navarro E, Canudas AM, Akerud P, Alberch J, Arenas E. Brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4/5 prevent the death of striatal projection neurons in a rodent model of Huntington's disease. J Neurochem. 2000b;75:2190–2199. doi: 10.1046/j.1471-4159.2000.0752190.x. [DOI] [PubMed] [Google Scholar]
- 39.Reichardt LF, Fariñas I. Neurotrophic factors and their receptors: roles in neuronal development and function. In: Cowan MW, editor. Molecular approaches to neuronal development. Oxford UP; New York: 1997. pp. 220–263. [Google Scholar]
- 40.Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington's disease. Proc Natl Acad Sci USA. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richfield EK, Maguire-Zeiss KA, Vonkeman HE, Voorn P. Preferential loss of preproenkephalin versus preprotachykinin neurons from the striatum of Huntington's disease patients. Ann Neurol. 1995;38:852–861. doi: 10.1002/ana.410380605. [DOI] [PubMed] [Google Scholar]
- 42.Rocamora N, Welker E, Pascual M, Soriano E. Upregulation of BDNF mRNA expression in the barrel cortex of adult mice after sensory stimulation. J Neurosci. 1996;16:4411–4419. doi: 10.1523/JNEUROSCI.16-14-04411.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samuel D, Pisano P, Forni C, Nieoullon A, Kerkerian-Le Goff L. Involvement of the glutamatergic metabotropic receptors in the regulation of glutamate uptake and extracellular excitatory amino acid levels in the striatum of chloral hydrate-anesthetized rats. Brain Res. 1996;739:156–162. doi: 10.1016/s0006-8993(96)00820-7. [DOI] [PubMed] [Google Scholar]
- 44.Shimohama S, Tamura Y, Akaike A, Tsukahara T, Ohara O, Watanabe S, Kimura J. BDNF pretreatment exerts a partially protective effect against glutamate-induced neurotoxicity in cultured rat cortical neurons. Neurosci Lett. 1993;164:55–58. doi: 10.1016/0304-3940(93)90856-g. [DOI] [PubMed] [Google Scholar]
- 45.Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 46.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 47.Timmusk T, Palm K, Lendahl U, Metsis M. Brain-derived neurotrophic factor expression in vivo is under the control of neuron-restrictive silencer element. J Biol Chem. 1999;274:1078–1084. [PubMed] [Google Scholar]
- 48.Venero JL, Vizuete ML, Revuelta M, Vargas C, Cano J, Machado A. Upregulation of BDNF mRNA and trkB mRNA in the nigrostriatal system and in the lesion site following unilateral transection of the medial forebrain bundle. Exp Neurol. 2000;161:38–48. doi: 10.1006/exnr.1999.7243. [DOI] [PubMed] [Google Scholar]
- 49.Xu B, Zang H, Ruff NL, Zhang YA, McConnell SK, Stryker MP, Reichardt LF. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000;26:233–245. doi: 10.1016/s0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]