

Abstract
Voltage-gated ion channels localized to dendritic membranes can shape signal processing in central neurons. This study describes the distribution and functional role of a high voltage-activating K+ channel in the electrosensory lobe (ELL) of an apteronotid weakly electric fish. We identify a homolog of the Kv3.3 K+ channel, AptKv3.3, that exhibits a high density of mRNA expression and immunolabel that is distributed over the entire soma–dendritic axis of ELL pyramidal cells. The kinetics and pharmacology of native K+ channels recorded in pyramidal cell somata and apical dendrites match those of AptKv3.3 channels expressed in a heterologous expression system. The functional role of AptKv3.3 channels was assessed using focal drug ejections in somatic and dendritic regions of an in vitro slice preparation. Local blockade of AptKv3.3 channels slows the repolarization of spikes in pyramidal cell somata as well as spikes backpropagating into apical dendrites. The resulting increase in dendritic spike duration lowers the threshold for a γ-frequency burst discharge that is driven by inward current associated with backpropagating dendritic spikes. Thus, dendritic AptKv3.3 K+ channels influence the threshold for a form of burst discharge that has an established role in feature extraction of sensory input.
Keywords: potassium channel, Kv3, dendritic spike, action potential, backpropagation, oscillatory discharge, repolarization, DAP, electrosensory
The kinetic properties and distribution of voltage-gated ion channels are important determinants of postsynaptic excitability in central neurons. A major influence on dendritic activity arises in cells that support an active “backpropagation” of Na+ spikes from the soma into the dendritic tree (Turner et al., 1991, 1994; Spruston et al., 1995; Magee et al., 1998; Golding et al., 1999). These spikes can secondarily activate other ion channels that augment membrane depolarizations or modify synaptic transmission. For example, Ca2+ currents activated by backpropagating spikes are important to the induction of synaptic plasticity, dendritic transmitter release, and coupling distal dendritic inputs to the soma (Chen et al., 1997; Magee and Johnston, 1997; Larkum et al., 1999). The amplitude of backpropagating spikes can be regulated in turn by dendritic K+ channels, as found in hippocampal pyramidal cells that exhibit a fivefold increase in the density of A-type K+ channels from the soma to distal apical dendrites (Hoffman et al., 1997). Given the wealth of information gained from molecular biology on a host of K+ channel subtypes (Coetzee et al., 1999), an understanding of dendritic activity will ultimately depend on identification of the distribution, kinetic properties, and molecular structures of the K+ channels that exhibit a dendritic distribution.
We are interested in the control of backpropagating Na+ spikes in pyramidal cells of the apteronotid electrosensory lateral line lobe (ELL). This sensory nucleus receives direct primary afferent input encoding modulations of external electric fields used by these animals for electrolocation (Berman and Maler, 1999). Signal processing by ELL pyramidal cells involves the generation of burst discharge for the purpose of feature extraction (Gabbiani et al., 1996). The burst response depends on depolarizing afterpotentials (DAPs) at the soma that arise from the long duration of Na+ spikes backpropagating into the dendrites (Turner et al., 1994). The DAP is potentiated during repetitive spike discharge through a frequency-dependent broadening of dendritic spikes that increases current flow to the soma and contributes to driving the burst depolarization (Lemon and Turner, 2000). The pivotal role of dendritic spike broadening in regulating DAP amplitude suggests that dendritic K+ channels could exert direct control over burst discharge by controlling the rate of dendritic spike repolarization.
We now report that an apteronotid homolog of the Kv3.3 K+ channel subtype is distributed over the entire soma–dendritic axis of ELL pyramidal cells and acts to repolarize Na+ spike discharge in both the soma and proximal apical dendrites (<150 μm). The ability of AptKv3.3 K+ channels to repolarize dendritic spikes allows them to contribute to the establishment of the threshold for γ-frequency oscillatory burst discharge.
MATERIALS AND METHODS
Kv3 amino acid sequences. Amino acid sequences for rat Kv3.1a,b, 3.2a–d, 3.4a,b, mouse Kv3.3a,b and Drosophila Shaw were obtained from the GenBank/European Molecular Biology Laboratory databases. Accession numbers are as follows: rKv3.1a(Y07521), rKv3.1b (M68880), rKv3.2a (A39402), rKv3.2b (M59211), rKv3.2c (M59313), rKv3.2d (S22703), mKv3.3a (Q63959), mKv3.3b (Q63959), rKv3.4a (X62841), hKv3.4b (M64676), Shaw (M32661).
Isolation of AptKv3.3 cDNA. We previously isolated by PCR a 124 bp DNA fragment spanning the pore and S6 domains of anApteronotus Kv3-type K+ channel gene, AptKChFr 3A (Rashid and Dunn, 1998). This fragment was used to probe an Apteronotus cDNA library (Bottai et al., 1998). Kv3 cDNAs were identified by sequencing both strands using the OpenGene Automated DNA Sequencing System (Visible Genetics, Toronto, Ontario, Canada). The sequences of overlapping clones were assembled and the open reading frames were determined using the LASERGENE software package (DNASTAR, Madison, WI). The translation start site for AptKv3.3 was designated as the first methionine codon downstream of an in-frame stop codon. The AptKv3.3 cDNA sequence has been submitted to GenBank (accession number AF308934).
The full coding region for AptKv3.3 was isolated by RT-PCR. PCR was performed on randomly primed brain cDNA using the GeneAmp XL PCR kit (Perkin-Elmer, Foster City, CA) with the following primers: Forward 5′(CAC TCG AGG CTC CCT CTA ATG CTC AGT T), Reverse 5′(CAT CTA GAC GCT CCA CGC TAC AAA A). The nucleotide sequence of the PCR product was confirmed by DNA sequence analysis. For in vitro expression,XhoI and XbaI restriction sites in the primers were used to directionally clone the cDNA into the vector pEUK-C1 (Clontech, Palo Alto, CA).
In situ hybridization. Apteronotus brain was fixed with 4% paraformaldehyde, and 10 μm cryostat sections were probed (Bottai et al., 1997) with an 35S-labeled RNA probe from the 3′ untranslated region of AptKv3.3 (nucleotide 2075–2482). After hybridization and washing, the slides were air-dried and exposed to x-ray film for 3 d, emulsion-dipped [1:1 dilution of NTB2 gel (Eastman Kodak, Rochester, NY) in 600 mm ammonium acetate] and exposed for 18 d at 4°C. After they were developed, the slides were counterstained in neutral red to permit the use of differential interference contrast (DIC) microscopy.
Immunocytochemistry. A C-terminal fragment of AptKv3.3 cDNA corresponding to amino acids 562–634 was subcloned in the GST fusion vector pGEX-4T-1 (Pharmacia Biotech, Uppsala, Sweden). The fusion protein was expressed in Escherichia coli strain DH5-α and purified from inclusion bodies as described (Frangioni and Neel, 1993). Antibodies were prepared in rabbits, depleted of GST immunoreactivity by adsorption to GST protein bound to Affigel 10 beads (Bio-Rad, Richmond, CA), and then affinity-purified by adsorption to AptKv3.3 fusion protein bound to Affigel 10 beads.
For Western blotting, Apteronotus brain proteins were prepared by homogenization of brain tissue in 50 mm Tris/HCl, pH 7.5, 0.25 msucrose, 25 mm KCl, 5 mmMgCl2, and 1 mmphenylmethylsulfonyl fluoride at 4°C. The homogenate was clarified by centrifugation (800 × g, 10 min), and then membranes were purified by centrifugation (100,000 × g, 60 min). Soluble and membrane fractions were fractionated by electrophoresis on a 7.5% polyacrylamide gel and transferred to polyvinylidene difluoride membranes. Membranes were incubated with the antibody (0.45 μg/ml) overnight at 4°C, and the immunoreactive proteins were detected with horseradish peroxidase-coupled secondary antibody and chemiluminescence (NEN Life Sciences, Boston, MA).
For immunohistochemistry, fish were perfused transcardially with 5 ml of PBS and 40 ml of cold 4% paraformaldehyde in PBS, and the brains were post-fixed overnight at 4°C. Coronal vibratome sections (40 μm) were cut and transferred to PBS (3 × 10 min) before they were incubated in blocking buffer (10% normal goat serum, 1% bovine serum albumin, and 0.2% Triton X-100 in PBS) for 2 hr at room temperature. Immunolabeling followed standard procedures using primary antibodies (anti-AptKv3.3; 0.9 μg/ml or mouse monoclonal anti-MAP2a,b; 1.0 μg/ml) and a 1:250 dilution of Oregon green-conjugated goat anti-rabbit IgG conjugate or rhodamine-conjugated goat anti-mouse IgG (Molecular Probes, Eugene, OR).
Heterologous expression of AptKv3.3. Human Embryonic Kidney (HEK) 293-tsA201 cells (Margolskee et al., 1993) were cotransfected with 10 μg AptKv3.3 vector DNA and 0.5 μg eGFP-C1 (Invitrogen, Carlsbad, CA) by the CaCl2 transfection procedure. After 24–48 hr, coverslips were transferred to an in vitro recording chamber on the stage of a Zeiss Axioskop.
ELL recording preparations. Patch recordings were obtained using a novel “Spread Print” preparation, and a conventionalin vitro slice preparation was used for intracellular recordings. The procedures for animal maintenance, anesthesia, brain dissection, and preparation of ELL tissue slices have been described previously (Turner et al., 1994, 1996). All chemicals for electrophysiological studies were obtained from Sigma (St. Louis, MO) unless indicated otherwise. A Tissue Print procedure developed byKotecha et al. (1997) was modified to generate a Spread Print preparation consisting of a partially dissociated thin tissue slice from adult tissue. Spread Prints were prepared by cutting 100 μm ELL slices on a vibratome in oxygenated (95% O2/5% CO2) artificial CSF (aCSF) consisting of (in mm): NaCl 124, KCl 2.0, KH2PO4 1.25, CaCl2 1.5, MgSO4 1.5, NaHCO3 24, and d-glucose 10, pH 7.4. Slices were transferred by spatula and floated onto the surface of a HEPES-buffered “print” medium consisting of (in mm): sucrose 218, NaHCO325, K-gluconate 3.25, MgCl2 4.5, CaCl2 0.1, d-glucose 10, Na pyruvate 1, pH 7.4 contained within a 35 mm Petri dish. The surface tension of this medium provided sufficient force to gradually dissociate the slice in all directions over a 10–15 min period. The extent of dissociation was monitored under a dissecting microscopic until the ELL pyramidal cell layer had just begun to spread. The dissociation was stopped when required by attaching the slice to a gelatin-coated (1.5%) 12-mm-round glass coverslip that was lowered to the medium surface on the tip of a spatula. The coverslip was then inverted under the medium surface and either transferred to the recording chamber or stored in this medium at 4°C for up to 5 hr.
The Spread Print procedure provided a rapid and enzymatic-free method to partially dissociate cells from adult tissue slices that retained an organotypic distribution to aid in identifying cells within a specific topographic map. Cell yield was very high, with excellent structural preservation in numerous cell types that were directly identified using DIC optics and infrared light transmission (DIC-IR). Cells exhibiting good structural definition had resting potentials between −50 and −70 mV and spontaneous TTX-sensitive spike discharge at −50 mV, which were comparable to previous recordings (Turner et al., 1994, 1996).
Recordings. Patch electrodes were constructed from borosilicate glass (Garner Instruments) (2.0 mm outer diameter, 1.2 mm inner diameter) using a Sutter P-87 microelectrode puller, and patch recordings were obtained using an Axopatch 200-A amplifier and PClamp 6 software (Axon Instruments, Foster City, CA) and with an electrolyte consisting of (in mm): KCl 140, HEPES 5, MgCl2 1, EGTA 5, Mg-ATP 1.5, pH 7.2. Electrode resistance ranged between 2 and 10 MΩ. Microelectrodes were constructed from fiber-filled borosilicate glass (1.5 mm O.D.; A-M Systems, Carlsborg, WA) using a Campden puller (Frederick Haer, Bowdoinham, MA). Intrasomatic and dendritic recordings were obtained in an in vitro slice preparation using an Axoclamp 2-A amplifier and CED software (Cambridge Electronic Design, Cambridge, UK). Microelectrodes were filled with 2 mK-acetate (80–110 MΩ). Average values are expressed as mean ± SD, and exponential fits were obtained using Microcal Origin software (Northampton, MA).
All pyramidal cell recordings were restricted to the centromedial segment (CMS) topographic map of the ELL. Dendritic recordings were restricted to the proximal 150 μm of apical dendrites to examine electrophysiological properties in the region over which Na+ spikes actively backpropagate (Turner et al., 1994). Antidromic discharge in the soma and apical dendrites was evoked by stimulating the plexiform layer using a bipolar electrode. In the Spread Print ELL preparation, AptKv3.3 currents were isolated in on-cell or outside-out recording configuration after washing out Ca2+-sensitive K+ channels as 5 mm EGTA in the electrolyte dialyzed the patch (<5 min).
Tissue slices and Spread Prints were perfused with oxygenated aCSF (see above). Transfected HEK cells were perfused with aCSF or in some cases with a medium consisting of (in mm): NaCl 140, HEPES 10, CaCl2 1, MgCl2 1, Glucose 10, KCl 5, pH 7.4. No detectable difference in current kinetics were observed between these two media. Drugs were bath-applied in the HEK cell and Spread Print preparations. All patch recordings of pyramidal cells were made in the presence of 1 μm TTX supplemented in some cases with 200 μmCd2+ and 0.1 mmCaCl2 in which MgCl2 and KCl were substituted for MgSO4 and KH2PO4, respectively. Drugs were focally ejected by applying one to five pressure pulses (10–14 psi, 100–200 msec) to the side port of an electrode holder containing a glass electrode with a tip broken back to ∼2 μm diameter (Turner et al., 1994). The carrier medium for pressure-ejected drugs consisted of (in mm): NaCl 148, KCl 3.75, CaCl21.5, MgCl2 1.5, HEPES 10, d-glucose 10, pH 7.4.
RESULTS
In the gymnotid electrosensory system, sensory input from electroreceptors is initially processed by pyramidal neurons of the medullary nucleus, the ELL. Pyramidal cell somata are positioned in a well defined cell layer, and they project extensive apical dendrites 600–800 μm into an overlying molecular layer (Maler, 1979). Outside-out patch recordings of K+channels were obtained from identified pyramidal cell somata (n = 31) and apical dendrites (n = 40) using a novel Spread Print slice preparation. The most frequently encountered K+ channel in both somata and apical dendrites up to 150 μm from the soma was a 20–25 pS channel (Fig. 1). Given the similarity in these channels in somatic and dendritic membranes, we describe their general properties without specific reference to recording location.
Fig. 1.

AptKv3.3 channels in ELL pyramidal cells.A, B, Outside-out patch recordings of K+ channels isolated from a pyramidal cell soma (A) and an apical dendrite 150 μm from the soma (B) in the presence of normal extracellular aCSF. A 7 sec depolarizing step from −80 to 0 mV produces a fast activation of K+ channels that subsequently inactivate over 1–2 sec to reveal a unitary conductance level for channels in the patch. C, An outside-out patch recording obtained from somatic membrane and stepped from a holding potential of −90 mV to the indicated potentials reveals K+ channels with a conductance of 24 pS. D, E, A macropatch K+ current in an outside-out recording isolated from a pyramidal cell apical dendrite (100 μm from soma) evoked by a depolarizing step from −100 to 0 mV for 7 sec. Outward current was blocked by 100 μm TEA (D).Inset shows single-channel recordings from another dendritic outside-out patch recording at 0 mV (100 μm from soma) with a substantial reduction of single-channel conductance by 100 μm TEA. E, After washout of TEA, outward current from the same dendritic patch in D is blocked by perfusion of 1 mm 4-AP. Currents in A,B, D, and E were leak-subtracted, and capacitance artifacts were removed by digital subtraction.
Patch formation isolates either single K+channels (one to seven channels per patch) or macropatch outward currents (more than seven single channels) of 10–120 pA for steps from −80 to 0 mV. There is an equal probability of obtaining macropatch currents from either somatic or dendritic membranes, suggesting a high density of expression in both soma and proximal apical dendrites (<150 μm). Step commands from −80 to 0 mV evoke immediate channel openings followed by inactivation over 1–2 sec to a lower open probability state (Fig. 1A,B). The rate of inactivation in macropatch recordings or ensemble averages of single-channel activity over 7 sec can be fit with a single exponential to τ = 330 ± 87 msec (n = 16). Significant recovery from inactivation requires a step command to potentials more negative than −60 mV for at least 2 sec, with the degree of recovery increasing with step potentials down to −120 mV. Recordings of single-channel activity at steady state reveal channel openings at membrane potentials more depolarized than −40 mV (Fig. 1C). Macropatch recordings of current are reduced by perfusion of <100 μm TEA (n = 6) (Fig.1D), revealing a high sensitivity to this K+ channel blocker. Analysis of single-channel amplitude reveals a TEA concentration-dependent decrease in the amplitude of transitions to the open state, with an IC50 = 78 μm (Fig.1D) (n = 5). Channel current in macropatches is also blocked by externally applied 4-AP at concentrations ≥200 μm (n = 5) (Fig. 1E).
These recordings indicate the presence of K+ channels in pyramidal cell somata and proximal apical dendrites that exhibit a high threshold for activation, a high sensitivity to TEA and 4-AP, and a conductance of ∼25 pS. All of these properties are consistent with the mammalian Kv3 class of voltage-dependent K+ channels (for review, see Rudy et al., 1999).
Molecular characterization of the Apteronotus Kv3.3 potassium channel
The properties of these K+ channels suggest the expression of one or more Kv3 channel subtypes in pyramidal cells. In a previous study, we amplified fragments specific for each of the four Kv K+ channel gene families, including Kv3, from Apteronotus genomic DNA (Rashid and Dunn, 1998). To identify the channel proteins responsible for the Kv3-like currents in pyramidal cells, Kv3-specific PCR fragments were used as probes to isolate cDNAs from an Apteronotus brain cDNA library. Five partial cDNAs encoding different Kv3 K+ channel sequences were recovered, and RNase protection studies indicated that one of these was expressed at high levels in ELL (data not shown). A full-length coding sequence for this channel predicts a protein sequence that extends for a length of 652 amino acids and is shown in Figure2A.
Fig. 2.

Molecular characterization of AptKv3.3.A, Alignment of predicted amino acid sequence of AptKv3.3 with that of the murine splice isoform mKv3.3b. The six transmembrane domains (S1–S6) and the pore domain (P) are indicated above the sequence. Also indicated are consensus sites for N-linked glycosylation (asterisks) and phosphorylation by protein kinase C (open squares), protein kinase A (filled square), and calcium/calmodulin-dependent protein kinase (filled ovals). Amino acid identities areshaded. B, Phylogenetic comparison of AptKv3.3 to members of the mammalian Kv3 family. The sequences used for comparison included splice isoforms of each Kv3 subtype and, with the exception of murine Kv3.3a and Kv3.3b, were from rat (see Materials and Methods). The phylogenetic tree demonstrates that AptKv3.3 is most related to mammalian Kv3.3. Analysis by the parsimony method was performed using the PROTPARS program in the Phylogeny Inference Package (PHYLIP) (Felsenstein, 1989). In this algorithm, the DrosophilaShaw K+ channel was used as the outgroup.
A phylogenetic comparison (Fig. 2B) and amino acid comparisons (Table 1) to the rodent Kv3 family members and their splice isoforms indicate that theApteronotus channel is most closely related to the Kv3.3 subtype. We therefore refer to the channel as AptKv3.3. The rodent Kv3.3 genes are subject to alternative RNA splicing in the gene segments encoding the intracellular C-terminal tail regions, giving rise to a series of alternatively spliced isoforms (mouse: mKv3.3a, mKv3.3b; rat: rKv3.3a, rKv3.3b, rKv3.3c). Sequence comparisons indicate that the C-terminal segment of AptKv3.3 is most similar to the carboxyl segment of mKv3.3b (Fig. 2A). The functional implications of this similarity to the carboxyl segment of mKv3.3b are unknown but may involve targeting of Kv3.3 channels to specific subcellular domains (Ponce et al., 1997).
Table 1.
AptKv3.3 amino acid sequence identities with mammalian Kv3 channels
| Subtype | % identity with Apt Kv3.3 |
|---|---|
| Kv3.1a | 58.7 |
| Kv3.1b | 64.9 |
| Kv3.2a | 57.4 |
| Kv3.2b | 61.8 |
| Kv3.2c | 57.9 |
| Kv3.2d | 57.5 |
| Kv3.3a | 63.8 |
| Kv3.3b | 71.3 |
| Kv3.4a | 60.6 |
| Kv3.4b | 57.4 |
Mammalian sequences are those used for the phylogenetic analysis (Fig. 2).
Within the central core of the channel sequence, each of the six transmembrane domains (S1–S6) and the pore domain are well conserved between Apteronotus and mammalian Kv3.3 (Fig.2A). The high sequence conservation of the S4, the S5, the pore, and the S6 domains suggests that the voltage dependencies and conductance properties of the fish channel should be similar to those reported for the rodent homologs. Outside of the transmembrane domains there is significantly less homology, one notable exception being the NAB domain (N-terminal A and B domains, AptKv3.3 amino acids 36–152), a segment of channel protein that determines Kv family-specific subunit assembly (Xu et al., 1995; Yu et al., 1996).
Two of the mammalian Kv3 subtypes, Kv3.3 and Kv3.4, contain N-terminal inactivation motifs and demonstrate fast channel inactivation when expressed in oocytes or cultured cells (Rudy et al., 1999). The amino-terminal sequence of AptKv3.3 fits closely with criteria established for fast N-type inactivation, including a string of 11 hydrophobic or uncharged residues followed by 8 hydrophilic amino acids containing four highly charged residues (K12, R14, K15, K19). These two regions together form the “ball” of the ball-and-chain model of inactivation (Hoshi et al., 1990). Although the N-terminal inactivation segment is conserved in AptKv3.3, the linker segment between the inactivation peptide and the NAB domain in AptKv3.3 (residues 20–35) is much shorter than the equivalent segment in rodent Kv3.3.
The primary sequence of AptKv3.3 suggests that it may be subject to regulation by protein phosphorylation. The AptKv3.3 sequence has consensus target sites for various protein kinases, including Ca2+-dependent phospholipid-sensitive protein kinase (PKC), cAMP-dependent protein kinase (PKA), and Ca2+/calmodulin-dependent protein kinase II (CamKII) (Fig. 2A). Seven of the nine consensus sequences for phosphorylation by PKC are conserved betweenApteronotus and mKv3.3b, including a site between S4 and S5, which is common to vertebrate K+ channels (Chandy and Gutman, 1995).
High levels of AptKv3.3 mRNA expression in electrosensory neurons
Preliminary RNase protection studies indicated that AptKv3.3 mRNA is present in brain but not in liver or skeletal muscle RNA samples (data not shown). To determine the specific cellular expression of AptKv3.3, we performed in situ hybridization analysis using a probe derived from the 3′ UTR of AptKv3.3 cDNA. The ELL has a well defined laminar organization, with the two principal classes of neurons distributed as a layer of pyramidal cells and a more ventral layer of granule cell interneurons. Figure3A–E illustrates that AptKv3.3 mRNA is expressed at high levels in both of these cell types. By comparison, mRNA is expressed at low levels in the overlying cerebellar structure, the eminentia granularis posterior (EGp). The levels of AptKv3.3 mRNA expression are similar for pyramidal and granule cells across each of four topographic maps (segments) of electroreceptor distribution in the ELL: the medial, centromedial, centrolateral, and lateral segments (Maler et al., 1991). The high levels of gene expression in ELL pyramidal cells identify AptKv3.3 as a probable candidate for the Kv3-like currents that dominate the patch-clamp recordings from pyramidal cell soma and dendrites (Fig.1).
Fig. 3.
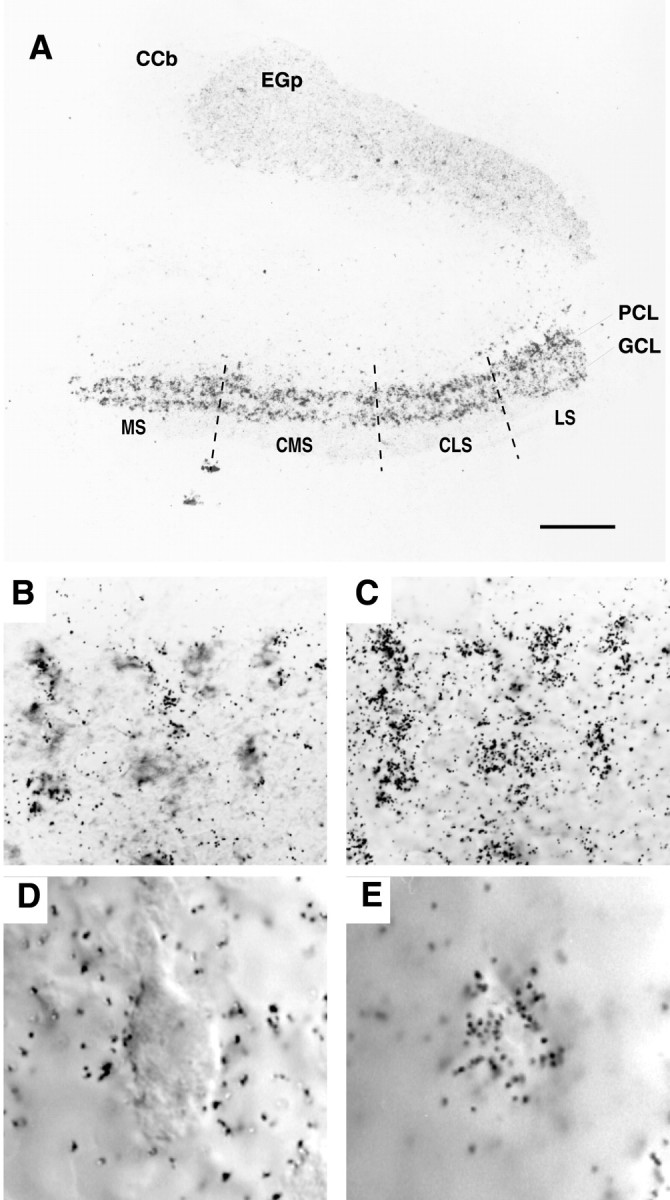
The expression of AptKv3.3 mRNA in the hindbrain. Tissue sections from hindbrain were hybridized with AptKv3.3 RNA probe. The distribution of silver grains over pyramidal and granule cell layers is indicated in A. B andC show that AptKv3.3 is expressed in all of the pyramidal cells. D and E illustrate localization of silver grains over the pyramidal cell somata.A, In situ hybridization of AptKv3.3 mRNA in the hindbrain at low power demonstrates prominent expression in the ELL and lighter expression in the adjacent caudal lobe of the cerebellum (eminentia granularis posterior; EGp) and the corpus cerebelli (CCb). The four topographic maps of the ELL are indicated: MS, medial segment;CMS, centromedial segment; CLS, centrolateral segment; LS, lateral segment. Label is dense over the entire extent of the ELL pyramidal cell (PCL) and granule cell (GCL) layers. Scale bar, 200 μm. B, A section of the pyramidal cell layer showing a cluster of pyramidal cell somata viewed under DIC optics. Scale bar, 25 μm. C, The position of silver grains in the micrograph shown in B when viewed at the plane of emulsion illustrates dense labeling positioned over individual pyramidal cell somata. D, A pyramidal cell viewed under DIC optics at higher magnification. Scale bar, 10 μm.E, Corresponding image as that shown in Dviewed at the plane of emulsion illustrates the restriction of grains to the somatic region of a pyramidal cell.
It should be noted that we have evidence for the additional expression of an AptKv3.1 mRNA in pyramidal cells (data not shown), providing the possibility for heteromeric channel formation between AptKv3.3 and AptKv3.1 proteins. However, AptKv3.1 mRNA is at a level ∼2% that of AptKv3.3 mRNA in the centromedial segment pyramidal cells examined here. Furthermore, we found no labeling in the ELL for two other Kv3 probes corresponding to putative analogs of mammalian Kv3.2 and 3.4. We can thus expect these pyramidal cell Kv3 channels to be composed predominantly of AptKv3.3 protein, an interpretation supported by the close match between the properties of native and expressed AptKv3.3 channels (Figs. 1, 5).
Fig. 5.

Expression of AptKv3.3 currents in HEK tsA201 cells. A, Whole-cell recording of currents expressed 1 d after AptKv3.3 cDNA transfection reveals an outward rectifying K+ current for command steps from −90 to 70 mV (10 mV steps after a 10 msec prepulse to −90 mV). AptKv3.3 K+ current exhibits a fast activation that reaches a characteristic early peak of ∼5 msec duration that subsequently relaxes to a steady-state current and fast deactivation on stepping back to −60 mV. Little steady-state inactivation is apparent after 60 msec. The current–voltage relationship indicates outward rectification, a high threshold for initial activation of −10 mV, and little saturation for steps up to 70 mV. B, Whole-cell AptKv3.3 currents are highly sensitive to micromole concentrations of externally applied TEA and 4-AP. C, An outside-out patch recording of AptKv3.3 single channels stepped to different steady-state potentials reveals a single channel slope conductance of 23 pS. D, An outside-out macropatch recording of AptKv3.3 current reveals a very similar response as found for whole-cell currents >100 msec (compare with A).E, An outside-out patch recording of AptKv3.3 single channels indicates a fast and maximal activation of channels over the initial 200 msec followed by inactivation during a 7 sec depolarizing command step from −100 to 0 mV. Capacitance artifacts inD and E were digitally subtracted.
ApKv3.3 protein localizes to dendrites and somata of ELL pyramidal cells
To investigate the subcellular distribution of AptKv3.3, a polyclonal antibody (α-AptKv3.3) was generated against the C-terminal region of AptKv3.3 (amino acids 562–634). In Western blot experiments, α-AptKv3.3 recognizes a single protein band of ∼87 kDa in brain membrane fractions with no signal in the corresponding soluble protein fraction (Fig. 4E). Comparison of this value with the predicted molecular weight of 72.8 kDa suggests that the channel is glycosylated at the three consensus sites for N-linked glycosylation located between transmembrane segments S1 and S2 (Fig. 2A).
Fig. 4.
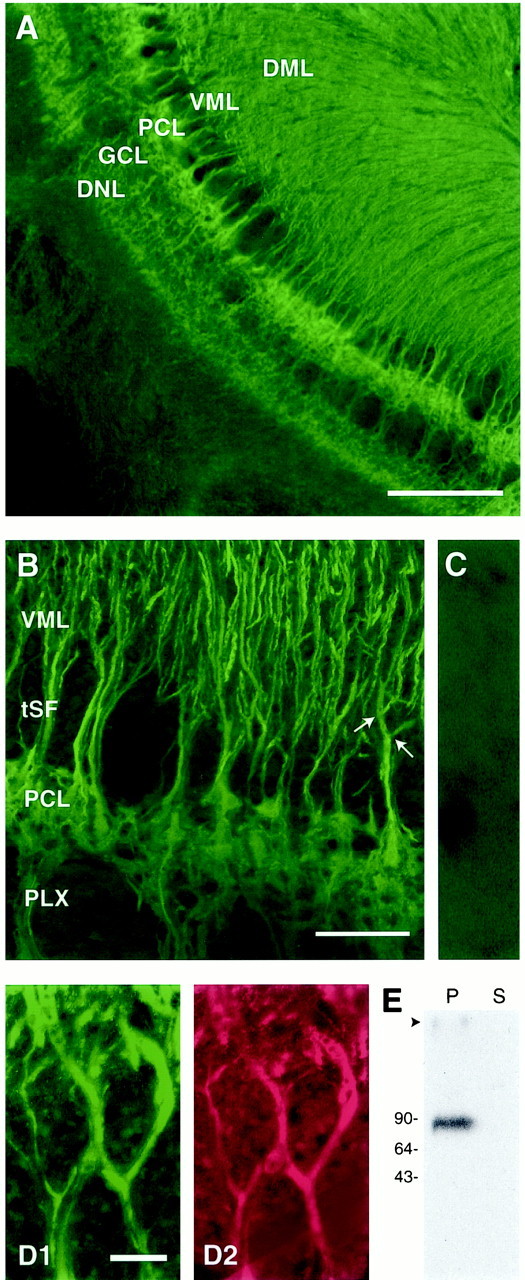
AptKv3.3 protein is localized to somata and dendrites of ELL pyramidal cells. Tissue sections of hindbrain were stained with α-AptKv3.3 antibody (A–D) or double-labeled with MAP-2 monoclonal antibody (D). The specificity of the α-AptKv3.3 antibody is indicated in a Western blot (E) and tissue section (C). A andB show that AptKv3.3 protein is distributed throughout the pyramidal cell somatic and dendritic domains. Colocalization of AptKv3.3 with MAP2 in apical dendrites is shown for pyramidal cells from the medial segment (D). A, Low-power micrograph of α-AptKv3.3-immunolabeled hindbrain. Intense label is seen throughout the ELL, particularly the pyramidal cell layer (PCL), granule cell layer (GCL), and deep neuropil layer (DNL). Note also the dense label in the ventral (VML) and dorsal (DML) molecular layers that overlie the PCL and contain pyramidal cell apical dendritic projections. Scale bar, 400 μm. B, Higher-magnification confocal image of the ELL pyramidal cell layer in the centrolateral segment illustrating immunolabeling of pyramidal cell somata and apical dendrites. Labeling of apical dendrites remains constant in intensity past primary and secondary branchpoints (indicated with arrows). Note the lack of immunolabel in the tractus stratus fibrosum (tSF) and plexiform (PLX) layers, which both contain dense axonal fascicles. Scale bar, 100 μm. C, A control section from centrolateral segment viewed at the magnification used forB. This section was treated with α-AptKv3.3 that had been preadsorbed with the AptKv3.3 fusion protein. Only a diffuse background signal is detected. D, High-magnification image of two medial segment apical dendrites colabeled with α-AptKv3.3 (D1) and MAP-2a,b (D2) antibodies. AptKv3.3 protein appears to be localized exclusively to MAP-2-containing dendritic structures. Scale bar, 10 μm.E, Western blot analysis demonstrates that α-AptKv3.3 recognizes a single protein of ∼87 kDa in the brain membrane fraction (P) but not the corresponding soluble protein fraction (S).
Immunohistochemical analysis reveals a striking distribution of AptKv3.3 protein in the ELL pyramidal and granule cell body layers, as well as throughout the entire extent of the molecular layer, where pyramidal and granule cells project extensive apical dendrites (Fig.4A,B). Control sections with the secondary antibody alone or α-AptKv3.3 preadsorbed with a 100-fold excess of fusion protein show only low levels of nonspecific background staining (Fig. 4C). Because our electrophysiological recordings focus on pyramidal cells (Fig. 1), we will restrict our cytochemical analysis primarily to pyramidal cell immunolabel.
Higher magnification reveals AptKv3.3 protein on pyramidal cell bodies (Fig. 4B), apical dendrites in the molecular layer (Fig. 4B,D1), and basilar dendrites that extend ventrally through the deep neuropil layer (Fig.4B). The high density of antigenic sites presents a continuous image of labeled structures, such that individual apical dendrites can be tracked from the soma, through the proximal dendritic shaft, and beyond secondary and tertiary dendritic branch points (Fig.4D1). In these sections, costaining with an antibody to the microtubule-associated protein MAP2 confirms the presence of AptKv3.3 out to the most distal aspects of the pyramidal cell apical dendrites (Fig. 4D2). Similarly, α-AptKv3.3 immunofluorescence labels individual basilar dendrites from the soma to the final extension of a basilar bush that receives primary afferent input 300–400 μm from the cell layer (Fig.4A,B).
Pyramidal cells extend axons that course medially in a plexiform layer immediately beneath the pyramidal cell layer. As apparent in Figure4B, AptKv3.3 immunoreactivity is absent in the axon fascicles of the plexiform layer, providing a stark contrast to the pyramidal cell basilar dendrites projecting through this region. Although the apparent lack of immunolabel of these structures is suggestive, further distinctions on axonal or presynaptic distributions need to be made at the ultrastructural level.
AptKv3.3 channels expressed in HEK cells resemble K+ channels in native pyramidal cells
To determine whether AptKv3.3 channel properties match those found in ELL pyramidal cells, we transfected HEK tsA201 cells with an expression vector for AptKv3.3 cDNA. Whole-cell recordings of transfected cells reveal an outward rectifying K+ current of 5–10 nA with a characteristic early peak that subsequently relaxed to a steady-state current within ∼5 msec (Fig.5A). AptKv3.3 whole-cell current was initially detected at a relatively high voltage (−20 to −10 mV) with a voltage for half-activation ofV0.5 = 15.6 ± 5.05 (Fig.5A) (n = 13). Activation and deactivation are fast, reaching the early peak of the outward current in 2.0 ± 0.64 msec for a command step from −90 to 50 mV (n = 27) and deactivating with τ = 0.6 ± 0.19 msec (n = 22) for voltage steps from 50 to −60 mV (Fig. 5A). AptKv3.3 currents are highly sensitive to both TEA and 4-AP, with substantial block obtained at concentrations >50 μm (Fig. 5B). Whole-cell currents exhibit little inactivation over 50–100 msec for voltage commands up to 70 mV (Fig. 5A). Command pulses of 1–7 sec invoked a voltage-dependent steady-state inactivation, although the voltage and time required to initiate this process were variable (data not shown).
Outside-out recordings reveal outward rectifying K+ channels with transitions to the open state detectable between −40 and 50 mV under steady-state conditions. The single-channel conductance calculated in either on-cell or outside-out recordings is 22.9 ± 0.53 pS (Fig. 5C) (n = 16). As found for whole-cell currents, AptKv3.3 single-channel conductance is reduced by low concentrations of TEA, with an IC50 = 40 μm(n = 4). Outside-out macropatch currents resemble whole-cell currents in exhibiting an early peak followed by a relaxation to a lower steady state with little subsequent inactivation during 50–100 msec step commands (Fig. 5D). Steady-state inactivation with longer duration pulses was more pronounced and more consistent in outside-out recordings than for whole-cell currents. An example is shown in Figure 5E, where inactivation is prominent over a 7 sec time frame for a step from −100 to 0 mV. Inactivation in macropatch recordings with depolarizing steps from −90 or −100 mV to 0 mV is fit by a single exponential of τ = 366 ± 84 msec (n = 9). Significant recovery from inactivation requires a return to holding potentials of at least −60 mV.
The voltage ranges for activation, single-channel conductance, and high TEA and 4-AP sensitivity of AptKv3.3 channels are in agreement with those reported for mammalian Kv3 channels (Vega-Saenz de Miera et al., 1992; Critz et al., 1993; Kanemasa et al., 1995; Rudy et al., 1999). Most importantly, these properties match the K+ channel recordings that we obtained in native ELL pyramidal cell somata and apical dendrites when recorded under the same ionic conditions (Fig. 1). Together with the cytochemical localization of AptKv3.3 (Fig. 4), our results provide compelling evidence that the AptKv3.3 channel subtype contributes prominently to the K+ channel activity recorded in pyramidal cell somata and apical dendrites.
AptKv3.3 channels are involved in spike repolarization and establishment of burst threshold in pyramidal cells
The functional significance of AptKv3.3 channels to spike discharge was assessed by focally ejecting TEA or 4-AP in the immediate vicinity of dendritic or somatic recordings in a conventional in vitro slice preparation, an approach that restricts drug ejections to a limited region of the cell axis (Turner et al., 1994). Focal ejections of either 1 mm TEA or 4-AP rapidly decreased the rate of spike repolarization in both somatic and dendritic locations (Fig. 6). This change in repolarization increases the half-width of somatic spikes by 46.5 ± 33% (n = 8) (Fig. 6A) and the half-width of dendritic spikes by 20.3 ± 9.8% (n = 6) (Fig. 6B), with a slight increase in spike amplitude in somatic recordings. It is important to note that diffusion and dilution of ejected drugs in the extracellular medium will result in effective concentrations that are substantially lower than the initial ejected medium. The immediate results obtained with 1 mm concentrations of these blockers thus indicate that K+ channels underlying spike repolarization are highly sensitive to TEA and 4-AP, as found for Kv3 K+ channels (Rudy et al., 1999). However, low concentrations of TEA and 4-AP can also affect large conductance (BK) Ca2+-activated K+ channels (Coetzee et al., 1999). Although BK K+ channels are present in both somatic and dendritic membranes of pyramidal cells, we have determined that BK channel conductance is reduced by bath-applied TEA only at concentrations >1 mm (E. Morales and R. W. Turner, unpublished observations). In addition, somatic and dendritic spikes proved insensitive to Cd2+ ejections (200–400 μm; n = 10), suggesting that BK K+ channels do not contribute to spike repolarization in pyramidal cells (data not shown). The Kv1 (Shaker) class of K+ channels is also sensitive to low concentrations of TEA (Coetzee et al., 1999). However, no effects are observed with focal ejection of 2 μmdendrotoxin in either somatic or dendritic regions (n = 5), suggesting that spike repolarization does not involve this class of K+ channels. These data strongly suggest that AptKv3.3 current contributes to the repolarization of both somatic and dendritic spikes in pyramidal cells. Nevertheless, it is important to note that spike half-widths could be increased in a dose-dependent manner by TEA or 4-AP concentrations of up to 5 mm, suggesting that additional K+ channel subtypes may contribute to spike repolarization.
Fig. 6.

Somatic and dendritic spike repolarization is highly sensitive to TEA and 4-AP. A, B, Schematic diagrams of pyramidal cells are shown to indicate the placement of a stimulating electrode in the plexiform layer for antidromic activation (double wires), intracellular recording electrodes (white fill), and pressure electrodes for focal drug ejections (shaded fill) in an in vitro ELL slice preparation. Focal pressure ejection of either TEA or 4-AP (1 mm) in the immediate region of separate (A) somatic or (B) dendritic recordings slows the rate of repolarization and increases the duration of antidromic spikes. Control and test responses are shown superimposed, with test responses identified as thick gray traces.
It has been established that ELL pyramidal cells discharge bursts of action potentials in vivo to encode specific features of electrosensory input, revealing a direct role in feature extraction (Gabbiani et al., 1996). These bursts arise through a process of “conditional spike backpropagation,” a newly recognized aspect of spike conduction that generates burst discharge in the γ-frequency range (20–80 Hz) (Lemon and Turner, 2000). Burst discharge is initiated when the long duration of a backpropagating dendritic Na+ spike generates a DAP at the soma (Turner et al., 1994). During repetitive discharge, dendritic spikes exhibit a frequency-dependent broadening that increases current flow back to the soma to potentiate the DAP. The increase in somatic depolarization eventually triggers a high-frequency spike doublet at the soma that exceeds the dendritic spike refractory period. As a result, spike backpropagation into dendrites abruptly fails, removing the dendritic depolarization driving the burst from one spike to the next, allowing a burst afterhyperpolarization (AHP) to terminate the burst (Lemon and Turner, 2000). A change in the rate of dendritic spike repolarization during repetitive discharge is thus a critical aspect of K+ channel function in pyramidal cells. Given the role for AptKv3.3 K+ channels in repolarizing dendritic spikes, one would predict that a reduction in dendritic AptKv3.3 conductance could directly modulate burst discharge by augmenting the current flow underlying the somatic DAP.
To test whether such a mechanism could affect burst generation, we examined the effects of focally ejecting TEA or 4-AP in dendritic regions on the pattern of spike output. In the first set of experiments, we recorded the antidromic somatic spike and DAP and current-evoked spike discharge when set below threshold for burst output (Fig. 7B). Focal ejections of 1 mm TEA or 4-AP in the dendritic region selectively increases the amplitude of the somatic DAP on average 116 ± 108% (n = 5; range, 41–274%) and immediately converts cell output from one of tonic to burst discharge (Fig. 7C). The lack of any change in somatic spike repolarization in these experiments confirms that drug actions were restricted to the dendritic region. We then examined the effects of these drugs on dendritic spike discharge by focally ejecting TEA (n = 6) or 4-AP (n = 5) in the immediate region of dendritic recordings. This slows dendritic spike repolarization and shifts current-evoked spike output from tonic discharge to an oscillatory series of spike bursts (Fig.7D–F). These studies reveal that reducing the ability of AptKv3.3 K+ channels to repolarize dendritic spikes immediately potentiates the somatic DAP and lowers the threshold for generating burst discharge.
Fig. 7.

Dendritic spike repolarization controls burst threshold. A–C, The effects of dendritic 4-AP ejection on somatic spike discharge. A, Schematic diagram of a pyramidal cell to indicate the placement of a stimulating electrode for antidromic activation (double wires), an intrasomatic recording electrode (white fill), and a pressure electrode for focal drug ejection of 2 mm 4-AP (shaded fill) in anin vitro ELL slice preparation. B,C, The effects of dendritic 4-AP ejection on somatic spike discharge. B, Control intrasomatic recordings of antidromic spike discharge and associated DAP (open arrow) and current-evoked spike discharge when set below threshold for generating oscillatory spike bursts. C, Focal ejection of 4-AP to apical dendrites selectively enhances the somatic DAP (open arrow), as shown by superimposition of the control and test antidromic response (test response shown bygray trace). The lack of any change in somatic spike repolarization confirms that the drug was restricted to the dendritic region. This is sufficient to convert cell output from a tonic to bursting pattern, as indicated by a repeating series of spike bursts. Burst period is indicated by solid arrows designating the occurrence of burst afterhyperpolarizations (burst AHPs). D, Schematic diagram of a pyramidal cell indicates an intradendritic recording in another pyramidal cell (white filled electrode) and pressure electrode for focal ejection of 2 mm 4-AP (shaded fill). E, F, The effects of dendritic 4-AP ejection on dendritic spike discharge.E, Control intradendritic recordings showing current-evoked spike discharge set below threshold for generating spike bursts. Insets to the left inE and F show expanded views of the first current-evoked spikes in control and test recordings (asterisks). F, Focal ejection of 4-AP in the dendritic region broadens the dendritic spike by slowing spike repolarization and shifts cell output from a tonic to bursting pattern.Solid arrows indicate the occurrence of burst AHPs that terminate each spike burst. Inset shows the control and test responses superimposed (test response shown by gray trace). Excitation after dendritic 4-AP results in burst discharge composed of a repeating series of spike doublets.
DISCUSSION
This study describes the distribution and functional roles of a Kv3 K+ channel subtype, AptKv3.3, in a principal sensory neuron. AptKv3.3 proves to be unique among the Kv3 K+ channel family in exhibiting an extensive localization to dendritic membranes. In dendrites, these channels act to repolarize spikes backpropagating from the somatic region, and in so doing, regulate the threshold for a type of burst discharge that has an established role in sensory processingin vivo.
Functional role of dendritic AptKv3.3 channels
K+ currents localized to dendritic membranes are increasingly recognized for their ability to shape signal processing (Hoffman et al., 1997). In only a few cases have specific channel subtypes been associated with dendritic K+ currents that have an identified role in modulating cell output (Magee et al., 1998). We have now shown that AptKv3.3 K+ channels are localized to both somata and apical dendrites of ELL pyramidal cells, a conclusion supported by both patch recordings and dense immunolabel over the entire soma–dendritic axis. Our data suggest that AptKv3.3 channels are distributed with essentially equivalent densities over the soma and proximal apical dendrites out to at least 150 μm, although further mapping will be required to rigorously establish channel densities. It is important to emphasize that the extensive dendritic distribution of AptKv3.3 channels reported here is entirely novel among the Kv3 class, because all previous immunolocalization studies have identified only somatic, axonal, and a limited extent of dendritic membranes (≈20 μm) as the distribution target for Kv3.1, 3.2, and 3.4 K+ channels (Weiser et al., 1995; Du et al., 1996; Perney and Kaczmarek, 1997; Sekirnjak et al., 1997). The distribution of Kv3.3 K+ channel proteins has not been examined previously.
In other cells, Kv3 channels contribute to spike repolarization and facilitate recovery from Na+ channel inactivation, helping to maintain high frequencies of spike discharge (Rudy et al., 1999). A similar role in spike repolarization is served by AptKv3.3 channels in ELL pyramidal cells at both the somatic and dendritic levels in ELL pyramidal cells. The most significant consequence of dendritic localization of AptKv3.3 channels is the control over a form of burst discharge known to be involved in feature extraction in vivo (Gabbiani et al., 1996). Through their ability to control dendritic spike repolarization, AptKv3.3 channels have a pronounced effect on burst discharge. Thus, focal dendritic ejections of TEA or 4-AP that block AptKv3.3 channels immediately increased DAP amplitude at the soma and shifted cell output from tonic to burst discharge. This demonstrates that the contribution by AptKv3.3 channels to repolarizing dendritic spikes establishes a specific threshold for γ-frequency burst discharge in pyramidal cells.
A characteristic of the burst response in ELL pyramidal cells is a progressive increase in the DAP resulting from a frequency-dependent broadening of dendritic spikes during repetitive discharge (Lemon and Turner, 2000). Although the mechanism underlying the change in dendritic spike duration remains to be determined, our data indicate that a decrease in dendritic AptKv3.3 currents would effectively broaden dendritic spikes. In this regard, it is interesting that Kv3.4 channel kinetics can be controlled by the degree of phosphorylation (Covarrubias et al., 1994) such that dephosphorylation is predicted to increase the rate of inactivation. If a similar action is found for AptKv3.3 channels, it would produce a state conducive to cumulative inactivation during repetitive discharge: decreasing the ability of AptKv3.3 channels to repolarize dendritic spikes.
We found that AptKv3.3 channels are distributed over the entire extent of the apical dendritic axis. This was somewhat unexpected for a K+ channel activated by high voltage because Na+ spike backpropagation is believed to occur only over the proximal third of the dendritic tree (Turner et al., 1994). The high threshold for activation of AptKv3.3 channels would appear to preclude a direct modulation of synaptic potentials. However, it is possible that under some conditions active membrane depolarizations may be triggered in more distal dendritic regions and be shaped by AptKv3.3 channels in a manner similar to dendritic K+ channels in other cells (Golding et al., 1999; Magee and Carruth, 1999). The functional role for AptKv3.3 channels in these distal dendritic regions will be an important area for future investigation.
Properties of AptKv3.3 channels
AptKv3.3 is the first member of the teleost Kv3 family of K+ channel α-subunits for which a complete amino acid sequence is known. There is remarkable sequence similarity with mammalian Kv3 channels, particularly within the six transmembrane domains and the pore domain. The fact that theXenopus Kv3.1 channel also shows high sequence similarity to mammalian Kv3.1 (Gurantz et al., 2000) confirms that the Kv3 amino acid sequences have been well maintained through vertebrate evolution. Phylogenetic analysis indicates that the apteronotid channel is most similar to the mammalian subtype Kv3.3, and moreover, the C terminus of AptKv3.3 is most similar to the mouse splice isoform mKv3.3b (Fig.2A). Interestingly, the C-terminal sequence of AptKv3.3 (-SIL) resembles the consensus sequences that bind to the PSD-95, Dlg-1, Zho-1 (PDZ) protein–protein interaction domains that are found on families of subcellular localizing proteins (Kim et al., 1995; Kim and Sheng, 1996; Songyang et al., 1997). It is possible that interactions with one of these PDZ domain proteins is responsible for the extensive dendritic targeting of AptKv3.3 channels.
A unique feature of mammalian Kv3 channels is their requirement for membrane voltages exceeding −20 mV for activation. This high-voltage dependence restricts Kv3 channel activation to depolarized potentials where they can mediate the repolarization of the action potentials without limiting the voltage-dependent conductances near resting potential (Kanemasa et al., 1995; Weiser et al., 1995; Du et al., 1996;Wang et al., 1998; Erisir et al., 1999). This role in spike repolarization is further assisted by the characteristically rapid activation and deactivation kinetics of Kv3 channels. AptKv3.3 channels expressed in heterologous cells display each of these key properties, with a high voltage for activation (approximately −20 mV) and fast activation–deactivation kinetics. Importantly, these properties were shared by the AptKv3.3 currents recorded in ELL pyramidal cell somata and apical dendrites, a comparison facilitated by recording under the same ionic conditions.
The primary distinction between mammalian Kv3 subtypes is the rate of steady-state inactivation, with the degree of Kv3.3 inactivation falling between the noninactivating Kv3.1 and 3.2 isoforms and the fast inactivating Kv3.4 channel (Rudy et al., 1999). The rate of AptKv3.3 inactivation during short step commands under whole-cell recording conditions in HEK cells (50 mV, 100 msec) was comparable to reported values for mammalian Kv3.3 (Vega-Saenz de Miera et al., 1994; Rae and Shepard, 2000). However, during longer step commands (1–7 sec) in whole-cell recording mode, the voltage dependence and rate of inactivation of AptKv3.3 was highly variable. In contrast, with outside-out patch recordings the rate of AptKv3.3 inactivation in response to similar steady-state commands was more consistent and also substantially increased. This may relate to the degree of washout of cytoplasmic constituents in the outside-out recording mode, reducing the ability of soluble second messengers to modulate the AptKv3.3 channel. Indeed, the rate of steady-state inactivation of AptKv3.3 channels isolated from HEK cells or pyramidal cells in outside-out recordings was essentially equivalent (Figs.1A,B,4E).
N-type inactivation of K+ channels depends on the presence of an inactivation motif at the amino terminus of the protein (Hoshi et al., 1990). This motif is conserved between AptKv3.3 and the two mammalian Kv3 channel subtypes that exhibit inactivation, Kv3.3 and Kv3.4. Specifically, the amino terminus of AptKv3.3 consists of a string of 11 hydrophobic or uncharged residues followed by a segment of 8 amino acids containing 4 highly charged residues, consistent with the proposed requirements for N-type inactivation (Hoshi et al., 1990; Zagotta et al., 1990). AptKv3.3 also contains a critical cysteine in the amino terminus (Fig. 2A, position 6) that has been shown to be important for inactivation in rodent Kv3.3 and Kv3.4 (Ruppersberg et al., 1991; Rudy et al., 1999). However, the portion of AptKv3.3 that links the inactivation motif to the rest of the channel, the so-called “chain” in the ball-and-chain model of inactivation, is much shorter than that found in mammalian Kv3.3. Although this would predict a faster rate of inactivation (Hoshi et al., 1990), our measured rate of AptKv3.3 inactivation is somewhat slower than mammalian channels when compared under equivalent recording conditions. The reason for this is unknown, but it indicates that the length of the linker peptide is not the limiting factor for N-type inactivation in AptKv3.3 channels and may well signify intracellular regulation of the inactivation mechanism.
We have established that dendritic AptKv3.3 channels contribute to determining the threshold for generating γ-frequency burst discharge in ELL pyramidal cells. Spike bursts are recorded in pyramidal cellsin vivo, and discharge is recorded in direct relation to relevant features of external sensory input (Gabbiani et al., 1996;Metzner et al., 1998). The known regulation of Kv3 K+ channels by second messengers provides a potential route by which modulation of AptKv3.3 channels could shape burst characteristics and contribute to feature detection during electrolocatory behavior in the live animal.
Footnotes
This research was supported by grants from the Medical Research Council to R.J.D. and R.W.T. and the National Engineering and Science Research Council to R.J.D. R.W.T. is an AHFMR Senior Scholar. We thank B. Ellis and S. Sinclair for technical assistance, E. Harvey-Girard and C. Doering for assistance with DNA transfections, and Rejean Munger and Len Maler for assistance with confocal microscopy.
Correspondence should be addressed to Dr. Robert J. Dunn, Department of Neurology, Montreal General Hospital, 1650 Cedar Avenue, Montreal, Quebec, Canada, H3G 1A4. E-mail:mc81@musica.mcgill.ca.
REFERENCES
- 1.Berman NJ, Maler L. Neural architecture of the electrosensory lateral line lobe: adaptations for coincidence detection, a sensory searchlight and frequency-dependent adaptive filtering. J Exp Biol. 1999;202:1243–1253. doi: 10.1242/jeb.202.10.1243. [DOI] [PubMed] [Google Scholar]
- 2.Bottai D, Dunn RJ, Ellis W, Maler L. N-methyl-d-aspartate receptor 1 mRNA distribution in the central nervous system of the weakly electric fish Apteronotus leptorhychus. J Comp Neurol. 1997;389:65–80. doi: 10.1002/(sici)1096-9861(19971208)389:1<65::aid-cne5>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 3.Bottai D, Maler L, Dunn RJ. Alternative RNA splicing of the NMDA receptor NR1 mRNA in the neurons of the teleost electrosensory system. J Neurosci. 1998;18:5191–5202. doi: 10.1523/JNEUROSCI.18-14-05191.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chandy KG, Gutman GA. Voltage gated channels. In: North RA, editor. Handbook of receptors and channels: ligand-gated and voltage-gated ion channels. CRC; Boca Raton, FL: 1995. pp. 1–71. [Google Scholar]
- 5.Chen WR, Midtgaard J, Shepherd GM. Forward and backward propagation of dendritic impulses and their synaptic control in mitral cells. Science. 1997;278:463–467. doi: 10.1126/science.278.5337.463. [DOI] [PubMed] [Google Scholar]
- 6.Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B. Molecular diversity of K+ channels. Ann NY Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- 7.Covarrubias M, Wei A, Salkoff L, Vyas TB. Elimination of rapid potassium channel inactivation by phosphorylation of the inactivation gate. Neuron. 1994;13:1403–1412. doi: 10.1016/0896-6273(94)90425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Critz SD, Wible BA, Lopez HS, Brown AM. Stable expression and regulation of a rat brain K+ channel. J Neurochem. 1993;60:1175–1178. doi: 10.1111/j.1471-4159.1993.tb03273.x. [DOI] [PubMed] [Google Scholar]
- 9.Du J, Zhang L, Weiser M, Rudy B, McBain CJ. Developmental expression and functional characterization of the potassium-channel subunit Kv3.1b in parvalbumin-containing interneurons of the rat hippocampus. J Neurosci. 1996;16:506–518. doi: 10.1523/JNEUROSCI.16-02-00506.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erisir A, Lau D, Rudy B, Leonard CS. Function of specific K(+) channels in sustained high-frequency firing of fast-spiking neocortical interneurons. J Neurophysiol. 1999;82:2476–2489. doi: 10.1152/jn.1999.82.5.2476. [DOI] [PubMed] [Google Scholar]
- 11.Felsenstein J. PHYLIP: phylogeny inference package (Version 3.2). Cladistics. 1989;5:164–166. [Google Scholar]
- 12.Frangioni JV, Neel BG. Solubilization and purification of enzymatically active glutathione S-transferase (pGEX) fusion proteins. Anal Biochem. 1993;210:179–187. doi: 10.1006/abio.1993.1170. [DOI] [PubMed] [Google Scholar]
- 13.Gabbiani F, Metzner W, Wessel R, Koch C. From stimulus encoding to feature extraction in weakly electric fish. Nature. 1996;384:564–567. doi: 10.1038/384564a0. [DOI] [PubMed] [Google Scholar]
- 14.Golding NL, Jung HY, Mickus T, Spruston N. Dendritic calcium spike initiation and repolarization are controlled by distinct potassium channel subtypes in CA1 pyramidal neurons. J Neurosci. 1999;19:8789–8798. doi: 10.1523/JNEUROSCI.19-20-08789.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gurantz D, Lautermilch NJ, Watt SD, Spitzer NC. Sustained upregulation in embryonic spinal neurons of a Kv3.1 potassium channel gene encoding a delayed rectifier current. J Neurobiol. 2000;42:347–356. [PubMed] [Google Scholar]
- 16.Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- 17.Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 18.Kanemasa T, Gan L, Perney TM, Wang LY, Kaczmarek LK. Electrophysiological and pharmacological characterization of a mammalian Shaw channel expressed in NIH 3T3 fibroblasts. J Neurophysiol. 1995;74:207–217. doi: 10.1152/jn.1995.74.1.207. [DOI] [PubMed] [Google Scholar]
- 19.Kim E, Sheng M. Differential K+ channel clustering activity of PSD-95 and SAP97, two related membrane-associated putative guanylate kinases. Neuropharmacology. 1996;35:993–1000. doi: 10.1016/0028-3908(96)00093-7. [DOI] [PubMed] [Google Scholar]
- 20.Kim E, Niethammer M, Rothschild A, Jan YN, Sheng M. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature. 1995;378:85–88. doi: 10.1038/378085a0. [DOI] [PubMed] [Google Scholar]
- 21.Kotecha SA, Eley DW, Turner RW. Tissue printed cells from teleost electrosensory and cerebellar structures. J Comp Neurol. 1997;386:277–292. [PubMed] [Google Scholar]
- 22.Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999;398:338–341. doi: 10.1038/18686. [DOI] [PubMed] [Google Scholar]
- 23.Lemon N, Turner RW. Conditional backpropagation generates burst discharge in a sensory neuron. J Neurophysiol. 2000;89:1519–1530. doi: 10.1152/jn.2000.84.3.1519. [DOI] [PubMed] [Google Scholar]
- 24.Magee J, Hoffman D, Colbert C, Johnston D. Electrical and calcium signaling in dendrites of hippocampal pyramidal neurons. Annu Rev Physiol. 1998;60:327–346. doi: 10.1146/annurev.physiol.60.1.327. [DOI] [PubMed] [Google Scholar]
- 25.Magee JC, Carruth M. Dendritic voltage-gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1999;82:1895–1901. doi: 10.1152/jn.1999.82.4.1895. [DOI] [PubMed] [Google Scholar]
- 26.Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- 27.Maler L. The posterior lobe of certain gymnotoid fish: quantitative light microscopy. J Comp Neurol. 1979;183:323–363. doi: 10.1002/cne.901830208. [DOI] [PubMed] [Google Scholar]
- 28.Maler L, Sas E, Johnston S, Ellis W. An atlas of the brain of the electric fish Apteronotus leptorhynchus. J Chem Neuroanat. 1991;4:1–38. doi: 10.1016/0891-0618(91)90030-g. [DOI] [PubMed] [Google Scholar]
- 29.Margolskee RF, McHendry-Rinde B, Horn R. Panning transfected cells for electrophysiological studies. Biotechniques. 1993;15:906–911. [PubMed] [Google Scholar]
- 30.Metzner W, Koch C, Wessel R, Gabbiani F. Feature extraction by burst-like spike patterns in multiple sensory maps. J Neurosci. 1998;18:2283–2300. doi: 10.1523/JNEUROSCI.18-06-02283.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perney TM, Kaczmarek LK. Localization of a high threshold potassium channel in the rat cochlear nucleus. J Comp Neurol. 1997;386:178–202. [PubMed] [Google Scholar]
- 32.Ponce A, Vega-Saenz de Miera E, Kentros C, Moreno H, Thornhill B, Rudy B. K+ channel subunit isoforms with divergent carboxy-terminal sequences carry distinct membrane targeting signals. J Membr Biol. 1997;159:149–159. doi: 10.1007/s002329900278. [DOI] [PubMed] [Google Scholar]
- 33.Rae JL, Shepard AR. Kv3.3 potassium channels in lens epithelium and corneal endothelium. Exp Eye Res. 2000;70:339–348. doi: 10.1006/exer.1999.0796. [DOI] [PubMed] [Google Scholar]
- 34.Rashid AJ, Dunn RJ. Sequence diversity of voltage-gated potassium channels in an electric fish. Mol Brain Res. 1998;54:101–107. doi: 10.1016/s0169-328x(97)00328-8. [DOI] [PubMed] [Google Scholar]
- 35.Rudy B, Chow A, Lau D, Amarillo Y, Ozaita A, Saganich M, Moreno H, Nadal MS, Hernandez-Pineda R, Hernandez-Cruz A, Erisir A, Leonard C, Vega-Saenz de Miera E. Contributions of Kv3 channels to neuronal excitability. Ann NY Acad Sci. 1999;868:304–343. doi: 10.1111/j.1749-6632.1999.tb11295.x. [DOI] [PubMed] [Google Scholar]
- 36.Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M. Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature. 1991;352:711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- 37.Sekirnjak C, Martone ME, Weiser M, Deerinck T, Bueno E, Rudy B, Ellisman M. Subcellular localization of the K+ channel subunit Kv3.1b in selected rat CNS neurons. Brain Res. 1997;766:173–187. doi: 10.1016/s0006-8993(97)00527-1. [DOI] [PubMed] [Google Scholar]
- 38.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 39.Spruston N, Schiller Y, Stuart G, Sakmann B. Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science. 1995;268:297–300. doi: 10.1126/science.7716524. [DOI] [PubMed] [Google Scholar]
- 40.Turner RW, Meyers DE, Richardson TL, Barker JL. The site for initiation of action potential discharge over the somatodendritic axis of rat hippocampal CA1 pyramidal neurons. J Neurosci. 1991;11:2270–2280. doi: 10.1523/JNEUROSCI.11-07-02270.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner RW, Maler L, Deerinck T, Levinson SR, Ellisman MH. TTX-sensitive dendritic sodium channels underlie oscillatory discharge in a vertebrate sensory neuron. J Neurosci. 1994;14:6453–6471. doi: 10.1523/JNEUROSCI.14-11-06453.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turner RW, Plant JR, Maler L. Oscillatory and burst discharge across electrosensory topographic maps. J Neurophysiol. 1996;76:2364–2382. doi: 10.1152/jn.1996.76.4.2364. [DOI] [PubMed] [Google Scholar]
- 43.Vega-Saenz de Miera E, Moreno H, Fruhling D, Kentros C, Rudy B. Cloning of ShIII (Shaw-like) cDNAs encoding a novel high-voltage-activating, TEA-sensitive, type-A K+ channel. Proc R Soc Lond B Biol Sci. 1992;248:9–18. doi: 10.1098/rspb.1992.0036. [DOI] [PubMed] [Google Scholar]
- 44.Vega-Saenz de Miera E, Weiser M, Kentros C, Lau D, Moreno H, Serodio P, Rudy B. Shaw-related K+ channels in mammals. In: Peracchia C, editor. Handbook of membrane channels. Academic; New York: 1994. pp. 41–78. [Google Scholar]
- 45.Wang LY, Gan L, Forsythe ID, Kaczmarek LK. Contribution of the Kv3.1 potassium channel to high-frequency firing in mouse auditory neurones. J Physiol (Lond) 1998;509:183–194. doi: 10.1111/j.1469-7793.1998.183bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weiser M, Bueno E, Sekirnjak C, Martone ME, Baker H, Hillman D, Chen S, Thornhill W, Ellisman M, Rudy B. The potassium channel subunit KV3.1b is localized to somatic and axonal membranes of specific populations of CNS neurons. J Neurosci. 1995;15:4298–4314. doi: 10.1523/JNEUROSCI.15-06-04298.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu J, Yu W, Jan YN, Jan LY, Li M. Assembly of voltage-gated potassium channels. Conserved hydrophilic motifs determine subfamily-specific interactions between the alpha-subunits. J Biol Chem. 1995;270:24761–24768. doi: 10.1074/jbc.270.42.24761. [DOI] [PubMed] [Google Scholar]
- 48.Yu W, Xu J, Li M. NAB domain is essential for the subunit assembly of both alpha-alpha and alpha-beta complexes of shaker-like potassium channels. Neuron. 1996;16:441–453. doi: 10.1016/s0896-6273(00)80062-8. [DOI] [PubMed] [Google Scholar]
- 49.Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]