

Abstract
Although vitamin D hormone (VDH; 1,25-dihydroxyvitamin D3), the active metabolite of vitamin D, is the major Ca2+-regulatory steroid hormone in the periphery, it is not known whether it also modulates Ca2+ homeostasis in brain neurons. Recently, chronic treatment with VDH was reported to protect brain neurons in both aging and animal models of stroke. However, it is unclear whether those actions were attributable to direct effects on brain cells or indirect effects mediated via peripheral pathways. VDH modulates L-type voltage-sensitive Ca2+ channels (L-VSCCs) in peripheral tissues, and an increase in L-VSCCs appears linked to both brain aging and neuronal vulnerability. Therefore, we tested the hypothesis that VDH has direct neuroprotective actions and, in parallel, targets L-VSCCs in hippocampal neurons.
Primary rat hippocampal cultures, treated for several days with VDH, exhibited a U-shaped concentration–response curve for neuroprotection against excitotoxic insults: lower concentrations of VDH (1–100 nm) were protective, but higher, nonphysiological concentrations (500–1000 nm) were not. Parallel studies using patch-clamp techniques found a similar U-shaped curve in which L-VSCC current was reduced at lower VDH concentrations and increased at higher (500 nm) concentrations. Real-time PCR studies demonstrated that VDH monotonically downregulated mRNA expression for the α1C and α1D pore-forming subunits of L-VSCCs. However, 500 nm VDH also nonspecifically reduced a range of other mRNA species. Thus, these studies provide the first evidence of (1) direct neuroprotective actions of VDH at relatively low concentrations, and (2) selective downregulation of L-VSCC expression in brain neurons at the same, lower concentrations.
Keywords: vitamin D, excitotoxicity, calcium channels, L-type, patch clamp, cell culture, hippocampus, calcitriol
Vitamin D hormone (VDH; 1,25-dihydroxyvitamin D3) is the biologically active metabolite of vitamin D and the major Ca2+-regulatory steroid hormone in peripheral tissues (DeLuca and Zierold, 1998; Norman, 1998; Brown et al., 1999). Despite its prominent role in regulating peripheral Ca2+ levels, however, little is known regarding the effects of VDH in the brain. Because receptors for VDH have been identified in multiple brain regions including the hippocampus (Stumpf et al., 1982; Stumpf and O'Brien, 1987;Veenstra et al., 1998; Prüfer et al., 1999), we postulated that VDH also regulates Ca2+-mediated processes in the brain. If so, then VDH might also be important for modulating neuronal death, in which Ca2+ homeostasis is well recognized to play a critical role (Rothman and Olney, 1987;Choi, 1988, 1992; Nicotera and Orrenius, 1998; Toescu, 1998). This possibility is supported indirectly by evidence that other peripheral steroid hormones, such as glucocorticoids and estrogens, can exert direct effects on the brain, modulate brain aging, and influence neuronal survival in response to insults (Landfield and Eldridge, 1994; Simpkins et al., 1994; McEwen and Sapolsky, 1995; Porter and Landfield, 1998; Zakon, 1998; Wise et al., 1999).
In addition, recent studies have shown that chronic peripheral treatment of rats with VDH retards the age-related decrease in neuronal density typically seen in rodent hippocampus (Landfield and Cadwallader-Neal, 1998) and protects against damage in a rodent model of stroke (Wang et al., 2000). Nevertheless, it is unclear whether these apparent neuroprotective effects of VDH are attributable to indirect actions (e.g., on peripheral Ca2+and PO4 regulation) or to direct actions on brain neurons.
If VDH does modulate Ca2+ homeostasis, one potential target for VDH may be the L-type voltage-sensitive Ca2+ channel (L-VSCC). Chronic VDH downregulates mRNA expression of the L-VSCC α1Csubunit in bone cells (Meszaros et al., 1996) and, conversely, can also increase L-VSCC current by a rapid, nongenomic action in the same cells (Caffrey and Farach-Carson, 1989; Takeuchi and Guggino, 1996). Furthermore, L-VSCCs may also play a role in neuronal survival. An increase in L-VSCC density has been implicated in hippocampal aging (Landfield et al., 1992; Moyer et al., 1992; Disterhoft et al., 1994;Thibault and Landfield, 1996) and in the increasing cell death seen with age in long-term hippocampal cultures (Porter et al., 1997). Moreover, although the NMDA receptor (NMDAR) appears to be the major Ca2+ source in excitotoxic cell death (Choi, 1992; Tymianski et al., 1993; Rajdev and Reynolds, 1994), there is also evidence that Ca2+ influx through L-VSCCs can contribute to excitotoxicity (Weiss et al., 1990;Uematsu et al., 1991; Faden and Salzman, 1992; Krieglstein et al., 1996; Stuiver et al., 1996; Geddes et al., 1997; Kimura et al., 1998).
Here we tested the hypothesis that VDH directly acts on neurons to modulate the expression of L-VSCCs and confer neuroprotection. Our results indicate that the Ca2+-regulatory functions of VDH extend to neurons and may play a role in neuronal survival.
MATERIALS AND METHODS
Preparation of hippocampal cultures. Modifications of established methods (Banker and Cowan, 1977) were used to prepare cell cultures from fetal rat hippocampi (Porter et al., 1997). All procedures were performed in accordance with the Society for Neuroscience Policy on the Use of Animals in Neuroscience and were approved by the Institutional Animal Care and Use Committee at the University of Kentucky. Fetuses (embryonic day 18) were removed from pregnant Fischer 344 or Sprague Dawley dams that had been first killed by CO2 and cervical dislocation. No differences were observed in cell cultures isolated from these two rat strains. Hippocampi were dissected out and placed in ice-cold Ca2+- and Mg2+-free HBSS. Tissue was then transferred to 10 ml of room temperature HBSS that contained 0.25% trypsin and 1 mm EDTA. After 10 min, hippocampi were washed three times with minimum essential medium (MEM; supplemented with 30 mm glucose) and then dispersed by repeated trituration. The cell suspension was diluted with MEM to a final concentration of 3–5 × 105 cells/ml. One milliliter of the diluted suspension was added to each poly-l-lysine-coated (100 μg/ml) plastic culture dish (35 mm; Corning, Corning, NY) that also contained 1 ml of MEM (with 10% fetal bovine and 10% horse sera) and had been preincubated overnight at 36°C in a humidified atmosphere of 5% CO2and 95% air. Culture dishes were then placed in an incubator, and on the next day half of the medium was removed and replaced with MEM and 10% horse serum (MEM/H). At 3 d in vitro (DIV), 5-fluoro-2′-deoxy-uridine (15 μg/ml) was added to inhibit glial proliferation (antimitotic), and uridine (35 μg/ml) was added to prevent the nonspecific inhibition of RNA synthesis by the antimitotic agent. To compensate for evaporation, 200 μl of 26 mm NaHCO3 was added to each dish at 10 DIV. All solutions, media, and sera were obtained from Life Technologies (Grand Island, NY) or Hyclone (Logan, UT).
Immunocytochemistry: vitamin D receptor labeling. Primary hippocampal cultures at 7 and at 14 DIV were rinsed three times in PBS and then fixed in PBS and 2% paraformaldehyde for 20 min at 4°C. After a series of PBS rinses, cells were permeabilized with 0.1% Triton X-100 (15 min), rinsed three times in PBS, and incubated in normal rabbit blocking serum (1:50, 30 min). Cultures were then incubated for 16 hr at 4°C in a 1:2000 dilution of antibody 4707 (Ab4707), made in goat, against the rat vitamin D receptor (VDR; kindly supplied by Dr. Nicholas Koszewski, Department of Internal Medicine, University of Kentucky; Langub et al., 2000). After rinses in PBS, cultures were incubated in a rabbit anti-goat biotinylated secondary antibody (1:200, 1 hr; Vector Laboratories, Burlingame, CA). The signal was amplified by avidin–biotin complex (Vector Laboratories) before staining with diaminobenzidine (DAB) and hydrogen peroxide. A negative control was performed by preadsorption of Ab4707 with the peptide used to generate the antibody and then adding this to the primary cultures (Fig. 1B).
Fig. 1.

Photomicrographs of VDR immunoreactivity in hippocampal cultures (7–14 DIV). A, Two representative pyramidal-shaped neurons positively stained for the VDR with Ab4707 (phase contrast optics). B, No cellular staining was observed when the VDR antibody was preincubated with its epitope.C, Cell culture double-labeled with antibodies to VDR and GFAP to determine cell-specific labeling of VDR. Most cells expressing the VDR (brown stain) were neurons, as indicated by the morphology and the lack of co-labeling with the glial marker GFAP (violet stain). D, Nuclei of astrocytes (arrow) showed either weak or no staining for the VDR. A pyramidal-shaped neuron in the field stained positively for VDR (arrowhead) is shown for comparison. Scale bars:A, B, D, 50 μm; C, 100 μm.
To determine whether glial cells in these cultures contain significant VDR, some cultures were sequentially double-labeled with Ab4707 and a monoclonal rat anti-glial fibrillary acidic protein (GFAP) antibody (clone 2.2b10; Zymed, San Francisco, CA). A rabbit blocking serum was applied (2%, 30 min) before cultures were incubated in anti-GFAP at 4°C for 16 hr (1:400). After rinses in PBS, cultures were incubated in a rabbit anti-rat biotinylated secondary antibody (1:200, 1 hr; Vector Laboratories), rinsed, and then incubated in solution that contained avidin–biotin complex. A Vector VIP substrate kit (violet to purple chromagen) was used to indicate anti-GFAP labeling in the cytoplasm, and DAB was used to show the VDR in the nucleus. A negative control was performed by incubating some cultures without anti-GFAP antibody.
VDH treatments. Either control vehicle (100% ethanol) or VDH (calcitriol, 1,25-dihydroxyvitamin D3;Biomol, Plymouth Meeting, PA) in vehicle was added to cultures at various times (see below) at concentrations ranging from 0.1 to 1000 nm. The final concentration of ethanol in all vehicle- or VDH-treated wells was 0.05%. Serum contains many hormones, including VDH. Before dilution in MEM, VDH concentrations in fetal bovine and horse sera were estimated by vendors to be 40 and 28 pm, respectively; therefore, ambient final concentrations were reduced by ∼10-fold. Unless otherwise stated (see below), cells were maintained under these serum-containing conditions.
In some studies, however, cultures were switched at 8 DIV from a serum-containing to a serum-free supplement (B27; Life Technologies) (Brewer et al., 1993) containing medium (MEM and 2% B27) before VDH treatment. The serum-free B27 supplement contains many nutrients, hormones, and antioxidants but lacks VDH.
Medium exchange toxicity studies. Complete medium exchange has been shown to be highly toxic to neurons in culture and can be blocked by inclusion of d-APV in the medium, indicating that the insult is glutamate-dependent (Driscoll et al., 1993; Ye and Sontheimer, 1998). At 15 DIV, cell culture medium was completely removed and replaced with fresh serum-containing medium (MEM/H). Neuronal survival was assessed 24 hr later.
NMDA and glutamate insults. Hippocampal neurons were treated with vehicle or VDH at 3, 6, and 8 DIV. Four to 6 hr after the last VDH treatment, glycine (10 μm) and then NMDA (100 μm) were added directly to the medium, and cultures were returned to the incubator for 10 min. The NMDA antagonistd-APV (100 μm) was added after the 10 min exposure to limit the excitotoxic insult. Cell survival was assessed 24 hr and 5 d later.
In some experiments, hippocampal cultures were subjected to excitotoxic insult with glutamate. Glycine (10 μm) and glutamate (5 μm) were added directly to the culture medium for 5 min. The medium was then aspirated and replaced with fresh medium containing MEM and 2% B27. Survival was assessed 24 hr after glutamate insult.
Viability assessment. Photomicrographs (35 mm) were taken on a phase contrast microscope at 100× magnification of fields at similar locations in each dish. Counts of viable neurons were performed on coded photomicrographs in a blind manner by two independent scorers, and the values were averaged. The accuracy of the counting procedure was validated in separate experiments in which we compared counts of phase-bright neuron bodies with counts of viable cells assessed by the fluorescent viability dye fluorescein diacetate (Novelli et al., 1988). In these parallel experiments, the difference between the counts obtained by the two separate methods was <5%. Our procedure appears highly accurate, because it assesses neuronal cell death specifically and is not confounded by the contribution of glial cell death in our co-culture system.
Electrophysiology: VDH treatment. Cultured rat hippocampal neurons, aged 14–16 DIV, were used for multichannel, cell-attached patch recording. Cultures were maintained as above until 8 DIV, when they were switched to a medium that contained a serum-free supplement (B27; Life Technologies). Cells were maintained under these conditions for ∼1 week before they were treated with VDH (5, 50, or 500 nm) for 24 hr and again 4–8 hr before recording. In a separate set of experiments, cells were exposed to VDH acutely (5 or 50 nm) by adding VDH either separately or in combination to the electrode and bath solutions. Recordings were obtained within 3–10 min after initial exposure.
Cell-attached patch recording. Standard multichannel cell-attached methods (Hamill et al., 1981) were used to record L-VSCC activity. Recording procedures were similar to those we have described previously (Thibault et al., 1993; Porter et al., 1997). The extracellular bath solution consisted of the following (in mm): 140 K-gluconate, 3 MgCl2, 10 glucose, 10 EGTA, and 10 HEPES. The pH was adjusted to 7.35 with KOH. This solution is typically used to “zero” the membrane potential (Fox et al., 1987) for recording VSCCs. The electrode solution consisted of (in mm): 90 choline chloride, 20 BaCl2, 10 tetraethylammonium (TEA)-Cl, and 10 HEPES, pH adjusted to 7.35 with TEA-OH. Sucrose was used to adjust the osmolarity of the bath and electrode solutions to 310 and 290 mOsm, respectively. To enhance L-VSCC current, electrodes also contained the L-VSCC dihydropyridine agonist Bay K8644 (500 nm) (Nowycky et al., 1985).
Patch-clamp electrodes, with resistances between 3.0 and 3.5mΩ, were pulled from glass capillary tubes (100 μl; Drummond Scientific, Broomall, PA) using a Flaming-Brown micropipette puller (model P-87; Sutter Instruments, Novato, CA). Immediately before use, electrode tips were coated with Sylgard (Dow Corning, Midland, MI) and then fire-polished (Narishige, Tokyo, Japan). Only recordings from electrodes that formed seals of at least 20 GΩ were included for analysis. An Axopatch-1D (Axon Instruments, Foster City, CA) was used for patch-clamp recording, and the pCLAMP6 software package (Axon Instruments) was used for data acquisition and analysis. Current records were filtered at 2 kHz and digitized at 5–8 kHz. Recordings were made at room temperature.
Leak and capacitive currents were subtracted from active current responses by using averaged currents from hyperpolarizing steps (−70 to −150 mV). Inward Ca2+ currents were assessed in a series of 15 depolarizing pulses elicited from −70 to +10 mV (100 msec duration) at 30 sec intervals. Ensemble averages were constructed from these 15 traces, and peak and total patch currents were then calculated for each patch from the ensemble average. The average patch current was obtained by dividing the integrated ensemble current area (total patch current) by the pulse duration. From the same patches, I–V series were obtained from each cell (10 mV increments of 100 msec duration from −70 to +30 mV at 30 sec intervals). Half-maximal activation voltages (V1/2) were derived by fitting data obtained from I–V relationships to the Boltzmann equation of the form y = total patch current/{1 + exp[(V1/2 − V)/k]}, where V is the maximal voltage from the I–Vrelationship, and k represents the steepness of the sigmoid curve.
Total patch current (I) is given byNPoi, where N = number of available channels, Po = probability of channel opening, and i = current amplitude of a single channel (Nowycky et al., 1985; Fox et al., 1987). With Bay K8644, the patch current is overwhelmingly dominated by L-type current, and Po for L-VSCC is extremely high (Nowycky et al., 1985; Bean, 1989; Fisher et al., 1990). To estimatei, the amplitudes of clearly resolvable single L-VSCC openings (≥3 msec) were measured from the I–V series. Because Po approaches 1.0 at maximally activating voltages in Bay K8644 (Thibault and Landfield, 1996), the method of maximal simultaneous openings was used to estimateN from the empirical determinations of i(N =Imax/i; Horn, 1991;Sigworth and Zhou, 1992). Imax, the maximal current for each patch, was determined from the largest instantaneous peak current in any of the15 depolarizing current pulses to +10 from −70 mV during the ensemble series (Thibault and Landfield, 1996; Porter et al., 1997). To estimate channel density, the number of channels per unit membrane area (N/μm2), the area of each patch was determined using the equation a = 12.6 (1/R + 0.018), where R is pipette resistance, anda is membrane area (square micrometers) (Sakmann and Neher, 1983). To control for variability in patch size between groups, only patch electrodes with similar resistances were used (3–3.5 mΩ).
RNA preparation and cDNA synthesis. Hippocampal neurons were plated into 24-well culture plates with equivalent cell numbers in each well and divided into four conditions: one vehicle control and three VDH concentrations. After VDH treatment, the total cellular contents of each well were used for RNA extraction and reverse transcription (RT). RNA was isolated using TRIzol reagents according to the manufacturer's protocol (Life Technologies). The purified RNA was redissolved in 10 μl RNase-free water and used for cDNA synthesis by RT. The RT was performed in a 50 μl reaction mixture containing 10 mm Tris buffer, pH 8.3, 50 mm KCl, 5 mm MgCl2, 1 mm each of dATP, dGTP, dCTP, and dTTP, 20 U of RNase inhibitor, 0.001 mm random hexamers and 50 U of murine leukemia virus reverse transcriptase. The reaction mixture was incubated at 42°C for 1 hr followed by 95°C for 10 min and then stored at −20°C until further PCR analysis.
Real-time quantitative PCR. mRNA was quantitated for α1C and α1D L-VSCC subunits (now also termed Cav1.2 and Cav1.3, respectively; Ertel et al., 2000), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and low molecular weight neurofilament (NFL). Quantitation of mRNA was performed using an Applied Biosystems (Foster City, CA) PRISM 7700 sequence detection system with TaqMan methods. This technique uses the 5′ nuclease activity of Taq DNA polymerase to generate a real-time quantitative DNA assay (Livak et al., 1995; Heid et al., 1996; Lie and Petropoulos, 1998; Freeman et al., 1999). Briefly, mRNA-specific oligonucleotide probes (TaqMan probe) with 5′-fluorescent reporter and 3′-quencher dyes were designed and used for the extension phase of the PCR. The degradation and release of the reporter dye (i.e., FAM) results in fluorescence at 518 nm, which is monitored during the complete amplification process.
Samples of cDNA (n = 10) from control and VDH groups were analyzed simultaneously (total of 40 samples) by real-time PCR, with each sample run in duplicate. The PCR mixture was prepared using the single-reporter real-time PCR protocol according to the manufacturer's instructions (Applied Biosystems PRISM 7700), and the PCR was run using the system software. For the α1C and α1D L-VSCC subunit messages, 0.2 μl of RT product for each sample was used as the template in a 50 μl reaction mixture, and each reaction was run in duplicate. The primers and the TaqMan probe for α1C were as follows: forward primer, 5′-CCGGAAGCCAGTGCATTTT-3′; reverse primer, 5′-TGGTGAAGATCGTGTCATTGACA-3′; and TaqMan probe, 5′6FAM-CCAAACAACAGGTTCCGCCTGCAGT-TAMRA-3′, which amplified an 88 bp region of the α1C mRNA (nucleotides 3377–3464; GenBank accession number M67516, rat brain). The primers and TaqMan probe for α1D were as follows: forward primer, 5′-GAAGAGGACGAGCCTGAGGTT-3′; reverse primer, 5′-TTTTCTCCTTCATGTTCAACTCTGA-3′; and TaqMan probe, 5′6FAMTGCTGGTCCCCGTCCTCGAAGA-TAMRA-3′, which amplified a 72 bp region of the α1D mRNA (nucleotides 3021–3092; GenBank accession number M57682, rat brain).
Messages for a cellular “maintenance” gene, GAPDH, and a neuronal marker gene, NFL, were also analyzed in VDH-treated samples to study the specificity of effects on expression patterns. The primers and TaqMan probe for GAPDH were as follows: forward primer, 5′-ACATGCCGCCTGGAGAAA-3′; reverse primer, 5′-AGCCCAGGATGCCCTTTAGT-3′; and TaqMan probe, [5′6FAM-CCCTCGGCCGCCTGCTTCATAMRA-3′, which amplified a 91 bp region of GAPDH (nucleotides 760–850; GenBank accession number M17701, rat brain). The primers for NFL were as follows: forward primer, 5′-CAGCAGAACAAGGTCCTGGAA-3′; reverse primer, 5′-GAAGCGGGAAGGCTCTGAGT-3′; and TaqMan probe, 5′6FAM-CTTCTGGCGCAGCACCAACAGCT-TAMRA-3′, which amplified a 69 bp region of NFL (nucleotides 408–476; GenBank accession number AF031880, rat brain).
mRNA quantitative analysis. For each message, real-time PCR quantitation was performed simultaneously for all conditions using the Applied Biosystems PRISM 7700 system. At the completion of the PCR reaction (total of 40 cycles), the amount of a target message in each sample was estimated from a threshold cycle number (CT), which is inversely correlated with the abundance of its initial mRNA level. TheCT was then converted to relative quantity of mRNA by using a standard curve calibrated according to the manufacturer's instructions. The standard curve was obtained by serial dilutions of known quantities of the target message. The PCR reaction was run simultaneously for the standard curve and the experimental samples.
Statistical analyses. Except where noted, all analyses were performed using ANOVA. For pair-wise comparisons between control and VDH-treated conditions, post hoc analyses were performed using Dunnett's multiple comparisons test.
RESULTS
Immunostaining of VDR
Cell-specific immunolabeling and morphological criteria are able to distinguish pyramidal neurons, nonpyramidal neurons, and glia in hippocampal cell cultures (Banker and Cowan, 1977; N. M. Porter, V. Thibault, and L. B. Brewer, unpublished observations). To determine whether neurons and/or glia express the VDR, hippocampal cultures were incubated in a monoclonal antibody to the VDR (Ab4707). VDR-positive cells were present in all cultures and appeared morphologically to be neuronal (Fig.1A). Most VDR-positive cells showed strong nuclear and cytoplasmic staining; however, some cells had very light staining, indicating variation in the level of VDR expression among neurons. To evaluate the specificity of VDR Ab4707 for neurons in our cultures, we performed double-labeling experiments using a monoclonal antibody to GFAP (a glial marker) along with the VDR antibody. Figure 1C shows numerous VDR-positive (brown stain) and GFAP-positive (violet stain) cells. Although some glia displayed very weak VDR labeling along the nuclear perimeter(Fig. 1D, arrow), the predominant VDR staining was not co-localized with the GFAP-stained cells. Therefore, despite a previous report of VDR labeling of glial cells (Neveu et al., 1994b), astrocyte VDR immunoreactivity was, for the most part, undetectable in our cultures.
Neurotoxicity Studies
Experiments in serum-containing medium
To more directly test the potential neuroprotective effects of VDH on cultured hippocampal neurons, we examined the effects of VDH in three excitotoxic cell death protocols: complete medium exchange (ME) and exposure to toxic concentrations of NMDA or glutamate. In the first series of studies, VDH-treated cultures were exposed to ME insult at 15 DIV, and neuronal survival was assessed 24 hr later. Representative photomicrographs show that cells treated with 5 and 50 but not 500 nm VDH were partially protected from the insult (Fig.2). Results indicated that the number of neurons in all treatment groups was equivalent before the insult, indicating that VDH did not increase survival by enhancing baseline neuronal density (Fig. 3A). Twenty-four hours after the ME insult, only 5% of the neurons in control cultures survived, whereas ∼50% of the neurons treated with 5 or 50 nm VDH survived the insult (Fig.3B). Treatment of cells with 500 nmVDH offered no neuroprotection, with survival approximating that of the control cultures.
Fig. 2.
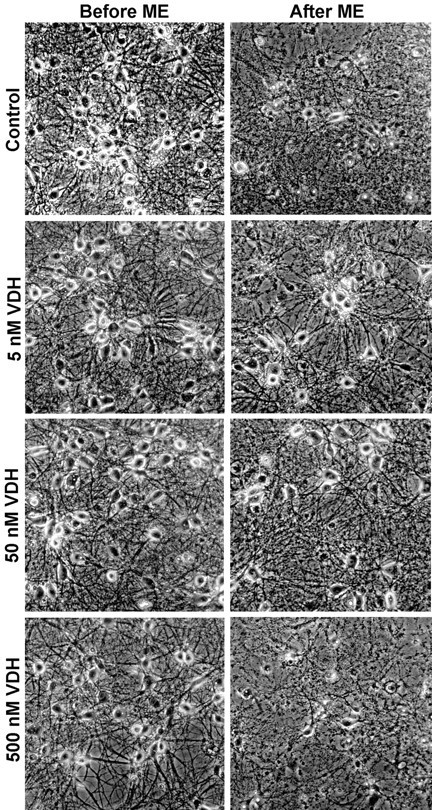
Representative photomicrographs of hippocampal neurons before an ME insult and again 24 hr after insult at 15 and 16 DIV, respectively. Cultures were chronically treated either with control vehicle (0.05% ethanol) or VDH (5, 50, and 500 nm) at 3, 6, 8, and 14 DIV. Few neurons survived the ME insult in control cultures; however, cultures treated with 5 and 50 nm VDH were partially protected from the insult. No protection was observed in cultures treated with the highest concentration of VDH, 500 nm.
Fig. 3.

VDH-treated hippocampal neurons were partially protected from ME insult. Quantitative results from the cultures in Figure 2 after ME insult of control (Cntl) and VDH-treated cells are shown. Survival was assessed from photographs taken at 15 DIV before ME insult and again 24 hr after insult.A, Chronic treatment with VDH did not alter the number of neurons before ME insult. B, The number of surviving neurons after ME insult was significantly greater in wells pretreated with 5 and 50 nm VDH (*p < 0.001 vs control). Results are mean ± SEM; n = 15–18 culture wells per group.
To confirm the results obtained with the ME insult, we next examined the effects of VDH treatment on toxic NMDA or glutamate exposure. Cell death can continue well beyond the initial 24 hr period after the insult (Weiss et al., 1990; Ankarcrona et al., 1995; Gwag et al., 1997). Therefore, we examined the survival of these same cultures 5 d later (at 16 DIV), as well, to determine whether the protective effects of VDH were long-lasting or limited only to the initial post insult period. To more closely identify neuroprotective concentrations of VDH, we also used a more-extensive range of concentrations. Cultures were chronically treated with VDH (0.1–1000 nm) and subjected to NMDA (100 μm) insult. Again, chronic treatment with VDH did not alter the number of hippocampal neurons before NMDA insult. At 24 hr after NMDA insult, survival was assessed and found to be significantly enhanced by twofold in cultures treated with 100 nm VDH (p < 0.05; Fig.4A, top panel). The survival of cultures treated with lower VDH concentrations (0.1, 1, and 10 nm) was increased to a similar degree but not quite significantly.
Fig. 4.
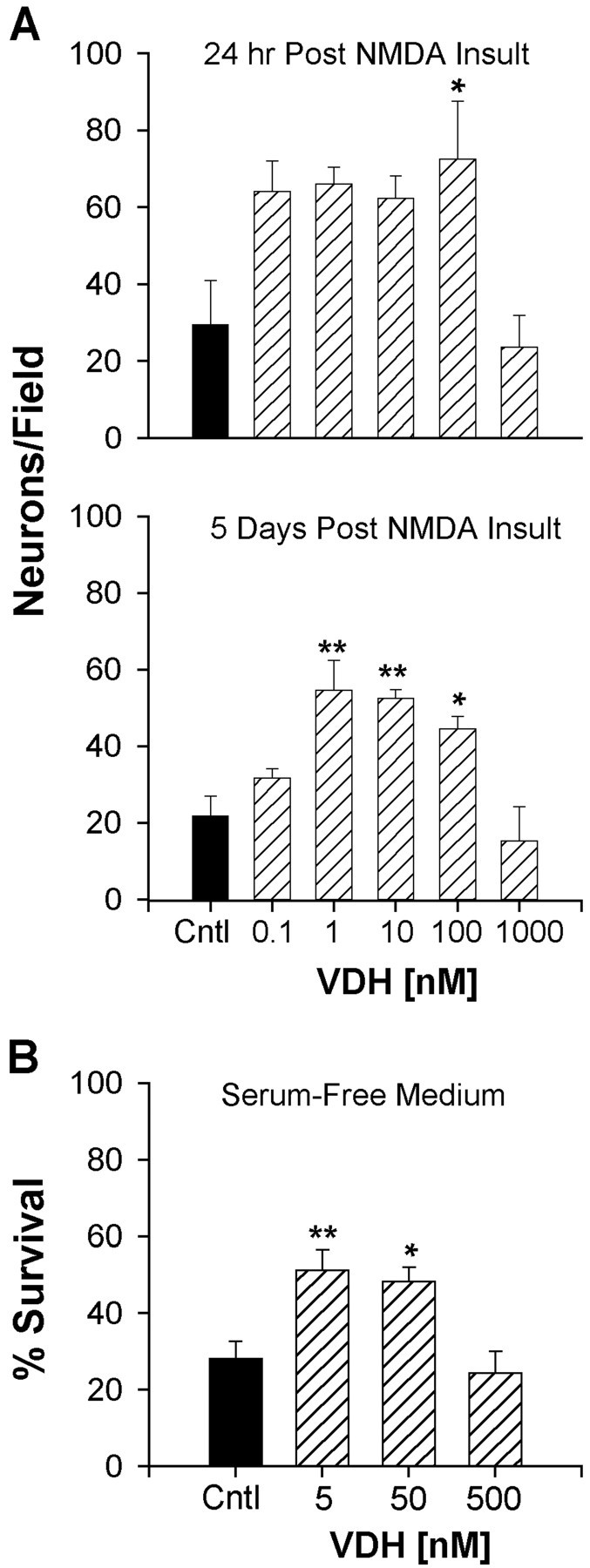
Survival of hippocampal neurons after insult with NMDA or glutamate in VDH-treated cultures. A, top panel, Cultures were treated with VDH at 3, 6, and 8 DIV and subjected to NMDA (100 μm) insult 6 hr after the last VDH treatment. Survival 24 hr after NMDA insult was significantly greater in cultures treated with 100 nm VDH (p < 0.05 vs control). A, bottom panel, Survival was reassessed in the same cultures 5 d after NMDA insult. Enhanced long-term survival of hippocampal neurons was found in cultures previously treated with 1, 10, and 100 nm VDH (*p < 0.05; **p < 0.01 vs control). Results are mean ± SEM; n = 4 culture wells/group. B, VDH added to cultures maintained in serum-free conditions (B27, lacking VDH). Hippocampal neurons in VDH-treated wells (5 and 50 nm) were partially protected from glutamate insult similar to experiments in serum-maintained cultures. Cultures were treated with VDH at 15 or 16 DIV for 24 hr and again 4–6 hr before glutamate insult (5 μm for 5 min). Survival was assessed 24 hr after insult. VDH treatment for 24–30 hr is sufficient to confer protection (*p < 0.05; **p < 0.01 vs control). Results are mean ± SEM;n = 8–12 culture wells per group.
As anticipated, additional cell death was observed in all groups 5 d after insult. However, after 5 d greater survival was found in cultures treated with 1, 10, and 100 nm VDH (Fig.4A, bottom panel), indicating that chronic pretreatment with lower concentrations of VDH had long-lasting neuroprotective effects. Again the highest concentration of VDH, 1000 nm, offered no neuroprotection at either 24 hr or 5 d after insult. Similar results were obtained when the these experiments were repeated using chronic treatment with 5, 50, and 500 nm VDH (concentrations identical to those in the ME experiments). Survival was assessed at 13 DIV (5 d after NMDA insult) and found to be enhanced at the lower concentrations but not at the highest of concentration of VDH (500 nm; data not shown).
Experiments in serum-free medium
The experiments above were performed in cells maintained in serum that contained very low concentrations of VDH (picomolar; see Materials and Methods). To rule out any contributions of VDH from the serum, we also examined the effects of VDH in cells maintained in a serum-free supplement (B27; Brewer et al., 1993). At 15 or 16 DIV, cultures were treated with VDH for 24 hr and again 4–6 hr before glutamate insult. Cell culture plates were marked, and survival was assessed from photomicrographs taken of the same area immediately before and 24 hr after the insult. Similar to previous experiments with cells maintained in serum (Figs. 3, 4A), VDH enhanced survival at lower concentrations (5 and 50 nm) but not at the highest concentration of 500 nm (Fig.4B). Furthermore, the results of this experiment were strikingly similar to those obtained for the ME studies. Taken together, these results indicate that the VDH present in our serum-containing cultures is unlikely to contribute to the effects observed, and that shorter treatments with VDH (24 hr) may be sufficient to induce neuroprotective effects.
VDH effects on L-VSCCs
Electrophysiology
Because our previous studies in B27 indicated that 24–30 hr treatments with VDH were sufficient to confer neuroprotection, cultures were treated in a similar manner for the subsequent electrophysiological studies. Using the cell-attached patch-clamp technique, L-VSCC activity was assessed in multichannel patches from the somata of control and VDH-treated neurons. Current traces from a representative control and a VDH-treated (50 nm) neuron illustrate reduced L-VSCC activity in the VDH-treated cell (Fig.5A). During the course of these studies, control and VDH recordings were obtained from different culture platings over several months. Therefore, currents in VDH-treated cells were expressed as a percentage of control. Because no difference in either peak current or average total current area was observed among control groups of different platings, all control cells were combined into one group (n = 89).
Fig. 5.
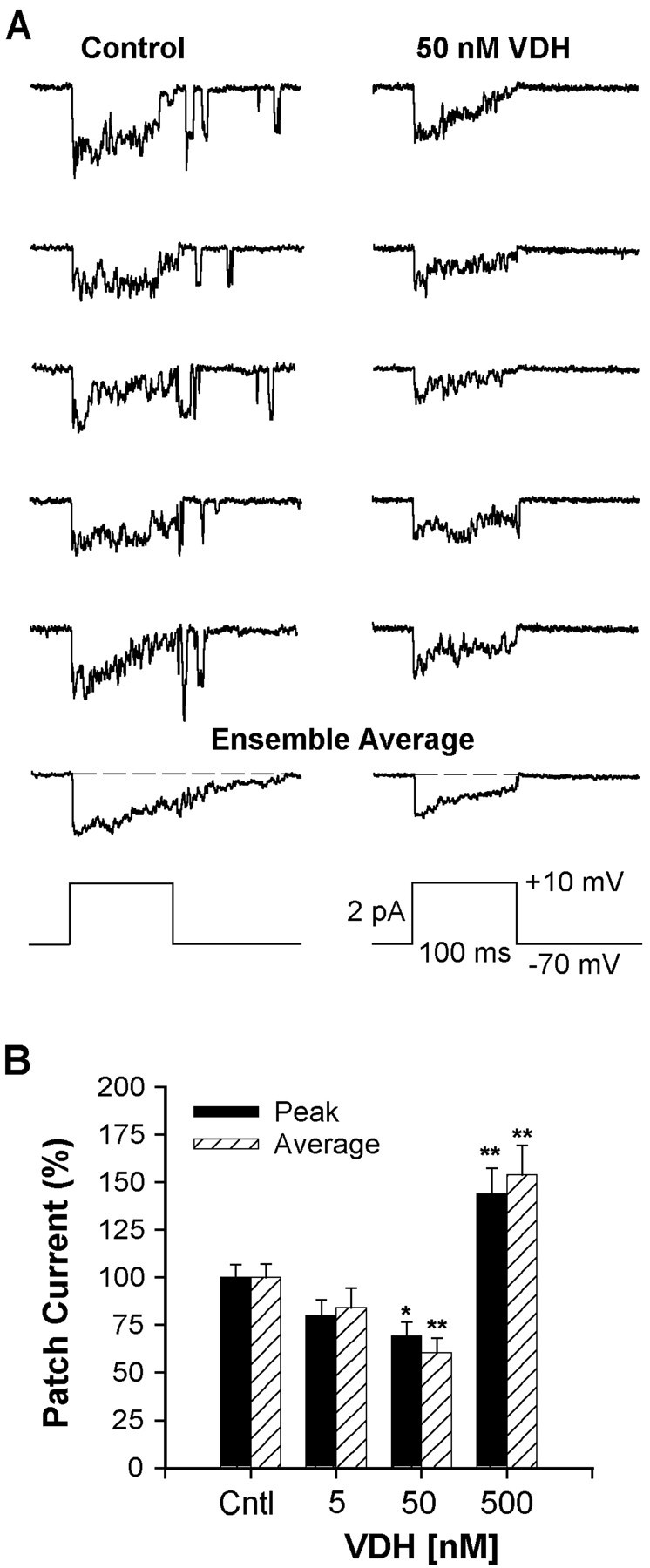
VDH reduced L-VSCC activity. A,Five representative traces of L-VSCC activity recorded in the cell-attached patch mode from a control (0.05% ethanol) and from a VDH-treated (50 nm, 24–30 hr) hippocampal neuron. Multichannel activity was evoked by depolarizations fromVh = −70 mV toVc = +10 mV. The ensemble average of the full 15 traces from each neuron is shown below the 5 traces. The voltage protocol is shown at the bottom.B, Relationship between VDH concentration and L-VSCC activity recorded from multichannel patches. Effects of VDH on peak and average current activity were determined as percentage of control. Treatment with 50 nm VDH for 24 hr significantly reduced both peak and average current activities (*p < 0.05; **p < 0.01, respectively). Reduction in current by 5 nm VDH was not significant. The highest concentration (500 nm) exerted an opposite action and increased L-VSCC activity. Results are mean ± SEM;n = 89 control neurons and 19–44 VDH-treated neurons.
Quantitative results of a concentration response to VDH are shown in Figure 5B. Both peak (*p < 0.05) and average (**p < 0.01) total patch ensemble currents were significantly reduced by ∼30–40% in cultures treated with 50 nm VDH. A similar but nonsignificant trend was seen in cells treated with 5 nm VDH. In contrast to the decrease in current seen at these lower concentrations, peak and total patch currents were significantly increased ∼45–55% (p < 0.01) in cells treated with the highest concentration of VDH (500 nm). These results indicate that VDH has a bimodal action on L-VSCCs in hippocampal neurons similar to that seen in the neurotoxicity studies. Furthermore, the concentrations of VDH that reduced L-VSCC activity (5 and 50 nm) were neuroprotective, whereas the highest concentration, which lacked neuroprotective effects (500 nm), increased L-VSCC activity.
In a subset of the cells studied, we measured I–Vrelationships to determine whether a relatively low concentration of VDH (50 nm) altered the voltage dependence of the L-VSCC. In these cells, VDH reduced average current amplitude without altering the voltage dependence of current activation (Fig.6A). Half-maximal activation voltages were comparable and were −17 and −13 mV, respectively, for control and VDH-treated cells. The voltage threshold for activation of L-VSCC current was similar in both groups (typically at −40 or −30 mV), as was the voltage at which maximal current occurred (between 0 and 10 mV).
Fig. 6.
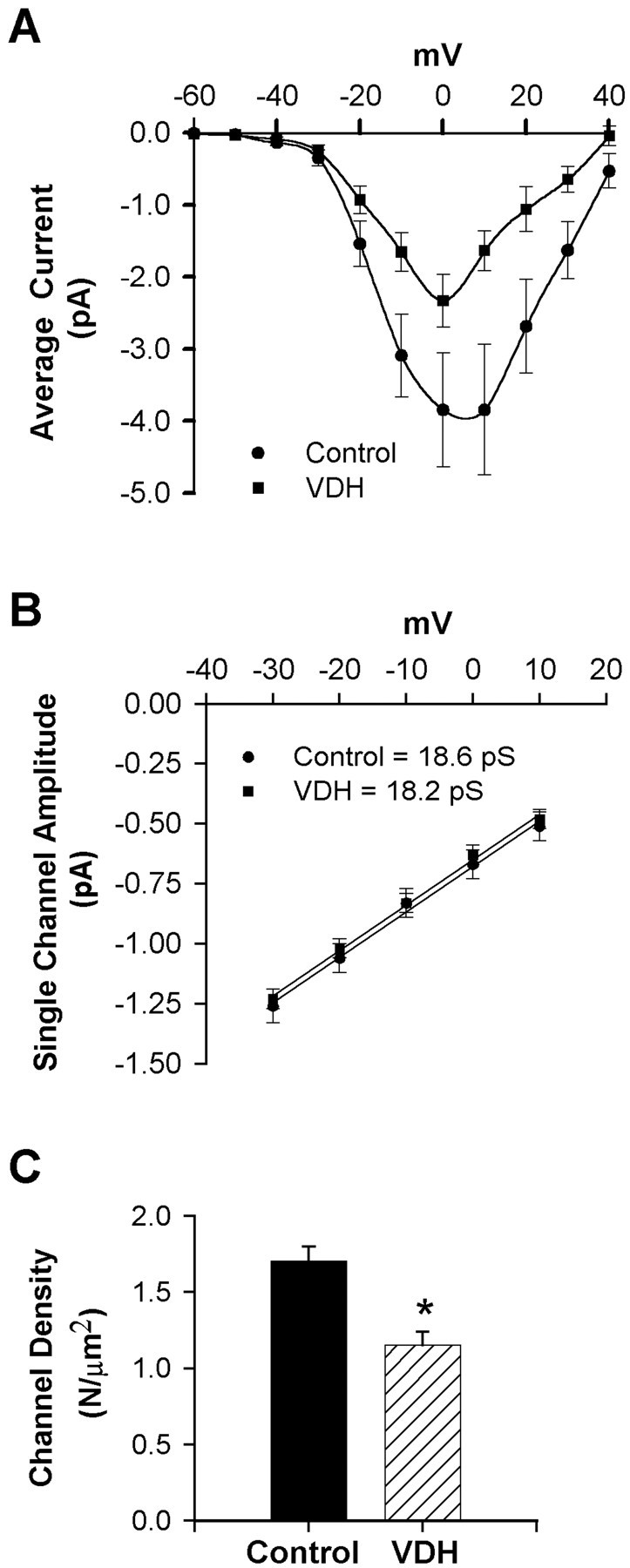
A, I–V series of average patch current from multichannel patches obtained from control or VDH-treated (50 nm, 24–30 hr) neurons. VDH reduced mean current amplitude without altering voltage dependence. Results are mean ± SEM B, Slope conductance of L-VSCCs was unaffected by VDH. Current amplitudes of clearly resolvable single-channel openings (of at least 3 msec duration) were measured from the I–V series. Mean slope conductance for each group was calculated from the average of individual slope conductances.C, Membrane density of functionally available L-VSCCs was reduced by VDH (p < 0.05). Channel density (N per square micrometer) was calculated using the method of maximal simultaneous openings. The membrane area (square micrometers) of a patch was calculated from pipette resistance (see Materials and Methods).
In addition, we examined single-channel properties of L-VSCCs to assess mechanisms underlying the decrease in current observed in cells treated with 50 nm VDH. Single-channel amplitude (i) was measured at multiple test voltages in patches with clearly resolvable openings of at least 3 msec duration from the I–V series and then averaged for each patch. Analyses of these results showed thati was not affected by VDH at any voltage. Furthermore, the average slope conductance was unaltered by VDH (∼18 pS in both groups; Fig. 6B). Using the estimated values ofi (Fig. 6B), the density of L-VSCCs (N per square micrometer) was determined by the method of maximal simultaneous openings and the individual values of pipette resistance. Our estimates of channel density indicated that 50 nm VDH reduced the membrane density of functionally available L-VSCCs by 37% (p < 0.05, t test; Fig. 6C).
A similar analysis was performed for cells treated with the highest concentration of VDH (500 nm), at which currents were increased (Fig. 5B). No differences were seen in the voltage dependence of currents in cells treated with 500 nm VDH. A trend toward increased channel density was observed in these cells (+23% over control), but this effect was not significant. In addition, slope conductance also was slightly larger in cells treated with 500 nm VDH (19.5 vs 18 pS for control), although this was also not statistically significant (data not shown). These equivocal results suggest that multiple alterations in channel properties may contribute to the increase in L-VSCCs observed in cells treated with the highest concentration of VDH. However, further studies will be needed to identify the specific mechanism of this effect.
Acute effects of VDH
The effects of acute treatment with VDH on L-VSCC activity were determined in a separate series of experiments. VDH, at a final concentration of 5 or 50 nm, was added to the electrode solution, to the bath, or to both. In some experiments, Bay K8644 was omitted from the electrode to ensure that the agonist did not block or mask possible acute effects of VDH. Patch recordings were typically obtained within 10 min of initial exposure. Acute exposure to 5 or 50 nm VDH, with or without Bay K8644, did not affect L-VSCC currents (data not shown). Mean values for control and VDH were within 10% for all conditions.
Effects of VDH on mRNA expression of L-VSCC subunits
To determine whether a change in L-VSCC gene expression could underlie the reduction in L-VSCC current and density, the mRNA levels for the two pore-forming subunits of the L-VSCC, α1C and α1D, and two control genes were assessed using real-time PCR. Hippocampal cultures were treated with vehicle or 5, 50, or 500 nm VDH before mRNA isolation. Figure 7 shows mRNA levels for the α1C and α1D subunits along with mRNA for the reference genes GAPDH and NFL. A representative PCR plot for α1C mRNA shows the kinetic curve of the amplification process as a function of cycle number (Fig.7A, inset). Treatment with 5 or 50 nm VDH selectively reduced mRNA levels only for L-VSCC subunits, whereas GAPDH and NFL mRNA were unaffected. mRNA levels for α1C and α1Dsubunits were reduced by ∼30% (p < 0.05) in 5 nm VDH-treated cells, and treatment with 50 nm VDH also significantly reduced α1C mRNA (p < 0.05; (Fig. 7A,B). The reduction in α1DmRNA at 50 nm VDH was consistent with the effects on α1C but was not significant. Treatment of hippocampal cultures with the highest concentration of VDH (500 nm) induced a still larger reduction in L-VSCC subunit mRNA content. However, at this concentration the effect was not selective for L-VSCC subunit mRNAs, and all measured mRNAs were substantially reduced (∼50%; p < 0.01; Fig.7A–D).
Fig. 7.
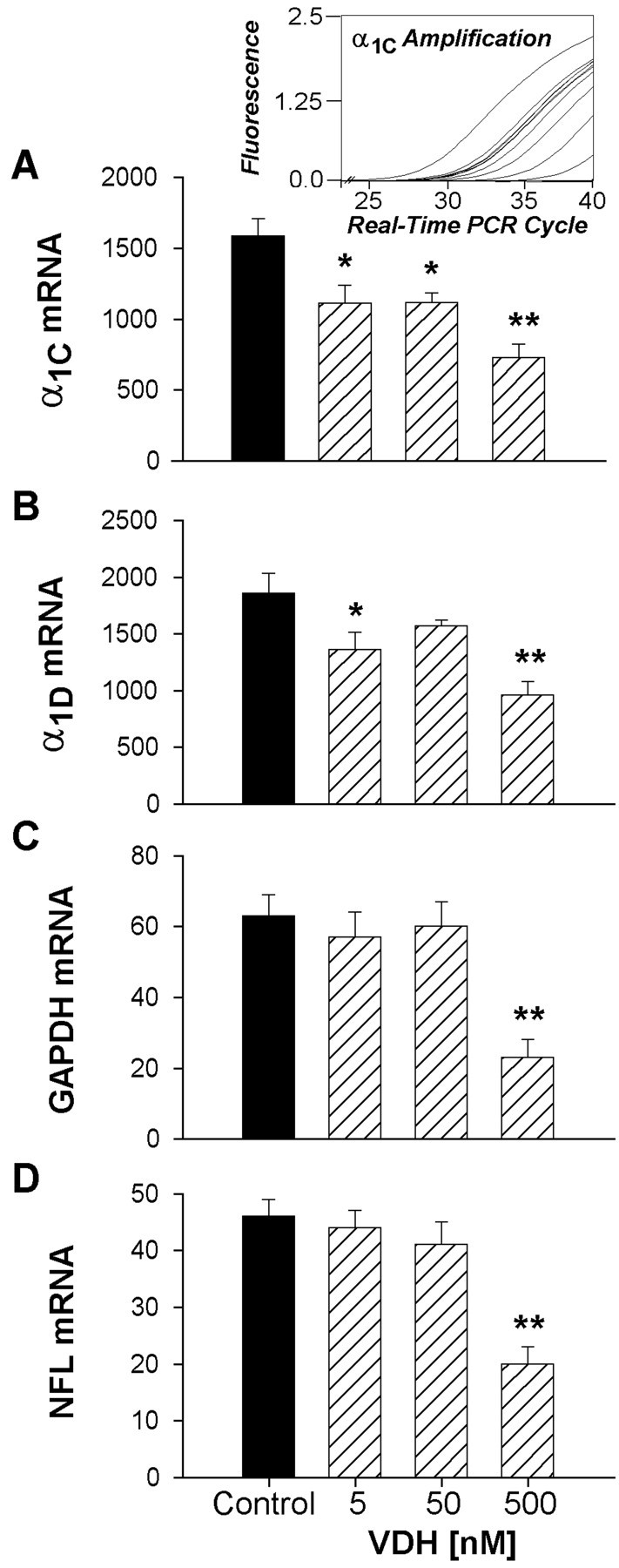
Low concentrations of VDH (5 and 50 nm) selectively reduced L-VSCC mRNA expression of the pore-forming α1C and α1D subunits. Quantitative real-time PCR was used to determine mRNA expression.A, inset, representative real-time PCR amplification plots for one row of α1C subunit samples over the entire kinetic curve. A–D, Quantitative results (mean ± SEM) for the four mRNA target species from control and VDH-treated cell culture samples estimated from a standard curve based on serial dilution of each mRNA species. Nonselective reduction of all messages was observed at the highest VDH concentration (500 nm). *p < 0.05; **p < 0.01 vs control; n = 10 samples per group.
DISCUSSION
The present studies identified two novel actions of relatively low concentrations of VDH on neurons: (1) a direct and highly consistent neuroprotective action against excitotoxic insults and (2) a decrease in both L-VSCC activity and mRNA levels of the corresponding pore-forming subunits (α1C and α1D) of the L-type channel. The VDH concentration–response curves for the neuroprotection and L-VSCC activity were bimodal and strikingly similar. Thus, both the neuroprotection and decrease in L-VSCC activity occurred at 5–50 nm VDH but not at the higher concentrations (500–1000 nm). The lower concentrations of VDH in this study are consistent with those that elicit VDH-dependent effects in other cell types (Neveu et al., 1994b; Meszaros et al., 1996; Alexianu et al., 1998). Moreover, because the KD of the VDR is ∼100 pm (Jones et al., 1998), the lower, neuroprotective concentrations used here are likely below those needed to saturate binding and thus may well represent specific actions mediated by the VDR.
The parallel VDH concentration curves for neuroprotection and L-VSCC current suggest that at least part of the neuroprotective effect of lower VDH concentrations may be mediated by actions on L-VSCC activity. In contrast, the treatment of hippocampal cultures with the higher concentrations of VDH (500–1000 nm) offered no protection to neurons exposed to a variety of excitotoxic insults, and concomitantly, increased rather than decreased L-VSCC current (Fig.5B). This concentration–response effect likely accounts for the failure of a previous study to see protection at 1000 nm VDH (Goodman et al., 1996). VDH has also been shown to have a direct toxic effect on motoneurons at high concentrations (Alexianu et al., 1998). Thus, the lack of neuroprotection seen at 500–1000 nm VDH in the present studies and the toxicity observed in motoneurons treated with high concentrations of VDH may somehow be related to increased L-VSCC current.
Relationship between VDH neuroprotection and L-VSCCs
Although it has been recognized for many years that the NMDAR is a major pathway for Ca2+ influx (MacDermott et al., 1986) and likely plays the major role in excitotoxic neuronal death (Rothman and Olney, 1987; Choi, 1988, 1992; Tymianski et al., 1993; Rajdev and Reynolds, 1994), a number of studies have shown that Ca2+ influx through L-VSCCs can also contribute to excitotoxicity (Weiss et al., 1990, Uematsu et al., 1991;Krieglstein et al., 1996; Stuiver et al., 1996; Kimura et al., 1998). Furthermore, indirect evidence is consistent with the view that L-VSCC activity is related to neuronal vulnerability. For example, hippocampal neuronal aging is associated with both increased vulnerability (Katzman and Saitoh, 1991; Gallagher et al., 1996) and elevated L-VSCC density (Thibault and Landfield, 1996; Herman et al., 1998). Somewhat analogously, cultured brain neurons become increasingly vulnerable to excitotoxicity with age in culture (Choi et al., 1987;Geddes et al., 1997; Cheng et al., 1999) and over the same age range exhibit increases in L-VSCCs (Porter et al., 1997; Blalock et al., 1999) as well as in expression of NMDAR NR2B subunit mRNA (Cheng et al., 1999).
Thus, L-VSCCs may somehow complement or interact with NMDARs in modulating excitotoxicity, particularly under conditions of less than maximal toxicity (see Choi, 1992, Discussion). If this is the case, then the parallel bimodal concentration curves for L-VSCC current and neuroprotection indicate that VDH may modulate neuronal vulnerability by regulating Ca2+ influx through VSCCs. However, the present studies do not, of course, rule out possible actions of VDH on other major Ca2+ sources in excitotoxicity (e.g., the NMDAR), and additional studies will be needed to test the specificity, or lack thereof, of VDH on multiple Ca2+ sources.
Mechanisms of vitamin D action on L-VSCC in neurons
In bone and skeletal muscle cells, VDH modulates Ca2+ current influx through L-VSCCs by two mechanisms (Norman et al., 1992). One involves a classic genomic pathway dependent on the binding of VDH to the nuclear VDR with resulting downregulation of L-VSCC mRNA (Meszaros et al., 1996). The other mechanism involves an increase of L-VSCC activity through a nongenomic pathway (Caffrey and Farach-Carson, 1989; Tornquist and Tashjian, 1989; Vazquez and de Boland 1993), possibly mediated via a plasma membrane VDR (Norman et al., 1992; Takeuchi and Guggino, 1996). The latter action may also occur in association with capacitative Ca2+ entry (Vazquez et al., 1998).
In contrast to the U-shaped effects on L-VSCC current and neuroprotection in the present studies, VDH down-regulated L-type subunit mRNA in a monotonic, concentration-dependent manner. At the lower concentrations (5–50 nm), this effect on mRNA expression was paralleled by decreased L-VSCC current and channel density, suggesting that genomic down-regulation underlies the decrease in available channels (Fass et al., 1999; Chen et al., 2000). This effect may be analogous to that in bone cells (Meszaros et al., 1996). Of course, further studies will be needed to determine whether VDH alters mRNA processing.
However, at the higher concentration (500 nm), L-VSCC current diverged from mRNA expression and instead was substantially greater than that of control. These results indicate that the effects of higher concentrations of VDH on L-VSCC current are mediated by a different mechanism and are independent of gene and mRNA expression. Multiple post-translational modifications (e.g., phosphorylation of L-VSCCs) can alter the density of available channels, and it seems likely that a non-VDR pathway may account for the increased L-VSCC current induced by high concentrations of VDH (Caffrey and Farach-Carson, 1989; Vazquez and de Boland, 1993).
Interestingly, at lower concentrations, VDH reduced L-VSCC subunit mRNA selectively compared with reference mRNAs (GAPDH and NFL), whereas at the highest VDH concentration there appeared to be a nonselective, generalized reduction of mRNA expression (Fig. 7A–D). Conceivably, this latter effect could reflect reduced cellular viability and could contribute to the lack of neuroprotection at this concentration. Such generalized reduction of mRNA expression could also be a factor in the antiproliferative actions of VDH in non-neuronal cells (Brown et al., 1999).
The present studies also do not rule out VDH influences mediated by other Ca2+ regulatory or non-Ca2+-dependent mechanisms in neurons. For example, several studies have found that levels of calbindin, as well as of other Ca2+-binding proteins (parvalbumin and calretenin), increase in specific brain areas after treatment with VDH (de Viragh et al., 1989; Alexianu et al., 1998), which could have implications for neuroprotection (Iacopino and Christakos, 1990; Mattson et al., 1991; Heizmann and Braun, 1992;Sutherland et al., 1992). In addition, VDH can protect against dymyelination (Cantorna et al., 1996; Garcion et al., 1997) and can upregulate trophic factors (NGF, GDNF, and NT-3) in glia and fibroblasts (Neveu et al., 1994a,b; Naveilhan et al., 1996; Musiol and Feldman, 1997; Wang et al., 2000). Nevertheless, the similarity of the bimodal effects of VDH on L-VSCC current and neuroprotection and the preferential localization of the VDR in neurons compared with glia (Fig. 1) strongly suggest that in the present studies, the neuroprotective actions of VDH were neuron-specific and mediated in part through reductions of L-VSCC expression.
Implications for function and disease
Although the CNS is not traditionally recognized as a target for VDH, the present studies appear to add a new class of steroids to those that have been found to directly modulate neuronal vulnerability (e.g., glucocorticoids and estrogens). Furthermore, they identify a novel molecular mechanism of VDH in neurons that has clear functional implications, beyond the question of vulnerability. L-VSCCs appear to play selective and major roles in the regulation of several key Ca2+-dependent neuronal processes, including neuronal excitability (Landfield et al., 1992; Moyer et al., 1992), gene expression (Bito et al., 1997; Finkbeiner and Greenberg, 1998), long-term potentiation (Teyler et al., 1995; Kapur et al., 1998), and long-term depression (Norris et al., 1998). Therefore, modulation of L-VSCCs by VDH may have a wide range of effects.
The direct neuroprotective actions of VDH seen here also may have specific health implications, particularly for the elderly. Recent epidemiological studies indicate that VDH deficiency is more prevalent than previously recognized, notably in sick adults (Thomas et al., 1998) and in the elderly (Lips et al., 1988; Gloth and Tobin, 1995;Jacques et al., 1997; Perry et al., 1999). Much of this deficiency may be attributable to lack of exposure to sunlight (McKenna, 1992; Perry et al., 1999) and insufficient intake of vitamin D (Gloth et al., 1995;Compston, 1998; Utiger, 1998), but other metabolic factors may contribute to the decline in VDH (Thomas et al., 1998; Utiger, 1998). Aging increases vulnerability of the brain and is the single greatest risk factor for Alzheimer's disease (Katzman and Saitoh, 1991). Moreover, aging and Alzheimer's disease appear to be associated with altered neuronal Ca2+ homeostasis (Gibson and Peterson, 1987; Khachaturian, 1989; Landfield et al., 1992;Disterhoft et al., 1994; Thibault et al., 1998; Verkhratsky and Toescu, 1998). Thus, the present results raise the possibility that an unrecognized consequence of inadequate VDH status in the elderly may be reduced endogenous neuroprotection and enhanced neuronal vulnerability. Conceivably, therefore, analogs of VDH with relatively enhanced CNS actions may potentially be useful for treating age-related or other neurodegenerative/neurotraumatic conditions.
Footnotes
This research was supported by National Institutes of Health Grants AG10836, AG04542, and RO3 AG14189 and Training Grant AG00242, and a grant from the Kentucky Spinal Cord and Head Injury Research Trust. We thank Elsie Barr, Jeanise Staton, and Ruth Wooten-Kee for excellent technical assistance, Dr. Eric Blalock for scientific discussion, and Judith Hower for editorial comments. The vitamin D receptor antibody was generously supplied by Dr. Nicholas Koszewski (Department of Internal Medicine, University of Kentucky).
Correspondence should be addressed to Dr. Nada M. Porter, University of Kentucky, Department of Pharmacology, MS-315 UKMC, Lexington, KY 40536-0084. E-mail: nadap@pop.uky.edu.
REFERENCES
- 1.Alexianu ME, Robbins E, Carswell S, Appel SH. 1α,25 Dihydroxyvitamin D3-dependent up-regulation of calcium binding proteins in motoneurons cells. J Neurosci Res. 1998;51:58–66. doi: 10.1002/(SICI)1097-4547(19980101)51:1<58::AID-JNR6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 2.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 3.Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- 4.Bean BP. Classes of calcium channels in vertebrate cells. Annu Rev Physiol. 1989;51:367–384. doi: 10.1146/annurev.ph.51.030189.002055. [DOI] [PubMed] [Google Scholar]
- 5.Bito H, Deisseroth K, Tsien RW. Ca2+-dependent regulation in neuronal gene expression. Curr Opin Neurobiol. 1997;7:419–429. doi: 10.1016/s0959-4388(97)80072-4. [DOI] [PubMed] [Google Scholar]
- 6.Blalock EM, Porter NM, Landfield PW. Decreased G-protein-mediated regulation and shift in calcium channel types with age in hippocampal cultures. J Neurosci. 1999;19:8674–8684. doi: 10.1523/JNEUROSCI.19-19-08674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 8.Brown AJ, Dusso A, Slatopolsky E. Vitamin D. Am J Physiol. 1999;277:F157–F175. doi: 10.1152/ajprenal.1999.277.2.F157. [DOI] [PubMed] [Google Scholar]
- 9.Caffrey JM, Farach-Carson MC. Vitamin D3 metabolites modulate dihydropyridine-sensitive calcium currents in clonal rat osteosarcoma cells. J Biol Chem. 1989;264:20265–20274. [PubMed] [Google Scholar]
- 10.Cantorna MT, Hayes CE, DeLuca HF. 1,25-Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc Natl Acad Sci USA. 1996;93:7861–7864. doi: 10.1073/pnas.93.15.7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen KC, Blalock EM, Thibault O, Kaminker P, Landfield PW. Expression of α1D subunit mRNA is correlated with L-type Ca2+ channel activity in single neurons of hippocampal “zipper” slices. Proc Natl Acad Sci USA. 2000;97:4357–4362. doi: 10.1073/pnas.070056097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng C, Fass DM, Reynolds IJ. Emergence of excitotoxicity in cultured forebrain neurons coincides with larger glutamate-stimulated [Ca(2+)](i) increases and NMDA receptor mRNA levels. Brain Res. 1999;849:97–108. doi: 10.1016/s0006-8993(99)01995-2. [DOI] [PubMed] [Google Scholar]
- 13.Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988;11:465–469. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- 15.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 16.Compston JE. Vitamin D deficiency: time for action. Evidence supports routine supplementation for elderly people and others at risk. Br Med J. 1998;317:1466–1467. doi: 10.1136/bmj.317.7171.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeLuca HF, Zierold C. Mechanisms and functions of vitamin D. Nutr Rev. 1998;56:S4–S10. doi: 10.1111/j.1753-4887.1998.tb01686.x. [DOI] [PubMed] [Google Scholar]
- 18.de Viragh PA, Haglid KG, Celio MR. Parvalbumin increases in the caudate putamen of rats with vitamin D hypervitaminosis. Proc Natl Acad Sci USA. 1989;86:3887–3890. doi: 10.1073/pnas.86.10.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Disterhoft JF, Moyer JR, Thompson LT. The calcium rationale in aging and Alzheimer's disease. Evidence from an animal model of normal aging. In: Disterhoft JF, Gispen WH, Traber J, Khachaturian ZS, editors. Calcium hypothesis of aging and dementia, Annals of the New York Academy of Sciences, Vol 747. New York Academy of Sciences; New York: 1994. pp. 382–406. [DOI] [PubMed] [Google Scholar]
- 20.Driscoll BF, Deibler GE, Law MJ, Crane AM. Damage to neurons in culture following medium change: role of glutamine and extracellular generation of glutamate. J Neurochem. 1993;61:1795–1800. doi: 10.1111/j.1471-4159.1993.tb09818.x. [DOI] [PubMed] [Google Scholar]
- 21.Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–535. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- 22.Faden AL, Salzman S. Pharmacological strategies in CNS trauma. Trends Pharmacol Sci. 1992;13:29–35. doi: 10.1016/0165-6147(92)90013-v. [DOI] [PubMed] [Google Scholar]
- 23.Fass DM, Takimoto K, Mains RE, Levitan ES. Tonic dopamine inhibition of L-type Ca2+ channel activity reduces alpha1D Ca2+ channel gene expression. J Neurosci. 1999;19:3345–3352. doi: 10.1523/JNEUROSCI.19-09-03345.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finkbeiner S, Greenberg ME. Ca2+ channel-regulated neuronal gene expression. J Neurobiol. 1998;37:171–189. [PubMed] [Google Scholar]
- 25.Fisher RE, Gray R, Johnston D. Properties and distribution of single voltage-gated calcium channels in adult hippocampal neurons. J Neurophysiol. 1990;64:91–104. doi: 10.1152/jn.1990.64.1.91. [DOI] [PubMed] [Google Scholar]
- 26.Fox AP, Nowycky MC, Tsien RW. Single-channel recordings of three types of calcium channels in chick sensory neurones. J Physiol (Lond) 1987;394:173–200. doi: 10.1113/jphysiol.1987.sp016865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freeman WM, Walker SJ, Vrana KE. Quantitative RT-PCR: pitfalls and potential. Biotechniques. 1999;26:112–125. doi: 10.2144/99261rv01. [DOI] [PubMed] [Google Scholar]
- 28.Gallagher M, Landfield PW, McEwen BS, Meaney MJ, Rapp PR, Sapolsky R, West MJ. Hippocampal neurodegeneration in aging (letter). Science. 1996;274:484–485. doi: 10.1126/science.274.5287.484. [DOI] [PubMed] [Google Scholar]
- 29.Garcion E, Nataf S, Berod A, Darcy F, Brachet P. 1,25-Dihydroxyvitamin D3 inhibits the expression of inducible nitric oxide synthase in rat central nervous during experimental allergic encephalomyelitis. Brain Res Mol Brain Res. 1997;45:255–267. doi: 10.1016/s0169-328x(96)00260-4. [DOI] [PubMed] [Google Scholar]
- 30.Geddes JL, Landfield PW, Porter NM (1997) Enhanced vulnerability to excitotoxicity in hippocampal neurons with age in culture. Soc Neurosci Abstr 23.
- 31.Gibson GE, Peterson C. Calcium and the aging nervous system. Neurobiol Aging. 1987;8:329–343. doi: 10.1016/0197-4580(87)90072-8. [DOI] [PubMed] [Google Scholar]
- 32.Gloth FM, III, Tobin JD. Vitamin D deficiency in older people. J Am Geriatr Soc. 1995;43:822–828. doi: 10.1111/j.1532-5415.1995.tb07059.x. [DOI] [PubMed] [Google Scholar]
- 33.Gloth FM, III, Gundberg CM, Hollis BW, Haddad JG, Jr, Tobin JD. Vitamin D deficiency in homebound elderly persons. JAMA. 1995;274:1683–1686. doi: 10.1001/jama.1995.03530210037027. [DOI] [PubMed] [Google Scholar]
- 34.Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid β-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 35.Gwag BJ, Koh JY, DeMaro JA, Ying HS, Jacquin M, Choi DW. Slowly triggered excitotoxicity occurs by necrosis in cortical cultures. Neuroscience. 1997;77:393–401. doi: 10.1016/s0306-4522(96)00473-3. [DOI] [PubMed] [Google Scholar]
- 36.Hamill OP, Marty E, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 37.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 38.Herman JP, Chen K-C, Booze R, Landfield PW. Up-regulation of α1D Ca2+ channel subunit mRNA expression in the hippocampus of aged F344 rats. Neurobiol Aging. 1998;19:581–587. doi: 10.1016/s0197-4580(98)00099-2. [DOI] [PubMed] [Google Scholar]
- 39.Heizmann CW, Braun K. Changes in Ca2+-binding proteins in human neurodegenerative disorders. Trends Neurosci. 1992;15:259–264. doi: 10.1016/0166-2236(92)90067-i. [DOI] [PubMed] [Google Scholar]
- 40.Horn R. Estimating the number of channels in patch recordings. Biophys J. 1991;60:433–439. doi: 10.1016/S0006-3495(91)82069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iacopino AM, Christakos S. Specific reduction of calcium-binding protein (28-kilodalton calbindin-D) gene expression in aging and neurodegenerative diseases. Proc Natl Acad Sci USA. 1990;87:4078–4082. doi: 10.1073/pnas.87.11.4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacques PF, Felson DT, Tucker KL, Mahnken B, Wilson PW, Rosenberg IH, Rush D. Plasma 25-hydroxyvitamin D and its determinants in an elderly population sample. Am J Clin Nutr. 1997;66:929–936. doi: 10.1093/ajcn/66.4.929. [DOI] [PubMed] [Google Scholar]
- 43.Jones G, Strugnell SA, DeLuca HF. Current understanding of the molecular actions of vitamin D. Physiol Rev. 1998;78:1193–1231. doi: 10.1152/physrev.1998.78.4.1193. [DOI] [PubMed] [Google Scholar]
- 44.Kapur A, Yeckel MF, Gray R, Johnston D. L-type calcium channels are required for one form of hippocampal mossy fiber LTP. J Neurophysiol. 1998;79:2181–2190. doi: 10.1152/jn.1998.79.4.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katzman R, Saitoh T. Advances in Alzheimer's disease. FASEB J. 1991;5:278–286. [PubMed] [Google Scholar]
- 46.Khachaturian ZS. The role of calcium regulation in brain aging: reexamination of a hypothesis. Aging. 1989;1:17–34. doi: 10.1007/BF03323872. [DOI] [PubMed] [Google Scholar]
- 47.Kimura M, Sawada K, Miyagawa T, Kuwada M, Katayama K, Nishizawa Y. Role of glutamate receptors and voltage-dependent calcium and sodium channels in the extracellular glutamate/aspartate accumulation and subsequent neuronal injury induced by oxygen/glucose deprivation in cultured hippocampal neurons. J Pharmacol Exp Ther. 1998;285:178–185. [PubMed] [Google Scholar]
- 48.Krieglstein J, Lippert K, Poch G. Apparent independent action of nimodipine and glutamate antagonists to protect cultured neurons against glutamate-induced damage. Neuropharmacology. 1996;35:1737–1742. doi: 10.1016/s0028-3908(96)00104-9. [DOI] [PubMed] [Google Scholar]
- 49.Landfield PW, Cadwallader-Neal L. Long-term treatment with calcitriol (1,25(OH)2 vitamin D3) retards a biomarker of hippocampal aging in rats. Neurobiol Aging. 1998;19:469–477. doi: 10.1016/s0197-4580(98)00079-7. [DOI] [PubMed] [Google Scholar]
- 50.Landfield PW, Eldridge JC. The glucocorticoid hypothesis of age-related hippocampal neurodegeneration: role of dysregulated intraneuronal calcium. In: deKloet ER, Azmitia CE, Landfield PW, editors. Brain corticosteroid receptors: studies on the mechanism, function, and neurotoxicity of corticosteroid action, Annals of the New York Academy of Sciences, Vol 747. New York Academy of Sciences; New York: 1994. pp. 351–364. [DOI] [PubMed] [Google Scholar]
- 51.Landfield PW, Thibault O, Mazzanti ML, Porter NM, Kerr DS. Mechanisms of neuronal death in brain aging and Alzheimer's disease: role of endocrine-mediated calcium dyshomeostasis. J Neurobiol. 1992;23:1247–1260. doi: 10.1002/neu.480230914. [DOI] [PubMed] [Google Scholar]
- 52.Langub MC, Reinhardt TA, Horst RL, Malluche HH, Koszewski NJ. Characterization of vitamin D receptor immunoreactivity in human bone cells. Bone. 2000;27:383–387. doi: 10.1016/s8756-3282(00)00335-5. [DOI] [PubMed] [Google Scholar]
- 53.Lie YS, Petropoulos CJ. Advances in quantitative PCR technology: 5′ nuclease assays. Curr Opin Biotechnol. 1998;9:43–48. doi: 10.1016/s0958-1669(98)80082-7. [DOI] [PubMed] [Google Scholar]
- 54.Lips P, Wiersinga A, van Ginkel FC, Jongen MJ, Netelenbos JC, Hackeng WH, Delmas PD, van der Vijgh WJ. The effect of vitamin D supplementation on vitamin D status and parathyroid function in elderly subjects. J Clin Endocrinol Metab. 1988;67:644–650. doi: 10.1210/jcem-67-4-644. [DOI] [PubMed] [Google Scholar]
- 55.Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K. Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl. 1995;4:357–362. doi: 10.1101/gr.4.6.357. [DOI] [PubMed] [Google Scholar]
- 56.MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 57.Mattson MP, Rychlik B, Chu C, Christakos S. Evidence for calcium-reducing and excito-protective roles for the calcium-binding protein calbindin-D28k in cultured hippocampal neurons. Neuron. 1991;6:41–51. doi: 10.1016/0896-6273(91)90120-o. [DOI] [PubMed] [Google Scholar]
- 58.McEwen BS, Sapolsky RM. Stress and cognitive function. Curr Opin Neurobiol. 1995;5:205–216. doi: 10.1016/0959-4388(95)80028-x. [DOI] [PubMed] [Google Scholar]
- 59.McKenna MJ. Differences in vitamin D status between countries in young adults and the elderly. Am J Med. 1992;93:69–77. doi: 10.1016/0002-9343(92)90682-2. [DOI] [PubMed] [Google Scholar]
- 60.Meszaros JG, Karin NJ, Akanbi K, Farach-Carson MC. Down-regulation of L-type Ca2+ channel transcript levels by 1,25-dihyroxyvitamin D3. Osteoblastic cells express L-type alpha1C Ca2+ channel isoforms. J Biol Chem. 1996;271:32981–32985. doi: 10.1074/jbc.271.51.32981. [DOI] [PubMed] [Google Scholar]
- 61.Moyer JR, Jr, Thompson LT, Black JP, Disterhoft JF. Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J Neurophysiol. 1992;68:2100–2109. doi: 10.1152/jn.1992.68.6.2100. [DOI] [PubMed] [Google Scholar]
- 62.Musiol IM, Feldman D. 1,25-Dihydroxyvitamin D3 induction of nerve growth factor in L929 mouse fibroblasts: effect of vitamin D receptor regulation and potency of vitamin D3 analogs. Endocrinology. 1997;138:12–18. doi: 10.1210/endo.138.1.4858. [DOI] [PubMed] [Google Scholar]
- 63.Naveilhan P, Neveu I, Wion D, Brachet P. 1,25-Dihydroxy vitamin D3, an inducer of glial cell line-derived neurotrophic factor. NeuroReport. 1996;7:2171–2175. doi: 10.1097/00001756-199609020-00023. [DOI] [PubMed] [Google Scholar]
- 64.Neveu I, Naveilhan P, Baudet C, Brachet P, Metsis M. 1,25-Dihydroxyvitamin D3 regulates NT-3, NT-4 but not BDNF mRNA in astrocytes. NeuroReport. 1994a;6:124–126. doi: 10.1097/00001756-199412300-00032. [DOI] [PubMed] [Google Scholar]
- 65.Neveu I, Naveilhan P, Jehan F, Baudet C, Wion D, DeLuca HF, Brachet P. 1,25-Dihydroxyvitamin D3 regulates the synthesis of nerve growth factor in primary cultures of glial cells. Brain Res Mol Brain Res. 1994b;24:70–76. doi: 10.1016/0169-328x(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 66.Nicotera P, Orrenius S. The role of calcium in apoptosis. Cell Calcium. 1998;23:173–180. doi: 10.1016/s0143-4160(98)90116-6. [DOI] [PubMed] [Google Scholar]
- 67.Norman AW. Receptors for 1alpha,25(OH)2D3: past, present, and future. J Bone Miner Res. 1998;13:1360–1369. doi: 10.1359/jbmr.1998.13.9.1360. [DOI] [PubMed] [Google Scholar]
- 68.Norman AW, Nemere I, Zhou LX, Bishop JE, Lowe KE, Maiyar AC, Collins ED, Taoka T, Sergeev I, Farach-Carson MC. 1,25(OH)2-vitamin D3, a steroid hormone that produces biologic effects via both genomic and nongenomic pathways. J Steroid Biochem Mol Biol. 1992;41:231–240. doi: 10.1016/0960-0760(92)90349-n. [DOI] [PubMed] [Google Scholar]
- 69.Norris CM, Halpain S, Foster TC. Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J Neurosci. 1998;18:3171–3179. doi: 10.1523/JNEUROSCI.18-09-03171.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Novelli A, Reilly JA, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- 71.Nowycky MC, Fox AP, Tsien RW. Long-opening mode of gating of neuronal calcium channels and its promotion by the dihydropyridine calcium agonist Bay K 8644. Proc Natl Acad Sci USA. 1985;82:2178–2182. doi: 10.1073/pnas.82.7.2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perry HM, III, Horowitz M, Morley JE, Patrick P, Vellas B, Baumgartner R, Garry PJ. Longitudinal changes in serum 25-hydroxyvitamin D in older people. Metabolism. 1999;48:1028–1032. doi: 10.1016/s0026-0495(99)90201-9. [DOI] [PubMed] [Google Scholar]
- 73.Porter NM, Landfield PW. Stress hormones and brain aging: adding injury to insult? Nat Neurosci. 1998;1:3–4. doi: 10.1038/196. [DOI] [PubMed] [Google Scholar]
- 74.Porter NM, Thibault O, Thibault V, Chen KC, Landfield PW. Calcium channel density and hippocampal cell death with age in long-term culture. J Neurosci. 1997;17:5629–5639. doi: 10.1523/JNEUROSCI.17-14-05629.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Prüfer K, Veenstra TD, Jirikowski GF, Kumar R. Distribution of 1,25-dihydroxyvitamin D3 receptor immunoreactivity in the rat brain and spinal cord. J Chem Neuroanat. 1999;16:135–145. doi: 10.1016/s0891-0618(99)00002-2. [DOI] [PubMed] [Google Scholar]
- 76.Rajdev S, Reynolds IJ. Glutamate-induced intracellular calcium changes and neurotoxicity in cortical neurons in vitro: effect of chemical ischemia. Neuroscience. 1994;62:667–679. doi: 10.1016/0306-4522(94)90468-5. [DOI] [PubMed] [Google Scholar]
- 77.Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor. Trends Neurosci. 1987;10:299–302. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- 78.Sakmann B, Neher E. Geometric parameters of pipettes and membrane patches. In: Sakmann B, Neher E, editors. Single channel recording. Plenum; New York: 1983. pp. 37–51. [Google Scholar]
- 79.Sigworth F, Zhou J. Ion channels. Analysis of nonstationary single-channel currents. In: Rudy B, Iverson LE, editors. Methods in enzymology, Vol 207. Academic; San Diego: 1992. pp. 746–762. [DOI] [PubMed] [Google Scholar]
- 80.Simpkins JW, Singh M, Bishop J. The potential role for estrogen replacement therapy in the treatment of the cognitive decline and neurodegeneration associated with Alzheimer's disease. Neurobiol Aging. 1994;15:S195–S197. doi: 10.1016/0197-4580(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 81.Stumpf WE, O'Brien LP. 1,25(OH)2-vitamin D3 sites of action in the brain. An autoradiographic study. Histochemistry. 1987;87:393–406. doi: 10.1007/BF00496810. [DOI] [PubMed] [Google Scholar]
- 82.Stumpf WE, Sar M, Clark SA, DeLuca HF. Brain target sites for 1,25-dihydroxyvitamin D3. Science. 1982;215:1403–1405. doi: 10.1126/science.6977846. [DOI] [PubMed] [Google Scholar]
- 83.Stuiver BT, Douma BRK, Bakker R, Nyackas C, Luiten PGM. In vivo protection against NMDA-induced neurodegeneration by MK-801 and nimodipine: combined therapy and temporal course of protection. Neurodegeneration. 1996;5:153–159. doi: 10.1006/neur.1996.0022. [DOI] [PubMed] [Google Scholar]
- 84.Sutherland MK, Somerville MJ, Yoong LKK, Bergeron C, Haussler MR, Craper DR, McLachlan DR. Reduction of vitamin D hormone receptor mRNA levels in Alzheimer as compared to Huntington hippocampus: correlation with calbindin-28k mRNA levels. Mol Brain Res. 1992;13:2339–2350. doi: 10.1016/0169-328x(92)90032-7. [DOI] [PubMed] [Google Scholar]
- 85.Takeuchi K, Guggino SE. 24R,25-(OH)2 vitamin D3 inhibits 1a,25-(OH)2 vitamin D3 and testosterone potentiation of calcium channels in osteosarcoma cells. J Biol Chem. 1996;27:33335–33343. doi: 10.1074/jbc.271.52.33335. [DOI] [PubMed] [Google Scholar]
- 86.Teyler TJ, Cavus I, Coussens C. Synaptic plasticity in the hippocampal slice: functional consequences. J Neurosci Methods. 1995;59:11–17. doi: 10.1016/0165-0270(94)00188-m. [DOI] [PubMed] [Google Scholar]
- 87.Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1017–1020. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- 88.Thibault O, Porter NM, Landfield PW. Low Ba2+ and Ca2+ induce a sustained high probability of repolarization openings in hippocampal L-type Ca2+ channels. Proc Natl Acad Sci USA. 1993;90:11792–11796. doi: 10.1073/pnas.90.24.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thibault O, Porter NM, Chen K-C, Blalock E, Kaminker P, Clodfelter G, Brewer L, Landfield PW. Calcium dysregulation in neuronal aging and Alzheimer's disease: history and new directions. Cell Calcium. 1998;24:417–433. doi: 10.1016/s0143-4160(98)90064-1. [DOI] [PubMed] [Google Scholar]
- 90.Thomas MK, Lloyd-Jones DM, Thadhani RI, Shaw AC, Deraska DJ, Kitch BT, Vamvakas EC, Dick IM, Prince RL, Finkelstein JS. Hypovitaminosis D in medical inpatients. N Engl J Med. 1998;338:777–783. doi: 10.1056/NEJM199803193381201. [DOI] [PubMed] [Google Scholar]
- 91.Toescu EC. Apoptosis and cell death in neuronal cells: where does Ca2+ fit in? Cell Calcium. 1998;24:387–403. doi: 10.1016/s0143-4160(98)90062-8. [DOI] [PubMed] [Google Scholar]
- 92.Tornquist K, Tashjian AH., Jr Dual actions of 1,25-dihydroxycholecalciferol on intracellular Ca2+ in GH4C1 cells: evidence for effects on voltage-operated Ca2+ channels and Na+/Ca2+ exchange. Endocrinology. 1989;124:2765–2776. doi: 10.1210/endo-124-6-2765. [DOI] [PubMed] [Google Scholar]
- 93.Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Uematsu D, Arki N, Greenberg JH, Sladky J, Reivich M. Combined therapy with MK-801 and nimodipine for protection of ischemic brain damage. Neurology. 1991;41:88–94. doi: 10.1212/wnl.41.1.88. [DOI] [PubMed] [Google Scholar]
- 95.Utiger RD. The need for more vitamin D. N Engl J Med. 1998;338:828–829. doi: 10.1056/NEJM199803193381209. [DOI] [PubMed] [Google Scholar]
- 96.Vazquez G, de Boland AR. Stimulation of dihydropyridine-sensitive Ca2+ influx in cultured myoblasts by 1,25(OH)2-vitamin D3. Biochem Mol Biol Int. 1993;31:677–684. [PubMed] [Google Scholar]
- 97.Vazquez G, de Boland AR, Boland RL. 1α,25-Dihydroxy-vitamin-D3-induced store-operated Ca2+ influx in skeletal muscle cells. J Biol Chem. 1998;273:33954–33960. doi: 10.1074/jbc.273.51.33954. [DOI] [PubMed] [Google Scholar]
- 98.Veenstra TD, Prüfer K, Koenissberger C, Brimijoin SW, Grande JP, Kumar R. 1,25-dihydroxyvitamin D3 receptors in the central nervous system of the rat embryo. Brain Res. 1998;804:193–205. doi: 10.1016/s0006-8993(98)00565-4. [DOI] [PubMed] [Google Scholar]
- 99.Verkhratsky A, Toescu EC. Calcium and neuronal ageing. Trends Neurosci. 1998;21:2–7. doi: 10.1016/s0166-2236(97)01156-9. [DOI] [PubMed] [Google Scholar]
- 100.Wang Y, Chiang YH, Su TP, Hayashi T, Morales M, Hoffer BJ, Lin SZ. Vitamin D(3) attenuates cortical infarction induced by middle cerebral arterial ligation in rats. Neuropharmacology. 2000;39:873–880. doi: 10.1016/s0028-3908(99)00255-5. [DOI] [PubMed] [Google Scholar]
- 101.Weiss JH, Hartley DM, Koh J, Choi DW. The calcium channel blocker nifedipine attenuates slow excitatory amino acid neurotoxicity. Science. 1990;247:1474–1477. doi: 10.1126/science.247.4949.1474. [DOI] [PubMed] [Google Scholar]
- 102.Wise PM, Smith MJ, Dubal DB, Wilson ME, Krajnak KM, Rosewell KL. Neuroendocrine influences and repercussions of the menopause. Endocr Rev. 1999;20:243–248. doi: 10.1210/edrv.20.3.0364. [DOI] [PubMed] [Google Scholar]
- 103.Ye ZC, Sontheimer H. Astrocytes protect neurons from neurotoxic injury by serum glutamate. Glia. 1998;22:237–248. doi: 10.1002/(sici)1098-1136(199803)22:3<237::aid-glia3>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 104.Zakon HH. The effects of steroid hormones on electrical activity of excitable cells. Trends Neurosci. 1998;21:202–207. doi: 10.1016/s0166-2236(97)01209-5. [DOI] [PubMed] [Google Scholar]