

Abstract
Photo-affinity labeling and mutagenesis studies have identified several amino acids that may contribute to the ligand binding domains of ligand-gated ion channels. These types of studies, however, only generate a one-dimensional, static description of binding site structure. In this study, we used the substituted cysteine accessibility method not only to identify binding pocket residues but also to elicit information about binding site dynamics and structure. Residues surrounding the putative loop C ligand binding domain of the GABAA receptor (β2V199 to β2S209) were individually mutated to cysteine, and the mutant subunits were coexpressed with wild-type α1subunits in Xenopus oocytes.N-biotinylaminoethyl methanethiosulfonate (MTSEA-biotin) reacts with cysteines introduced at positions G203, S204, Y205, P206, R207, and S209. This accessibility pattern is not consistent with either an α-helix or β-strand. Instead, G203–S209 seems to form a water-accessible extended coil, whereas V199–T202 appears to buried in the protein or membrane. Coapplication of either GABA or the competitive antagonist SR-95531 significantly slows MTSEA-biotin modification of cysteines introduced at positions S204, Y205, R207, and S209, demonstrating that these residues line and face into the GABA binding pocket. MTSEA-biotin reaction rates reveal a steep accessibility gradient from G203–S209 and suggests that the binding pocket is a deep narrowing cleft. Pentobarbital activation of the receptor significantly slows MTSEA-biotin modification of cysteines at S204, R207, and S209, suggesting that the binding site may constrict during gating.
Keywords: GABA, GABAA receptor, binding site, substituted cysteine accessibility method, cysteine mutagenesis, agonist efficacy, protein structure
GABAAreceptors share their fundamental structure and functional properties with an evolutionarily related superfamily of ligand-gated ion channels (LGICs) that also includes nicotinic acetylcholine (nACh), 5-HT3, glycine, and GABAC receptors (Ortells and Lunt, 1995). For these receptors, neurotransmitter binding induces allosteric changes in the protein that result in channel opening and fast synaptic response. A complete understanding of the function of these receptors will not only require detailed structural information regarding the protein domains involved in agonist binding, transduction, and gating but will also necessitate knowledge of the relative movements of these domains, and their constituent amino acid residues, when the receptor undergoes transitions from the unliganded, closed state to the fully liganded, open state.
Although high-resolution crystal structures of liganded and unliganded receptors may ultimately help us to answer these questions, this data has proven to be notoriously elusive, and precise information regarding the structure and dynamics of the GABA binding domain remains scarce. Methods such as photo-affinity labeling and site-directed mutagenesis have led to the identification of several individual amino acid residues on the α and β subunits that may contribute to the ligand binding pocket (Sigel et al., 1992; Amin and Weiss, 1993; Smith and Olsen, 1994; Westh-Hansen et al., 1997). This work, and parallel work on the closely related nicotinic acetylcholine receptor, indicates that agonist binding takes place at inter-subunit interfaces (Czajkowski et al., 1993) and may be coordinated by residues from at least six polypeptide loops designated loop A–loop F (Corringer et al., 2000).
One means of extracting more detailed information about the secondary structure, solvent accessibility, and dynamics of a protein domain is the substituted cysteine accessibility method (SCAM) (Javitch et al., 1995; Xu and Akabas, 1996; Wilson and Karlin, 1998; Basiry et al., 1999). SCAM entails the mutation of a residue to cysteine and the subsequent observation of the functional effect (if any) caused by reaction of the introduced cysteine with a sulfhydryl reactive reagent (Karlin and Akabas, 1998). Previously, we used SCAM on the F64 region (loop D) of the GABAA α1subunit to define the secondary structure of this region as a β-strand and identified α1F64, α1R66, and α1S68 as residues likely to line the GABA binding pocket (Boileau et al., 1999).
In the present study, we performed SCAM analysis on residues β2V199–S209, which comprise the putative loop C domain of the GABA binding pocket. These experiments identified four residues that face into the GABA binding pocket: S204, Y205, R207, and S209. Residues that influence GABA affinity but are not part of the pocket were also identified: F200, S201, T202, and G203. Accessibility and rate of reaction studies indicate that loop C has an extended conformation that may traverse the GABA binding pocket from its rim to its depths. Finally, we demonstrate that this region of the binding pocket experiences structural rearrangements consistent with a constriction of the binding pocket during pentobarbital-mediated gating of the receptor.
MATERIALS AND METHODS
Site-directed mutagenesis. The β2 cysteine mutant constructs were made by recombinant PCR, which has been described previously (Kucken et al., 2000). Cysteine substitutions were made in the rat β2 subunit at positions V199, F200, S201, T202, G203, S204, Y205, P206, R207, L208, and S209 (Fig.1). The β2cysteine mutants were subcloned into pGH19 (Liman et al., 1992;Robertson et al., 1996) for expression in Xenopus laevisoocytes. All β2 cysteine mutants were verified by double-stranded DNA sequencing. The β2cysteine mutants have been named, using the single letter code, as wild-type residue, residue number, and mutated residue.
Fig. 1.

Alignment of loop C domains from different LGICs. The β2 loop C domain of the GABA binding site is aligned with homologous domains from the benzodiazepine binding site of the GABAA α1 subunit, the acetylcholine binding site of the nicotinic acetylcholine receptor α1 subunit, and the glycine binding site of the glycine receptor α1subunit. Residues that have been predicted to be in or near the binding pocket by photo-affinity labeling or mutagenesis are shown inbold (Galzi and Changeux, 1994). Residues in β2 that were mutated to cysteines are denoted by aCabove the wild-type residue.
Expression in oocytes and voltage-clamp analysis. Oocytes from Xenopus laevis were prepared and injected with cRNA as described previously (Boileau et al., 1998). GABAA receptor rat α1, β2, or β2 cysteine mutants in pGH19 were expressed by injection of cRNA into oocytes at 20 ng of each RNA species per oocyte, except for β2-T202C, which was injected at 100 ng α1/100 ng β2-T202C per oocyte. RNA concentrations were determined by measuring absorbance at 260 nm and confirmed by observation of ethidium staining of RNA run out on agarose gels. The oocytes were maintained in ND96 (in mm: 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, and 5 HEPES, pH 7.4) supplemented with 100 μg/ml gentamicin and 100 μg/ml BSA for 2–14 d and used for electrophysiological recordings.
Oocytes under two-electrode voltage-clamp (Vhold of −80 mV) were perfused continuously with ND96 recording solution at a rate of 5 ml/min. The bath volume was 200 μl. Drugs and reagents were dissolved in ND96, except for N-biotinylaminoethyl methanethiosulfonate (MTSEA-biotin), which was made as a stock solution in DMSO and diluted to working concentrations in ND96. [DMSO] was ≤1% in final solutions and did not affect GABAA receptor properties. Standard two-electrode voltage-clamp recording was performed using a GeneClamp 500 (Axon Instruments, Foster City, CA) interfaced to a computer with a Digidata 1200 (Axon Instruments). Electrodes were filled with 3 m KCl and had a resistance of 0.5–1.5 MΩ. Data acquisition and analysis were performed using pClamp (Axon Instruments).
EC50 analysis. To compensate for slow drift in current responses to GABA application (IGABA) and pentobarbital application (Ipentobarbital), dose–response trials were performed by applying a low concentration of agonist (EC2–EC7) just before the test concentration of agonist. Before curve fitting, currents evoked by each test concentration were normalized to the corresponding low-concentration current. Full dose–response curves were measured for each cell tested, and the resulting data were fit to the following equation: I = Imax/(1 + (EC50/[A])n), whereI is the peak response to a given concentration of agonist,Imax is the maximum current, EC50 is the concentration of agonist that evokes a current half the maximum, [A] is the concentration of GABA, andn is the Hill coefficient.
IC50 analysis. SR-95531 IC50 values were measured using a protocol in which an application of a fixed concentration of GABA was immediately followed by coapplication of the same concentration of GABA and a test concentration of SR-95531. For each SR-95531 concentration, inhibition was calculated as IGABA + SR-95531/IGABA. Full inhibition curves were measured for each cell tested, and the resulting data were fit to the following equation: inhibition = 1 − 1/(1 + (IC50/[Ant])n), where IC50 is the concentration of antagonist that blocks half of IGABA, [Ant] is the concentration of antagonist, and n is the Hill coefficient. Ki values were calculated using the Cheng–Prusoff/Chou equation (Cheng and Prusoff, 1973; Chou, 1974): Ki = IC50/(1 + [A]/EC50), where [A] is the concentration of GABA used, and EC50 is the GABA-EC50 for the mutant in question.
Measurement of MTSEA-biotin effects. All oocytes were tested for stability of IGABA before addition of MTSEA-biotin (Toronto Research Chemicals Inc., North York, Canada) by applying a 5 sec pulse of GABA every 10 min until the peak currents varied by <3% from one trial to the next. Stability was usually obtained after three to six trials (30–60 min). GABA concentrations ranged between EC30 and EC60. After the GABA response stabilized, we bath applied freshly diluted MTSEA-biotin (2 mm) for 2 min, washed for 5 min, and then recordedIGABA at the same concentration used before MTSEA-biotin treatment. The covalent effect of MTSEA-biotin was calculated as (IGABA-post/IGABA-pre) − 1.
Rate of reaction assays. The rate at which MTSEA-biotin covalently modified introduced cysteines was determined by observing the effects of sequential applications of MTSEA-biotin onIGABA. The protocol was as follows: apply GABA (EC30–EC50) for 5 sec, wash for 30 sec, apply MTSEA-biotin for 5–20 sec, wash for 2.5 min, and repeat sequence (see Fig. 4). This protocol was repeated until the reaction was complete (IGABA no longer changed). To accommodate for the disparate rates at which MTSEA-biotin reacts with the various mutants, the concentration and time of MTSEA-biotin application was varied as follows: G203C, 1 μm, 5 sec; S204C, 1 μm, 10 sec; Y205C, 200 μm, 10 sec; P206C, 200 μm, 20 sec; R207C, 200 μm, 20 sec; and S209C, 1 mm, 10 sec. The effects of agonists and antagonists on reaction rates were assayed by coapplying GABA (EC60–EC80), SR-95531 (IC90–IC95), or pentobarbital (50 μm or 1 mm) with the MTSEA-biotin.
Fig. 4.
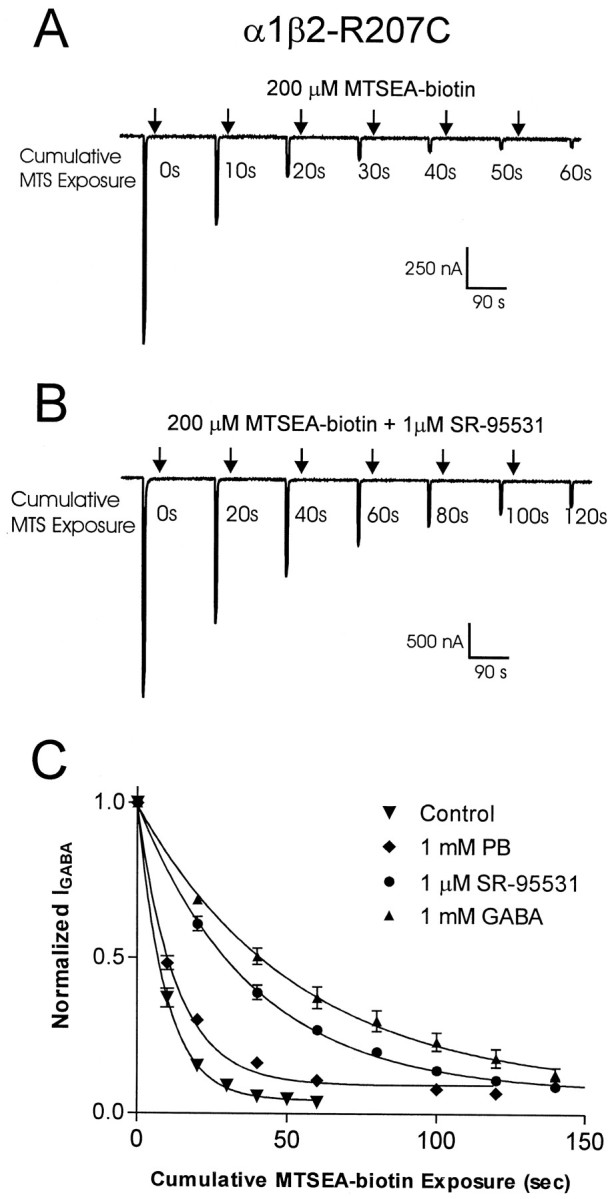
Measurement of MTSEA-biotin reaction rates.A, B, Examples of traces recorded during experiments measuring the reaction rate of MTSEA-biotin with α1β2-R207C receptors. Downward deflections represent inward current elicited by a 5 sec application of 300 μm GABA (≈EC50).Arrows indicate either 10 sec application of 200 μm MTSEA-biotin (A) or a 20 sec coapplication of MTSEA-biotin plus 1 μm SR-95531 (B). C, NormalizedIGABA plotted as a function of cumulative time of MTSEA-biotin exposure. Single exponential curve fits illustrate the effect of various compounds on the reaction rate of MTSEA-biotin with α1β2-R207C receptors. Data points are normalized to the current measured at t = 0 and are presented as mean ± SEM. PB, Pentobarbital.
The data gathered with the rate of reaction protocol was plotted asIGABA versus cumulative time of MTSEA-biotin exposure The pseudo-first-order rate constant (k) was determined by fitting the plotted data to a single exponential decay equation: y = (span − span × e−kt) + plateau, where span = max − plateau. The second-order rate constant (k2) was determined by dividing the pseudo-first-order rate constant by the concentration of MTSEA-biotin used (Pascual and Karlin, 1998). To verify the accuracy of our protocol, k2 was determined at two different concentrations of MTSEA-biotin for several of the mutants.
Statistical analysis. When determining EC50, IC50, ork2, complete data sets were obtained from individual oocytes. Curve fitting was subsequently performed on the data from each oocyte, and the resultant parameters were used in statistical analysis. Statistical analysis for significant differences was performed by one-way ANOVA with Dunnett's post hoc test for multiple independent samples. In the case of EC50 and IC50 results, analysis for significance was performed using log values. All curve fits and statistical analysis were performed using Prism software (GraphPad Software Inc., San Diego, CA).
RESULTS
Cysteine mutation of the β2 loop C region
Mutations at β2-Y205 and β2-T202 cause large shifts in EC50-GABA values of α1β2mut and α1β2mutγ2GABAA receptors but have no effect on direct activation of receptors by pentobarbital (Amin and Weiss, 1993), indicating that these residues may contribute to the ligand binding pocket. These residues align with putative ligand binding domains of the nACh α subunit (Dennis et al., 1988) and the glycine receptor α subunit (Vandenberg et al., 1992), and this region has been termed loop C (Corringer et al., 2000). To fully evaluate the contribution of the loop C region to ligand binding and gating in the GABAA receptor, 11 cysteine mutants were made at positions V199, F200, S201, T202, G203, S204, Y205, P206, R207, L208, and S209 of the β2 subunit (Fig. 1). The mutant β2 subunits were then coexpressed with wild-type α1 subunits in Xenopusoocytes and physiologically characterized using the two-electrode voltage-clamp technique.
All of the mutant subunits assembled into functional α1β2mut receptors. Mean maximal responses to GABA ranged from 1 to 10 μA and did not differ significantly from wild type (data not shown). GABA dose–response analysis of the mutant receptors revealed six residues that cause shifts in EC50-GABA values when mutated to cysteine, demonstrating that EC50-GABA is exquisitely sensitive to perturbation of this domain. The F200C, S201C, or R207C mutations caused 70- to 300-fold shifts in EC50-GABA values relative to wild type, whereas the T202C, G203C, and Y205C mutations resulted in 4800-to 18,000-fold increases in EC50-GABA values (Fig.2A, Table1). All of these mutations (with the exception of R207C) also caused significant shifts in the IC50 values of the competitive antagonist SR-95531 (12- to 100-fold increases), but none of the mutations had a significant effect on the EC50 values for direct activation of the receptor by pentobarbital (Table 1). Notably, the mutations that reduced EC50-GABA have much smaller effects on the apparent affinity of SR-95531. This could be attributable to the fact that SR-95531, a much larger molecule than GABA, may enjoy extra binding interactions that make it more tolerant to a single point mutation within the binding pocket.
Fig. 2.
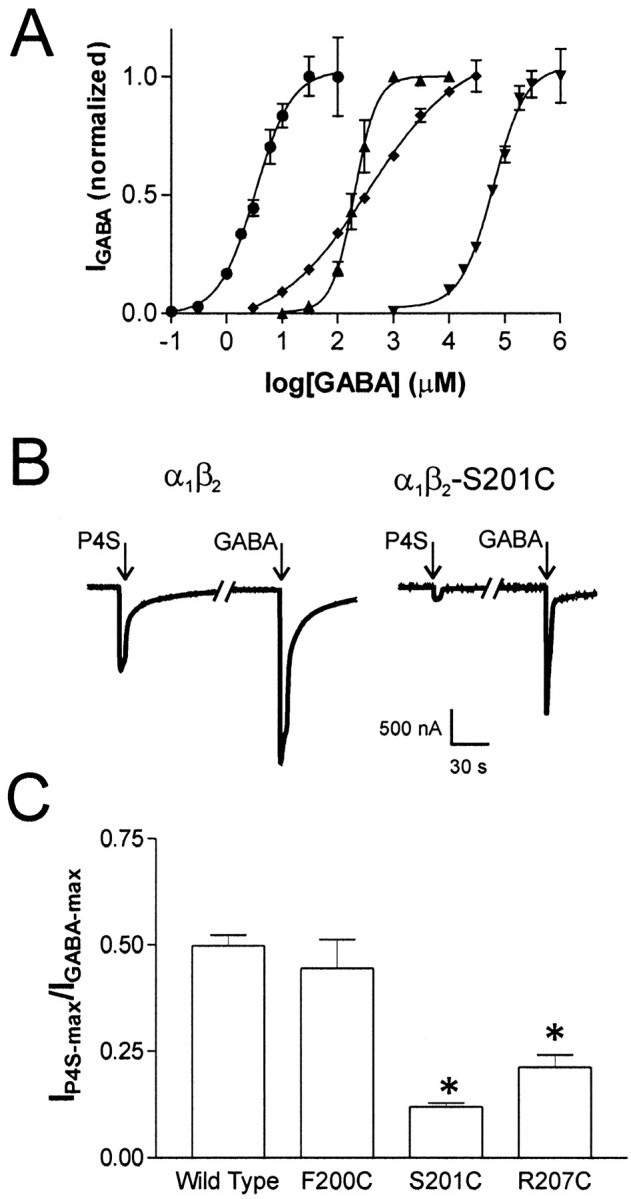
GABA dose–response curves and P4S currents.A, GABA dose–response relationships for wild-type α1β2 receptors (●) and three representative mutants: α1β2-R207C (▴), α1β2-S201C (♦), and α1β2-Y205C (▾). Data were fit by nonlinear regression as described in Materials and Methods. All data points are normalized to Imax-GABA and are shown as mean responses ± SEM from four or more cells.B, Current traces recorded from oocytes expressing wild type or α1β2-S201C. Arrowsindicate a 5 sec application of saturating P4S (wild type, 1 mm; S201C, 10 mm) or GABA (wild type, 1 mm; S201C, 100 mm). Line breakin current trace represents 5 min wash with ND96. C, Bar graph denoting P4S efficacy of wild-type and mutant receptors asImax-P4S/Imax-GABAwhere values given as mean ± SEM follow: α1β2, 0.50 ± 0.03,n = 4; α1β2-F200C, 0.45 ± 0.06, n = 4; α1β2-S201C, 0.12 ± 0.01,n = 3; and α1β2-R207C, 0.21 ± 0.02, n = 4. * p< 0.01 indicates values that are significantly different from wild type calculated using a one-way ANOVA with a Dunnett's post hoc test.
Table 1.
Apparent affinites of wild-type and mutant receptors for GABA, SR-95531, and pentobarbital
| Receptor | GABA | SR-95531 | PB | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50(μm) | nH | n | wt/mut | Ki(μm) | n | wt/mut | EC50(μm) | n | wt/mut | |
| α1β2 Wild type | 4.3 ± 1.2 | 1.1 | 8 | 1.0 | 0.12 ± 0.02 | 8 | 1.0 | 570 ± 66 | 5 | 1.0 |
| α1β2-V199C | 3.1 ± 0.4 | 1.4 | 4 | 0.7 | 0.08 ± 0.01 | 3 | 0.6 | 577 ± 195 | 5 | 1.0 |
| α1β2-F200C | 1292 ± 170* | 1.1 | 4 | 300 | 4.34 ± 2.6* | 3 | 36.2 | 712 ± 96 | 3 | 1.2 |
| α1β2-S201C | 725 ± 160* | 0.5* | 4 | 170 | 1.42 ± 0.3* | 3 | 11.8 | 1038 ± 425 | 3 | 1.8 |
| α1β2-T202C | 61100 ± 10900* | 1.0 | 5 | 14000 | 1.90 ± 0.4* | 3 | 15.8 | 690 ± 170 | 3 | 1.2 |
| α1β2-G203C | 20480 ± 6990* | 1.0 | 10 | 4800 | 4.67 ± 0.4* | 4 | 38.9 | 1088 ± 70 | 3 | 1.9 |
| α1β2-S204C | 1.5 ± 0.3 | 1.1 | 6 | 0.3 | 0.05 ± 0.03 | 5 | 0.4 | 427 ± 86 | 5 | 0.7 |
| α1β2Y205C | 78000 ± 7070* | 1.4 | 5 | 18000 | 12.40 ± 1.5* | 3 | 103.3 | 800 ± 42 | 4 | 1.4 |
| α1β2-P206C | 2.6 ± 1.0 | 1.2 | 4 | 0.6 | 0.53 ± 0.13 | 6 | 4.4 | 347 ± 46 | 4 | 0.6 |
| α1β2-R207C | 310 ± 30* | 1.3 | 4 | 70 | 0.11 ± 0.02 | 4 | 0.9 | 480 ± 72 | 4 | 0.8 |
| α1β2-L208C | 2.7 ± 0.5 | 1.5 | 5 | 0.6 | 0.07 ± 0.02 | 3 | 0.6 | 327 ± 77 | 5 | 0.6 |
| α1β2-S209C | 5.4 ± 1.1 | 1.1 | 3 | 1.3 | 0.11 ± 0.01 | 3 | 0.9 | 583 ± 105 | 3 | 1.0 |
EC50 and Ki values are presented as mean ± SEM. An asterisk indicates that the value is significantly different from wild type (p < 0.01). PB, Pentobarbital; wt, wild type; mut, mutant.
Determining agonist efficacy in cysteine mutants
Receptor occupancy and gating of LGICs can be most simply described by the model represented in Scheme 1 (del Castillo and Katz, 1957).

In this model, the microscopic affinity for agonist is represented by the dissociation constant (KD) and agonist efficacy is represented by E, where E is the ratio of the number of fully liganded receptors that are open to the number of fully liganded receptors that are closed (Colquhoun, 1998). When using highly efficacious agonists (E > 10), changes in efficacy have little effect on maximum current and can be difficult or impossible to detect. For instance, if using an agonist with E = 20, a mutation that causes a twofold reduction of efficacy will only produce a 5% change in Imax. To determine whether the cysteine mutations that shift EC50-GABA also cause shifts in efficacy, experiments were performed using piperidine-sulfonic acid (P4S), which acts as a partial agonist (E ≈ 1) at α1 containing GABAA receptors (Krogsgaard-Larsen et al., 1980;O'Shea et al., 2000).
In oocytes expressing wild-type α1β2 receptors, the ratio of current elicited by a saturating concentration of P4S to current elicited by a saturating GABA concentration (Imax-P4S/Imax-GABA) was 0.50 (Fig. 2B,C). In oocytes expressing α1β2-F200C receptors, theImax-P4S/Imax-GABAratio (0.45) was not significantly different from wild type, indicating that this mutation has no effect on agonist efficacy. However, oocytes expressing α1β2-S201C and α1β2-R207C receptors had significantly reducedImax-P4S/Imax-GABAratios of 0.12 and 0.20, respectively (Fig.2B,C). These results demonstrate that mutation of either β2-S201 or β2-R207 to cysteine reduces agonist efficacy at the GABA binding site. A reduction in efficacy can also result in a reduction of the Hill coefficient (Colquhoun, 1998) and may explain why α1β2-S201C receptors have a significantly reduced Hill coefficient (nH) for GABA (Fig.2A, Table 1). It was not possible to test the remaining mutants that caused EC50-GABA shifts (T202C, G203C, and Y205C) for changes in efficacy because their severely reduced affinities require concentrations of GABA near or above 1 m to elicit maximal responses.
Reaction of introduced cysteines with MTSEA-biotin
One of the caveats of SCAM analysis is that the data gathered describes the structure of a mutant receptor that may not be the same as the structure of a wild-type receptor. Because of this, the results of SCAM studies are most reliable if the introduced mutations do not cause large changes in the functional properties of the receptor. Unfortunately, in domains that are functionally significant, even small changes in structure can translate into noticeable changes in receptor behavior. This seems to be the case for the region in question (β2V199–S209) in which 6 of the 11 mutations caused significant changes in EC50-GABA values. However, the fact that none of the mutations significantly affected direct activation by pentobarbital and no significant difference inImax-GABA was detectable suggests that the global structure of the receptor was not altered by any of the cysteine mutations, and it is likely that the changes in EC50-GABA represent small local effects. In addition, cysteine substitution of five of the residues mutated had no discernable effect on any of the receptor properties that we assayed, making them ideal candidates for this study.
Reaction of wild-type α1β2GABAA receptors with the sulfhydryl-specific reagent MTSEA-biotin caused no significant change in GABA-mediated current (Fig. 3). Therefore, if MTSEA-biotin treatment alters IGABA in a mutant receptor, we assume that MTSEA-biotin has modified the introduced cysteine. MTSEA-biotin treatment significantly decreasedIGABA in 6 of the 11 mutant receptors tested (Fig. 3). For each affected mutant,IGABA was inhibited as follows: G203C, −36 ± 9%; S204C, −26 ± 5%; Y205C, −98 ± 1%; P206C, −47 ± 13%; R207C, −55 ± 8%; S209C, −85 ± 1% (mean ± SEM; % inhibition = 100 × [(IGABA-post MTSEA-biotin/IGABA-pre MTSEA-biotin) − 1]). Because six of the seven consecutive residues from G203–S209 reacted with MTSEA-biotin, the accessibility pattern of this region is not predictive of an α-helix or β-strand and, therefore, the region is likely to be a turn or random coil. MTSEA-biotin treatment had no significant effect onIGABA from V199C-, F200C-, S201C-, T202C-, and L208C-containing receptors. Because we cannot detect reaction of MTSEA-biotin with any residue from V199C to T202C, it seems likely that this region is buried in the hydrophobic core of the subunit, but it is also possible that these residues react with MTSEA-biotin without affectingIGABA.
Fig. 3.
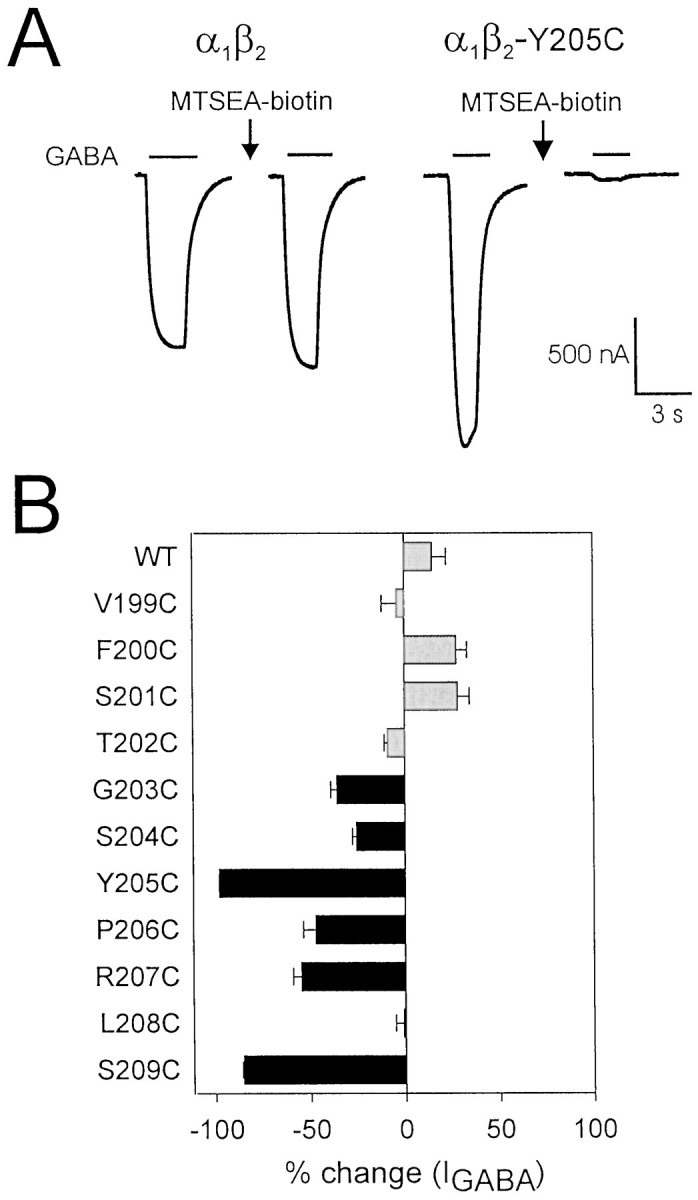
Effects of MTSEA-biotin on wild-type and mutant GABAA receptors. A, Representative current traces demonstrating the effect of MTSEA-biotin treatment (2 mm, 2 min) on currents from wild-type and Y205C-containing receptors. For wild-type traces, [GABA] is 3 μm, and for Y205C traces, [GABA] is 30 mm. B, Effect of MTSEA-biotin treatment on all mutants shown as % change = ([IGABA-post MTSEA-biotin/IGABA-pre MTSEA-biotin] − 1) × 100. Results represent the mean ± SEM of at least three experiments. Black bars indicate that the percent change is significantly different from wild type (p < 0.01). Gray barsindicate no significant difference from wild type (p > 0.05).
Observation of reaction of an introduced sulfhydryl with a methanethiolsulfonate (MTS) reagent can also provide information about the dimensions of the binding site crevice. MTSEA-biotin is composed of two distinct structural domains: a flexible tail ∼14 Å long and 2.5 Å in diameter, and a 4 × 5 Å planar head group. The reactive disulfide is near the end of the tail, ∼12 Å from the head group. Therefore, any residue that reacts with MTSEA-biotin must be accessible via an aqueous pathway >2.5 Å in diameter and <12 Å deep. GABA is a linear molecule ∼6 Å long and 3 Å in diameter, and the dimensions of SR-95531 are ∼16 Å long and 6 Å in diameter.
Measurement of MTSEA-biotin reaction rates
The rate at which MTSEA-biotin reacts with a cysteine side chain is determined by the physical environment of the sulfhydryl group (e.g., steric hindrance to reaction) and the ionization of the sulfhydryl group, which depends on the local dielectric constant and the local electrostatic potential. Thus, a residue in a relatively open, aqueous environment will display a faster rate of reaction than a residue in a relatively restrictive, nonpolar environment (Pascual and Karlin, 1998). To acquire insight into the physicochemical environment of the loop C domain of the GABA binding site, we determined the reaction rate of MTSEA-biotin with each of the accessible introduced cysteines (G203C–R207C and S209C).
Reaction rates were measured by serial presentations of a GABA test pulse (EC30–EC60), followed by a 5–20 sec application of MTSEA-biotin. This protocol was repeated until IGABA plateaued, and the data were plotted as IGABA versus cumulative time of exposure to MTSEA-biotin. Single exponential decay curves were fit to the data, and second-order rate constants (k2) for MTSEA-biotin were calculated (Fig. 4A; see Materials and Methods). The measured k2 values span three orders of magnitude (Table 2). The fastest reaction rate was recorded for the most N-terminal residue tested (G203C, 258,000 M−1s−1), and the rates steadily declined with slowest reaction rate recorded at the most C-terminal residue tested (S209C, 120 M−1 s−1). This steep rate gradient implies that this stretch of amino acids starts in a region in which the sulfhydryl group is in an aqueous environment that is highly accessible to MTSEA-biotin and steadily progresses into a much less accessible cleft.
Table 2.
Summary of second-order rate constants for reaction of MTSEA-biotin with introduced sufhydryls
| Receptor | Control | GABA | SR-95531 | 1 mm PB | 50 μm PB | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| k2(M−1s−1) | n | k2(M−1s−1) | n | k2(M−1s−1) | n | k2(M−1s−1) | n | k2(M−1s−1) | n | |
| α1β2-G203C | 257,600 ± 32,700 | 3 | 184,400 ± 14,300 | 3 | 317,700 ± 19,500 | 3 | 579,200 ± 9900* | 3 | 400,100 ± 7300* | 3 |
| α1β2-S204C | 12,060 ± 980 | 5 | 6050 ± 980* | 5 | 3735 ± 310* | 3 | 5656 ± 1180* | 3 | 14,990 ± 3300 | 3 |
| α1β2-Y205C | 955 ± 150 | 5 | 410 ± 15* | 4 | 285 ± 5* | 3 | 870 ± 40 | 3 | ND | 0 |
| α1β2-P206C | 276 ± 41 | 4 | 715 ± 70* | 3 | 398 ± 16 | 3 | 385 ± 25 | 3 | 352 ± 17 | 3 |
| α1β2-R207C | 585 ± 40 | 6 | 100 ± 9* | 4 | 135 ± 10* | 3 | 370 ± 32* | 3 | 690 ± 65 | 3 |
| α1β2-S209C | 120 ± 2 | 4 | 67 ± 11* | 4 | 40 ± 7* | 3 | 77 ± 8* | 3 | 125 ± 3 | 3 |
Second-order rate constants (k2) were calculated by dividing psuedo-first-order rate constants (k1; see Materials and Methods) by the concentration of MTSEA-biotin used during rate experiments, which were as follows: G203C, 1 μm; S204C, 10 μm; Y205C, P206C, and R207C, 200 μm; and S209C, 1 mm. Concentrations of GABA and SR-95531 present during the MTSEA-biotin reaction, which varied according to the affinity of the mutant receptor for each compound, were always between EC60–IC80 for GABA and IC90–IC95 for SR-95531. An asterisk indicates that the rate is significantly different from control (p < 0.01). PB, Pentobarbital; ND, not determined. Values are mean ± SEM.
Identification of binding site residues
To identify residues in loop C that line the GABA binding pocket, we measured the second-order rate constant (k2) for reaction of MTSEA-biotin with each accessible introduced cysteine both in the presence of GABA and in the presence of SR-95531. SR-95531, a classical competitive agonist for GABA, binds within the GABA binding pocket. Although evidence suggests that SR-95531 may also allosterically modulate the GABAA receptor (Uchida et al., 1996; Ueno et al., 1997), it does not activate the receptor and clearly does not induce the same change in receptor structure as GABA. Therefore, if the rate at which MTSEA-biotin reacts with an introduced cysteine is slowed by both SR-95531 and GABA, then it is likely that both compounds are sterically interfering with the reaction and that the sulfhydryl side chain is facing into the GABA binding pocket. GABA (at EC60–EC80 concentrations) and SR-95531 (at IC90–IC95concentrations) significantly slowed the reaction rate of MTSEA-biotin with cysteines introduced at positions S204, Y205, R207, and S209 (Fig.5, Table 2). Therefore, these residues face into the GABA binding pocket.
Fig. 5.

Summary of effect of GABA, SR-95531, and pentobarbital on the rate at which MTSEA-biotin modifies introduced cysteines. Second-order rate constants were calculated for each reaction, and for each mutant, the rates were normalized to the control rate (rate measured when no other compound is present). *p < 0.01 indicates that rate is significantly different from control rate. All data represent the mean ± SEM of at least three experiments.
State-dependent changes of binding site conformation
According to Scheme 1, receptor activation has two distinct steps, binding of agonist and isomerization of the receptor from the closed to open state. The closed-to-open transition involves a global allosteric rearrangement of the receptor that not only opens a gate but also changes the structure of the binding pocket. We examined gating-related structural changes of the GABA binding pocket by measuring the effect of the barbiturate pentobarbital on k2values for the MTSEA-biotin reaction.
Pentobarbital directly activates the GABAAreceptor but does not bind at the same location as GABA (Ito et al., 1996). Because the single channel conductances of GABAA receptors activated by GABA and pentobarbital are similar (Jackson et al., 1982; Akk and Steinbach, 2000), it is likely that the open states of receptors activated by either of these compounds have similar conformations. Therefore, if the rate at which MTSEA-biotin reacts with an introduced cysteine is altered in the presence of pentobarbital, we infer a gating-related structural rearrangement of the binding pocket. Pentobarbital at EC50–EC70 significantly slowed the reaction rate of MTSEA-biotin with cysteines at positions S204, R207, and S209 and significantly increased the rate at position G203 (Fig. 5, Table 2).
In addition to directly activating the GABAAreceptor, pentobarbital potentiates GABA currents by binding to a site that is presumably separate from the site responsible for direct activation (Ito et al., 1996). Because the allosteric modulatory site has a higher apparent affinity for pentobarbital than the direct activation site (Thompson et al., 1996), rate changes induced by 1 mm pentobarbital could be attributable to its action at either of these sites. To determine whether pentobarbital effects on the MTSEA-biotin reaction rates were mediated by the high-affinity modulatory site, we measured the reaction rate in the presence of 50 μm pentobarbital, a concentration that robustly potentiates IGABA but causes little direct activation of the receptor. Pentobarbital (50 μm) had no effect on the MTSEA-biotin reaction with cysteines introduced at positions S204, R207, and S209 (Fig. 5, Table 2). This result confirms the hypothesis that the ability of 1 mm pentobarbital to decrease the MTSEA-biotink2 values at these positions is attributable to activation of the receptor. Therefore, these residues, which we have demonstrated to be facing into the binding pocket, undergo a change in environment during gating.
The only reaction rate that was affected by a modulatory concentration of pentobarbital was for G203C, in which 50 μmpentobarbital caused an increase in k2(Fig. 5, Table 2). This is direct evidence that allosteric modulation of IGABA by pentobarbital involves a structural rearrangement near the GABA binding pocket that makes G203C more accessible.
The rate of reaction of MTSEA-biotin with a cysteine introduced at position P206 was significantly increased in the presence of GABA. Pentobarbital and SR-95531 had no significant effect onk2 for MTSEA-biotin at P206C (Fig. 5, Table 2). An increase in reaction rate can be interpreted as GABA binding resulting in an allosteric response that causes P206C to be in a more accessible environment. Because pentobarbital has no effect on the rate of reaction at this position, this allosteric response must be specific to rearrangements concomitant with GABA binding.
DISCUSSION
SCAM analysis of the β2V199–S209 (loop C) region of GABAA receptor identified several amino acid residues that face into the GABA binding pocket and mediate agonist affinity (KD) and efficacy. In addition, we provide evidence that the ligand binding pocket is a deep narrowing structure that constricts during gating.
Mutations that affectKD-GABA
Mutation to cysteine causes significant shifts in EC50-GABA for six residues: F200C, S201C, T202C, G203C, Y205C, and R207C. According to the model shown in Scheme 1, shifts in EC50-GABA can be caused by changes in affinity of the closed receptor for GABA (KD) and/or changes in the ability of GABA to induce opening of the receptor (efficacy or E). Determining which of these parameters is responsible for mutation-induced EC50 shifts is difficult (Colquhoun, 1998). However, for receptors that require the binding of two ligands for efficient opening and have relatively lowE values, a large increase in EC50that is not accompanied by a significant reduction inImax can be attributed to a reduction in KD (Amin and Weiss, 1993; Anson et al., 1998). In fact, for the GABAA receptor, a 50-fold increase in EC50-GABA would have to be accompanied by a >99% reduction inImax-GABA for the shift in the dose–response curve to be caused solely by changes in efficacy. Because none of the mutations in this study cause significant reductions in Imax-GABA and the shifts in EC50 values range from 70-fold (R207C) to 18,000-fold (Y205C), it is clear that the increases in EC50-GABA values reflect, at least in part, a reduction in ligand affinity (KD) at the GABA binding site. Although Imaxcomparisons between different mutant receptors are problematic because of poor control of expression levels, we feel confident that we would detect a >99% reduction inImax-GABA. Moreover, five of the six mutations that shift EC50-GABA (all except R207C) also significantly reduce affinity for SR-95531, further suggesting that mutation of F200, S201, T202, G203, Y205, and R207 to cysteine alters the microscopic binding affinity of ligands at the GABA binding site.
When mutation of an amino acid disrupts agonist affinity, it has been used as evidence that the residue in question is located in the binding pocket. This, however, is not proof. Our result, that there is no detectable reaction of MTSEA-biotin with cysteines introduced at the positions F200–T202, suggests that these side chains are not facing into the water-accessible GABA binding pocket. Rather, these residues are likely to be buried in the protein or membrane lipid. Caution, however, must be taken with the interpretation of the accessibility results. The possibility that MTSEA-biotin modifies an introduced cysteine without affecting IGABA must also be considered. However, it is unlikely that addition of the large biotin moiety would have no discernable affect onIGABA if F200C, S201C, and T202C actually face into the binding pocket. Thus, although mutation of F200, S201, and T202 to cysteine results in large shifts in EC50-GABA, we believe these residues are not lining the GABA binding pocket.
In contrast, the large shifts in EC50-GABAvalues caused by mutation of Y205 and R207 likely reflect disruptions of residues that line the binding site. Because MTSEA-biotin reacts with cysteines at positions S204, Y205, R207, and S209 and both GABA and SR95531 significantly slow their modification, we believe that these residues face into, and are part of, the GABA binding pocket.
In addition to reducing KD-GABA, cysteine substitution at positions S201 or R207 also causes significant reductions in receptor efficacy. The fact that these mutations disrupt both agonist affinity and agonist efficacy suggests that this entire region may mediate local (i.e., in or near the binding pocket) allosteric transitions that translate agonist binding into channel opening.
Structure of the GABA binding pocket
Assessment of the accessibility of introduced cysteines to reaction with MTSEA-biotin reveals direct structural information about loop C of the GABA binding pocket. MTS reagents react 109–1010times faster with ionized sulfhydryl groups than they do with protonated sulfhydryls (Roberts et al., 1986). Therefore, an introduced cysteine that reacts with an MTS reagent is likely to be oriented with its side chain in an aqueous environment in which ionization of the sulfhydryl is more probable (Pascual and Karlin, 1998).
Patterns of accessibility can be used to discern the secondary structure of a region. For example, after mutation to cysteine, alternating residues of the α1 loop D domain are accessible to MTSEA-biotin, indicating that the region is a β-strand (Boileau et al., 1999). Here we show that six of seven sequential cysteine mutants in the β2 loop C domain (G203C–R207C and S209C) are available for reaction with MTSEA-biotin. This accessibility pattern does not suggest a regular secondary structure, indicating that the region in question may be an extended coil or loop. This result agrees with secondary structure predictions for the N-terminal domain of the nACh receptor in which the loop C region is predicted to be a coil (Le Novère et al., 1999).
Additional structural information emerges from the rates at which the introduced cysteines react with MTSEA-biotin. Two main factors influence these rates: (1) ionization of the sulfhydryl side chain, which is more likely in an aqueous environment, and (2) steric hindrance (i.e., how difficult is it for the MTSEA-biotin molecule to physically approach and interact with the sulfhydryl group). The fast reaction rate measured for G203C (k2≈ 250,000 M−1 s−1) indicates that the side chain of this residue is in an aqueous and sterically unrestricted environment such as would exist at the mouth of the binding pocket. The >2000-fold slower reaction rate measured for S209C (k2 ≈ 120 M−1 s−1) indicates that the side chain of this residue is poorly ionized (in a relatively hydrophobic environment), located in a sterically confined region, or both. These are the conditions one might expect to find near the deepest point of the binding pocket. Significantly, the reaction rates for the introduced cysteines between G203 and S209 sequentially decline, almost continually, with progression along the peptide chain (Fig. 6, Table 2). This rate gradient is highly suggestive of a protein domain that traverses an aqueous pocket from its rim to its depths. This type of structure correlates with the water-filled tunnels in the nACh receptor identified by electron microscopy (Miyazawa et al., 1999). We hypothesize that, in the GABAA receptor, at least a portion of these tunnels lie at an α/β interface. The fact that none of the introduced cysteines before G203 appear to react with MTSEA-biotin suggests that the polypeptide chain may turn at this glycine (a residue that allows for maximum flexibility) and dive into the hydrophobic core of the protein or the lipid membrane.
Fig. 6.

Graphic summary of results. Barsindicate which residues fall into each of the following categories:Mediates KD-GABA, mutation of this residue alters microscopic affinity for GABA; Mediates Efficacy, mutation of this residue reduces efficacy of P4S;Accessible to MTS, we can detect reaction of MTSEA-biotin with a cysteine introduced at this position; In Binding Pocket, the rate at which MTSEA-biotin reacts with a cysteine introduced at this residue is slowed by the presence of both GABA and SR-95531. Relative Rxn. Rate, The height of thebar is scaled to the log ofk2 for each mutant with MTSEA-biotin.
Structural rearrangements involved in receptor gating
It has been speculated that the allosteric transition underlying gating of LGICs is primarily from quaternary rearrangements of the N-terminal domains of subunits with little change in tertiary or secondary structure (Corringer et al., 2000). The results presented here suggest that activation of the receptor involves movement of the α1 and β2 domains of the GABA binding site toward each other. Three residues that face into the binding pocket (S204, R207, and S209) experience reductions in accessibility to MTSEA-biotin during gating. This is exactly the result we would expect if convergence of two subunits were to decrease the volume of the binding pocket, making it more difficult for MTSEA-biotin to interact with residues in the pocket. Interestingly, GABA causes an increase in accessibility to MTSEA-biotin at position P206. We envision that P206C faces away from the binding pocket and, when GABA binds the area near P206, becomes more accessible to MTSEA-biotin.
Overall role of loop C in GABA binding and gating
Some of the results presented in this study are graphically summarized in Figure 6. This region appears to consist of two structurally distinct domains. The C-terminal residues of this domain (S204–S209) predominantly line the GABA binding pocket and are in an aqueous environment. The N-terminal residues (V199–T202) do not appear to be in an aqueous environment and thus are not part of the binding pocket. G203 seems to be a transition residue between these two domains in that it is easily modified by MTSEA-biotin (i.e., in an aqueous environment), but its modification is not slowed by GABA or SR95531 and thus does not seem to be facing into the binding pocket.
Functionally, the roles of the two structural domains seem to converge. The exquisite sensitivity ofKD-GABA to perturbation of this entire region implies both domains are critically involved in maintaining the structural integrity of the binding pocket. Additionally, both domains contain residues (S201 and R207) that, when mutated to cysteine, cause reductions in agonist efficacy, implying that they may be part of the allosteric mechanism coupling binding to channel opening. The result that a cysteine introduced at position G203 experiences an environmental change in the presence of modulatory concentrations of pentobarbital indicates that the loop C region also responds to GABAA allosteric modulators. Finally, a role for this region in receptor gating is demonstrated by the fact that side chains at positions S204, R207, and S209 experience a change in environment concomitant with gating of the receptor. Thus, the loop C region of the GABA binding site contains dynamic elements that respond to both modulators and channel activation. The agonist-mediated binding site movements may be the initial trigger that drives channel opening.
Footnotes
This work was supported in part by National Institute of Neurological Disorders and Stroke Grants NS10579 (to D.W.) and NS34727 (to C.C.). C.C. is a recipient of the Burroughs Welcome Fund New Investigator Award in the Basic Pharmacological Sciences. We thank Amy Kucken, Jeremy Teissére, Dr. Andrew Boileau, Dr. Meyer Jackson, and Dr. Matt Jones for critical reading of this manuscript.
Correspondence should be addressed to Dr. Cynthia Czajkowski, University of Wisconsin, Department of Physiology, Room 197 MSC, 1300 University Avenue, Madison, WI 53706. E-mail:czajkowski@physiology.wisc.edu.
REFERENCES
- 1.Akk G, Steinbach JH. Activation and block of recombinant GABA(A) receptors by pentobarbitone: a single-channel study. Br J Pharmacol. 2000;130:249–258. doi: 10.1038/sj.bjp.0703335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amin J, Weiss DS. GABAA receptor needs two homologous domains of the beta-subunit for activation by GABA but not by pentobarbital. Nature. 1993;366:565–569. doi: 10.1038/366565a0. [DOI] [PubMed] [Google Scholar]
- 3.Anson LC, Chen PE, Wyllie DJA, Colquhoun D, Schoepfer R. Identification of amino acid residues of the NR2A subunit that control glutamate potency in recombinant NR1/NR2A NMDA receptors. J Neurosci. 1998;18:581–589. doi: 10.1523/JNEUROSCI.18-02-00581.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basiry SS, Mendoza P, Lee PD, Raymond LA. Agonist-induced changes in substituted cysteine accessibility reveal dynamic extracellular structure of M3–M4 loop of glutamate receptor GluR6. J Neurosci. 1999;19:644–652. doi: 10.1523/JNEUROSCI.19-02-00644.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boileau AJ, Kucken AM, Evers AR, Czajkowski C. Molecular dissection of benzodiazepine binding and allosteric coupling using chimeric gamma-aminobutyric acidA receptor subunits. Mol Pharmacol. 1998;53:295–303. doi: 10.1124/mol.53.2.295. [DOI] [PubMed] [Google Scholar]
- 6.Boileau AJ, Evers AR, Davis AF, Czajkowski C. Mapping the agonist binding site of the GABAA receptor: evidence for a β-strand. J Neurosci. 1999;19:4847–4854. doi: 10.1523/JNEUROSCI.19-12-04847.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 8.Chou T. Relationships between inhibition constants and fractional inhibition in enzyme-catalyzed reactions with different numbers of reactants, different reaction mechanisms, and different types and mechanisms of inhibition. Mol Pharmacol. 1974;10:235–247. [PubMed] [Google Scholar]
- 9.Colquhoun D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corringer PJ, Le Novère N, Changeux JP. Nicotinic receptors at the amino acid level. Annu Rev Pharmacol Toxicol. 2000;40:431–458. doi: 10.1146/annurev.pharmtox.40.1.431. [DOI] [PubMed] [Google Scholar]
- 11.Czajkowski C, Kaufmann C, Karlin A. Negatively charged amino acid residues in the nicotinic receptor delta subunit that contribute to the binding of acetylcholine. Proc Natl Acad Sci USA. 1993;90:6285–6289. doi: 10.1073/pnas.90.13.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.del Castillo J, Katz B. Interaction at endplate receptors between different choline derivatives. Proc R Soc Lond B Biol Sci. 1957;146:369–381. doi: 10.1098/rspb.1957.0018. [DOI] [PubMed] [Google Scholar]
- 13.Dennis M, Giraudat J, Kotzyba-Hibert F, Goeldner M, Hirth C, Chang JY, Lazure C, Chretien M, Changeux JP. Amino acids of the Torpedo marmorata acetylcholine receptor alpha subunit labeled by a photoaffinity ligand for the acetylcholine binding site. Biochemistry. 1988;27:2346–2357. doi: 10.1021/bi00407a016. [DOI] [PubMed] [Google Scholar]
- 14.Galzi JL, Changeux JP. Neurotransmitter-gated ion channels as unconventional allosteric proteins. Curr Opin Struct Biol. 1994;4:554–565. [Google Scholar]
- 15.Ito T, Suzuki T, Wellman SE, Ho IK. Pharmacology of barbiturate tolerance/dependence: GABAA receptors and molecular aspects. Life Sci. 1996;59:169–195. doi: 10.1016/0024-3205(96)00199-3. [DOI] [PubMed] [Google Scholar]
- 16.Jackson MB, Lecar H, Mathers DA, Barker JL. Single channel currents activated by γ-aminobutyric acid, muscimol, and (−)-pentobarbital in cultured mouse spinal neurons. J Neurosci. 1982;2:889–894. doi: 10.1523/JNEUROSCI.02-07-00889.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Javitch JA, Fu D, Chen J, Karlin A. Mapping the binding-site crevice of the dopamine D2 receptor by the substituted-cysteine accessibility method. Neuron. 1995;14:825–831. doi: 10.1016/0896-6273(95)90226-0. [DOI] [PubMed] [Google Scholar]
- 18.Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 19.Krogsgaard-Larsen P, Falch E, Schousboe A, Curtis DR, Lodge D. Piperidine-4-sulphonic acid, a new specific GABA agonist. J Neurochem. 1980;34:756–759. doi: 10.1111/j.1471-4159.1980.tb11211.x. [DOI] [PubMed] [Google Scholar]
- 20.Kucken AM, Wagner DA, Ward PR, Boileau JA, Czajkowski C. Identification of benzodiazepine binding site residues in the gamma2 subunit of the gamma-aminobutyric acid(A) receptor. Mol Pharmacol. 2000;57:932–939. [PubMed] [Google Scholar]
- 21.Le Novère N, Corringer PJ, Changeux JP. Improved secondary structure predictions for a nicotinic receptor subunit: incorporation of solvent accessibility and experimental data into a two-dimensional representation. Biophys J. 1999;76:2329–2345. doi: 10.1016/S0006-3495(99)77390-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- 23.Miyazawa A, Fujiyoshi Y, Stowell M, Unwin N. Nicotinic acetylcholine receptor at 4.6 A resolution: transverse tunnels in the channel wall. J Mol Biol. 1999;288:765–786. doi: 10.1006/jmbi.1999.2721. [DOI] [PubMed] [Google Scholar]
- 24.Ortells MO, Lunt GG. Evolutionary history of the ligand-gated ion-channel superfamily of receptors. Trends Neurosci. 1995;18:121–127. doi: 10.1016/0166-2236(95)93887-4. [DOI] [PubMed] [Google Scholar]
- 25.O'Shea SM, Wong LC, Harrison NL. Propofol increases agonist efficacy at the GABA(A) receptor. Brain Res. 2000;852:344–348. doi: 10.1016/s0006-8993(99)02151-4. [DOI] [PubMed] [Google Scholar]
- 26.Pascual JM, Karlin A. State-dependent accessibility and electrostatic potential in the channel of the acetylcholine receptor. Inferences from rates of reaction of thiosulfonates with substituted cysteines in the M2 segment of the alpha subunit. J Gen Physiol. 1998;111:717–739. doi: 10.1085/jgp.111.6.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts DD, Lewis SD, Ballou DP, Olson ST, Shafer JA. Reactivity of small thiolate anions and cysteine-25 in papain toward methyl methanethiosulfonate. Biochemistry. 1986;25:5595–5601. doi: 10.1021/bi00367a038. [DOI] [PubMed] [Google Scholar]
- 28.Robertson GA, Warmke JM, Ganetzky B. Potassium currents expressed from Drosophila and mouse eag cDNAs in Xenopus oocytes. Neuropharmacology. 1996;35:841–850. doi: 10.1016/0028-3908(96)00113-x. [DOI] [PubMed] [Google Scholar]
- 29.Sigel E, Baur R, Kellenberger S, Malherbe P. Point mutations affecting antagonist affinity and agonist dependent gating of GABAA receptor channels. EMBO J. 1992;11:2017–2023. doi: 10.1002/j.1460-2075.1992.tb05258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith GB, Olsen RW. Identification of a [3H]muscimol photoaffinity substrate in the bovine gamma-aminobutyric acidA receptor alpha subunit. J Biol Chem. 1994;269:20380–20387. [PubMed] [Google Scholar]
- 31.Thompson SA, Whiting PJ, Wafford KA. Barbiturate interactions at the human GABAA receptor: dependence on receptor subunit combination. Br J Pharmacol. 1996;117:521–527. doi: 10.1111/j.1476-5381.1996.tb15221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uchida I, Cestari IN, Yang J. The differential antagonism by bicuculline and SR95531 of pentobarbitone-induced currents in cultured hippocampal neurons. Eur J Pharmacol. 1996;307:89–96. doi: 10.1016/0014-2999(96)00156-2. [DOI] [PubMed] [Google Scholar]
- 33.Ueno S, Bracamontes J, Zorumski C, Weiss DS, Steinbach JH. Bicuculline and gabazine are allosteric inhibitors of channel opening of the GABAA receptor. J Neurosci. 1997;17:625–634. doi: 10.1523/JNEUROSCI.17-02-00625.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vandenberg RJ, Handford CA, Schofield PR. Distinct agonist- and antagonist-binding sites on the glycine receptor. Neuron. 1992;9:491–496. doi: 10.1016/0896-6273(92)90186-h. [DOI] [PubMed] [Google Scholar]
- 35.Westh-Hansen SE, Rasmussen PB, Hastrup S, Nabekura J, Noguchi K, Akaike N, Witt MR, Nielsen M. Decreased agonist sensitivity of human GABA(A) receptors by an amino acid variant, isoleucine to valine, in the alpha1 subunit. Eur J Pharmacol. 1997;329:253–257. [PubMed] [Google Scholar]
- 36.Wilson GG, Karlin A. The location of the gate in the acetylcholine receptor channel. Neuron. 1998;20:1269–1281. doi: 10.1016/s0896-6273(00)80506-1. [DOI] [PubMed] [Google Scholar]
- 37.Xu M, Akabas MH. Identification of channel-lining residues in the M2 membrane-spanning segment of the GABA(A) receptor alpha1 subunit. J Gen Physiol. 1996;107:195–205. doi: 10.1085/jgp.107.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]