

Abstract
Munc13 proteins form a family of three, primarily brain-specific phorbol ester receptors (Munc13-1/2/3) in mammals. Munc13-1 is a component of presynaptic active zones in which it acts as an essential synaptic vesicle priming protein. In contrast to Munc13-1, which is present in most neurons throughout the rat and mouse CNS, Munc13-3 is almost exclusively expressed in the cerebellum. Munc13-3 mRNA is present in granule and Purkinje cells but absent from glia cells. Munc13-3 protein is localized to the synaptic neuropil of the cerebellar molecular layer but is not found in Purkinje cell dendrites, suggesting that Munc13-3, like Munc13-1, is a presynaptic protein at parallel fiber–Purkinje cell synapses. To examine the role of Munc13-3 in cerebellar physiology, we generated Munc13-3-deficient mutant mice. Munc13-3 deletion mutants exhibit increased paired-pulse facilitation at parallel fiber–Purkinje cell synapses. In addition, mutant mice display normal spontaneous motor activity but have an impaired ability to learn complex motor tasks. Our data demonstrate that Munc13-3 regulates synaptic transmission at parallel fiber–Purkinje cell synapses. We propose that Munc13-3 acts at a similar step of the synaptic vesicle cycle as does Munc13-1, albeit with less efficiency. In view of the present data and the well established vesicle priming function of Munc13-1, it is likely that Munc13-3-loss leads to a reduction in release probability at parallel fiber–Purkinje cell synapses by interfering with vesicle priming. This, in turn, would lead to increases in paired-pulse facilitation and could contribute to the observed deficit in motor learning.
Keywords: exocytosis, secretion, synaptic vesicle, priming, phorbol ester, unc-13
Synaptic vesicles are the key organelles in neurotransmitter release at synapses. In the presynaptic terminal of a nerve cell, synaptic vesicles pass through a complex cycle of membrane fusion and fission reactions that govern the release process (Südhof, 1995). Vesicles are probably generated by budding from early endosomes. They are loaded with transmitter, translocate to the plasma membrane, and dock at the active zone, an electron-dense presynaptic membrane region to which the release process is restricted. Docked vesicles have to be primed to a fusion-competent state before a rise in the intracellular calcium concentration can trigger fusion and secretion of the transmitter content (Zucker, 1996). After fusion, vesicular membrane and protein components are retrieved by clathrin-mediated endocytosis and recycled directly or via early endosomes (Cremona and De Camilli, 1997; Klingauf et al., 1998; Murthy and Stevens, 1998).
Munc13 proteins constitute a family of brain-specific molecules (Munc13-1, Munc13-2, and Munc13-3) with homology toCaenorhabditis elegans Unc-13 (Brose et al., 1995). A splice variant of Munc13-2 is also expressed outside of the CNS (Song et al., 1998) (A. Betz and N. Brose, GenBank accession number AF159706). Munc13 proteins contain a phorbol ester-binding C1domain and two C2 domains (Brose et al., 1995). The most abundant isoform in rat brain, Munc13-1, is specifically targeted to presynaptic active zones (Betz et al., 1998). It binds to syntaxin, a component of the synaptic vesicle fusion apparatus (Betz et al., 1997), to DOC2 (Orita et al., 1997), as well as to several other synaptic proteins (for review, see Brose et al., 2000), and acts as a phorbol ester-dependent enhancer of neurotransmitter secretion when overexpressed in Xenopus neuromuscular junctions (Betz et al., 1998). Loss of Munc13-1 in deletion mutant mice leads to early postnatal death because of an arrest of the synaptic vesicle cycle of neurons in the CNS. A detailed analysis of primary hippocampal neurons from Munc13-1-deficient mice demonstrated that synaptic vesicles in mutant synapses cannot acquire fusion competence, resulting in a depletion of readily releasable vesicles and a functional shutdown of synaptic activity. Mutant synapses contain normal numbers of docked vesicles that are unable to fuse in response to physiological stimuli (Augustin et al., 1999b), demonstrating that Munc13-1 is an essential synaptic vesicle priming protein in neurons. Interestingly, this Munc13-1 mutant phenotype is restricted to excitatory–glutamatergic neurons, whereas inhibitory–GABAergic nerve cells are unaffected by the Munc13-1 deficiency (Augustin et al., 1999b). Moreover, only 90% of synapses of any given excitatory–glutamatergic neuron appear to be dependent on Munc13-1, whereas the remaining 10% function normally and are apparently independent of Munc13-1-mediated vesicle maturation (Augustin et al., 1999b). Complementing observations on Munc13-1-deficient mice and further supporting the conclusion that Munc13-1 is a secretory vesicle priming protein, overexpression of Munc13-1 in adrenal chromaffin cells leads to a dramatic increase in priming of chromaffin granules (Ashery et al., 2000).
Munc13-1 is expressed in all regions of the rat CNS (Augustin et al., 1999a). In contrast, Munc13-2 and Munc13-3 exhibit strikingly differential expression patterns in which Munc13-2 is only present in rostral brain regions, including cerebral cortex and CA regions of the hippocampus, whereas Munc13-3 expression is restricted to the cerebellum (Augustin et al., 1999a). Thus, neurons in the rat brain coexpress Munc13-1 with either Munc13-2 or Munc13-3, depending on the brain region under examination. In view of this differential expression pattern, it is possible that the restriction of the Munc13-1 mutant phenotype to a subpopulation of cells or synapses is caused by the fact that one of the other Munc13 isoforms compensates for the Munc13-1 loss in Munc13-1-independent synapses.
Evidently, Munc13-1 is essential for normal function of most excitatory synapses in the CNS. To determine whether other Munc13 isoforms perform comparably important roles, we generated deletion mutant mice that lack the cerebellar presynaptic Munc13-3 protein. A detailed analysis showed that, despite normal cerebellar morphology, synaptic transmission at parallel fiber–Purkinje cell synapses is impaired in mutant mice. This physiological deficit is paralleled by compromised motor learning. We conclude that Munc13-3 regulates synaptic transmission at parallel fiber–Purkinje cell synapses in the cerebellum. The impairment in synaptic transmission at these synapses after Munc13-3 loss likely contributes to the observed motor learning deficits in Munc13-3 mutants.
MATERIALS AND METHODS
Munc13-3 deletion mutant mice. For the generation of a Munc13-3 targeting vector, we used genomic clones pMunc13-3/13 and pMunc13-3/14 (isolated from a 129SVJ mouse genomic library in λFIX II; Stratagene, La Jolla, CA), which contain multiple exons of the murine Munc13-3 gene. In the targeting vector, two of these exons representing bp 3108–3265 of the rat Munc13-3 cDNA (GenBank accession number U24072) were replaced by a neomycin resistance cassette. The vector also contained two copies of the herpes simplex virus thymidine kinase gene. Upon homologous recombination of the targeting vector with the Munc13-3 gene, the insertion of the neomycin resistance cassette results in a deletion that represents amino acid residues 1014–1066 (upstream of the C1 domain) and a shift in the open reading frame in the mature mRNA. The linearized targeting vector was electroporated into embryonic stem cells (E14; gift of Dr. K. Rajewski, Köln, Germany). Southern blotting analysis of 70 clones resistant to neomycin and 1-(2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)-5-iodouracil revealed two positive clones that were injected into blastocysts (Biological Research Laboratories, Füllinsdorf, Switzerland). One clone generated highly chimeric mice that transmitted the mutation through the germ line. Homozygous mutants were obtained by interbreeding of animals heterozygous for the Munc13-3 mutation. After germ line transmission of the mutation had been verified by Southern blotting, offspring of heterozygous interbreedings were routinely genotyped by PCR. Homozygous mutations were confirmed by immunoblotting with an antibody against the N terminus of Munc13-3 (Augustin et al., 1999a). The mutation was outbred into C57BL/6 background for more than seven generations. For all experiments, Munc13-3-deficient mice were obtained by interbreeding of animals heterozygous for the Munc13-3 mutation. Only wild-type littermates were used as controls in all experiments.
Histological analysis and quantitative Western blotting. In situ hybridization was done as described previously (Augustin et al., 1999a). For that purpose, the following oligonucleotide was chosen from the exon sequences of the murine Munc13-3 gene (corresponding to bp 1731–1772 of rat Munc13-3 cDNA): AGGAAAGGAGGGAGCTCCCATTAGTTCGACAGAAAAACTGGC. For immunocytochemistry, 10 μm cryosections were prepared from unfixed mouse brains and immunostained according to Rickmann and Wolff (1995) using affinity-purified antibodies to Munc13-3 (Augustin et al., 1999a) at 0.5–1 μg/ml. Histological sections of paraffin-embedded brains (3 μm) were stained with cresyl violet. Quantitative Western blotting of wild-type and mutant cerebella was performed with125I-labeled secondary antibodies according to published procedures (Augustin et al., 1999b).
Whole-cell voltage-clamp recordings of cerebellar Purkinje cells. Patch-clamp experiments were performed on Purkinje cells in thin slices of the cerebellum following standard procedures (Hamill et al., 1981; Edwards et al., 1989). Sagittal slices of cerebellum (150 μm) were prepared from 10- to 11-d-old mice (for the measurement of spontaneous IPSCs) or from 18- to 21-d-old mice (for the measurement of EPSCs) as described previously (Edwards et al., 1989). Slices were maintained at 37°C in a continuously bubbled (95% O2, 5% CO2) solution (in mm: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 1 MgCl2, and 25 glucose). After 60 min recovery, slices were placed in the recording chamber and superfused with the above solution at room temperature. Recordings were performed in the presence of 15 μm bicuculline (to block inhibitory synapses during the measurement of EPSCs) or in the presence of 10 μm6-cyano-7-nitroquinoxaline-2,3-dione (to block excitatory synapses during the measurement of spontaneous IPSCs). TTX (1 μm) was present during all measurements of spontaneous postsynaptic currents. Purkinje cells were selected using an upright microscope (Axioscope; Zeiss, Oberkochen, Germany) with a 63× water-immersion lens (Zeiss). Electrodes were pulled from borosilicate glass capillaries and filled with a solution containing (in mm):145 CsCl, 10 HEPES, 10 EGTA, 1 CaCl2, 2 MgCl2, 4 Na2-ATP, and 0.4 Na3-GTP, adjusted to pH 7.3 with CsOH. All experiments were performed using the whole-cell configuration of the patch-clamp technique with borosilicate pipettes (resistance of 2.5–4 MΩ when filled with an intracellular solution; see above). Ionic currents were recorded with an EPC-9 patch-clamp amplifier (Heka Elecktronik, Lambrecht, Germany). The pipette access resistance was compensated. All cells were studied at a holding potential of −70 mV. Stimulation and on-line data acquisition were performed using the PULSE program (version 8.11; Heka Elektronik) on a Macintosh computer (Apple Computers, Cupertino, CA). For stimulation of parallel fibers, a glass pipette with 5–10 μm tip diameter filled with standard saline was used. Square pulses (duration, 0.2 msec; amplitude, 1–10 V) were applied for focal stimulation. All experiments were performed at room temperature.
Ultrastructural analysis. Mice were anesthetized with ether and perfused transcardially with 30 ml of PBS, followed by 50 ml of fixative containing 2% paraformaldehyde–2.5% glutaraldehyde in PBS. The brain was removed, and the cerebellum was cut into pieces not larger than 2 mm3. The tissue blocks were osmicated for 90 min using 1% osmium tetroxide in water, rinsed in water, dehydrated in an ascending gradient of alcohols followed by propylene oxide, and left for 12 hr in propylene oxide/Epon (1:1). After polymerization in Epon 812 for 48 hr at 60°C, ultrathin sections were cut and stained with uranyl acetate and lead citrate. The sections were examined on a Zeiss EM109 electron microscope at 20,000-fold magnification. Randomly chosen areas of the cerebellar molecular layer from two +/+ and two −/− animals were photographed and analyzed. To determine synapse densities, 42 photographs (124 synapses) from two independent +/+ cerebella (79 and 45 synapses per animal), and 47 photographs (142 synapses) from two independent −/− cerebella (80 and 62 synapses per animal) were examined. For the ultrastructural comparison, 24 synapses from two independent +/+ cerebella (13 and 11 synapses per animal) and 25 synapses from two independent −/− cerebella (14 and 11 synapses per animal) were analyzed. Synapses for ultrastructural analysis were selected randomly. However, images of suboptimal quality with respect to staining, contrast, ultrastructural detail, or photo reproduction were excluded.
Behavioral analysis. A rotating rod apparatus (TSE-Systems, Bad Homburg, Germany) was used to measure the ability of mice to improve motor performance during repeated exposure to the apparatus (Pekhletski et al., 1996). Before the first training session, all mice were habituated to the apparatus by placing them on the rod rotating at 5 rpm (three times for 2 min sessions). The training consisted of 22 sessions: three sessions per day with a 90 min intersession interval and three trials per session with 5 sec intertrial interval. The rotation speed was set initially at 5 rpm, and the length of each trial was 30 sec (i.e., the maximum cumulative training duration in a session was 90 sec). If a mouse stayed on the rotating rod for a total cumulative duration of at least 60 sec during the three trial session, the speed was increased by 3 rpm for the next session. If a mouse accumulated <60 sec on the rod during the three trial session but >30 sec, no speed increase was given. If a mouse could not accumulate 30 sec of on-rod time, the speed was decreased by 3 rpm at the next session. The rotation speed the mice were able to master according to these definitions (learning to criterion) was recorded and analyzed via a two-way repeated-measure ANOVA with factors genotype and session (the repeated factor). The genotype groups were compared with post hoc Tukey's honestly significant difference (HSD) tests by sessions. An additional group of untrained mice that had never been exposed to the apparatus was also tested on the rotating rod at 40 rpm to analyze whether naïve mutant mice had a preexisting performance deficit that could be detected only at higher rotation speeds or in more demanding tasks.
RESULTS
Deletion mutations in the Munc13-3 gene were generated by homologous recombination in mouse embryonic stem cells (Fig.1A,B). Recombinant stem cells were injected into blastocysts. These were used to produce chimeric mice that transmitted the mutated gene through the germ line. Munc13-3-deficient mice were obtained at the predicted Mendelian frequency by interbreeding of animals heterozygous for the Munc13-3 mutation (38 +/+, 71 +/−, and 37 −/− in 146 analyzed animals). Although mutant mice did not express Munc13-3 protein (Figs.1C, 2E), they exhibited no obvious physical or behavioral abnormalities and had a normal life expectancy under our laboratory conditions. Both male and female Munc13-3 mutants were fertile and had normal reproductive capabilities. Litter sizes resulting from interbreeding of homozygous Munc13-3-deficient animals were indistinguishable from those obtained with wild-type controls. The obvious lack of a pronounced phenotypic change in Munc13-3-deficient mice is in contrast to deletion mutations in the Munc13-1 gene that result in an almost complete arrest of transmitter release from glutamatergic neurons and early postnatal lethality (Augustin et al., 1999b).
Fig. 1.

Targeting strategy and identification of the Munc13-3 deletion mutation in mice.A, Structure of the targeted region of the murine Munc13-1 gene, targeting vector, and mutated gene resulting from homologous recombination. Exons are indicated by black boxes (numbers give corresponding base pairs in the rat Munc13-3 cDNA), and the location of the probe used for Southern analysis of genomic DNA digested with BamHI is indicated by a hatched bar. The location of the neomycin resistance gene (Neo) and two copies of the HMV thymidine kinase gene (TK) in the targeting vector are indicated by gray boxes.BglII/BamHI indicates fused site. The positions of the BamHI sites upstream of theProbe and at the very 3′ end of the gene representation are not part of genomic clones used for the construction of the targeting vector and were not mapped. B, Southern analysis of genomic DNA from different Munc13-3 genotypes. DNA was digested with BamHI, separated electrophoretically, blotted onto nylon filters, and probed with the probe shown above. Bands representing the wild-type (WT) and knock-out (KO) allele are indicated. C, Western blot analysis of cerebellum homogenates from different Munc13-3 genotypes. Brain homogenates were separated by SDS-PAGE, blotted onto nitrocellulose filters, and probed with an antibody directed against the N terminus of Munc13-3 (Augustin et al., 1999b). Note the complete absence of Munc13-3 in knock-out brain (arrowhead).
Fig. 2.
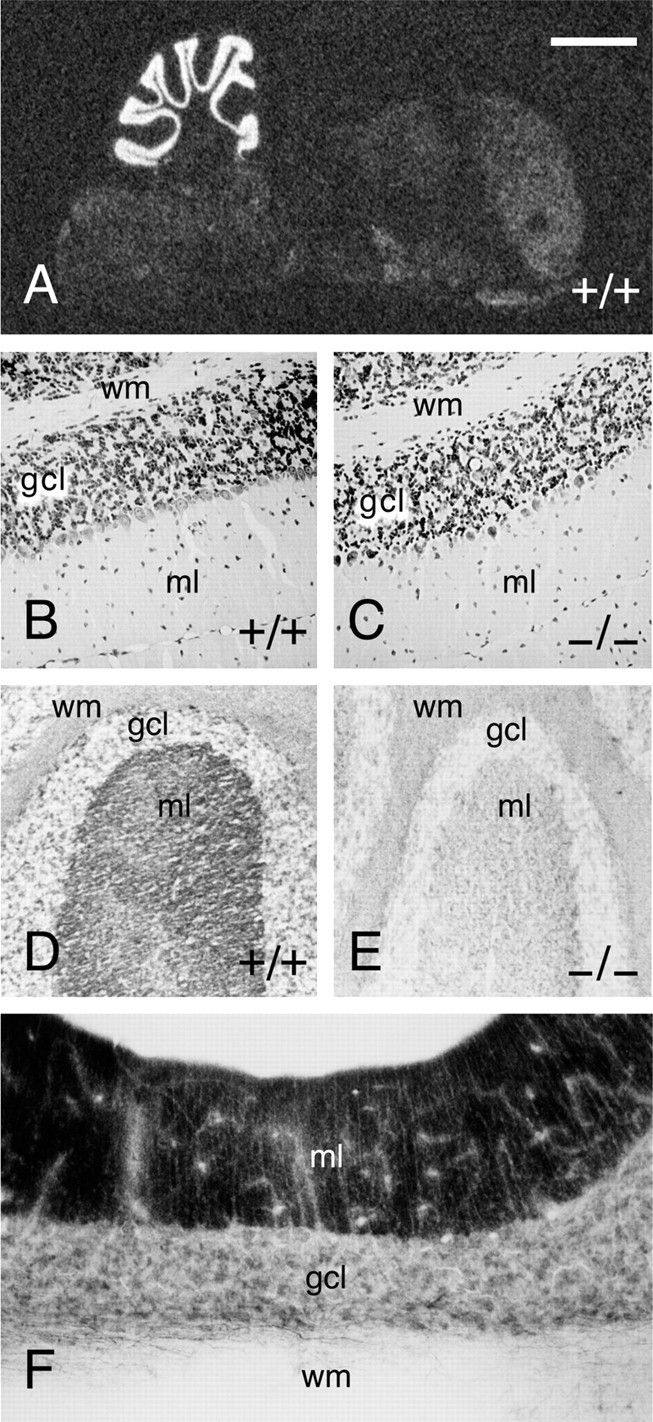
Normal cytoarchitecture in Munc13-3-deficient mice. A, In situhybridization for Munc13-3 in mouse brain. Like in rat, Munc13-3 mRNA expression is almost exclusively restricted to the cerebellum in mouse brain. B, C, Cresyl violet stainings of paraffin sections through wild-type and Munc13-3-deficient cerebellum. Note the normal cytoarchitecture and cell density in Munc13-3-deficient cerebellum. D, Frozen section through wild-type cerebellum stained with an antibody specific to Munc13-3 (Augustin et al., 1999b). Munc13-3 is localized to the molecular layer of the cerebellum, which mainly contains synaptic neuropil with parallel fiber–Purkinje cell synapses. E, Frozen section through Munc13-3 mutant cerebellum, demonstrating the complete absence of Munc13-3 protein and thus the specificity of the immunostaining procedure. F, Staining for Munc13-1 protein in rat cerebellum using a specific monoclonal antibody according to published procedures (Betz et al., 1998). wm, White matter;gcl, granule cell layer; ml, molecular layer. Scale bar: A, 2 mm; B–E, 100 μm; F, 75 μm.
In mouse, Munc13-3 is specifically expressed in the cerebellum (Fig.2A) (for corresponding data on rat, see Augustin et al., 1999a). Within the cerebellum, Munc13-3 mRNA is most abundant in the granule cell layer (Fig. 2A) (for corresponding data on rat, see Augustin et al., 1999a). Accordingly, Munc13-3 protein is almost exclusively present in the molecular layer in which the parallel fiber axons of granule cells terminate on dendrites of Purkinje neurons (Fig. 2D,E) (for corresponding data on rat, see Augustin et al., 1999a). The localization of Munc13-3 protein coincides exactly with that of Munc13-1 in cerebellum (Fig. 2F). In view of this highly specific localization of Munc13-3, we concentrated our analyses on the protein composition, structure, and function of the cerebellum.
Quantitative analyses of synaptic protein expression levels in cerebella from Munc13-3-deficient mice revealed no obvious abnormalities. Homozygous mutant cerebella were devoid of Munc13-3 protein (Figs. 1C, 2D,E; Table 1). In contrast, Munc13-3 expression in heterozygous mutants was indistinguishable from wild-type controls (Table 1). Moreover, expression levels of other Munc13 isoforms and 29 selected synaptic proteins were not significantly different from wild-type controls (Table 1).
Table 1.
Levels of synaptic proteins in Munc13-3-deficient cerebella
| Protein level (% of wild-type control) | |
|---|---|
| Munc13-1* | 104 ± 2 |
| Munc13-2 | 94 ± 4 |
| Munc13-3 | 0 |
| α-SNAP | 81 ± 5 |
| Amphiphysin | 95 ± 10 |
| Arf6 | 105 ± 4 |
| Complexin I* | 110 ± 4 |
| Complexin II* | 107 ± 1 |
| Doc2α | 87 ± 2 |
| Dynamin | 105 ± 3 |
| GDI | 95 ± 8 |
| Mint I | 114 ± 4 |
| Msec7 | 101 ± 4 |
| Munc18-1 | 87 ± 5 |
| Neuroligin 1/3 | 105 ± 7 |
| NMDA RI | 101 ± 4 |
| NSF | 99 ± 10 |
| PSD95 | 83 ± 4 |
| Rab3A* | 108 ± 7 |
| Rabphilin | 86 ± 8 |
| SAP102 | 111 ± 5 |
| SNAP25 | 88 ± 6 |
| SV2B | 117 ± 4 |
| SV2C | 87 ± 5 |
| Synapsin I | 89 ± 5 |
| Synapsin II | 109 ± 3 |
| Synaptobrevin II | 91 ± 8 |
| Synaptogyrin* | 104 ± 5 |
| Synaptophysin | 100 ± 8 |
| Synaptoporin* | 87 ± 10 |
| Synaptotagmin I | 92 ± 5 |
| Syntaxin IA/B | 104 ± 6 |
Data are given as mean ± SEM (n = 3;
n = 5). Munc13-3 levels in hetergozygous mutant cerebella were not significantly different from wild-type controls (113 ± 6; n = 3), indicating a lack of a gene dosage effect.
Anatomical and morphological analyses of adult offspring from heterozygous interbreedings revealed no overt abnormalities in Munc13-3-deficient animals. In particular, the overall structure of the cerebellum, and that of other brain regions, was indistinguishable from wild-type controls, and paraffin sections of cerebella from adult wild-type and mutant mice stained with cresyl violet showed a distribution and density of nerve cells that is typical for this age (Fig. 2B,C).
Electrophysiological analyses of synaptic transmission in Munc13-3-deficient mice were performed in cerebellar slices from postnatal days 18–21. Because Munc13-3 is mainly expressed in cerebellar granule cells that target the protein to their presynaptic parallel fiber axon terminals (Fig.2D,E) (Augustin et al., 1999a), we analyzed parallel fiber–Purkinje cell synapses. For that purpose, we stimulated parallel fibers with an extracellular stimulation electrode and made simultaneous patch-clamp recordings of evoked excitatory postsynaptic responses from Purkinje cells in the whole-cell configuration. To detect subtle changes in synaptic transmitter release after Munc13-3 loss, we used a paired-pulse facilitation paradigm. The degree of paired-pulse facilitation can be used as an indirect measure for changes in presynaptic transmitter release (Zucker, 1989). Apart from a superexcitability period of parallel fiber axons at short interstimulus intervals, paired-pulse facilitation reflects the fact that, at synapses with rather low release probability, transmitter release in response to two immediately subsequent stimuli differs in size, with the second response being larger than the first. Decreases in release probability result in a concomitant increase in paired-pulse facilitation. Paired-pulse facilitation was examined in cerebellar Purkinje cells from mutant and wild-type mice at 10 different interstimulus intervals, ranging from 50 to 400 msec (Fig.3A,B). At interstimulus intervals of up to 200 msec, a significant enhancement of the mean paired-pulse facilitation ratio was detected in cerebellar slices lacking Munc13-3 (Fig.3A,B), suggesting a reduction in release probability from mutant synapses compared with wild-type controls. In contrast, frequency and amplitude of spontaneous excitatory postsynaptic responses of Purkinje cells were not affected by the Munc13-3 mutation (data not shown). In several deletion mutants of presynaptic proteins, short-term plasticity during high-frequency stimulation is altered. For example, in the case of Rab3A mutants, repetitive stimulation of Rab3A-deficient hippocampal synapses at 14 Hz leads to an accelerated rundown of synaptic transmission (Geppert et al., 1994). To examine whether similar changes in high-frequency synaptic transmission are caused at parallel fiber–Purkinje cell synapses by Munc13-3 loss, we stimulated these synapses at 14 Hz. Unlike hippocampal neurons, we found that neither wild-type nor mutant Purkinje cells show synaptic depression at this stimulation frequency (Fig. 3C).
Fig. 3.
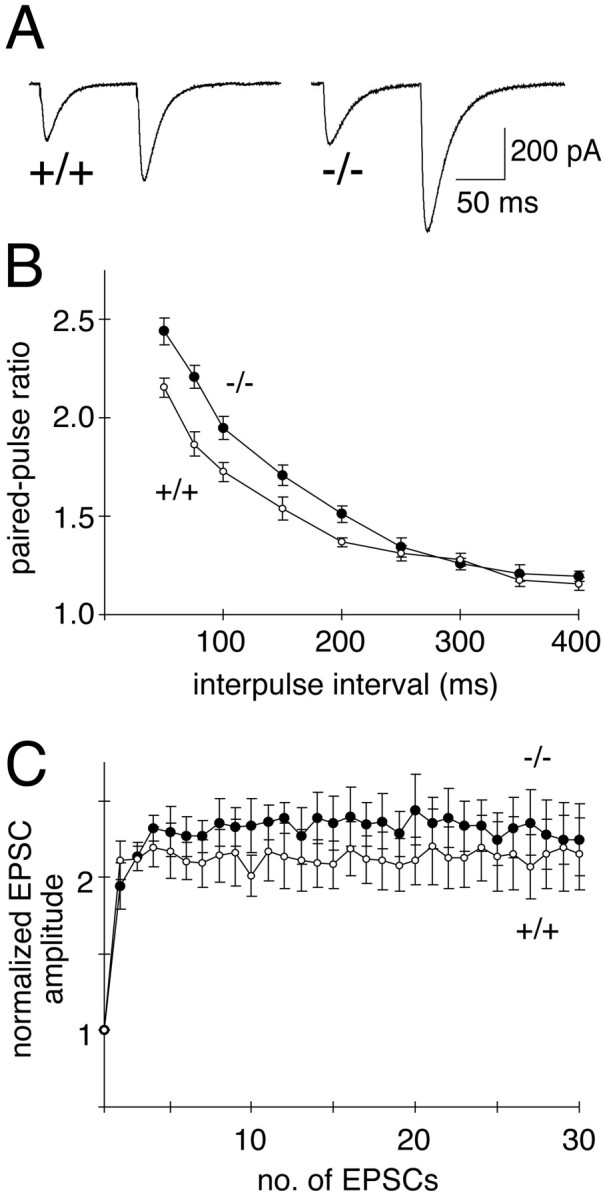
Increased paired-pulse facilitation in parallel fiber–Purkinje cell synapses in Munc13-3-deficient mice.A, Sample synaptic responses taken from wild-type (+/+) and Munc13-3-deficient (−/−) Purkinje cells after paired stimulation (75 msec interval) of parallel fibers. Note the increase in paired-pulse facilitation in mutant cells. B, Magnitude of the paired-pulse facilitation as a function of interstimulus intervals. Ratios of the second to first EPSC amplitude (mean ± SEM) from wild-type (open circles; 8 animals, 17 cells) and mutant (filled circles; 9 animals, 19 cells) mice are plotted as a function of interstimulus interval. Note significant increases in the paired-pulse facilitation ratio in Munc13-3-deficient Purkinje cells (p < 0.05) for intervals up to 200 msec. C, Amplitudes of successive EPSCs plotted as a function of stimulus number during repetitive stimulation at 14 Hz from wild-type (open circles; 9 animals, 9 cells) and mutant (filled circles; 10 animals, 10 cells) animals. No depression is observed in mutant or wild-type cells. Error bars indicate SE.
Our electrophysiological analysis of synaptic transmission at parallel fiber–Purkinje cell synapses clearly demonstrated that Munc13-3 modulates synaptic transmission at these glutamatergic synapses. The finding that synaptic vesicle priming by Munc13-1 is essential only for excitatory–glutamatergic synapses in hippocampal primary neurons led to the idea that other Munc13 isoforms (e.g., Munc13-3) might play similarly essential roles in inhibitory–GABAergic synapses (Augustin et al., 1999b). In principle, the mild phenotypic changes in Munc13-3-deficient mice (see also our behavioral analysis described below) excludes the possibility that Munc13-3 is essential for inhibitory–GABAergic neurotransmission in the cerebellum. We substantiated this conclusion by analyzing spontaneous miniature inhibitory postsynaptic responses of Purkinje cells in cerebellar slices from 10- to 11-d-old mice. We found that frequency, amplitude, and rise time of spontaneous miniature inhibitory postsynaptic responses were indistinguishable between Munc13-3-deficient and wild-type Purkinje cells. This is in contrast to glutamatergic transmission in Munc13-1 deletion mutants in which an arrest at the vesicle priming step leads to a strong decrease in the frequency of spontaneous excitatory miniature postsynaptic responses (Augustin et al., 1999b). Our data suggest that synaptic vesicle priming at GABAergic synapses terminating on Purkinje cells is not strongly affected by Munc13-3 loss (Fig. 4).
Fig. 4.

Unaltered spontaneous inhibitory neurotransmission in Purkinje cells from Munc13-3-deficient cerebellum. General features of miniature inhibitory synaptic currents of Munc13-3-deficient Purkinje cells. A, Examples for inhibitory synaptic responses in mutant (right) and control (left) cells. Experimental recordings consisted of six sweeps per cell, each with 10 sec length. B, Isolated single events from A. The thick gray lines show the averaged miniature IPSCs. C–E, Frequency, amplitude, and rise time of miniature IPSCs remain unimpaired in Munc13-3-deficient mice. Four-hundred eighty single events from six cells (wild-type) and 540 single events from seven cells (mutant), respectively, were used to compile these histograms. Error bars indicate SEM.
To determine whether the observed changes in synaptic transmission at parallel fiber axon terminals is paralleled by changes in synaptic morphology, we performed a detailed ultrastructural analysis of wild-type and Munc13-3-deficient cerebella. We found that the density and ultrastructure of synapses in the molecular layer of mutant cerebella was indistinguishable from that of wild-type controls. In particular, the number of synapses per squared micrometer of section, the number of vesicles per synapse, the size of active zones, and the number of docked vesicles per micrometer active zone were not affected by the Munc13-3 mutation (Fig.5).
Fig. 5.

Normal ultrastructure of synapses in Munc13-3-deficient cerebellum. Two brains per genotype were analyzed. For the determination of synapse densities, 42 photographs (124 synapses; 79 and 45 synapses per animal, respectively) from +/+, and 47 photographs (142 synapses; 80 and 62 synapses per animal, respectively) from −/− cerebella were examined. For the ultrastructural comparison, 24 +/+ (13 and 11 synapses per animal, respectively) and 25 −/− (14 and 11 synapses per animal, respectively) synapses were analyzed. Error bars indicate SEM.
As mentioned above, Munc13-3-deficient mice were indistinguishable from their wild-type littermates in terms of gross cage behavior. In addition, mutant mice reproduced normally and were perfectly able to care for their offspring. To determine in more detail whether the observed functional changes in synaptic transmission at parallel fiber–Purkinje cell synapses cause a detectable alteration of motor learning in mutant mice, we studied motor performance and learning of mutant mice on a rotating rod. In initial experiments, we examined the falling latency of naïve, untrained mice at a rotating speed of 40 rpm. We found no significant differences between wild-type and Munc13-3-deficient mice (Fig.6A), suggesting that no preexisting motor or sensory defects are present in these mutants. Rather, homozygous mutants performed slightly better than their wild-type littermates. However, a detailed analysis of motor learning using a 22 session training paradigm during which mice were required to cope with an increasingly difficult motor task revealed a significant motor learning deficit in Munc13-3 mutant mice (Fig.6B). Repeated-measure ANOVA demonstrated a significant genotype × session interaction (F(21,210) = 15.12; p< 0.0001). Tukey's HSD multiple comparison tests showed that, after the 11th trial, motor learning of mutant mice was impaired significantly compared with wild-type controls (p < 0.05). In contrast, learning curves of mutant mice and wild-type controls were virtually identical during the first seven trials and not significantly different in trials 8–11. Thus, all mice improved similarly with training at lower rotation speeds, but Munc13-3-deficient animals exhibited significant deficits in motor learning at higher rotation speeds. This demonstrates that the deficiency of synaptic transmission at parallel fiber–Purkinje cell synapses of Munc13-3-deficient mice is paralleled by (or even causing) a deficiency in motor learning.
Fig. 6.

Deficits in motor learning in Munc13-3-deficient mice. A, Falling latency of naïve wild-type and mutant mice measured with the rotating rod apparatus at 40 rpm. Note that falling latency was not significantly affected by the lack of Munc13-3. Mean values are given (n = 11). Error bars indicate SEM.B, Behavioral analysis of wild-type (+/+) and mutant (−/−) mice in the rotating rod motor learning task.Points represent the mean of the rotation speed learned to criterion (n = 11). Error bars indicate SEM. Note that wild-type mice achieved a significantly (p < 0.05) higher rotation speed than mutants compared from the 11th session onward.
DISCUSSION
Deletion mutations in the murine Munc13-1 gene cause a perinatally lethal phenotype (Augustin et al., 1999b). The analysis of mutant mice resulted in the identification of Munc13-1 as an essential synaptic vesicle maturation protein. One of the most striking features of the Munc13-1-deficient phenotype was that this essential role of Munc13-1 appeared to be restricted to excitatory–glutamatergic neurons, whereas inhibitory–GABAergic neurons remained unaffected by Munc13-1 loss.
One possible explanation for the surprising transmitter specificity of the Munc13-1 priming function was that other Munc13 isoforms (Munc13-2 or Munc13-3) substitute for Munc13-1 in Munc13-1-independent synapses (Augustin et al., 1999b). The present study suggests that Munc13-3, the only Munc13 isoform that is coexpressed with Munc13-1 in the cerebellum (Augustin et al., 1999a), is not essential for inhibitory–GABAergic neurotransmission–vesicle priming in the cerebellum. First, the overall mild phenotype of mice lacking Munc13-3 is not compatible with an essential role of Munc13-3 in inhibitory–GABAergic neurotransmission in the cerebellum because a much more pronounced phenotype would be expected if inhibitory neurotransmission was blocked in these mice to the extent observed in Munc13-1-deficient glutamatergic hippocampal neurons. Second, amplitude, rise time, and frequency of spontaneous inhibitory postsynaptic responses in Purkinje cells are normal in Munc13-3-deficient mice (Fig. 4). This indicates that synaptic vesicle priming at inhibitory synapses terminating on Purkinje cells is not strongly affected by Munc13-3 loss and certainly not to the same extent observed in Munc13-1-deficient glutamatergic neurons in which vesicle priming is completely arrested and, as a consequence, the frequency of spontaneous release events is reduced by 90% (Augustin et al., 1999b). However, based on the present data, we cannot exclude more subtle effects of Munc13-3 loss on GABAergic transmission.
Although Munc13-3 alone is not an essential vesicle priming protein, it is still possible that a more complex compensatory interaction between Munc13 isoforms is responsible for the striking transmitter specificity of phenotypic changes after deletion of the Munc13-1 gene. Essentially, neurons in the brain, including GABAergic cells in the cerebellum, coexpress Munc13-1 with either Munc13-2 or Munc13-3 (Augustin et al., 1999a) (I. Augustin and N. Brose, unpublished observations). Thus, the redundant expression of Munc13-2 in hippocampus (which is devoid of Munc13-3) may be able to compensate for Munc13-1 loss specifically in GABAergic cells but not in glutamatergic cells of this brain region. In cerebellum, Munc13-1 may be able to primarily compensate loss of Munc13-3, the only other isoform expressed here. Alternatively, multiple and partly transmitter-specific synaptic vesicle maturation mechanisms may be in effect. One of these may be dependent on Munc13 isoforms and restricted to excitatory–glutamatergic neurons, whereas others are mediated by as yet unidentified proteins. To ultimately resolve the problem of transmitter specificity of Munc13 action in future studies, it will be necessary to examine inhibitory GABAergic cells in double mutants. Here, the ideal system would be the hippocampus of Munc13-1/Munc13-2 double mutants, which will be available soon. Unfortunately, the problem cannot be addressed in Munc13-1/Munc13-3 double mutants because the cerebellum, which is the only brain region in which the two proteins are coexpressed, is not developed at the time when such mutants die (i.e., at birth).
The present data demonstrate that Munc13-3 is an important regulator of synaptic transmission at parallel fiber–Purkinje cell synapses. In our electrophysiological analysis of Munc13-3 mutants, we observed a significant increase in paired-pulse facilitation at parallel fiber–Purkinje cell synapses (Fig. 3) with normal synapse morphology (Fig. 5). This phenotype may reflect a decreased release probability in mutant synapses and therefore represent a mild variant of the phenotype observed in Munc13-1. In Munc13-1 mutants, vesicle maturation in Munc13-1-dependent synapses is completely arrested, leading to total depletion of the pool of readily releasable vesicles and, as a result, to a complete block of transmitter release (Augustin et al., 1999b). In contrast, vesicle maturation in Munc13-3 mutants may be inhibited only partially. This may then lead to small reductions of the readily releasable vesicle pool and the synaptic release probability, resulting in the observed increase in paired-pulse facilitation. Essentially, this hypothesis implies that Munc13-3 acts at a similar step of the synaptic vesicle pathway as does Munc13-1, albeit with much less efficiency. This view is supported by the facts that the two proteins are colocalized to parallel fiber axon terminals in the cerebellum (Fig. 2) (Augustin et al., 1999a) and that Munc13-3 is much less abundant than Munc13-1 in rat cerebellum (Brose et al., 1995; Augustin et al., 1999a) (N. Brose and I. Augustin, unpublished observations). An additional argument in favor of a common priming role of Munc13-1 and Munc13-3 is the observation that the two proteins are structurally highly conserved in their C-terminal two-thirds, the so-called R-region (Augustin et al., 1999a; Koch et al., 2000), in which they both contain a functionally intact syntaxin binding site (Augustin et al., 1999a).
The cerebellum plays a crucial role in the execution of smooth and skilled movements as well as in more complex behavioral phenomena, such as motor learning. Central to cerebellar function is a highly ordered neuronal network in the cerebellar cortex, with Purkinje cells projecting to deep cerebellar nuclei as the only output neurons. Purkinje cells are thought to have an integrative function in the control of motor coordination and learning. They have two major sources of afferent information: (1) climbing fibers originating from the inferior olive, and (2) parallel fibers originating from cerebellar granule cells, which receive their inputs from several nuclei of the brainstem and neurons of the spinocerebellar tract. Numerous studies on spontaneous and induced mutations in mice demonstrate that an intact cerebellar network is essential for normal motor coordination. For example, migration defects or cell loss observed in the cerebella of a number of mutants are paralleled by strong ataxia (e.g.,reeler, staggerer, and others) (for review, seeMullen et al., 1997). Also, more subtle developmental and functional defects in Purkinje cell inputs, such as persistent multiple climbing fiber innervation after deletion of metabotropic glutamate receptor 1 (mGluR1), Gαq, PKCγ, or phospholipase C-β4 (for review, see Offermanns, 2000), may contribute to pronounced ataxia, indicating that an extremely accurate organization and function of Purkinje cell inputs is important for normal cerebellar function.
In addition to mutants with obvious changes in cerebellar cytoarchitecture and severe ataxia, a number of mutant mouse strains have been described that exhibit deficits in complex motor learning tasks despite normal cerebellar cytoarchitecture–ultrastructure and normal motor coordination in the cage environment. These include GFAP (Shibuki et al., 1996), adenylate cyclase 1 (AC1) (Storm et al., 1998), mGluR4 (Pekhletski et al., 1996), calbindin (Airaksinen et al., 1997), and calretinin (Schiffmann et al., 1999) knock-outs. In all of these mutant strains, changes in cerebellar physiology were shown to parallel motor learning deficits. The observed physiological changes were quite diverse but invariably involved inputs or characteristics of Purkinje cells, including impaired long-term depression (GFAP; Shibuki et al., 1996) and long-term potentiation (AC1; Storm et al., 1998) at synapses between granule and Purkinje cells, a reduction in paired-pulse facilitation ratio at parallel fiber–Purkinje cell synapses (mGluR4;Pekhletski et al., 1996), and changes in Purkinje cell firing behavior (Calretinin; Schiffmann et al., 1999) or Ca2+-transients (Calbindin; Airaksinen et al., 1997). In all these cases, the described physiological aberrations are likely contributors to the concomitant motor learning deficits, although a direct causal relationship between the observed physiological changes and the corresponding motor learning deficits was never demonstrated (mainly because such causal interdependence between physiology and behavior is extremely difficult to prove unequivocally).
In summary, previous analyses of cerebellar mutants with mild physiological and behavioral aberrations suggest that many different types of physiological changes at the level of Purkinje cells and their inputs would cause deficits in motor coordination or motor learning. Consequently, the unique increase in paired-pulse facilitation ratio that we observed at parallel fiber–Purkinje cell synapses of Munc13-3-deficient mice (Fig. 3) should result in a concomitant deficit in motor coordination or learning. Indeed, we found that the changes in synaptic transmission at parallel fiber–Purkinje cell synapses of Munc13-3-deficient cerebella are paralleled by a distinct motor learning deficit of mutant mice (Fig.6B). Although this deficit is not detectable in the normal cage environment, it becomes clearly apparent during the behavioral analysis of complex motor learning using a rotating rod. Here, Munc13-3-deficient mice exhibit significantly impaired motor learning when required to adapt to high rotation speeds during a 22 session motor learning paradigm (Fig. 6B), indicating that a learning or training phase is required to reveal the motor deficit in Munc13-3-deficient mice. At present, we cannot completely rule out that effects of the Munc13-3 deletion on basic motor performance, sensory systems, habituation, or motivation contributed to the behavioral phenotype described here and remained undetected in our test system. However, the fact that naïve mutant mice showed normal falling latencies at a rotation speed of 40 rpm (Fig.6A) would indicate that the Munc13-3 deficiency does not result in basic motor or sensory deficits. Rather, the present data suggest that the regulation of neurotransmission, in particular short-term plasticity, at the parallel fiber–Purkinje cell synapse by Munc13-3 is important for the acquisition of complex motor skills. Because our electrophysiological search was not exhaustive, it is possible that other changes that were not detected in our analysis (e.g., subtle alterations in inhibitory synaptic transmission) also contribute to the motor deficit in Munc13-3 mutants. A very similar phenotype with normal gross behavior and falling latency from the rotating rod, but impaired paired-pulse facilitation and motor learning, was observed in mice lacking the mGluR4 subtype of metabotropic glutamate receptor (Pekhletski et al., 1996). Interestingly, the direction of change in paired-pulse facilitation ratios was different between Munc13-3 mutants (increase) and mGluR4 mutants (decrease), although the behavioral consequences of the mutations were strikingly similar. Thus, together with data on other cerebellar mutants discussed above, our observations support the view that different types of alterations in parallel fiber–Purkinje cell neurotransmission can contribute to motor learning defects.
Footnotes
This work was supported by German Research Foundation Grants SFB406/A1 (to N.B.) and SFB406/A10 (to H.A.K. and J.W.H.) and by the Max-Planck-Society (N.B.). N.B. is a Heisenberg-Fellow of the German Research Foundation. We thank C. Rosenmund for helpful discussions and C. Pouzat for the program for analysis of spontaneous synaptic currents. We are particularly grateful to all members of the Max-Planck-Institut für experimentelle Medizin Animal Facility for help with mouse colonies, L. Kolb, J. Ficner, and R. Schubert for artwork, S. Wenger for excellent technical assistance, and F. Benseler and I. Thanhäuser for DNA synthesis and sequencing.
Drs. Augustin and Korte contributed equally to this work
Correspondence should be addressed to Nils Brose, Max-Planck-Institut für experimentelle Medizin, AG Molekulare Neurobiologie, Hermann-Rein-Straβe 3, D-37075 Göttingen, Germany. E-mail: brose@em.mpg.de.
REFERENCES
- 1.Airaksinen MS, Eilers J, Garaschuk O, Thoenen H, Konnerth A, Meyer M. Ataxia and altered dendritic calcium signaling in mice carrying a targeted null mutation of the calbindin D28k gene. Proc Natl Acad Sci USA. 1997;94:1488–1493. doi: 10.1073/pnas.94.4.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashery U, Varoqueaux F, Voets T, Betz A, Thakur P, Koch H, Neher E, Brose N, Rettig J. Munc13-1 acts as a priming factor for large dense-core vesicles in bovine chromaffin cells. EMBO J. 2000;19:3586–3596. doi: 10.1093/emboj/19.14.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Augustin I, Betz A, Herrmann C, Jo T, Brose N. Differential expression of two novel Munc13 proteins in rat brain. Biochem J. 1999a;337:363–371. [PMC free article] [PubMed] [Google Scholar]
- 4.Augustin I, Rosenmund C, Südhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999b;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- 5.Betz A, Benseler F, Okamoto M, Brose N. Direct interaction of the rat unc-13 homologue Munc13-1 with the N terminus of syntaxin. J Biol Chem. 1997;272:2520–2526. doi: 10.1074/jbc.272.4.2520. [DOI] [PubMed] [Google Scholar]
- 6.Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Südhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- 7.Brose N, Hofmann K, Hata Y, Südhof TC. Mammalian homologues of C. elegans unc-13 gene define novel family of C2-domain proteins. J Biol Chem. 1995;270:25273–25280. doi: 10.1074/jbc.270.42.25273. [DOI] [PubMed] [Google Scholar]
- 8.Brose N, Rosenmund C, Rettig J. Regulation of neurotransmitter release by Unc-13 and its homologues. Curr Opin Neurobiol. 2000;10:303–311. doi: 10.1016/s0959-4388(00)00105-7. [DOI] [PubMed] [Google Scholar]
- 9.Cremona O, De Camilli P. Synaptic vesicle endocytosis. Curr Opin Neurobiol. 1997;7:323–330. doi: 10.1016/s0959-4388(97)80059-1. [DOI] [PubMed] [Google Scholar]
- 10.Edwards FA, Konnerth A, Sakmann B, Takahashi TA. A thin slice preparation for patch-clamp recordings from neurons of the mammalian central nervous system. Pflügers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- 11.Geppert M, Bolshakov VY, Siegelbaum SA, Takei K, De Camilli P, Hammer RE, Südhof TC. The role of Rab3A in neurotransmitter release. Nature. 1994;369:493–497. doi: 10.1038/369493a0. [DOI] [PubMed] [Google Scholar]
- 12.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 13.Klingauf J, Kavalali ET, Tsien RW. Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature. 1998;394:581–585. doi: 10.1038/29079. [DOI] [PubMed] [Google Scholar]
- 14.Koch H, Hofmann K, Brose N. Definition of Munc13-homology-domains and characterization of a novel ubiquitously expressed Munc13 isoform. Biochem J. 2000;349:247–253. doi: 10.1042/0264-6021:3490247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullen RJ, Hamre KM, Goldowitz D. Cerebellar mutant mice and chimeras revisited. Perspect Dev Neurobiol. 1997;5:43–55. [PubMed] [Google Scholar]
- 16.Murthy VN, Stevens CF. Synaptic vesicles retain their identity through the endocytic cycle. Nature. 1998;392:497–501. doi: 10.1038/33152. [DOI] [PubMed] [Google Scholar]
- 17.Offermanns S. Mammalian G-protein function in vivo: new insights through altered gene expression. Rev Physiol Biochem Pharmacol. 2000;140:63–133. doi: 10.1007/BFb0035551. [DOI] [PubMed] [Google Scholar]
- 18.Orita S, Naito A, Sakaguchi G, Maeda M, Igarashi H, Sasaki T, Takai Y. Physical and functional interactions of DOC2 and Munc13 in Ca2+-dependent exocytotic machinery. J Biol Chem. 1997;272:16081–16084. doi: 10.1074/jbc.272.26.16081. [DOI] [PubMed] [Google Scholar]
- 19.Pekhletski R, Gerlai R, Overstreet LS, Huang X-P, Agopyan N, Traverse Slater N, Abramow-Newerly W, Roder JC, Hampson DR. Impaired cerebellar synaptic plasticity and motor performance in mice lacking the mGluR4 subtype of metabotropic glutamate receptor. J Neurosci. 1996;16:6364–6373. doi: 10.1523/JNEUROSCI.16-20-06364.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rickmann M, Wolff JR. S-100 protein expression in subpopulations of neurons of rat brain. Neuroscience. 1995;67:977–991. doi: 10.1016/0306-4522(94)00615-c. [DOI] [PubMed] [Google Scholar]
- 21.Schiffmann SN, Cheron G, Lohof A, d'Alcantara P, Meyer M, Parmentier M, Schurmans S. Impaired motor coordination and Purkinje cell excitability in mice lacking calretinin. Proc Natl Acad Sci USA. 1999;96:5257–5262. doi: 10.1073/pnas.96.9.5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shibuki K, Gomi H, Chen L, Bao S, Kim JJ, Wakatsuki H, Fujisaki T, Fujimoto K, Katoh A, Ikeda T, Chen C, Thompson RF, Itohara S. Deficient cerebellar long-term depression, impaired eyeblink conditioning, and normal motor coordination in GFAP mutant mice. Neuron. 1996;16:587–599. doi: 10.1016/s0896-6273(00)80078-1. [DOI] [PubMed] [Google Scholar]
- 23.Song Y, Ailenberg M, Silverman M. Cloning of a novel gene in the human kidney homologous to rat munc13s: its potential role in diabetic nephropathy. Kidney Int. 1998;53:1689–1695. doi: 10.1046/j.1523-1755.1998.00942.x. [DOI] [PubMed] [Google Scholar]
- 24.Storm DR, Hansel C, Hacker B, Parent A, Linden DJ. Impaired cerebellar long-term potentiation in type I adenylyl cyclase mutant mice. Neuron. 1998;20:1199–1210. doi: 10.1016/s0896-6273(00)80500-0. [DOI] [PubMed] [Google Scholar]
- 25.Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 26.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]
- 27.Zucker RS. Exocytosis: a molecular and physiological perspective. Neuron. 1996;17:1049–1055. doi: 10.1016/s0896-6273(00)80238-x. [DOI] [PubMed] [Google Scholar]