

Abstract
The α1β2γ2 is the most abundant subtype of the GABAA receptor and is localized in many regions of the brain. To gain more insight into the role of this receptor subtype in the modulation of inhibitory neurotransmission, we generated mice lacking either the α1 or β2 subunit. In agreement with the reported abundance of this subtype, >50% of total GABAA receptors are lost in both α1−/− and β2−/− mice. Surprisingly, homozygotes of both mouse lines are viable, fertile, and show no spontaneous seizures. Initially half of the α1−/− mice died prenatally or perinatally, but they exhibited a lower mortality rate in subsequent generations, suggesting some phenotypic drift and adaptive changes. Both adult α1−/− and β2−/− mice demonstrate normal performances on the rotarod, but β2−/− mice displayed increased locomotor activity. Purkinje cells of the cerebellum primarily express α1β2γ2 receptors, and in electrophysiological recordings from α1−/− mice GABA currents in these neurons are dramatically reduced, and residual currents have a benzodiazepine pharmacology characteristic of α2- or α3-containing receptors. In contrast, the cerebellar Purkinje neurons from β2−/− mice have only a relatively small reduction of GABA currents. In β2−/− mice expression levels of all six α subunits are reduced by ∼50%, suggesting that the β2 subunit can coassemble with α subunits other than just α1. Our data confirm that α1β2γ2 is the major GABAA receptor subtype in the murine brain and demonstrate that, surprisingly, the loss of this receptor subtype is not lethal.
Keywords: GABAA receptor, mouse, cerebellum, radioligand, benzodiazepine, inhibitory current, locomotor activity, rotarod
The GABAergic system is the major contributor of the inhibitory tone throughout the CNS. GABAA receptors are ligand-gated ion channels that exist as a number of different subtypes. The GABAA receptor is a pentameric structure, which is formed by the coassembly of subunit polypeptides that exist in a large multigene family (McKernan and Whiting, 1996; Barnard et al., 1998). There are at least 16 different members of the GABAA receptor gene family, including 6α, 3β, 3γ, δ, ε, θ, and π subunits (Whiting et al., 1999). The GABAA receptor genes are differentially expressed both temporally and spatially throughout the mammalian brain. For example the α2, α3, and β3 subunits are the major α and β subunits in the fetal brain, respectively, whereas the α1 and β2 subunits are mainly expressed after birth (Zhang et al., 1991; Laurie et al., 1992b). Therefore, in the adult brain the α1β2γ2 subtype is the major subtype accounting for ∼43% of all GABAA receptors, whereas the remaining receptors are made up mostly by α2- and α3-containing receptors together with other combinations of quantitatively more minor GABAA receptor subtypes (McKernan and Whiting, 1996).
Several mouse strains lacking individual GABAAreceptor subunits have been generated to study the physiological role of GABAergic system in the living organism. Mice lacking the γ2 subunit die shortly after birth (Gunther et al., 1995), whereas heterozygotes have a normal life expectancy and demonstrate neophobia in a novel environment (Crestani et al., 1999). The lethality of the γ2−/− mice can be rescued by transgenic overexpression of either the γ2S or γ2L subunit isoforms of the GABAAreceptor indicating that both γ2 subunit splice variants can substitute for each other (Baer et al., 2000; Wick et al., 2000). Mice lacking the β3 subunit of the GABAA receptor have cleft palate, epilepsy, and many behavioral characteristics of Angelman syndrome (Culiat et al., 1995; Homanics et al., 1997; DeLorey et al., 1998). Most of the β3−/− mice die as neonates, but the survivors, which are runted until weaning, can achieve normal body size by adulthood. In contrast, mice lacking the α6 subunit of the GABAA receptor, which is expressed exclusively in cerebellar granule cells, have no major phenotypic abnormalities (Jones et al., 1997). Expression of the δ subunit is inhibited in the α6−/− mice, suggesting that both subunits form functional GABAA receptor subtypes in the cerebellar granule cells. Finally, mice deficient for the δ subunit are viable but show attenuated sensitivity to neuroactive steroids and epileptic seizures (Mihalek et al., 1999).
GABAA receptors are the site of action of a number of clinically important drugs, including benzodiazepines, barbiturates, and anesthetics (Sieghart, 1995; Whiting et al., 1995). The α1β2γ2 subtype is of particular interest in this context because it comprises the major benzodiazepine binding site in the brain. We addressed the question about the physiological role of this receptor subtype by generating mice lacking either the α1 or β2 subunit of the GABAA receptor which are thought to primarily coassemble to form the α1β2γ2 subtype.
MATERIALS AND METHODS
Generation of α1−/− mice. The GABAA receptor α1 gene-targeting vector was constructed from the same genomic 129/SvEv λ fixII clone, which has been used for the introduction of the α1H101R mutation (McKernan et al., 2000). However, for the complete gene knock-out, exon 4 was disrupted at the MscI restriction site by cloning the 1.2 kb BstBI+MscI and the 7 kbEcoRV+BamHI DNA fragment blunt-ended into the targeting vector. A phosphoglycerate kinase I (PGK) neo and a thymidine kinase (TK) cassette were also engineered blunt-ended into the loxP site containing targeting vector. After linearization with NotI the targeting vector was introduced into AB2.2 embryonic stem (ES) cells (Lexicon Genetics) as described (Soriano et al., 1991; Rosahl et al., 1993, 1995). Homologous recombinants were identified by PCR using the primers 5′-ATTAATGGAGAGTGTGGTAATCTTT-3′ and 5′-GGATGCGGTGGGCTCTATGGCTTCTGA-3′ and were further confirmed by genomic Southern blotting. Correctly targeted ES cell clones were injected into C57BL6 blastocysts, and one of three clones gave rise to highly chimeric males, which transmitted the targeted allele into the germ line. A colony of homozygous and wild-type control animals were established and used for the present study. Some α1−/− mice were crossed with a cre-transgenic mouse (Schwenk et al., 1995) and further interbred to establish α1 homozygotes, which no longer contained the neomycin resistance gene marker.
Mice lacking the β2 subunit were generated in a similar way. A 17.5 kb genomic λ/FixII clone containing exons 6, 7, and 8 of the β2 subunit was subcloned into pBluescript via the NotI sites. An 8.5 kb SalI DNA fragment as a long arm and a 1.25 kbHpaI + FspI DNA fragment as a short arm were cloned into the PGK neo and TK containing modified pBS246 plasmid (T. W. Rosahl and K. L. Hadingham, unpublished observations) resulting in the deletion of exons 6 and 7 in the targeting vector. After linearization of the targeting vector with NotI and introduction into AB2.2 ES cells, homologous recombinants were identified using the following PCR primers: 5′-ACCAGTCTGGACCATGAGTTCCCA-3′ and 5′-GGATGCGGTGGGCTCTATGGCTTCTGA-3′ One of three injected ES cell clones gave rise to a chimera transmitting the gene disruption into the germ line. A colony of β2−/− mice containing the neo gene and some −neo gene by crossing with the deleter mice were generated as for the α1−/− mice.
Radioligand binding and biochemical analysis. Radioligand binding assays with [3H]Ro15–1788 (87 Ci/mmol; NEN Life Sciences), [3H]Ro15–4513 (21.7 Ci/mmol; NEN Life Sciences) in the presence or absence of 10 μmdiazepam and [3H]muscimol (19.1 Ci/mmol; NEN Life Sciences) were performed on membrane preparations as previously described (Quirk et al., 1994; Sur et al., 1998, 1999a).
Autoradiographic studies of the convulsant binding site of GABAA receptors were performed using 8 nm[35S]t-butylbicyclophosphorothionate (TBPS) on coronal sections of wild-type and knock-out mouse brains cut at a thickness of 12–16 μm. Sections were washed in 50 mm Tris—citrate and 200 mm NaBr, pH 7.4, buffer for 10 min and then incubated in the same buffer containing [35S]TBPS or [35S]TBPS plus 10 μmpicrotoxin for nonspecific binding for 90 min at room temperature. Slides were washed twice for 5 min in cold buffer, rinsed in distilled water, and exposed to film for 48 hr.
Autoradiographic analyses of the different benzodiazepine-binding sites were done as previously described (Turner et al., 1991; Sur et al., 1999b) with 2 nm[3H]Ro15–1788 (labels α1βγ2, α2βγ2, and α3βγ2 subtypes), 4 nm[3H]L-655,708 plus 10 μmzolpidem (labels α5βγ2 subtype), and 8 nm[3H]Ro15–4513 plus 20 μmdiazepam (labels α4βγ2 and α6βγ2 subtypes) on coronal sections (12–16 μm) from two to four mice per genotype. After 3–8 weeks exposure, autoradiograms were analyzed with a Micro Computer Imaging Device M2 imaging system (Imaging Research, St. Catharines, Ontario, Canada).
Immunoprecipitation studies with selective α1, α2, and α3 antibodies were performed on solubilized receptors as previously described (McKernan et al., 1991) using a 10 nmconcentration of [3H]Ro15–1788.
For Western blot analyses of 30 μg of protein were loaded on a 4–12% Bis-Tris gel (Novex, San Diego, CA). Proteins were then transferred to nitrocellulose membrane (Hybond-C; Amersham Pharmacia Biotech, Little Chalfont, UK) by semidry blotting, and the presence of α1 and α3 subunit was detected by incubation with specific rabbit anti-α1 (10 μg/ml) and anti-α3 (7 μg/ml) antibodies and the ECL detection system (Amersham Pharmacia Biotech).
Data analyses and statistics were performed with GraphPad Prism (San Diego, CA).
Electrophysiology. Cerebellum was removed from α1−/−, β2−/−, and wild-type mice at postnatal days 11–17, and the vermal layer was isolated and placed into ice-cold oxygenated dissociation media containing (in mm): 82Na2SO4, 30 K2SO4, 5 MgCl2, 10 HEPES buffer, and 10 glucose at pH 7.4. Tissue was then stirred for 7 min in 10 ml of dissociation media containing 3 mg/ml of protease XXIII (Sigma) at 37°C. The tissue was then washed in warmed oxygenated dissociation media containing 1 mg/ml bovine serum albumin and 1 mg/ml trypsin inhibitor and maintained under oxygenating conditions at room temperature in Tyrode's solution (in mm: 150 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 10 glucose, pH 7.4). Tissue was withdrawn as needed and triturated with a fire-polished Pasteur pipette to liberate individual cells. Cells were plated onto a glass coverslip and left to settle for at least 30 min before use. Purkinje cell bodies were identified by their characteristic size and morphology. Cells could be used for up to 5 hr after preparation.
Glass coverslips containing the dissociated cells were placed in a perspex recording chamber on the stage of a Nikon Diaphot inverted microscope. Cells were perfused continuously with artificial CSF (aCSF) containing (in mm): 149 NaCl, 3.25 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 11 d-glucose, d(+)-sucrose, pH 7.4, and observed with phase-contrast optics. Fire-polished patch pipettes were pulled on a WZ, DMZ-Universal puller (Zeitz-Instruments, Munich, Germany) using conventional 120TF-10 electrode glass. Pipette tip diameter was ∼1.5–2.5 μm, with resistances ∼4 MΩ. The intracellular solution contained (in mm): 130 CsCl, 10 HEPES, 10 BAPTA-Cs, 5 ATP-Mg, 0.1 leupeptin, and 1 MgCl2, with 100 μmNaCO3, pH-adjusted to 7.3 with CsOH and 320–340 mOsm. Cells were voltage-clamped at −60 mV using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Drug solutions were applied to the cells via a multibarrel drug delivery system, which could pivot the barrels into place using a stepping motor. This ensured rapid application and washout of the drug. Drugs were applied to the cell for 5 sec with a 30 sec washout period between applications. Allosteric potentiation of GABAA receptors was measured relative to a GABA EC20 determined for each cell to account for differences in GABA potency. Modulators were pre-equilibrated for 30 sec before coapplication of GABA.
Behavioral analysis. All mice tested were in a 50% C57BL6, 50% 129SvEv background. They were housed in groups under standard 12 hr light/dark cycle and had ad libitum access to standard mouse diet and water. Two- to 3-month-old F4 generation mice were tested for motor coordination on the rotarod, and 6-month-old F5 mice were used to determine spontaneous locomotor activity.
Motor coordination was assessed using the rotarod test. Wild-type (n = 9), α1−/− (n = 7), and β2−/− (n = 6) mice were first trained until they could remain on a rotarod revolving at 16 rpm for three consecutive 120 sec trials. The next day the mice were placed back on the rotarod for a single trial at 18 rpm (maximum duration 120 sec). The duration a mouse could remain on the rotarod was recorded, and the mouse was then returned to its home cage. The speed of the rotarod was then increased to 21 rpm, and all mice were given another test trial. This process was repeated for rotarod speeds of 24, 27, 30, 33, and 36 rpm. Spontaneous locomotor activity was measured using individual perspex activity chambers (215 × 270 × 210 mm) equipped with two parallel infrared beams running across each end of the base of the chamber. Naive α1−/− and wild-type mice (n = 10 per group) were placed in the activity cages, and cage crosses (i.e., consecutive breaks of each beam) were measured for 1 hr. In a separate experiment naive β2−/− and wild-type mice (n = 12 per group) were also assessed for spontaneous locomotor activity for 1 hr. Data were analyzed using a repeated measures ANOVA, and then individual time bins were examined with a two-way ANOVA.
RESULTS
Generation of α1−/− and β2−/− mice
Exon 4 of the α1 subunit gene of the GABAAreceptor was disrupted, and exons 6 and 7 of the β2 subunit gene were deleted in mice by the homologous recombination technique (Rosahl et al., 1993, 1995; McKernan et al., 2000) (Fig.1a–h). A colony of α1−/− and β2−/− mice were bred and analyzed, whereas the neomycin resistance gene flanked by loxP sites (Fig. 1c,g, floxed) remained in the targeted locus. These α1 or β2−/− [+neo] mice were kept in a ∼50% C57BL6–50% 129SvEv genetic background. Some homozygotes of the F3 generation were crossed with the crecontaining “deleter” mouse (Schwenk et al., 1995) to remove the neo gene in the offspring (Fig. 1d,h). This resulted in a change of the genetic background to ∼75% C57BL6–25% 129SvEv. The heterozygous offspring was interbred to produce α1 or β2−/− [−neo] and wild-type control mice. Northern blot results using a 45 mer antisense oligonucleotide specific for exon 4 upstream of the gene disruption confirmed complete absence of α1 mRNA in both the α1−/− [+neo] and α1−/− [−neo] mice (Fig. 1i). Similarly, a probe for the 5′ part of the β2 subunit gene demonstrated lack of any β2 mRNA in the β2−/− [+neo] and β2−/− [-neo] mice. No major changes in the expression of the α3, α6, β3, and γ2 genes were found in either knock-out mouse lines regardless of the presence or absence of the neo gene (Fig.1i) (data not shown). Therefore, all experiments presented in Figures 1j–6 were performed on α1−/− or β2−/− mice containing the neomycin resistance gene. The complete loss of α1 protein in α1−/− [+neo] mice was further substantiated by the absence of a major 50 kDa band on Western blots (Fig. 1j). Absence of the β2 subunit protein in the β2−/− mice could not be confirmed by Western blotting because no antibody specific for the β2 subunit was available.
Fig. 1.
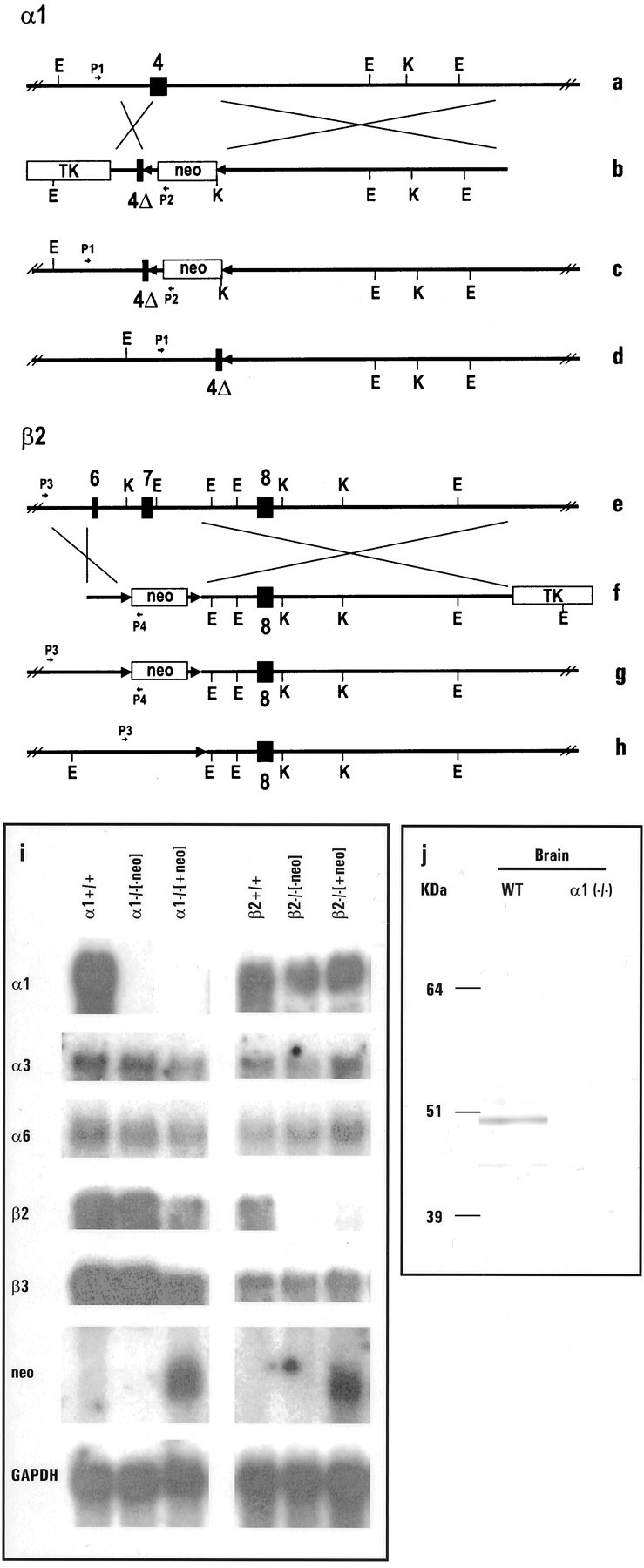
Generation and validation of α1 and β2 −/− mice. a, b, e,f, Schematic representation of the WT α1 (a, b) and β2 allele (e, f) of the GABAA receptor and the corresponding targeting vectors.4, 6, 7, Exons 4, 6, and 7, respectively; 4Δ, partial deletion of exon 4;neo, neomycin resistance gene; TK,thymidine kinase gene; E, EcoRI restriction site;K, KpnI restriction site; P1, P2, P3, P4, PCR primers for detecting the mutant α1 and β2 allele, respectively. c,g, Targeted allele after homologous recombination for α1 and for β2 allele, and after cre-mediated removal of the neomycin resistance gene (d, h), respectively. i, Northern blot results using 45 mer oligonucleotides for the α1, α3, α6, β2, and β3 subunit genes of the GABAA receptor and neomycin resistance (neo) and glyceraldehyde-3-phosphate dehydrogenase gene as probes; 5 μg of poly(A+) selected RNA was loaded per lane; α1+/+ and β2+/+: wild-type control samples; α1−/−[-neo] and β2−/−[-neo]: samples from α1 and β2 homozygotes lacking the neo gene in their genome; α1−/−[+neo] and β2−/−[+neo]: samples from α1 and β2 homozygotes with the neo gene in their genome. j, Western blot demonstrating the absence of α1 polypeptide in the brain of α1−/− mice.
Pharmacological characterization of GABAA receptors in α1 −/− and β2 −/− mice
Autoradiography with [35S]TBPS, a radioligand that binds to a site putatively located to the channel pore of all GABAA receptors (Luddens and Korpi, 1995), confirmed a large and widespread loss of GABAAreceptors in cortex (−66 and −46%), septum (−53 and −60%), caudate putamen (−40 and −37%), globus pallidus (−72 and −74%), hippocampus (−53 and −27%), thalamus (−65 and −63%), and cerebellum (−67 and −71%) of both α1−/−and β2−/− mice, respectively (Fig. 2). These losses were further demonstrated by the 66 and 58% reduction in the total number of GABAA receptors, as measured by the binding of [3H]muscimol to membranes from the brains of α1−/− and β2 −/− mice (Table1). Saturation experiments revealed a large reduction in [3H]Ro15–1788 binding sites in the forebrain of α1−/− (−57 ± 4%;n = 3) and β2−/− (−51 ± 4%;n = 4) compared with wild-type mice with no change in the affinity of the radioligand (Tables 1, 2). The expression of [3H]Ro15–1788 binding sites was also reduced in the cerebellum of α1−/− (−67 ± 3%;n = 3) and β2−/− (−71 ± 5%;n = 4) animals. Surprisingly the reduction of [3H]Ro15–1788 binding in the cerebellum of α1−/− mice was less than expected because the α1βγ2 subtype is reported to be the main benzodiazepine-sensitive GABAA receptor in this brain region, accounting for 90 ± 3% (n = 4) of total [3H]Ro15–1788 sites, as determined by immunoprecipitation with an α1-specific antisera. Additional immunoprecipitation experiments with an α3 subunit-specific antisera indicated that in wild-type mice the remaining cerebellar [3H]Ro15–1788 binding sites (14 ± 3%; n = 3) are contributed by α3βγ2 receptors. These observations pointed toward a putative upregulation of α3 subunit containing GABAA receptors in the cerebellum of α1−/− mice (see below). Radioligand inhibition binding studies with [3H]Ro15–1788 and a number of benzodiazepine site compounds revealed no significant (Student's t test, unpaired, two-tailed) changes in the pharmacology of the remaining GABAA receptors in both α1−/− and β2−/− mouse lines (Table 2) with the exception of the complete loss of high-affinity binding sites for zolpidem, an α1-selective compound (Sieghart, 1995), in α1−/− mice whereas high-affinity zolpidem binding sites still accounted for 49% of total [3H]Ro15–1788 sites in β2−/− brains (Table 2).
Fig. 2.
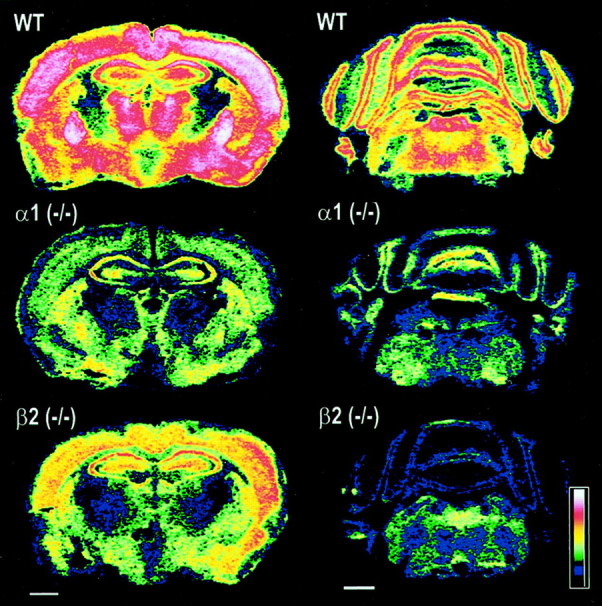
Loss of GABAA receptors in the forebrain and cerebellum of α1 −/− and β2 −/− mice. Color-coded autoradiograms for [35S]TBPS (8 nm) binding to sections of mouse brain revealed a widespread reduction of GABAA receptors. Major losses are observed in cortex (−66 and −46%), globus pallidus (−72 and −74%), thalamus (−65 and −63%), and cerebellum (−67 and −71%) for example of α1 −/− and β2 −/− mice, respectively. Scale bars, 1 mm.
Table 1.
Amount (fmol/mg protein) of GABAA receptors and benzodiazepine binding sites in WT, α1−/−, and β2−/− mice determined by nonlinear regression analysis of saturation data sets
| Wild type | α1−/−1-a | β2−/−1-b | |
|---|---|---|---|
| Muscimol | 3014 ± 274 | 1011 ± 241 | 1275 ± 210 |
| Ro15-1788: | |||
| Forebrain (F3) | 2677 ± 174 | 1160 ± 219 | 1326 ± 143 |
| Cerebellum (F3) | 1212 ± 123 | 419 ± 18 | 339 ± 34 |
Note that Ro15-1788 values for α1−/− derived from F3 generation mice. Data are the mean ± SEM of three or four experiments.
All Bmax are different from wild-type p < 0.003 (t test; unpaired, two-tailed).
All Bmax are different from wild-type p < 0.002 (t test; unpaired, two-tailed).
Table 2.
Pharmacological characterization of GABAAreceptors in WT, α1 (−/−), and β2 (−/−) mice
| Kd*/Ki(nm) | |||
|---|---|---|---|
| Wild type | α1 (−/−) | β2 (−/−) | |
| Muscimol* | 30 ± 5 | 16 ± 6 | 41 ± 18 |
| Ro15-1788* | 2.4 ± 0.6 | 1.9 ± 0.3 | 1.8 ± 0.4 |
| Flunitrazepam | 9.7 ± 2.2 | 5.1 ± 1.4 | 13 ± 3 |
| Ro15-4513 | 12 ± 3 | 9.6 ± 2.7 | 10 ± 3 |
| Zolpidem | |||
| H.A. | 35 ± 12 | n.d. | 23 ± 7 |
| % H.A. | 62 ± 15 | n.d. | 49 ± 14 |
| L.A. | 784 ± 312 | 770 ± 358 | 727 ± 267 |
Data are the mean ± SEM of three to five experiments.Kd values were determined from nonlinear regression analysis of full saturation curves. Student's ttest (unpaired, two-tailed) indicated that compounds have a similar affinity for GABAA receptors in wild-type than in α1−/− or β2−/− mice, respectively. n.d., Not detected.
Electrophysiological analyses of GABAAreceptors in Purkinje neurons of α1−/− and β2−/− mice
Cerebellar Purkinje neurons have been shown to express a limited repertoire of GABAA receptor subunits, predominantly consisting of α1β2γ2 (Laurie et al., 1992a; Persohn et al., 1992; Fritschy and Mohler, 1995). These cells then provided an ideal candidate for the study of the effects of elimination of the α1 or the β2 subunits by gene targeting. Whole-cell patch-clamp recordings were made from dissociated cerebellar Purkinje neurons isolated from both the α1−/− and β2−/− mice and compared with those of wild-type littermates. Recordings from wild-type neurons revealed the presence of robust GABA-mediated currents in all cells tested with a mean amplitude of 2982 ± 271 nA (n = 30) (Fig.3a) in response to 1 mm GABA application. GABA EC20 currents were significantly enhanced by the benzodiazepine modulators chlordiazepoxide (148 ± 8% at 3 μm; n = 20) and the α1-selective compound zolpidem (182 ± 13% at 100 nm;n = 17) (Fig. 3a,b). In contrast, 23 of 52 of the cells from the α1−/− mice did not produce a response to 1 mm GABA. In those cells that did respond to GABA, these currents were significantly reduced in amplitude (mean of 52 cells, 257 ± 54 pA). Comparing measured EC20 values in these cells also demonstrated a significant decrease in the potency of GABA in the α1−/− cells [3.8 ± 0.3 μm (20) in wild-type and 19.0 ± 4.6 μm (5) in α1−/−]. Of those cells with GABA currents large enough to measure an EC20 response, five were studied using the benzodiazepine modulators. The potentiation by chlordiazepoxide was identical to that in wild-type animals (138 ± 35%), however the effect of 100 nm zolpidem was markedly reduced (34 ± 17%), suggesting that these receptors contained either α2 or α3 subunits or are formed from β2γ2 alone. Similar experiments using the β2−/− animals revealed a different profile. In contrast to the α1−/− mice, every cell responded to GABA, however the mean current amplitude was reduced compared with wild-type (1234 ± 144 pA; n = 13). As with the α1−/− mice the potentiation of the GABA currents of Purkinje cells from β2−/− mice by chlordiazepoxide was similar to wild-type (104 ± 17%; n = 6), however there was marked potentiation produced by 100 nm zolpidem (108 ± 15%;n = 5). The effect of zolpidem was significantly reduced compared with wild-type mice, suggesting that the receptors present contain a mixed population of α1βxγ2 and α2/α3βxγ2. To determine the β-isoform present in the receptors in the β2−/− mice, the effects of the β-subunit-selective agents loreclezole and etomidate were investigated. These compounds selectively potentiate receptors containing a β2 or β3 subunit, but not β1-containing receptors (Wafford et al., 1994; Hill-Venning et al., 1997). Robust potentiation of the GABA EC20 by both agents was observed in both the wild-type and β2−/− mice (Fig.4), suggesting that the remaining receptors expressed in Purkinje contained predominantly β3 subunits.
Fig. 3.

Electrophysiological analysis of GABA currents recorded from cerebellar Purkinje neurons of wild-type and α1−/− and β2−/− mice. a, Example recordings from wild-type (WT), α1−/−, and β2−/− mice, of the response to 1 mm GABA, and potentiation of submaximal, EC20 currents by 3 μm chlordiazepoxide or 100 nm zolpidem (this concentration would distinguish α1 from other subtypes). Amplitude is indicated by the scale bar in each case, and drugs were applied as shown by the bar above each response. B, Mean current amplitude in response to 1 mm GABA from isolated Purkinje neurons; nnumber is shown in parentheses above each column and includes all cells tested including unresponsive cells.c, Mean potentiation of GABA EC20 by 3 μm chordiazepoxide; n number is shown inparentheses above each column. d, Mean potentiation of GABA EC20 by 100 nm zolpidem;n number is shown in parentheses above each column.
Fig. 4.

Effects of loreclezole, etomidate, and pentobarbital on GABA receptors from WT and β2−/− Purkinje neurons.a, Recording from WT and β2−/− neurons showing a GABA response to 1 mm GABA followed by the potentiation of a GABA EC20 response by 3 μm loreclezole, 30 μm etomidate, and 100 μm pentobarbital. Amplitude is indicated by the scale bar, and drugs were applied as shown by the bar above each response. b–d, Mean potentiation of the GABA EC20 response by 3 μm loreclezole (b), 30 μmetomidate (c), and 100 μm pentobarbital (d); n number is shown inparentheses above each column.
Behavioral phenotype of α1−/− and β2−/− mice
After breeding of α1 heterozygotes of the F1 generation, α1 −/− mice were found to be under-represented in the F2 generation, accounting for only 13.4% of all offspring (29 α1−/− of 217 pups in total). The surviving α1 homozygotes appeared to have lower body weights and were less well groomed before weaning, but improved their appearance with age. Therefore, the behavioral analysis focused on adult animals only. The β2−/− mice were represented in a Mendelian manner in the F2 generation (23 of 95 animals in total) and had no obvious phenotypical abnormalities.
A neurological screen was initially performed on adult F2 mice to determine whether there were any major neurological deficits in these animals. Neither α1−/− nor β2−/− mice displayed any major deficits on beam balancing and swimming ability tests (data not shown). Two-month-old α1−/− were smaller and had lower body weights (∼30%) than wild-type controls, and this difference persisted until at least 3 months of age. α1−/− mice were also observed to have a tremor when handled, but this did not impair their ability to perform motor tasks. β2−/− mice had normal body weights (data not shown).
A more detailed assessment of motor coordination was performed using the rotarod. A two-factor repeated measures ANOVA indicated that both α1−/− and β2−/− mice were as capable of remaining on the rotarod as wild-type controls (genotype:F(2,19) = 0.05; p = 0.95) (Fig. 5a). As expected, performance declined as the revolution speed increased (speed:F(6,114) = 22.73; p < 0.00005), but there were no deficits seen in either of the knock-out lines. α1−/− mice displayed a similar level of spontaneous locomotor activity and exploration compared with wild-types (Fig.5b) when placed in the novel environment of activity chambers (genotype: F(1,18) = 0.02;p = 0.66; repeated measures ANOVA). They also habituated to this environment over a similar time scale as wild-types (time × genotype interaction:F(29,522) = 0.90; p = 0.51). In contrast, β2−/− animals (Fig. 5c) exhibited a much higher level of activity in this test (genotype:F(1,22) = 10.63; p = 0.0036), although they did habituate to a similar degree as wild-types (time × genotype interaction:F(29,638) = 0.72; p = 0.86).
Fig. 5.
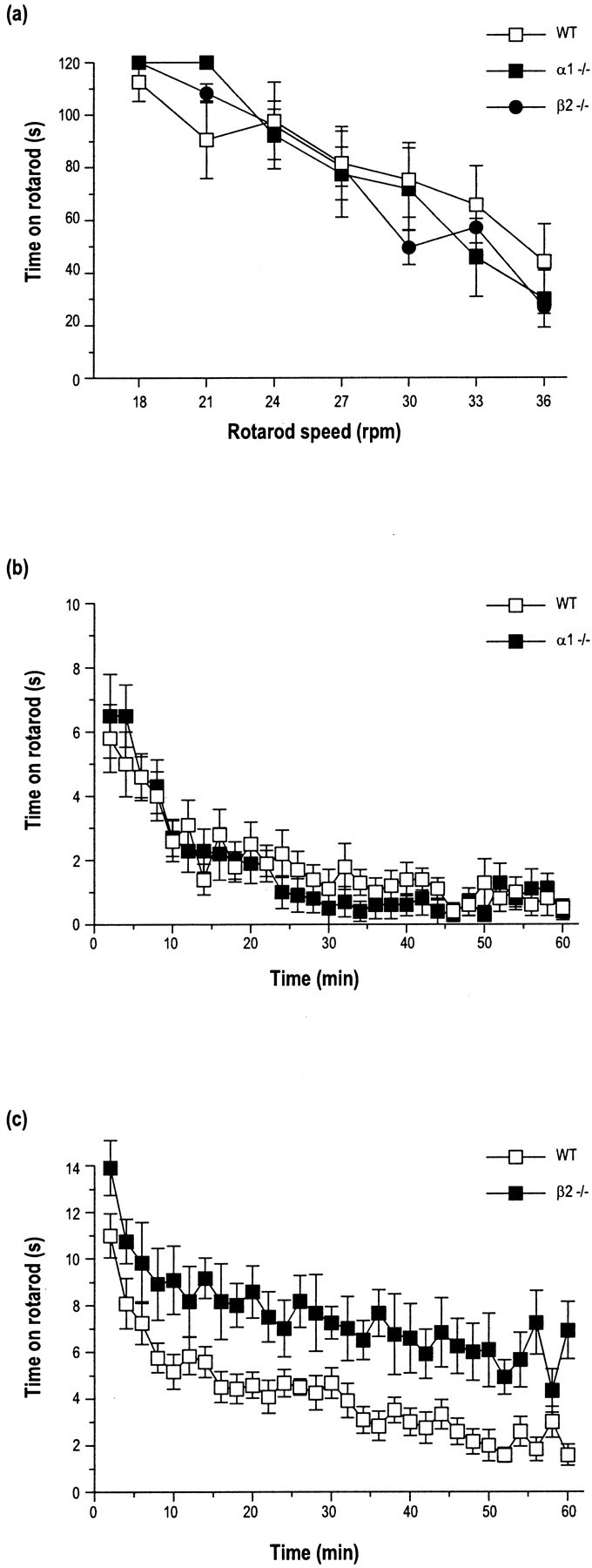
Behavioral evaluation of α1 and β2 −/− mice.a, The duration a mouse remained walking on the rotarod revolving at different speeds (18–36 rpm) decreased as the speed was increased. α1 and β2 −/− mice did not differ from wild-type mice at any speed examined. Data are the means ± SEM;n = 6–9. The spontaneous locomotor activity of α1 −/− mice (b) did not differ from wild-type mice. In contrast, β2 −/− mice (c) showed a marked increase in activity (p < 0.005) compared with wild-type mice. Data are the mean number of cage crosses in 2 min time bins ± SEM; n = 10–12.
Generation-dependent litter size and upregulation of α2 and α3 subunits in α1−/− mice
To establish an α1−/− mouse line, homozygotes of the F2 generation were interbred using a variety of breeding pairs and avoiding any brother–sister matings. Similarly, β2−/− and wild-type control lines originating from their F2 littermates were established. However, there were some indications that the α1−/− surviving pups appeared to be phenotypically less affected in the F4 and F5 generation in comparison with the initial findings of the α1 homozygotes in the F2 generation. Therefore, we analyzed the number of pups in each litter for the α1−/− mouse line in detail. The litter size from wild-type matings remained constant at approximately seven pups per litter between the F3 and F5 generations (Table3). In contrast, F2 α1−/− matings produced significantly smaller litters (approximately three pups per litter), but litter size increased with increasing generation number.
Table 3.
Litter size increases across several generations
| Genotype | Generation | ||
|---|---|---|---|
| F3 | F4 | F5 | |
| WT | 6.96 ± 0.65 (23)3-a | 6.13 ± 0.77 (15) | 8.35 ± 0.50 (34) |
| α1 −/− | 3.06 ± 0.40 (16) | 4.46 ± 0.54 (22) | 6.46 ± 0.71 (28)b |
Litter size (Mean number of pups ± s.e.m.).
,bp < 0.05 compared with F3 α1 −/−; numbers in brackets indicate number of litters analyzed.
The possibility that this generation-dependent phenotypic change is paralleled by modification in GABAA receptor expression was investigated with [3H]Ro15–1788 saturation binding, a radioligand that would likely detect upregulation or downregulation of GABAA receptors (Fig.6a,b). Indeed, in the cerebellum of α1−/− mice, a generation-dependent reduction in the amount of GABAA receptors was observed. Specifically, the loss of total [3H]Ro15–1788 binding sites determined by Scatchard analyses was increased from generation F2 (49%;n = 1) to generation F3 (67 ± 3%;n = 3). The reduction in generation F3 was significantly different (p < 0.001; Student'st test) from generation F5 (87 ± 1%;n = 4) which was equivalent to the amount of wild-type cerebellar α1 receptors sensitive to Ro15–1788 (90 ± 3%;n = 4), as determined by immunoprecipitation with an α1-specific antibody (Fig. 6a). The last observation indicated that in F5 generation mice there is no longer an upregulation of benzodiazepine-sensitive GABAA receptors. A similar trend was demonstrated in forebrains of α1−/− mice where reduction in [3H]Ro15–1788 sites increased from 45% (n = 1) in generation F2 to 57 ± 4% (n = 3) in generation F3, and finally reached 66 ± 2% (n = 4) in generation F5. In this F5 generation, no upregulation of [3H]Ro15–1788 sites occurred because the recorded reduction was similar to the number of wild-type forebrain α1 receptors sensitive to Ro15–1788 (67 ± 3%;n = 4) determined by immunoprecipitation as well as the proportion of high-affinity zolpidem binding sites (62 ± 15%;n = 4) found in wild-type mouse (Fig. 6b,Table 2). To determine which GABAA receptor subtype was responsible for this upregulation in the F3 generation, we focused our attention on α2 and α3 subunits that are the most abundantly expressed subunits after the α1 subunit in adult brain and also the predominant embryonic isoforms (Laurie et al., 1992b). In cerebellum, Western blot analyses revealed a marked increased in the expression of α3 subunit in α1−/− compared with wild-type mice (Fig. 6c). The faint band observed in wild-type cerebellum is in agreement with α3 receptors accounting for 14 ± 3% (n = 3) of total [3H]Ro15–1788 binding sites in this brain region. Furthermore, quantitative immunoprecipitation experiments allowed us to demonstrate that an α3 subunit-specific antisera precipitated all (92 ± 8%; n = 4) available [3H]Ro15–1788 binding sites (Fig.6c). These results indicated that upregulation of α3 subunit in the cerebellum of α1−/− is solely responsible for the increase in total [3H]Ro15–1788 sites observed in F3 generation. The contribution of α2 and α3 subunits in the upregulation of [3H]Ro15–1788 binding sites in the forebrain of α1−/− mice was investigated by quantitative immunoprecipitation. Results showed that α2 and α3 subunits account for 17 ± 3% (n = 4) and 14 ± 4% (n = 4) of total [3H]Ro15–1788 sites in wild-type mice, respectively, and their respective contribution increased to 56 ± 7% (n = 4) and 45 ± 7% (n = 4) in α1−/− animals. Given that the total number of [3H]Ro15–1788 sites in the forebrain of F3 generation α1−/− mice has been shown to represent 43% of that in wild-type mice (Table 1), a net increase of 42% in the expression of both α2 and α3 subunits was estimated (Fig. 6d). Putative changes in the expression of minor populations of GABAA receptors were also investigated by various pharmacological approaches. Quantitative autoradiography with [3H]L-655,708, a α5 subtype-selective ligand, revealed no change in expression of this subunit in CA1-CA3 fields of hippocampus (96% of wild-type), dentate gyrus (99% of wild-type), and endopiriform nucleus (99% of wild-type) (Table4). Similar autoradiography experiments with [3H]Ro15–4513 plus 20 μm diazepam showed no changes in the expression of α4βγ2 receptors in the thalamus, hippocampus, and cortex of α1−/− mice. However, binding experiments with 40 nm [3H]Ro15–4513 plus 10 μm diazepam on cerebellar membranes from F5 generation α1−/− mice demonstrated a significant reduction (−38 ± 10%; n = 4) in the expression of α6 receptors (Table 4). This reduction did not result from the presence of the neomycin cassette in the targeted gene because a similar (p > 0.42; Student's t test) 26 ± 10% (n = 4) reduction in α6 subunit expression was determined in the cerebellum of F6 generationcre-excised α1−/− [−neo] mice.
Fig. 6.

Generation-dependent upregulation of α2 and α3 subunits in cerebellum and forebrain of α1−/− mice.a, The loss of [3H]Ro15–1788 binding sites increased in cerebellum (left panel) and forebrain (right panel) of α1−/− mice from generation F2 to F5 to finally reach the proportion of [3H]Ro15–1788 sites immunoprecipitated by a selective α1 antibody from wild-type membrane (filled bars). ***p < 0.001; Student's t test. b, Evidence for an upregulation of α3 subunit in the cerebellum of α1−/− mice. Western blot showing an increase in α3 subunit expression in knock-out mice compared with wild-type (left panel). Quantitative immunoprecipitation with a α3 antibody demonstrated that α3 subunit-containing GABAAreceptors account for 14 ± 3% (mean ± SEM;n = 3) and 92 ± 8% (mean ± SEM;n = 4) of total [3H]Ro15–1788 binding sites in the cerebellum of wild-type and α1−/− mice, respectively (right panel). c, Quantitative immunoprecipitation demonstrated a 42% increase in the expression of α2 and α3 subunit-sensitive [3H]Ro15–1788 binding sites in the forebrain of α1−/− compared with wild-type mice.
Table 4.
Variation in the expression of various GABAAreceptor subtypes in α1−/− and β2−/− mice
| Subtype | α1−/− | β2−/− |
|---|---|---|
| α1βγ2/3 | −100% | −60% |
| α2βγ2/3 | 0% | −48% |
| α3βγ2/3 | 0% | −69% |
| α4βγ2/3 | 0% | −60% |
| α5βγ2/3 | 0% | −39% |
| α6βγ2/3 | −38% | −62% |
For α1−/− mice, data are derived from F5 generation animals. In α1−/− and β2−/− mice, data correspond to α1, α2, α3 expression in forebrain, α4 expression in cortex, thalamus, and hippocampus, α5 expression in hippocampus and endopiriform nucleus, and α6 expression in cerebellum. The data were obtained through various methodological approaches such as immunoprecipitation (α1–3 in β2−/−), autoradiography (α4, α5), Western blot (α1), and radioligand binding (α1–4 and α6).
Effect of deletion of the β2 subunit gene on the expression of the various α subunits
To determine whether the compensatory changes in subunit expression observed in α1−/− mice were because of the loss of α1 subunit or the loss of the α1β2γ2 receptor per se, the expression of the various α subunits was investigated in the β2−/− mice. Immunoprecipitation experiments indicated that α1, α2, and α3 subunits represented 54 ± 12% (n = 4), 18 ± 5% (n = 4), and 11 ± 1% (n = 4) of total forebrain [3H]Ro15–1788 binding sites from β2−/− mice, respectively. These proportions equated to a reduction by 60, 48, and 69% of α1, α2, and α3 subunit-containing receptors (Table 4), given the fact that in β2−/− brains the [3H]Ro15–1788 binding sites represent only 49% (Table 1) of the wild-type complement. Quantitative autoradiography using the α5 subtype-selective ligand [3H]L-655,708 revealed an overall 39% reduction in the expression of α5 subunit with losses of 33% in the hippocampus and 53% in the endopiriform nucleus. Similar experiments with [3H]Ro15–4513 plus 20 μm diazepam revealed significant losses of α4βγ2 receptors in various regions such as the cortex (−55%), the hippocampus (−50%), and the thalamus (−59%). Radioligand binding experiments on mice forebrain with 40 nm[3H]Ro15–4513 plus 10 μm diazepam confirmed these autoradiographic measures and demonstrated an overall 60 ± 7% (n= 5) decrease in α4-containing receptors (Table 4). Finally, the expression of diazepam-insensitive [3H]Ro15–4513 receptors in the cerebellum of β2−/− mice was reduced by 62 ± 6% (n = 5) (Table 4). This downregulation of α6 receptors was again independent of the presence of the neomycin cassette in the targeted gene because diazepam-insensitive [3H]Ro15–4513 sites were also decreased by 80 ± 3% (n = 2) in the cerebellum of β2−/− [−neo] mice.
DISCUSSION
Mouse lines lacking functional GABAAreceptor subunits α1 and β2 have been generated to further our understanding of the role of the GABAergic system in the control of inhibitory neurotransmission in the CNS. As expected, a widespread elimination of GABAA receptors was observed with brain regions having the strongest expression of the α1 and β2 subunit experiencing the highest loss of receptors. The consequences of these disruptions in the GABAergic system on behavior and regulation of subunit expression and assembly are discussed.
Lack of major phenotypic deficiency in adult α1−/− and β2−/− mice
In both knock-out mouse lines ∼60% of the total number of GABAA receptors were lost, and it was therefore surprising to find that adult α1−/− and β2−/− mice do not experience major phenotypic abnormalities or spontaneous seizures. Indeed, targeted disruptions of the β3 or γ2 subunit of the GABAA receptor result in a similar substantial loss of GABAA receptors, but lethality and serious epileptic seizures have been reported for both β3- and γ2-deficient mice (Gunther et al., 1995; Homanics et al., 1997). The balanced reduction in the β2−/− mice of receptors containing each of the six α subunits could explain the seemingly normal appearance of the β2−/− mice. These observations also allude to the putative existence of spare receptors among GABAA receptor subtype populations. Adult α1−/− mice, but not β2−/− mice, did exhibit tremor when handled, despite a wide codistribution in the brain of α1 and β2 subunit and a similar loss in the total number of GABAA receptors. It would be of interest to generate an α1/β2 double knock-out mouse, but the close linkage of these two genes makes this an impractical task given current technologies. Although it is likely that there are multiple anatomical and physiological substrates responsible for this phenotypic difference, the different responsiveness to GABA of α1−/− and β2−/− Purkinje cells may be a contributing factor. GABA currents were almost absent in the Purkinje cells of α1−/−, but only halved in those of β2−/− mice compared with wild-type animals. This is in agreement with the predominant expression of α1 subunit in Purkinje neurons, whereas there is equivalent expression of β2 and β3 subunits (Fritschy et al., 1992; Wisden et al., 1992; Fritschy and Mohler, 1995). However, neither the presumed incomplete inhibitory control of the main cerebellar output, nor the pronounced reduction of α1 and α6 subunits in granule cells of α1−/− and β2−/− had a detectable impact on the motor capacity of the knock-out mice. Similarly, α6 −/−/δ deficient mice, in which the cerebellum contains only half the number of GABAA receptors, have no impairment of motor skills (Jones et al., 1997; Nusser et al., 1999). Altogether, these genetic-based observations indicate that synaptic integration in granule and Purkinje cells is apparently preserved in cerebellum expressing only α1 or α6 or a mix of α1 and α6 (as in β2−/− mice) subunits. Moreover they suggest that α1 and α6 subunits have some functional overlap in addition to common synaptic localization at the surface of granule cells (Nusser et al., 1998). The availability of these different strains of mice should prove very useful to dissect the role of GABAergic inhibition in cerebellar physiology.
Generation-dependent change in phenotype and subunit regulation
Initially, the α1−/− mice displayed a severely compromised phenotype, with only half of them surviving after birth and a variable phenotype in the survivors. A similar perinatal lethality has also been observed in GABAA receptor δ subunit knock-out mice (Mihalek et al., 1999). However, the litter size of α1−/− mice increases in successive generations, which may be the result from selective breeding of the α1 homozygotes, which have a less compromised phenotype and therefore exhibit higher rates of reproduction. Interestingly, the increase in the litter size and a subjectively milder phenotype in the later generations correlates with decreased upregulation of the α2/3 subunit. However, it should be noted that this upregulation of α2/3 subunit was incomplete because ∼45% of GABAA receptors were still missing in the brains of mice from the F2 generation. At present it is not clear how this process originates and is regulated, nor why the α2/3 upregulation results in a subjectively more severe phenotype. A possible explanation would be that several regulatory events take place in α1−/− mice to compensate for their inability to switch on α1 subunit around the time of birth (Laurie et al., 1992b). Initially, retention of embryonically highly expressed α2 and α3 subunits (Laurie et al., 1992b) could be one event of this adaptive process and that it is then lost in later generations because of putative detrimental effects on animal development. At the molecular level, one could speculate that expression of membrane potential regulating ion channels is changed in a way that relatively normal neuronal inhibition can occur in the absence of α1 subunit-containing GABAA receptors (Brickley et al., 2001). In such a context, overexpression of α2 and α3 subunit could boost inhibition to a level that impairs normal physiological development. Interestingly, the weight and size of the brain of α1−/− mice from F3 generation was somewhat smaller (∼15%) than in wild-type controls. Although such adaptive phenomena limit our assessment of the role of the targeted protein, their future analysis may provide insights into neuronal regulatory mechanisms as well as on the role of compensatory proteins.
Assembly of GABAA receptor subunits
Selective subunit knock-out mice represent an ideal system to study the abundance and assembly of subunit that generates native GABAA receptors (Fritschy et al., 1997; Jones et al., 1997; Nusser et al., 1999). Combining data from analysis of both α1−/− and β2−/− mice confirms that the combination α1β2γ2/3 is the predominant isoform accounting for 40% of total benzodiazepine-sensitive GABAAreceptors in mouse brain (McKernan et al., 1991; McKernan and Whiting, 1996). A specific decrease of α6 subunit protein by ∼30% was found in the cerebellum of both adult α1−/− [+neo] and [−neo] mice in the later generations (Table 4). Unlike in the case for the α6 −/− mice (Uusi-Oukari et al., 2000), the presence of the neomycin resistance gene in the targeted locus does not seem to have a major influence on the expression level of its neighboring GABAA receptor genes in the β2−α6−α1−γ2 gene cluster (Russek, 1999) in the α1 deficient mice, as documented by normal transcriptional expression of the β2, α6, and γ2 genes. The coassembly of α1 and α6 subunits is known in rat cerebellum (Pollard et al., 1995; Khan et al., 1996; Jechlinger et al., 1998) where α1α6βγ2 complexes may represent ∼40% of diazepam-insensitive Ro15–4513 binding sites (Khan et al., 1996). Thus, the loss of α6 subunit in α1−/− mice could be explained by the existence of a partnership in which α1 subunit is required for proper expression of α1α6βγ and/or α1α6βδ. Such a possibility would be reminiscent of the α6/δ partnership (Jones et al., 1997) and support the view that in vivo α subunits may play an important role in the assembly of GABAA receptors (Fritschy et al., 1997; Jones et al., 1997).
The reduction of both total GABAA receptors and [3H]Ro15–1788 binding sites in both the forebrain and cerebellum of β2−/− mice is consistent with expression levels of this subunit in rat brain (Benke et al., 1994; Li and DeBlas, 1997) and support the conclusion that β2 is an abundant neuronal β subunit and there was no significant compensatory upregulation of β1 or β3 subunit. The β2−/− mice demonstrated a rather ubiquitous association of β2 subunit with α1–6 subunits in agreement with previous biochemical studies (Benke et al., 1994;Jechlinger et al., 1998) and the codistribution of β2 and α1–6 subunits in most brain regions (Persohn et al., 1992; Wisden et al., 1992).
In conclusion, biochemical and pharmacological analyses of α1−/− and β2−/− mice demonstrated that the subunit combination α1β2γ2 constitutes the most abundant GABAAreceptor subtype in rodent brain, and the β2 subunit can coassemble with all α subunits. The loss of half of total GABAA receptors in both knock-out mouse lines and the subsequent disruption of GABA-mediated inhibition resulted in only mild behavioral deficits in the adult animals. Future studies will focus on the GABA-mediated synaptic transmission and the compensatory mechanisms responsible for the seemingly normal behavior of the α1−/− and β2−/− mice.
Footnotes
C.S., K.W., and D.R. contributed equally to different aspects of this work.
Correspondence should be addressed to Dr. Thomas W. Rosahl, Merck Sharp and Dohme Neuroscience Research Center, Terlings Park, Eastwick Road, Harlow, Essex, CM20 2QR, UK. E-mail: thomas_rosahl@merck.com.
REFERENCES
- 1.Baer K, Essrich C, Balsiger S, Wick MJ, Harris RA, Fritschy J-M, Luscher B. Rescue of γ2 subunit-deficient mice by transgenic overexpression of the GABAA receptor γ2S or γ2L subunit isoforms. Eur J Neurosci. 2000;12:2639–2643. doi: 10.1046/j.1460-9568.2000.00159.x. [DOI] [PubMed] [Google Scholar]
- 2.Barnard EA, Skolnick P, Olson RW, Mohler H, Sieghart W, Biggio G, Braestrup C, Bateson AN, Langer SZ. International Union of Pharmacology. XV. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. Pharmacol Rev. 1998;50:291–313. [PubMed] [Google Scholar]
- 3.Benke D, Fritschy J-M, Trzeciak A, Bannwarth W, Mohler H. Distribution, prevalence, and drug binding profile of γ-aminobutyric acid type A receptor subtypes differing in the β-subunit variant. J Biol Chem. 1994;269:27100–27107. [PubMed] [Google Scholar]
- 4.Brickley SG, Revilla V, Cull-Candy SG, Wisden W, Farrant M. Adaptive regulation of neuronal excitability by a voltage-independent K+ conductance. Nature. 2001;409:88–92. doi: 10.1038/35051086. [DOI] [PubMed] [Google Scholar]
- 5.Crestani F, Lorez M, Baer K, Essrich C, Benke D, Laurent JP, Belzung C, Fritschy J-M, Luscher B, Mohler H. Decreased GABAA receptor clustering results in enhanced anxiety and a bias for threat cues. Nat Neurosci. 1999;2:833–839. doi: 10.1038/12207. [DOI] [PubMed] [Google Scholar]
- 6.Culiat CT, Stubbs L, Woychik RP, Russell LB, Johnson DK, Rinchik EM. Deficiency of the β3 subunit of the type A γ-aminobutyric acid receptor causes cleft palate in mice. Nat Genet. 1995;11:344–346. doi: 10.1038/ng1195-344. [DOI] [PubMed] [Google Scholar]
- 7.DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD, Olson RW. Mice lacking the β3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci. 1998;18:8505–8514. doi: 10.1523/JNEUROSCI.18-20-08505.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fritschy J-M, Mohler H. GABAA receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- 9.Fritschy J-M, Benke D, Mertens S, Oertel WH, Bachi T, Mohler H. Five subtypes of type A γ-aminobutyric acid receptors identified in neurons by double and triple immunofluorescence staining with subunit-specific antibodies. Proc Natl Acad Sci USA. 1992;89:6726–6730. doi: 10.1073/pnas.89.15.6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fritschy JM, Benke D, Johnson DK, Mohler H, Rudolph U. GABAA-receptor α-subunit is an essential prerequisite for receptor formation in vivo. Neuroscience. 1997;81:1043–1053. doi: 10.1016/s0306-4522(97)00244-3. [DOI] [PubMed] [Google Scholar]
- 11.Gunther U, Benson J, Benke D, Fritschy J-M, Reyes G, Knoflach F, Crestani F, Aguzzi A, Arigoni M, Lang Y, Bluethmann H, Mohler H, Luscher B. Benzodiazepine-insensitive mice generated by targeted disruption of the γ2 subunit gene of γ-aminobutyric acid type A receptors. Proc Natl Acad Sci USA. 1995;92:7749–7753. doi: 10.1073/pnas.92.17.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill-Venning C, Belelli D, Peters JA, Lambert JJ. Subunit-dependent interaction of the general anaesthetic etomidate with the gamma-aminobutyric acid type A receptor. Br J Pharmacol. 1997;120:749–756. doi: 10.1038/sj.bjp.0700927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Homanics GE, DeLorey TM, Firestone LL, Quinlan JJ, Handforth A, Harrison NL, Krasowski MD, Rick CEM, Korpi ER, Makela R, Brilliant MH, Hagiwara N, Ferguson C, Snyder K, Olsen RW. Mice devoid of γ-aminobutyrate type A receptor β3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc Natl Acad Sci USA. 1997;94:4143–4148. doi: 10.1073/pnas.94.8.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jechlinger M, Pelz R, Tretter V, Klausberger T, Sieghart W. subunit composition and quantitative importance of hetero-oligomeric receptors: GABAA receptors containing α6 subunits. J Neurosci. 1998;18:2449–2457. doi: 10.1523/JNEUROSCI.18-07-02449.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones A, Korpi ER, McKernan RM, Pelz R, Nusser Z, Makela R, Mellor JR, Pollard S, Bahn S, Stephenson FA, Randall AD, Sieghart W, Somogyi P, Smith AJH, Wisden W. Ligand-gated ion channel subunit partnerships: GABAA receptor α6 subunit gene inactivation inhibits δ subunit expression. J Neurosci. 1997;17:1350–1362. doi: 10.1523/JNEUROSCI.17-04-01350.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khan ZU, Gutierrez A, DeBlas AL. The α1 and α6 subunits can coexist in the same cerebellar GABAA receptor maintaining their individual benzodiazepine-binding specificities. J Neurochem. 1996;66:685–691. doi: 10.1046/j.1471-4159.1996.66020685.x. [DOI] [PubMed] [Google Scholar]
- 17.Laurie DJ, Seeburg PH, Wisden W. The distribution of thirteen GABA-A receptor subunit mRNAs in the rat brain. II. Olfactory bulb and cerebellum. J Neurosci. 1992a;12:1063–1076. doi: 10.1523/JNEUROSCI.12-03-01063.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laurie DJ, Seeburg PH, Wisden W. The distribution of thirteen GABA-A receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J Neurosci. 1992b;12:4151–4172. doi: 10.1523/JNEUROSCI.12-11-04151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li M, DeBlas AL. Coexistence of two β subunit isoforms in the same γ-aminobutyric acid type A receptor. J Biol Chem. 1997;272:16564–16569. doi: 10.1074/jbc.272.26.16564. [DOI] [PubMed] [Google Scholar]
- 20.Luddens H, Korpi ER. GABA antagonists differentiate between recombinant GABAA/benzodiazepine receptor subtypes. J Neurosci. 1995;15:6957–6962. doi: 10.1523/JNEUROSCI.15-10-06957.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKernan RM, Whiting PJ. Which GABAA receptor subtypes really occur in the brain? Trends Neurosci. 1996;19:139–143. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- 22.McKernan RM, Quirk K, Prince R, Cox PA, Gillard NP, Ragan CI, Whiting P. GABAA receptor subtypes immunopurified from rat brain with α subunit-specific antibodies have unique pharmacological properties. Neuron. 1991;7:667–676. doi: 10.1016/0896-6273(91)90379-e. [DOI] [PubMed] [Google Scholar]
- 23.McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, Farrar S, Myers J, Cook G, Ferris P, Garrett L, Bristow L, Marshall G, Macaulay A, Brown N, Howell O, Moore KW, Carling RW, Street LJ, Castro JL, Ragan CI, Dawson GR, Whiting PJ. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nat Neurosci. 2000;3:587–592. doi: 10.1038/75761. [DOI] [PubMed] [Google Scholar]
- 24.Mihalek RM, Banerjee PK, Korpi ER, Quinlan JJ, Firestone LL, Mi Z-P, Lagenaur C, Tretter V, Sieghart W, Anagnostaras G, Sage JR, Fanselow MS, Guidotti A, Spigelman I, Li Z, DeLorey TM, Olsen RW, Homanics GE. Attenuated sensitivity to neuroactive steroids in γ-aminobutyrate type A receptor delta subunit knock-out mice. Proc Natl Acad Sci USA. 1999;96:12905–12910. doi: 10.1073/pnas.96.22.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nusser Z, Sieghart W, Somogyi P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci. 1998;18:1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nusser Z, Ahmad Z, Tretter V, Fuchs K, Wisden W, Sieghart W, Somogyi P. Alterations in the expression of GABAA receptor subunits in cerebellar granule cells after the disruption of the α6 subunit gene. Eur J Neurosci. 1999;11:1685–1697. doi: 10.1046/j.1460-9568.1999.00581.x. [DOI] [PubMed] [Google Scholar]
- 27.Persohn E, Malherbe P, Richards JG. Comparative molecular neuroanatomy of cloned GABAA receptor subunits in the rat CNS. J Comp Neurol. 1992;326:192–216. doi: 10.1002/cne.903260204. [DOI] [PubMed] [Google Scholar]
- 28.Pollard S, Thompson CL, Stephenson FA. Quantitative characterization of α6 and α1α6 subunit-containing native γ-aminobutyric acidA receptors of adult rat cerebellum demonstrates two α subunits per receptor oligomer. J Biol Chem. 1995;270:21285–21290. doi: 10.1074/jbc.270.36.21285. [DOI] [PubMed] [Google Scholar]
- 29.Quirk K, Gillard NP, Ragan CI, Whiting PJ, McKernan RM. γ-aminobutyric acid type A receptors in the rat brain can contain both γ2 and γ3 subunits, but γ1 does not exist in combination with another γ subunit. Mol Pharmacol. 1994;45:1061–1070. [PubMed] [Google Scholar]
- 30.Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC, Sudhof TC. Short term synaptic plasticity is altered in mice lacking synapsin I. Cell. 1993;75:661–670. doi: 10.1016/0092-8674(93)90487-b. [DOI] [PubMed] [Google Scholar]
- 31.Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Sudhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- 32.Russek SJ. Evolution of GABAA receptor diversity in the human genome. Gene. 1999;227:213–222. doi: 10.1016/s0378-1119(98)00594-0. [DOI] [PubMed] [Google Scholar]
- 33.Schwenk F, Baron U, Rajewsky KA. cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acid Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sieghart W. Structure and pharmacology of γ-aminobutyric acidA receptor subtypes. Am Soc Pharmacol Exp Ther. 1995;47:181–234. [PubMed] [Google Scholar]
- 35.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteoporosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 36.Sur C, Quirk K, Dewar D, Atack JR, McKernan RM. Rat and human hippocampal α5 subunit containing γ-aminobutyric acid-A receptors have α5β3γ2 pharmacological characteristics. Mol Pharmacol. 1998;54:928–933. doi: 10.1124/mol.54.5.928. [DOI] [PubMed] [Google Scholar]
- 37.Sur C, Fresu L, Howell O, McKernan RM, Atack JR. Autoradiographic localization of α5 subunit-containing GABAA receptors in rat brain. Brain Res. 1999a;822:265–270. doi: 10.1016/s0006-8993(99)01152-x. [DOI] [PubMed] [Google Scholar]
- 38.Sur C, Farrar SJ, Kerby J, Whiting PJ, Atack JR, McKernan RM. Preferential coassembly of α4 and δ subunits of the γ-aminobutyric acidA receptor in rat thalamus. Mol Pharmacol. 1999b;56:110–115. doi: 10.1124/mol.56.1.110. [DOI] [PubMed] [Google Scholar]
- 39.Turner DM, Sapp DW, Olsen RW. The benzodiazepine/alcohol antagonist Ro15–4513: Binding to a GABAA receptor subtype that is insensitive to diazepam. J Pharmacol Exp Ther. 1991;257:1236–1242. [PubMed] [Google Scholar]
- 40.Uusi-Oukari M, Heikkila J, Sinkkonen ST, Makela R, Hauer B, Homanics GE, Sieghart W, Wisden W, Korpi ER. Long-range interactions in neuronal gene expression: evidence from gene targeting in the GABAA receptor β2−α6−α1−γ2 subunit gene cluster. Mol Cell Neurosci. 2000;16:34–41. doi: 10.1006/mcne.2000.0856. [DOI] [PubMed] [Google Scholar]
- 41.Wafford KA, Bain CJ, Quirk K, McKernan RM, Wingrove PB, Whiting PJ, Kemp JA. A novel allosteric modulatory site on the GABAA receptor β subunit. Neuron. 1994;12:775–782. doi: 10.1016/0896-6273(94)90330-1. [DOI] [PubMed] [Google Scholar]
- 42.Whiting PJ, McKernan RM, Wafford KA. Structure and pharmacology of vertebrate GABAA receptor subtypes. Int Rev Neurobiol. 1995;38:95–138. doi: 10.1016/s0074-7742(08)60525-5. [DOI] [PubMed] [Google Scholar]
- 43.Whiting PJ, Bonnert TP, McKernan RM, Farrar S, LeBourdelles B, Heavens RP, Smith DW, Hewson L, Rigby MR, Sirinathsinghji DJS, Thompson SA, Wafford KA (1999) Molecular and functional diversity of the expanding GABAA receptor gene family. Ann NY Acad Sci: 645–653. [DOI] [PubMed]
- 44.Wick MJ, Radcliffe RA, Bowers BJ, Mascia MP, Luscher B, Harris RA, Wehner JM. Behavioural changes produced by transgenic overexpression of γ2L and γ2S subunits of the GABAA receptor. Eur J Neurosci. 2000;12:2634–2638. doi: 10.1046/j.1460-9568.2000.00160.x. [DOI] [PubMed] [Google Scholar]
- 45.Wisden W, Laurie DJ, Monyer HM, Seeberg PH. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon and mesencephalon. J Neurosci. 1992;12:1040–1062. doi: 10.1523/JNEUROSCI.12-03-01040.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang J-H, Sato M, Tohyama M. Different postnatal ontogenic profiles of neurons containing β (β1, β2 and β3) subunit mRNAs of GABAA receptor in the rat thalamus. Dev Brain Res. 1991;58:289–292. doi: 10.1016/0165-3806(91)90017-d. [DOI] [PubMed] [Google Scholar]