

Abstract
Mutations were identified in the Cu/Zn superoxide dismutase gene (SOD1) in ∼15% of patients with familial amyotrophic lateral sclerosis. Transgenic animals expressing mutant SOD1 in all tissues develop an ALS-like phenotype. To determine whether neuron-specific expression of mutant SOD1 is sufficient to produce such a phenotype, we generated transgenic animals carrying the G37R mutation that is associated with the familial form of ALS (FALS), which is driven by the neurofilament light chain promoter. The transgenic animals express high levels of the human SOD1 protein in neuronal tissues, especially in the large motor neurons of the spinal cord, but they show no apparent motor deficit at up to 1.5 years of age. Our animal model suggests that neuron-specific expression of ALS-associated mutant human SOD1 may not be sufficient for the development of the disease in mice.
Keywords: amyotrophic lateral sclerosis, superoxide dismutase, mutant SOD1, transgenic mice, neuronal expression, cell death, neurofilament promoter
Amyotrophic lateral sclerosis (ALS) is a late onset neurodegenerative disorder characterized by the death of large motor neurons in the cerebral cortex and spinal cord (Tandan and Bradley, 1985). The familial form of ALS (FALS) accounts for ∼10% of cases and usually is transmitted as an autosomal dominant trait (Mulder et al., 1986). Mutations in Cu/Zn superoxide dismutase gene (SOD1), a ubiquitously expressed and highly conserved metalloenzyme involved in the detoxification of free radicals, are responsible for ∼15% of FALS (Rosen et al., 1993; Fridovich, 1995;Pramatarova et al., 1995).
In vitro studies showed that some mutants had a reduced specific activity whereas others retained full activity (Borchelt et al., 1994). The wide variety of enzymatic activities and the absence of motor dysfunction in transgenic SOD1 null mice make loss of function an unlikely pathogenic mechanism of FALS and suggest that mutant SOD1 toxicity arises from a gain rather than from a loss of function (Reaume et al., 1996).
Consistent with this model, transgenic mice overexpressing FALS-associated SOD1 mutants (G93A, G37R, and G85R) developed a rapidly progressive disease with lower motor neuron degeneration that is reminiscent of human ALS (Gurney et al., 1994; Dal Canto and Gurney, 1995; Wong et al., 1995; Mourelatos et al., 1996; Bruijn et al., 1997). In G37R or G93A animals high levels of mutant SOD1 expression were required to develop the disease, and the total SOD1 enzyme activity was increased 4- to 14-fold. In these models the age of onset of disease and neuronal populations that were affected depended on the levels of mutant enzyme. On the other hand, transgenic animals expressing the G85R mutation developed a rapidly progressing fatal disease, astrocytic inclusions that stained positive for SOD1, and ubiquitin as well as SOD1-positive aggregates in motor neurons, despite modest levels of expression (Bruijn et al., 1997).
Although these animal models implicate mutant SOD1 in the development of motor neuron degeneration, many questions remain, particularly with respect to the mechanism of disease pathogenesis. One critical question is whether cellular damage arises from direct motor neuron toxicity or whether indirect toxic effects, for example via the astrocytes as suggested by the G85R transgenics, contribute to neuronal cell death. A recent study showed that expression of mutant mouse G86R SOD1 cDNA in the astrocytes of transgenic mice was not sufficient to cause an ALS-like motor deficit (Gong et al., 2000).
To test whether motor neuron expression of mutant SOD1 is sufficient for disease development, we generated transgenic animals carrying a human SOD1 cDNA with the G37R FALS-associated mutation that is driven by the neurofilament light chain promoter. We found that, in contrast to observations in transgenic lines overexpressing FALS-associated SOD1 mutations in a constitutive manner, neuron-specific expression does not cause significant motor neuron cell death.
MATERIALS AND METHODS
Construction of transgenic mice expressing wild-type and mutant G37R human SOD1. Total human RNA was extracted from lymphoblasts by homogenization in guanidinium thiocyanate and ultracentrifugation through a cesium chloride cushion for 20 hr at 35,000 rpm. Then the RNA pellet was washed, resuspended in water, and kept at −20°C (Chirgwin et al., 1979). RT-PCR was performed on 2 μg of RNA, using the primers SOD1A, 5′-AAG TCG ACA AGC TTT GCG TCG TAG TCT CCT GCA-3′, and SOD1B, 5′-GCC CTC GAG AAG CTT TTT TTT AAG ATT ACA GTG-3′, which introduce a HinDIII restriction site. The cDNA was cloned and sequenced with a Sequenase kit (Amersham Life Science, Arlington Heights, IL). We introduced the mutation G37R by PCR site-directed mutagenesis (Tomic et al., 1990). The vector carrying the neurofilament light chain promoter (pGCHNFL) was generously provided by Dr. J. P. Julien (McGill University, Montreal, Canada).
Transgenic animals were generated via standard microinjection techniques that used hybrid mouse embryos (C57BL/6J × C3H/HeJ) (Brinster et al., 1981). To test for transgene transmission, we isolated DNA from mouse tail; we performed Southern blotting by using the human SOD1 cDNA as probe. The animals were housed in a pathogen-free environment, were checked on a regular basis for microbial infections, and also were examined for motor dysfunction (general motility and activity, grooming, holding by the tail, hanging to a rod; Collard and Julien, 1995).
Expression analysis. Mice (6–7 months of age) were killed by cervical dislocation; the dissected tissues (brain, spinal cord, heart, liver, kidney, spleen, muscle, and testis) were frozen rapidly in liquid nitrogen and then stored at −80°C until used for RNA or protein extraction.
Multiple tissues RT-PCR was performed on 1 μg of total DNase-treated RNA, with an oligo-dT primer, using the kit from PerkinElmer Life Sciences (Norwalk, CT). PCR was performed for 35 cycles at 94°C for 1 min, 60°C for 30 sec, and 72°C for 45 sec, using primers SOD1A and SOD1B, and amplifying specifically the human SOD1 cDNA. RT-PCR was performed also, in parallel, for 35 cycles of 94°C for 1 min, 62°C for 30 sec, and 72°C for 45 sec, using primers amplifying the GAPDH cDNA (5′-CCT TTC ATT GAC CTC AAC TAC ATG G-3′ and 5′-AGT CTT CTG GGT GGC AGT GAT GG-3′) (Hyman Friedman et al., 1996).
Protein was extracted by homogenization of the tissues in lysis buffer (1% NP-40 and 1 mm PMSF in PBS), and centrifugation for 20 min at 13,000 rpm at 10°C. The soluble protein was quantified by the Lowry method and diluted in loading buffer (15% glycerol, 5% SDS, 80 mm Tris-HCl, pH 6.8, 5% β-mercapto-ethanol, and 0.01% bromophenol blue). Each sample (100 μg) was run on a 12% SDS/glycine polyacrylamide gel according to Laemmli and then transferred electrophoretically on a nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany). Human SOD1 (20–40 ng) was used as a positive control (Sigma, St. Louis, MO). The blot was blocked in 2% nonfat milk/0.1% Tween 20 in PBS and probed with a sheep anti-human SOD1 antibody diluted 1:500 in the blocking buffer (Biodesign, Kennebunkport, ME). The immunodetection was performed with an HRP-labeled secondary antibody, and the detection was done with the Renaissance kit (PerkinElmer Life Sciences). For the protein quantification serial dilutions of each sample (10–150 μg) as well as diluted hSOD1 (0.6–40 ng) were run on 12% SDS/glycine polyacrylamide gels, transferred on nitrocellulose, probed, and developed as previously described; densitometry analysis was performed with the Is-1000 Digital Imaging System from Alpha Innotech (San Leandro, CA).
Generation and characterization of the LAP2-10AP anti-human SOD1 antibody. Antibodies were raised against a region of the human SOD1 protein (hSOD1) spanning amino acids 18–37, which displays low homology with the mouse SOD1 protein (mSOD1; 50% identity). A synthetic peptide (IINFEQKESNGPVKVWGSIK+C) was cross-linked to keyhole limpet hemocyanin (KLH), emulsified in Freund's adjuvant, and used to immunize rabbits (1 mg in complete Freund's adjuvant for the first injection, followed every 4–6 weeks by 0.5 mg in incomplete Freund's adjuvant). Rabbit serum was affinity-purified on a column, and aliquots were kept frozen at −20°C. The affinity-purified hSOD1 antibody (LAP2-10AP) was characterized by immunoblotting. Human SOD1 (Sigma) and protein extracts from spinal cord of transgenic animals overexpressing the human SOD1 as well as of nontransgenic animals were run on a 12% SDS/glycine polyacrylamide gel and electrotransferred on nitrocellulose. The blots were blocked in 2% nonfat milk/0.1% Tween 20 in PBS and then incubated with either preimmune serum (LAP2-1) or hSOD1 antibody or with peptide-preabsorbed hSOD1 antibody diluted 1:500 in the blocking buffer. The immunodetection was done by incubating the blots with a biotinylated goat anti-rabbit antibody, followed by incubation with streptavidin–HRP according to the instructions from the manufacturer; final detection was made with the Renaissance kit (PerkinElmer Life Sciences).
Immunocytochemical analysis and morphometry. Mice (8 months old) were anesthetized with an intraperitoneal injection of 4% chloral hydrate and perfused intracardially with PBS, followed by 4% paraformaldehyde, pH 7.4, in 0.1 m sodium phosphate buffer. Spinal cords were removed, post-fixed for 1 hr, and then frozen in Tissue-Tek OCT embedding compound (Sakura Finetek, Torrance, CA).
Immunocytochemistry was performed on 30-μm-thick floating sections. Sections were quenched first in 0.3% H2O2 for 30 min, permeabilized in 1% Tween 20 for 10 min, blocked in 3% normal goat serum for 3 hr, and then incubated in the primary hSOD1 antibody (LAP2-10AP) for 16 hr at 4°C. The immunoreactivity was visualized with diaminobenzidine after amplification of the signal with the TSA-indirect reagent (PerkinElmer Life Sciences) and Vectastain ABC Elite (Vector Laboratories, Burlingame, CA).
To determine the percentage of hSOD1-positive cells in the spinal cords that were analyzed, we photographed multiple sections, circled the hSOD1-positive cells, and determined their relative surface area with an Image 1 Analyzer (Universal Imaging, West Chester, PA).
Motor function measurements. Motor capacity was assessed by measuring the grip strength of the animals. The data were analyzed by ANOVA.
Microscopic analysis and quantification of motor neuron degeneration. Mice (11–12 months old) were anesthetized with an intraperitoneal injection of 4% chloral hydrate and perfused with PBS, followed by 3% glutaraldehyde, pH 7.4, in 0.1 m sodium phosphate buffer. Ventral roots from levels L4 and L5 were removed, post-fixed for 16 hr, incubated in 1% osmium tetroxide, dehydrated in a graded series of ethanol, and processed for embedding in Epon. Thick sections (1 μm) were stained with 1% toluidine blue and photographed. The axons in cross sections of the ventral roots were circled on the photographs, and the sizes were determined with an Image 1 Analyzer (Universal Imaging). The measurements were grouped by size and analyzed by an ANOVA test.
RESULTS
Transgenic lines expressing wild-type and FALS-associated G37R mutant human SOD1
Transgenic mice were created by using a 10 kb construct carrying a human SOD1 cDNA that is driven by a neurofilament light chain promoter system, which contains some genomic regulatory elements destined to ensure adequate levels and localization of expression (Fig.1,1) (Charron et al., 1995). Two different constructs were used, one containing a normal human SOD1 cDNA and one carrying a FALS-associated mutation G37R (GGA→AGA) (Pramatarova et al., 1995). Founder animals were identified by Southern blotting of tail DNA; three lines were established, one carrying the normal cDNA (SOD1-4305) and two lines carrying the mutant cDNA (G37R-3156 and G37R-4012).
Fig. 1.

1, Diagram of the construct used to generate the transgenic animals. The pGCHNFL vector contains the promoter region, some enhancing intronic sequences, and the 3′-untranslated region (3′-UTR) of the neurofilament light chain gene. 2, RT-PCR and Western blots show expression of the human transgene in nervous tissue both at the mRNA and protein levels. 2A, Multiple tissue RT-PCR showing expression of the human SOD1 cDNA. 2B, Same as2A but using primers amplifying the GAPDH cDNA.2C, Western blots probed with an anti-SOD1 antibody recognizing both human and mouse proteins, showing expression of the hSOD1 in nervous tissue exclusively. mSOD1, Mouse SOD1;hSOD1, human SOD1.
For each line, RNA was extracted from brain, spinal cord, heart, liver, kidney, spleen, muscle, and testis and treated with DNase to avoid DNA contamination and false positives. RT-PCR showed expression of the transgene in brain and spinal cord (Fig. 1,2A), as expected. The signal detected in certain other tissues like the heart, kidney, and spleen may result from transcription caused by the integration site of the transgene.
Expression of the transgene at the protein level was assessed by Western blotting of total soluble protein from brain, spinal cord, heart, liver, kidney, spleen, muscle, and testis. The blots were probed with a polyclonal antibody (Biodesign) recognizing both human (hSOD1) and mouse (mSOD1) proteins. All three lines show expression of the hSOD1 exclusively in brain and spinal cord, but not in any other tissue (Fig. 1,2C). The hSOD1 concentration is highest in the spinal cord where the large neurons are concentrated.
To quantify the amount of hSOD1 in the spinal cords, we ran serial dilutions of each brain and spinal cord extract as well as known amounts of commercial hSOD1 protein. The amount of human protein was estimated by densitometry (Table 1). For the SOD1-4305 line (normal SOD1 control) the amount of protein in the total brain extract was too small to be quantified reliably by our method.
Table 1.
hSOD1 concentrations in total brain and spinal cord of transgenic animals
| Transgenic line | Brain (ng/100 μg) | Spinal cord (ng/100 μg) |
|---|---|---|
| G37R-3156 | 11.7 ± 1.1 (n = 3) | 17.9 ± 6.4 (n = 4) |
| G37R-4012 | 4.6 ± 1.3 (n = 3) | 24.8 ± 4.6 (n = 3) |
| SOD1-4305 | NA | 6.35 ± 0.7 (n = 2) |
Localization and quantification of transgene expression: high levels of human SOD1 expressed in large motor neurons
To determine the cell-specific expression of hSOD1 in the transgenic mice, we raised antisera to the human SOD1 protein by using a peptide displaying low homology with the mouse SOD1 protein; i.e., 10 of 20 amino acids are human-specific. Serum was harvested and affinity-purified against the peptide that was used for the immunization. The preimmune serum and the affinity-purified hSOD1 antibody were tested on immunoblots. The hSOD1 antibody detects the hSOD1 (from total cell extracts and from the commercially purified) and does not cross-react with the mouse SOD1 (Fig.2). Preabsorption of the hSOD1 antibody with the peptide abolishes the hSOD1 reactivity.
Fig. 2.

Characterization of the human SOD1 antibody.A, Western blot probed with preimmune serum.B, Western blot probed with the human SOD1 antibody, which specifically recognizes the hSOD1, but not the mSOD1, protein.C, Western blot probed with peptide-preabsorbed human SOD1 antibody. h/mSOD1, Protein extract from spinal cord of transgenic animal overexpressing hSOD1; mSOD1, protein extract from spinal cord of nontransgenic animal;hSOD1, human SOD1 (Sigma).
Immunocytochemistry was performed on samples from each transgenic line as well as on normal nontransgenic animals. Using the human-specific SOD1 antibody, we were able to show that the hSOD1 protein is expressed specifically in a subset of large neurons concentrated in the ventral horns of the spinal cords of transgenic animals (Fig.3).
Fig. 3.
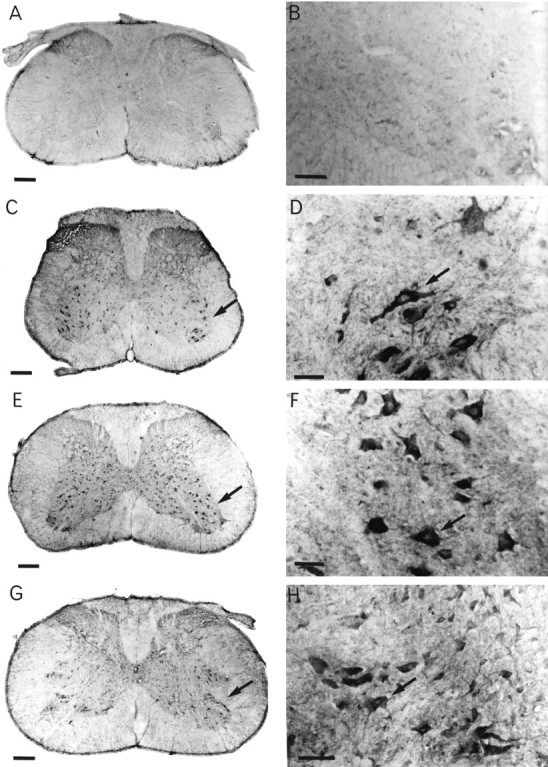
Human SOD1 is expressed in large neurons in the ventral horn of the spinal cord in transgenic animals. A, B, Normal nontransgenic animal. C, D, Transgenic line G37R-3156. E, F, Transgenic line G37R-4012.G, H, Transgenic line SOD1-4305. Scale bars: A, C, E, G, 200 μm; B, 100 μm; D, F, H, 50 μm.
Because only a proportion of cells in the spinal cord expresses hSOD1, we undertook to determine specifically the amount of hSOD1 per motor neuron. We morphometrically estimated what percentage of the total surface area of the spinal cord is occupied by hSOD1-positive cells. Depending on the different lines, between ∼2 and 5% of the surface area of the spinal cord stained positively for hSOD1 (Table2).
Table 2.
Estimated hSOD1 concentration in the hSOD1-positive cells in the spinal cords of transgenic animals
| Line | hSOD1 in whole spinal cord2-a | Positive cells per spinal cord2-b section (%) | hSOD1 per positive cell2-c | Human/mouse SOD1 ratio | Age of onset of disease |
|---|---|---|---|---|---|
| G37R-3156 | 17.9 ng/100 μg | 2.2 | 8.1 ng/μg | 4.3 | No disease |
| G37R-4012 | 24.8 ng/100 μg | 4.7 | 5.3 ng/μg | 2.8 | No disease |
| SOD1-4305 | 6.3 ng/100 μg | 2.7 | 2.3 ng/μg | 1.2 | No disease |
| G37R29 | NA | 1002-d | NA | 4–52-c | 6–8 months2-d |
| G93A-G1 | NA | É1002-e | 4.1 ng/μg2-e | 2.22-d | 5 months2-e |
The human SOD1 concentration in mouse spinal cord was estimated from Western blots.
Surface area occupied by cells staining positive for human SOD1 in mouse spinal cord was estimated from immunostained sections of spinal cord of transgenic animals, using the LAP2-10AP antibody.
The human SOD1 concentration per total protein in the hSOD1-positive cell in the spinal cord of transgenic animals as estimated from the results from Western blotsa and from immunocytochemistryb.
Data as published (Wong et al., 1995).
Data as published (Gurney et al., 1994).
Total hSOD1 in spinal cords was quantified by Western blotting (see Table 1), and a “corrected” concentration of hSOD1 in those specific hSOD1-positive cells in the spinal cords of the transgenic animals was determined (Table 2), ranging from 2.3 to 8.1 ng/μg of total protein. These amounts of human SOD1 are equivalent to a ratio of 4.3 of human-to-mouse SOD1 in the highest expressing line. This level is comparable with the values reported by other groups in transgenic animals expressing the G37R or G93A mutations in a constitutive manner and that show an ALS-like phenotype (Table3) (Gurney et al., 1994; Wong et al., 1995).
Table 3.
Summary of previously published SOD1 transgenic models
| Transgenic line | h/m SOD1 ratio | Onset of disease | Reference |
|---|---|---|---|
| h-SOD1 | 1–10 | No disease | (Epstein et al., 1987) |
| (Ceballos-Picot et al., 1991) | |||
| (Gurney et al., 1994) | |||
| (Wong et al., 1995) | |||
| (Bruijn et al., 1997) | |||
| m-SOD1 null | No SOD1 | No disease | (Reaume et al., 1996) |
| h-G37R | 10–12 | 3.5–4 months | (Wong et al., 1995) |
| 4–5 | 6–8 months | (Wong et al., 1995) | |
| h-G85R | 1 | 8–10 months | (Bruijn et al., 1997) |
| 0.2 | 12–14 months | (Bruijn et al., 1997) | |
| m-G86R | 1 | 3–4 months | (Ripps et al., 1995) |
| m-G86R | Restricted to astocytes | No disease | (Gong et al., 2000) |
| h-G93A | 4 | 4–6 months | (Gurney et al., 1994) |
h, Human SOD1; m, mouse SOD1 transgene.
Absence of motor degeneration in G37R transgenic animals
The survival of the transgenic animals was similar in all lines, with 97% of the mice from line G37R-3156 (n = 41) living for >1 year, 96% of line G37R-4012 (n = 46) and 82% of line SOD1-4305 (n = 11). Furthermore, 85% of lines G37R-3156 and G37R-4012 and 73% of the mice from line SOD1-4305 survived 1.5 years or older.
The transgenic animals were examined on a weekly basis for general health (grooming, general activity) but showed no apparent motor dysfunction at up to 1.5 years of age. More specifically, there was no significant difference in performance between the transgenic and nontransgenic animals subjected to grip strength tests (Fig.4). We have compared toluidine blue-stained spinal cord sections from 1-year-old animals and found no significant differences among the different lines (data not shown). Because our animals did not show any gross motor dysfunction, they also were tested for axonal degeneration, often an early sign of neuronal degeneration. One well documented approach is to quantify the size and number of the motor neuron axons in the ventral root of the spinal cord of the mice. Animals aged between 11 and 12 months (“middle” aged) were used to avoid confusion with normal age-related neurodegeneration. Both L4 and L5 ventral roots were examined and showed no apparent degeneration in our transgenic animals (Fig.5, 1). The size and number of axons were determined, and no significant differences were seen between the transgenic and nontransgenic animals (Fig.5B).
Fig. 4.

Limb grip strength measurement. There is no significant difference in the performance of transgenic versus nontransgenic animals.
Fig. 5.
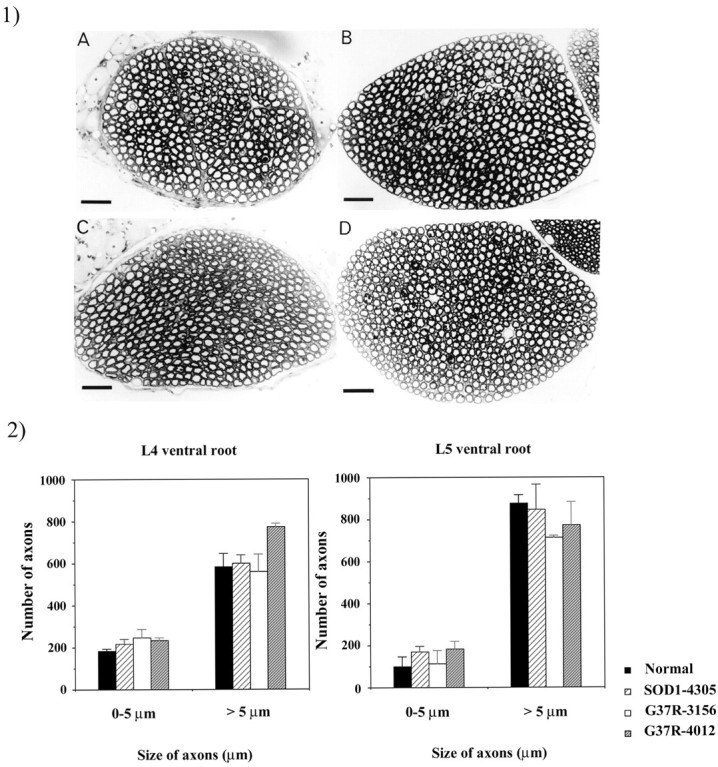
Size of motor neurons in L4 and L5 ventral roots.1, Photographs of ventral root axons of 1-year-old normal and transgenic animals. 1A, Nontransgenic animal.1B, Line SOD1-4305. 1C, Line G37R-3156.1D, Line G37R-4012. Scale bar, 100 μm.2, Axon counts in L4 and L5 ventral roots of 1-year-old normal and transgenic animals. 1, 2, No significant differences are seen between the normal and transgenic animals.
DISCUSSION
The neurofilament light chain promoter allowed us to express high levels of human SOD1 specifically in large motor neurons of the spinal cord of transgenic animals. Surprisingly, these animals did not develop a neurodegenerative disease. Other groups have shown previously that the same G37R mutant, when expressed ubiquitously under the SOD1 promoter in mice, leads to a disorder characterized by weakness of the limbs, muscle wasting, and paralysis, with pathological changes reminiscent of human ALS (Wong et al., 1995). At the ultrastructural level motor neurons in the ventral horn of the spinal cord as well as in the brainstem were degenerating with cytoplasmic vacuoles in dendrites, proximal axons, and perikarya, containing degenerating and swollen mitochondria. The age at onset of disease in these mice appeared to be dependent on the amount of mutant protein, often expressed as the human-to-mouse SOD1 ratio, and occurred as early as 3.5 months of age for the high expressors, with ratios of 12:1 in the spinal cord; most importantly, for lines expressing levels similar to our transgenics (4–5:1), the disease developed at 6–8 months of age, with a significant loss of motor neurons by 20 weeks of age. Our model, which expressed similar high levels of human mutant protein (4.3:1 ratio) in motor neurons, the cells most affected in humans and mice, appeared healthy at >18 months of age. The absence of phenotype in our animals expressing the mutant protein in only a subset of neurons suggests that the localization of the transgene expression is important for neurodegeneration.
There are two possible explanations for the absence of phenotype in our animals. The first is that insufficient levels of transgene expression were achieved. It is well established that the levels of mutant transgene expression are crucial for determining the age at onset of the disease in transgenic animals. This raises the concern that our animals might not be expressing high enough levels of mutant protein. Although we estimated that the mutant human SOD1 levels in individual motor neurons of the spinal cord of our animals were comparable with the levels achieved in the other animal models, which did develop motor dysfunction, our methods of dosage may have overestimated the human SOD1 levels, or the published ratios may have been underestimated. When the SOD1 gene is regulated by its own promoter, the protein is expressed at high levels throughout the entire life of the animals, whereas the NFL promoter is downregulated slightly later in life (Kuchel et al., 1997). The damage caused by mutant SOD1 is thought to be cumulative, but it is probably also important to have high levels of the defective protein later in life when the damage starts to be visible. In our model this should not be the cause of the absence of phenotype because the G37R mutant, when expressed at a 4–5:1 of human-to-mouse ratio, induces the disease very early in the life of transgenic animals, long before the NFL age-related downregulation occurs. Therefore, our mice should have achieved the desired levels of expression at the appropriate time for the disease to develop.
Another important point to consider is that there might be a threshold of human mutant protein for the disease to appear, which varies according to the mutation. For example, there seems to be a lower threshold for the G85R mutant (0.2–1:1 human-to-mouse ratio) and a higher one for G37R mutant (4–5:1 ratio). This threshold also could differ according to the localization of the expression (ubiquitous vs neuronal), which might result in the need to reach a higher threshold for neuron-specific expression to provoke the disease in a similar time frame.
The second explanation for the absence of phenotype in our mice is that the presence of the mutant protein in motor neurons might not be sufficient to lead to the development of a neurodegenerative disease. Widespread expression of the mutant SOD1 might be necessary for the development of the disease. One possible pathogenic model that requires extra-neuronal expression of the mutant protein is suggested by the G85R mouse model. In these animals the first signs of damage were shown in the astrocytes (Bruijn et al., 1997). In parallel with the neuronal loss and astrocytic inclusions, at end stage the G85R mice showed a 50% decrease in the astrocyte-specific glutamate transporter GLT-1, which is the major glutamate transporter in the spinal cord. This model suggests that astrocyte expression with loss of astrocytic function and perhaps loss of glutamate uptake may be important in disease development. The recent report by Gong et al. (2000), who found that astrocytic expression of mutant SOD1 leads to astrocytic changes, supports the hypothesis that these cells may be involved in the motor neuron cell death that is seen in this model of ALS.
The importance of glutamate toxicity was suggested by experiments of nonselective blockage of glutamate transporters, which lead to pathological abnormalities reminiscent of neurodegenerative diseases with cellular swellings and vacuolation of mitochondria and endoplasmic reticulum (Choi, 1988; Ikonomidou et al., 1996; Rothstein et al., 1996). A selective decrease in glutamate transporters has been observed in multiple animal models of motor neuron disease (Mnd, G85R). At the ultrastructural level the pathology seen in the animal models of ALS (G37R, G93A) is very similar to the neurodegeneration induced by excitotoxins (agonists of glutamate transporters) (Ikonomidou et al., 1996). More significantly, a decrease in glutamate transporter GLT-1 and an increase in extracellular glutamate in the CSF were shown in ALS patients (Rothstein et al., 1990, 1992, 1995). These data suggest that glutamate, via the astrocytes, might induce/propagate/aggravate the motor neuron-specific degeneration seen in FALS and in the animal models of the disease. This mechanism could explain our observation that transgenic animals expressing high levels of mutant human SOD1 in motor neurons do not develop the disease. Additional experiments need to be performed to test this hypothesis.
We have generated transgenic animals carrying a human SOD1 gene with the G37R mutation associated with FALS and an ALS-like phenotype in transgenic mice. The neurofilament light chain promoter was used successfully to express specifically the mutant protein in neurons, the cells that are depleted in ALS patients and in animal models of ALS. Our transgenic animals express high levels of the mutant human SOD1 protein in nervous tissue, especially in the large neurons of the spinal cord, which degenerate in ALS but remain healthy at up to 1.5 years of age, suggesting that mutant SOD1 expression in other cell types may contribute to motor neuron loss that is caused by mutant SOD1 toxicity in mice.
Footnotes
This work was supported by the Muscular Dystrophy Association, the Muscular Dystrophy Association of Canada, the Medical Research Council of Canada, and the Fonds de Recherche en Santé du Québec. Scholarships to A.P. were provided by the Fonds pour la Formation de Chercheurs et l'Aide à la Recherche and the McGill University Faculty of Medicine. The production of transgenic mice was supported by the Canadian Network of Centres of Excellence. We thank Dr. Jean Pierre Julien for providing the pGCHNFL vector and Dr. Philip Wong for the G37R29 transgenic line that was used as a positive control in Figure 2. We are especially grateful to Dr. Diane Merry for helping with the grip strength measurements.
Correspondence should be addressed to Guy A. Rouleau, Centre for Research in Neuroscience, Montreal General Hospital, 1650 Cedar Avenue, Montreal, Quebec, H3G 1A4, Canada. E-mail:mi32@musica.mcgill.ca.
A. Pramatarova's present address: Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892.
REFERENCES
- 1.Borchelt DR, Lee MK, Slunt HS, Guarnieri M, Xu ZS, Wong PC, Brown RH, Jr, Price DL, Sisodia SS, Cleveland DW. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc Natl Acad Sci USA. 1994;91:8292–8296. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brinster RL, Chen HY, Trumbauer M, Senear AW, Warren R, Palmiter RD. Somatic expression of herpes thymidine kinase in mice following injection of a fusion gene into eggs. Cell. 1981;27:223–231. doi: 10.1016/0092-8674(81)90376-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 4.Ceballos-Picot I, Nicole A, Briand P, Grimber G, Delacourte A, Defossez A, Javoy-Agid F, Lafon M, Blouin JL, Sinet PM. Neuronal-specific expression of human copper/zinc superoxide dismutase gene in transgenic mice: animal model of gene dosage effects in Down's syndrome. Brain Res. 1991;552:198–214. doi: 10.1016/0006-8993(91)90084-9. [DOI] [PubMed] [Google Scholar]
- 5.Charron G, Guy LG, Bazinet M, Julien JP. Multiple neuron-specific enhancers in the gene coding for the human neurofilament light chain. J Biol Chem. 1995;270:30604–30610. doi: 10.1074/jbc.270.51.30604. [DOI] [PubMed] [Google Scholar]
- 6.Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 7.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 8.Collard JF, Julien JP. A simple test to monitor the motor dysfunction in a transgenic mouse model of amyotrophic lateral sclerosis. J Psychiatry Neurosci. 1995;20:80–86. [PMC free article] [PubMed] [Google Scholar]
- 9.Dal Canto MC, Gurney ME. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu/Zn SOD, and in mice overexpressing wild-type human SOD: a model of familial amyotrophic lateral sclerosis (FALS). Brain Res. 1995;676:25–40. doi: 10.1016/0006-8993(95)00063-v. [DOI] [PubMed] [Google Scholar]
- 10.Epstein CJ, Avraham KB, Lovett M, Smith S, Elroy-Stein O, Rotman G, Bry C, Groner Y. Transgenic mice with increased Cu/Zn superoxide dismutase activity: animal model of dosage effects in Down syndrome. Proc Natl Acad Sci USA. 1987;84:8044–8048. doi: 10.1073/pnas.84.22.8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- 12.Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20:660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu/Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 14.Hyman Friedman HC, Jelsma TN, Bray GM, Aguayo AJ. A distinct pattern of trophic factor expression in myelin-deficient nerves of Trembler mice: implications for trophic support by Schwann cells. J Neurosci. 1996;16:5344–5350. doi: 10.1523/JNEUROSCI.16-17-05344.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikonomidou C, Qin Qin Y, Labruyere J, Olney JW. Motor neuron degeneration induced by excitotoxin agonists has features in common with those seen in the SOD-1 transgenic mouse model of amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1996;55:211–224. doi: 10.1097/00005072-199602000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Kuchel GA, Poon T, Irshad K, Richard C, Julien JP, Cowen T. Decreased neurofilament gene expression is an index of selective axonal hypotrophy in aging. NeuroReport. 1997;8:799–805. doi: 10.1097/00001756-199702100-00044. [DOI] [PubMed] [Google Scholar]
- 17.Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu/Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci USA. 1996;93:5472–5477. doi: 10.1073/pnas.93.11.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mulder DW, Kurland LT, Offord KP, Beard CM. Familial adult motor neuron disease: amyotrophic lateral sclerosis. Neurology. 1986;36:511–517. doi: 10.1212/wnl.36.4.511. [DOI] [PubMed] [Google Scholar]
- 19.Pramatarova A, Figlewicz DA, Krizus A, Han FY, Ceballos-Picot I, Nicole A, Dib M, Meininger V, Brown RH, Rouleau GA. Identification of new mutations in the Cu/Zn superoxide dismutase gene of patients with familial amyotrophic lateral sclerosis. Am J Hum Genet. 1995;56:592–596. [PMC free article] [PubMed] [Google Scholar]
- 20.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr, Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 21.Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzy RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Hanes H, Rouleau GA, Gusella JS, Horvitz HR, Brown RH. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 23.Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1990;28:18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- 24.Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- 25.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 26.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knock-out of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 27.Tandan R, Bradley WG. Amyotrophic lateral sclerosis. Part 1. Clinical features, pathology, and ethical issues in management. Ann Neurol. 1985;18:271–280. doi: 10.1002/ana.410180302. [DOI] [PubMed] [Google Scholar]
- 28.Tomic M, Sunjevaric I, Savtchenko ES, Blumenberg M. A rapid and simple method for introducing specific mutations into any position of DNA leaving all other positions unaltered. Nucleic Acids Res. 1990;18:1656. doi: 10.1093/nar/18.6.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]