

Abstract
Delta power, a measure of EEG activity in the 1–4 Hz range, in slow-wave sleep (SWS) is in a quantitative and predictive relationship with prior wakefulness. Thus, sleep loss evokes a proportional increase in delta power, and excess sleep a decrease. Therefore, delta power is thought to reflect SWS need and its underlying homeostatically regulated recovery process. The neurophysiological substrate of this process is unknown and forward genetics might help elucidate the nature of what is depleted during wakefulness and recovered during SWS. We applied a mathematical method that quantifies the relationship between the sleep–wake distribution and delta power to sleep data of six inbred mouse strains. The results demonstrated that the rate at which SWS need accumulated varied greatly with genotype. This conclusion was confirmed in a “dose–response” study of sleep loss and changes in delta power; delta power strongly depended on both the duration of prior wakefulness and genotype. We followed the segregation of the rebound of delta power after sleep deprivation in 25 BXD recombinant inbred strains by quantitative trait loci (QTL) analysis. One “significant” QTL was identified on chromosome 13 that accounted for 49% of the genetic variance in this trait. Interestingly, the rate at which SWS need decreases did not vary with genotype in any of the 31 inbred strains studied. These results demonstrate, for the first time, that the increase of SWS need is under a strong genetic control, and they provide a basis for identifying genes underlying SWS homeostasis.
Keywords: EEG delta power, slow-wave activity, sleep deprivation, homeostatic regulation of non-REM sleep, simulation of Process S, BXD recombinant-inbred mouse strains, QTL, Dps1, Dps2, Dps3, forward genetics
Slow oscillations in the delta frequency range (1–4 Hz) are characteristic of the EEG during slow-wave sleep (SWS) (i.e., non-REM sleep in humans). Delta oscillations reflect synchronized burst-pause firing patterns of hyperpolarized thalamocortical and corticothalamic neurons (Steriade et al., 1993; McCormick and Bal, 1997; Steriade, 1999). Activity in the delta frequency range can be quantified as delta power by Fourier analysis. Delta power is negatively correlated with the response to arousing stimuli (Neckelmann and Ursin, 1993) and SWS fragmentation (Franken et al., 1991a) and thus can be seen as a measure of SWS intensity. Delta power is also in a quantitative and predictive relationship with prior sleep and wakefulness in mammals, including humans. Sleep loss evokes an increase in delta power during subsequent SWS that is proportional to the loss (Tobler and Borbély, 1986;Dijk et al., 1987), excess sleep results in an attenuation of delta power (Werth et al., 1996), and delta power decreases over the course of a sleep period, independent of the circadian phase at which sleep is initiated (Dijk and Czeisler, 1995). These and other observations have been interpreted as evidence that SWS is a restorative and homeostatically regulated behavior and that delta power reflects the need for SWS (Borbély, 1982; Daan et al., 1984). The dynamics of this homeostatically regulated process, referred to as Process S (Borbély, 1982), have been studied extensively, and mathematical simulations that quantify the relationship between the sleep–wake distribution and delta power predicted the time course of delta power remarkably well (Franken et al., 1991b; Achermann et al., 1993). However, the neurophysiological substrate of what is restored by SWS (and what is depleted in its absence) is unknown.
Several features of the normal EEG are among the most heritable traits in humans (Beijsterveldt and Boomsma, 1994); however, little progress has been made in identifying the underlying genes. Only for a low-voltage waking EEG variant has linkage with a discrete region of chromosome 20 been established (Anokhin et al., 1992). By comparing several inbred strains of mice, we have identified several EEG features that are under strong genetic control (Franken et al., 1998). For one of those, the frequency of the theta rhythm, we established a single gene mode of inheritance (Tafti et al., 1998) and recently identified its genomic localization (our unpublished results). We also observed strain differences in the rebound of delta power after a sleep deprivation (Franken et al., 1999). Following the segregation of this trait in recombinant offspring should allow identification of genomic regions containing genes that modify the accumulation of a need for SWS. Ultimately, the identification of such genes will yield important information on the neurophysiological substrate of Process S.
We used a computational method that quantifies the relationship between the changes observed in delta power and the sleep–wake distribution in six inbred strains of mice. This method separates the effects of sleep–wake patterns, which varied greatly between strains, from the effects of different dynamics of Process S on delta power. The analysis suggests genetic differences in the rate at which SWS need accumulates. We verify this empirically in a “dose–response” study in which mice were subjected to sleep deprivations of varying duration. Finally, we provide a preliminary mapping of genes that modify this trait in recombinant offspring.
MATERIALS AND METHODS
The methods concerning the recording and analysis of the EEG in mice have been described in detail elsewhere (Franken et al., 1998,1999). The experimental protocols were approved by the local veterinary office (Office Vétérinaire Cantonal de Genève) and the ethical committee of the University of Geneva. Experimental animals were adult male mice obtained from Jackson Laboratory (Bar Harbor, ME), with the exception of 129/OLA mice in experiment 1 that were bred locally. All mice were individually housed in an experimental room under a 12 hr light/dark cycle (lights on at 8:00 A.M.). Food and water were available ad libitum. Animals were kept under these conditions for at least 18 d before the experiment. EEG and electromyogram (EMG) electrodes were implanted under deep pentobarbital anesthesia. Mice were allowed 10–14 d of recovery from surgery and habituation before the experiments.
The EEG and EMG signals were recorded continuously for the entire duration of the experiments. Both signals were amplified, filtered, and analog-to-digital converted. The EEG signal was subjected to Fast-Fourier Transform yielding power spectra between 0 and 25 Hz using a 4 sec window. The behavior in each of these 4 sec epochs was classified as SWS, paradoxical sleep (PS), or wakefulness by visual inspection of the EEG and EMG signals. The present analyses concern the mean power in the delta band (1–4 Hz), referred to as delta power, and its relation to the sleep–wake distribution.
Experiment 1: simulation of Process S. Data obtained in six inbred strains [AKR/J (AK), BALB/cByJ (C), C57BL/6J (B6), C57BR/cdJ (BR), DBA/2J (D2), and 129/Ola (129); n = 7 per strain] contributed to this analysis. The age at the first recording day ranged from 71 to 87 d, and body-weight ranged from 24 to 35 gm. EEG and EMG signals were recorded continuously for two consecutive 24 hr periods, starting at lights on (8:00 A.M.). Day one was considered red baseline (BSL). On day two, starting at lights on, mice were sleep-deprived (SD) for 6 hr by handling. The remaining 18 hr were considered recovery (REC). Data on strain differences in the amount and distribution of the behavioral states and EEG spectra obtained in these mice have been published elsewhere (Franken et al., 1998, 1999).
The simulation procedure was similar to the approach used previously in the rat (Franken et al., 1991b, 1993, 1995), with the exception that for the present analyses, absolute delta power values were simulated instead of linearly transformed values. This required the estimation of the asymptotes of the two exponential functions from the data, whereas in the previous simulations, these asymptotes were arbitrarily set to 0 and 1. Furthermore, in the present approach, the initial value (S0) was derived from the sleep–wake distribution and the time constants and thus was no longer a free-parameter (see below).
Within each animal, on the basis of the sequence of the 4 sec behavioral state scores that constitute the 48 hr recording, the time course of Process S was calculated iteratively by assuming that it increases according to an exponential saturating function (Eq.1) during epochs scored as wakefulness or PS and decreases according to an exponential function (Eq. 2) during epochs scored as SWS (see Fig. 1a):
| Equation 1 |
| Equation 2 |
St+1 andSt are values of S for consecutive 4 sec epochs (t = 0–43,200 epochs), τi is the time constant of the increasing exponential saturating function with an upper asymptote (UA), τdis the time constant of the decreasing exponential function with a lower asymptote (LA), and dt is the time step of the iteration (4 sec). Within each mouse, the two asymptotes were derived from the distribution of delta power values of all 4 sec epochs of both recording days scored as either SWS or PS (see Fig.1b). Because PS is characterized by the absence of synchronized EEG activity in the delta range, the intersection of the relative delta power distributions in PS and SWS was chosen as theLA. The choice of the UA is based on the assumption that as a response to prolonged wakefulness, the median power in a given SWS episode cannot exceed the 99% level of the distribution of 4 sec values. The initial value of S at time 0 (S0 at lights on of baseline) with which the iteration started was determined by assuming that the baseline represented a steady state, i.e., the value obtained at the end of the 24 hr baseline period equals the value reached at the end of the day preceding baseline. For a given combination of time constants, the value of S reached after 24 hr is independent ofS0 (see Fig. 1c). Therefore, the simulation was first started with an arbitraryS0 value (i.e., the mean SWS delta power in baseline). The value reached after 24 hr baseline was taken as the “real” value for S0, and the simulation was then restarted for the entire 48 hr recording period. Thus, only the two time constants were free parameters, and these were optimized in the simulation.
Fig. 1.

Illustration of the assumptions and parameter estimation of the simulation of Process S. a, The time course of S was calculated iteratively on the basis of the sequence of the 4 sec scores of the behavioral states wakefulness (W), slow-wave sleep (SWS), and paradoxical sleep (PS) and was assumed to increase during W and PS and to decrease during SWS, according to Equations 1and 2 (see Materials and Methods), respectively. S varies between an upper (UA) and lower asymptote (LA;dashed lines). b, These asymptotes were derived from the relative frequency distribution of delta power for 4 sec epochs scored as PS or SWS during the 48 hr recording. The 99% level of the SWS distribution (gray area) was chosen as the UA; the intercept of the PS and SWS distributions was chosen as the LA. c, As the initial S value (S0) at light onset of the baseline day, the value ofS obtained at the end of the baseline dark period (black horizontal bar on top) was used. This value was not affected by the values with which the iteration started, illustrated by starting at either the UA (curve 1) or the LA (curve 2). Curve 3starts at the S0 used in the final simulation for this mouse.Black bars at the bottom mark SWS episodes during which S decreases. d, Contour plot of the mean square of differences (DIF2) between simulated and empirical data as a function of the time constants for the increase (Ti) and the decrease (Td) of Process S. Numbers that label the contour lines indicate the number of times the DIF2 was larger than the least DIF2 (i.e., 44 μV4) obtained at Ti = 6.9 and Td = 1.5 hr.e, Final simulation of Process S (solid line) for one individual mouse of the C strain. The estimation of the parameters for this animal is illustrated inb–d. Process S was fitted to the absolute median values of delta power (gray circles) reached in SWS episodes of >5 min. Dark horizontal bars on top indicate the 12 hr dark periods.
The values for Process S obtained by this approach were compared directly with the delta power values observed in sustained SWS episodes (>5 min). The median delta power of all 4 sec epochs scored as SWS in a given SWS episode was taken to best represent delta power reached at the time of episode midpoint. The number of SWS episodes selected did not differ among strains [one-way ANOVA factor “strain”:p = 0.2; AK (84 ± 4), C (97 ± 3), B6 (91 ± 6), Br (77 ± 3), D2 (79 ± 5), 129 (81 ± 8); mean ± SEM; n = 7 per strain]. The goodness of fit between simulated (i.e., the level of Process S reached at each SWS episode midpoint) and empirical data was assessed by calculating the mean of the square of the differences and Pearson's correlation coefficient (r). The fit was optimized by minimizing the mean square of the differences for a range of τi (1–25 hr, step-size 0.12 hr) and τd (0.1–5 hr, step-size 0.025 hr) values, i.e., the simulation was run for 40,000 different combinations of time constants for each mouse. Within these ranges of time constants and the constraints of the model one unique solution was obtained for each mouse (see Fig. 1d). The time constants with which the best fit was obtained were used to assess differences between strains.
Further statistical comparisons were made by comparing simulated and empirical delta power at specific times of the experiment after both values were expressed as a percentage of the individual mean delta power in SWS over the last 4 hr of the baseline light period. This transformation was conducted to correct for individual differences in the absolute power. Mean values of S and delta power were calculated over consecutive 45 min intervals within each individual and then among the seven individuals of each strain. Intervals in which fewer than four animals contributed to the mean were omitted from the figure and the t tests evaluating differences between empirical and simulated data. To follow Process S through the entire experiment including the SD, 15 min mean values of S were calculated regardless of behavioral state.
Experiment 2: a sleep deprivation dose–response curve. The outcome of experiment 1 was verified by subjecting AK and D2 mice to SDs of varying length (n = 6 per strain; body weight, 29.3 ± 1.1 gm; range, 23.4–35.4 gm; age, 91 d at recording day 1). EEG and EMG signals were recorded continuously for nine consecutive 24 hr periods, starting at lights on (8:00 A.M.). The first 2 d were considered baseline (baselines 1 and 2). On days 3, 4, and 5, starting 7 hr after lights on (3:00 P.M.), mice were sleep deprived by handling for 70, 35, and 140 min, respectively. Day 7 was again considered baseline (baseline 3). On day 8, mice were sleep deprived for 9 hr starting at lights on (8:00 A.M.). After noticing that the first 35 min SD on day 4 actually resulted in 71 min of wakefulness, we repeated this SD on day 9, the day after the 9 hr SD, expecting that the residual increased sleep drive from the long SD would shorten sleep latency. Sleep latency, SD duration, and SWS delta power were significantly attenuated compared with day 4 (analysis not shown), and the two SDs were therefore treated as two distinct “doses.”
Including the 6 hr SD of experiment 1, six SD doses were obtained per strain. The SDs were scheduled to end within the third quarter of the light period so that the subsequent rebound in delta power was not confounded by the high and changing values of delta power present in the initial half of the baseline light period or by possible circadian factors. We point out here that the 6 hr and 9 hr SDs were initiated at light onset when Process S (and delta power) was higher (∼153%; i.e., S0 in Table 1) compared with the shorter SDs (∼103%) starting 7 hr later. With the simulation, it can be estimated that delta power after 6 hr SD would have been 13 and 26% (for AK and D2, respectively) lower, and after 9 hr SD would have been 6 and 15% lower, if these SDs had been initiated 7 hr later.
Table 1.
Parameters describing the time course of Process S
| τi (hr) | τd(hr) | UA (%) | LA (%) | S0 (%) | DIF2(%2) | r | |
|---|---|---|---|---|---|---|---|
| AK | 5.3 (0.3) | 1.9 (0.1) | 335 (7) | 47 (3) | 148 (4) | 408 (85) | 0.92 |
| C | 7.6 (1.1) | 1.6 (0.2) | 290 (5) | 55 (3) | 109 (6) | 245 (35) | 0.90 |
| B6 | 8.0 (0.5) | 1.8 (0.2) | 303 (13) | 54 (2) | 160 (16) | 194 (29) | 0.92 |
| Br | 7.7 (0.8) | 1.5 (0.1) | 357 (18) | 65 (2) | 178 (15) | 379 (62) | 0.93 |
| D2 | 12.6 (1.6) | 1.8 (0.3) | 293 (15) | 70 (2) | 157 (8) | 282 (103) | 0.88 |
| 129 | 8.8 (1.4) | 1.6 (0.2) | 321 (10) | 60 (3) | 155 (7) | 283 (51) | 0.89 |
| p | 0.003 | 0.7 | 0.002 | <0.0001 | 0.0006 | 0.3 | 0.4 |
The time course of Process S that yielded the best fit with delta power was defined by the time constant for the increase τi with upper asymptote UA, and the time constant for the decrease τd with lower asymptote LA. τi, UA, LA, and S0, the level of S at time 0, varied with genotype [mean (SEM); p values were obtained with a one-way ANOVA factor strain; Tukey's range test, τi: D2 = 129 = B6 = Br > 129 = B6 = Br = C = AK; UA: Br = AK = 129 > AK = 129 = B6 = D2 = C; LA: D2 = Br = 129 > Br = 129 = C = B6 > C = B6 = AK; S0: Br = B6 = D2 = 129 = AK > C; p < 0.05; n = 7 per strain]. UA, LA, S0, and DIF2 are all expressed as a percentage of the mean SWS delta power in last 4 hr of the baseline light period. Individual r values were Fisher-Z transformed before averaging.
Some mice were spontaneously awake before the SD. This time and the time it took to initiate sleep after the end of the SD (i.e., sleep latency) were added to the total time awake in the dose–response curve. Therefore the SD durations deviated from the intended durations. Nevertheless, for convenience, the duration for which the animals were handled will be used to indicate SD dose.
The response to the SDs was measured by averaging delta power over the first 225 4 sec epochs scored as SWS after the onset of recovery sleep. All values were contrasted to the prevailing level of SWS delta power in the last 4 hr of the baseline light periods (=100%) within each individual mouse. For the SDs on days 3, 4, and 5, mean delta power over the last 4 hr of the light periods of baselines 1 and 2 was used as a reference; for the SDs on days 8 and 9, that of baseline 3; and for the 6 hr SD, that of the preceding baseline day.
During the SD, special care was taken to mark all 4 sec epochs with EEG delta waves even if the EMG did not decrease to the low levels normally associated with SWS. This was observed especially toward the end of the two longest SDs. The amount of SWS thus obtained is higher than in previously published SD studies, but because we assume that cerebral activity is important in sleep regulation, this might represent a more reliable estimate of the amount of SWS.
The effects of spontaneous awakenings of varying duration on subsequent delta power were analyzed within the last 4 hr of the three baseline light periods. During these periods, delta power reached lowest values, and no changes in its overall level were observed. Therefore, the effects of waking episodes on delta power in subsequent SWS episodes could be assessed without the confounding effects of the varying “background” levels of delta power associated with its normal decline during the initial part of a baseline light period (Franken et al., 1999) and of potential circadian factors. Waking bouts (range, 3–51 min) were selected that did not contain a single 4 sec epoch of sleep and were followed by >8 min of sleep. Waking bouts were divided into three categories according to their duration: <12 min, 12–24 min, and >24 min. The mean number of bouts in each category were for AK: 4.3, 4.7, and 4.6, respectively; for D2 they were 3.3, 3.0, and 3.0 (n = 6 per category per strain). Delta power in a sleep bout after a given waking bout was calculated over a minimum of 75 and a maximum of 225 4 sec epochs scored as SWS. Delta power values were averaged for the three categories and related to the average time awake for each category.
Decreased rates for Process S were derived from the observed data by determining the decrease of delta power over the first 1000 4 sec epochs (1.1 hr) after recovery onset after the six SDs that contributed to the dose–response curve and by following the time course of delta power in the initial 6 hr after the 6 hr SD. In both analyses, the delta power data represent means over consecutive 225 4 sec epochs (15 min) scored as SWS expressed as a percentage of the same reference discussed above. For the analyses presented in Figure 5, the delta power difference between the initial 15 min of SWS of recovery (d1) at timet1, and its value (d2) reached in the last 15 min of SWS of a 1.1 hr period at t2, was expressed as a function of d1. Botht1 andt2 represent the mean times at which the 225 SWS epochs that contributed occurred and differed in time by 0.65 ± 0.2 hr (n = 74). The time constant (τd) of this exponential function was calculated according to Equation 3 for each combination ofd1 andd2 (n = 37 per strain):
| Equation 3 |
Combinations for which eitherd2 ord1 was less than LA were omitted from the analyses (AK: n = 32; D2:n = 36). As LA, the level ofd1 at which the delta decrease is 0 according to the linear relationship betweend1 and (d1−d2) was taken (1O2% for both AK and D2; see Fig. 5). For the analyses presented in Figure 6, the parameters describing the exponential decrease in delta power after the 6 hr SD were estimated with a nonlinear fitting procedure (method, Gauss–Newton; procedure, NLIN; SAS/STAT software, SAS Institute, Cary, NC) according to Equation 4:
| Equation 4 |
where S0 is the estimated level of delta power at recovery sleep onset (t = 0) andt is the time elapsed.
Fig. 5.

The decrease in SWS delta power during recovery depends on its initial level. Left panel, As in Figure 3but now for each SD, both the mean delta power in the initial 15 min of SWS (d1) and the last 15 min of SWS of the first 1.1 hr after the SD (d2) are plotted (mean ± SEM; n = 6 per strain, except for 6 hr SD;n = 7). Right panel, Individual combinations of the delta power decrease (d1 −d2) and initial delta power (d1). Linear regression analysis demonstrated that the decrease strongly depended ond1 for both strains [AK: (d1 −d2) = 0.47 ·d1 − 48;r2 = 0.89; p< 0.0001; D2: (d1 −d2) = 0.49 ·d1 − 50;r2 = 0.87; p< 0.0001; n = 37 per strain]. These linear relationships define exponential functions with time constants of 1.3 ± 0.1 hr for AK (n = 32) and 1.1 ± 0.1 hr for D2 (n = 36; p = 0.2;t test; see Materials and Methods). Symbols are as in Figure 3.
Fig. 6.

Decrease in SWS delta power during the first 6 hr of recovery sleep after a 6 hr SD. Delta power data (mean ± SEM;n = 7 per strain) are taken from experiment 1. Each value represents the mean over consecutive 225 4 sec epochs scored as SWS and is plotted at the mean time after recovery sleep onset.Stars at the bottom indicate significant strain differences in delta power (t tests;p < 0.05). The two pairs of lines delineate the average results (±1 SEM) of the individually performed nonlinear regression analyses assuming an exponential decrease with time in delta power. The function is determined by the time constant (τd; p = 0.8), the initial value at recovery sleep onset (S0;p < 0.0001), and the asymptote (LA;p = 0.6; t tests AK vs D2;n = 7 per strain; values in Table 4). Symbols are as in Figure 3.
Experiment 3: Quantitative trait loci (QTL) analysis of delta power at sleep onset after enforced and spontaneous periods of wakefulness. Data from 25 BXD/Ty recombinant inbred (RI) strains (n = 114, 4–6 per strain; weight, 25.8 ± 0.3 gm; range, 18.3–33.7 gm; age, 94 ± 1 d; range, 67–142 d) and their two progenitor strains B6 and D2 (n = 7 per strain; same individuals as in experiment 1) contributed to this analysis. The experimental protocol was identical to that in experiment 1. As in experiment 2, the response to a 6 hr SD was determined by averaging delta power in the first 225 4 sec epochs scored as SWS after the onset of recovery sleep.
We also determined delta power in the first 225 4 sec epochs scored as SWS after the onset of the major sleep period(s) during baseline. Both the onset and the end of the major sleep periods were individually determined according to previously published criteria (Franken et al., 1999). In short, SWS time was determined over 2 hr intervals that were offset 15 min to produce a running average. This variable was then expressed as a fraction of the mean amount of SWS over the 24 hr baseline. Consecutive 15 min intervals in which this value was >1 constituted a sleep period.
QTL analysis was used to identify genomic regions containing genes that may modulate the delta power rebound after SD and the delta power at sleep onset in baseline. For this initial mapping, point-wise or nominal correlations between the strain distribution pattern (SDP) of the quantitative trait (i.e., delta power) and the genotypic SDPs of the 788 MIT markers polymorphic between B6 and D2 mice and typed in the BXD-RI strains (alleles set to 0 and 1 for the B6 and D2 genotypes, respectively) were performed. The BXD MIT marker map was kindly provided by Dr. Robert W. Williams (Williams et al., 2001). In testing multiple markers in a genome-wide scan, one will obtain significant correlations just by chance (type-1 error). To correct for this, genome-wide probability thresholds can be established using a permutation algorithm in which trait data are 10,000 times randomly reassigned among the RI strains (Churchill and Doerge, 1994). For each permutation, the single best correlation statistic is recorded generating an empirical probability distribution for the pvalues of the correlation between the randomized trait and genotype. The genome-wide significance thresholds for false positive rates are set to 0.63, 0.05, and 0.001 for “suggestive,” “significant,” and “highly significant” linkage, respectively, according to Lander and Kruglyak (1995).
In the three experiments, all main effects of factors “strain,” “SD dose,” “waking-bout duration,” and “time-of-experiment” on the various variables were assessed with ANOVA statistics. Whenever significant (p < 0.05), post hoccomparisons were performed with t tests or, when more than two levels per main factor were compared, Tukey's studentized multiple range tests to control the experiment-wise error rate. Linear and nonlinear regression analyses were used to quantify relationships between SD length or wake-bout duration and subsequent SWS delta power and between time elapsed since recovery sleep onset and delta power. All statistical analyses were performed using SAS/STAT software.
RESULTS
Simulation of Process S
An example of the time course of Process S, derived from the behavioral state sequence, and of delta power reached in SWS episodes is depicted in Figure 1e. This example shows that the model can predict the data with great accuracy in individual recordings. In addition, it illustrates that the relationship between wakefulness and delta power in undisturbed baseline conditions and during recovery from a SD are similar; i.e., the spontaneous 6 hr waking bout in baseline results in a level of delta power in subsequent SWS comparable to that after the 6 hr SD.
The overall performance of the model was good in that the main features of the time course of delta power such as the exponential decrease during the main sleep period and the increase after the 6 hr SD and subsequent rapid decline were reliably reproduced (Fig.2). This conclusion was supported by highly significant correlation coefficients (r) for all strains (Table 1), which indicates that a large portion of the variance in delta power (77–85%) can be explained by the distribution of SWS. The three-way ANOVA with factors “method”' (simulation vs empirical), “strain,” and “time” indicated that the fit did not vary according to genotype (Table 1) but that it did interact with time (factor method, p = 0.053; factors strain and time, p < 0.0001; interactions method × strain, p = 0.60; method × time, p = 0.027; strain × time,p < 0.0001). Post hoc analysis within strains identified only occasional intervals for which Process S and delta power differed (Fig. 2), but when data from the six strains were combined, a tendency was observed in both baseline and recovery that the simulated values were higher than the observed data in the first half of the dark period. During this period, mice of all strains were predominantly awake (84% of recording time), and delta power measured in the little SWS that occurred might not reliably reflect SWS need. Alternatively, the strong circadian drive for wakefulness at this time might directly modulate delta power expression.
Fig. 2.
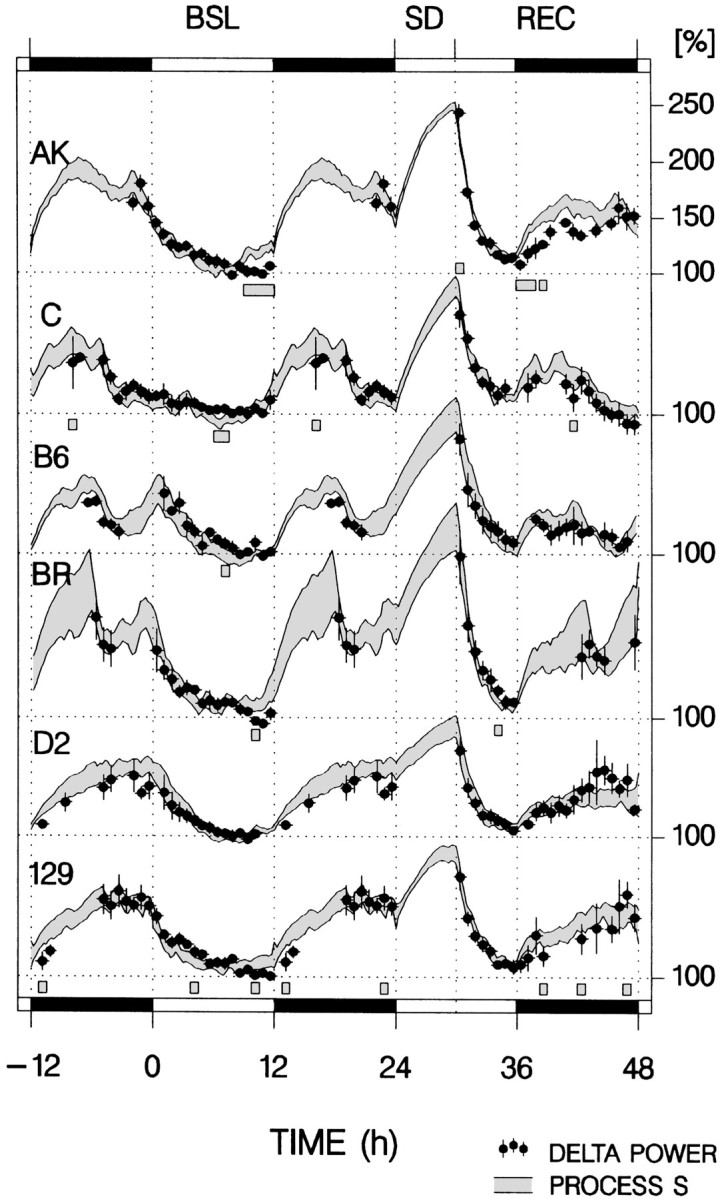
Mean time course over 48 hr of empirical and simulated delta power for six inbred strains of mice in baseline (BSL) and recovery (REC) from a 6 hr sleep deprivation (SD). Black dotsindicate mean (±SEM) delta power averaged over 45 min intervals (n = 4–7 per interval; see Materials and Methods).Gray areas delimit the mean ± SEM range for the simulated Process S for consecutive 15 min intervals. Forty-five minute intervals for which delta power and S significantly differed are indicated by gray bars underneath the curves of each strain (paired t tests; p < 0.05).Black horizontal bars mark the dark periods. Data for the BSL dark period are plotted twice to illustrate the dark-to-light transition. Delta power and S values are both expressed as a percentage of the mean SWS delta power in the last 4 hr of the BSL light period.
The most conspicuous outcome of the simulation is the large strain differences observed for the rate at which SWS need increases (τi), with the largest difference between strains AK and D2 (Table 1). The difference between a time constant of 5.3 hr in AK and 12.6 hr in D2 translates into an initial threefold larger increase in delta power for AK for a given period of wakefulness (estimation based on the parameters listed in Table 1). As wakefulness progresses and delta power further approaches the upper asymptote, this difference reduces to a 2.3-fold larger increase after 6 hr and, ultimately, would stabilize to a 1.4-fold increase according to the upper asymptotes estimated for the two strains. The dynamic range of Process S (i.e., the distance between the two asymptotes) also varied between strains (Table 1). This can affect the outcome of the analysis because to maintain a good fit with the data, a decrease of the upper asymptote will be countered by an increase in the rate of increase (i.e., shorter τi). However, the longest τi was obtained in D2, the strain with the second lowest upper asymptote. Raising the upper asymptote in D2 to the level determined for AK mice while maintaining a similar good fit with the data resulted in a further lengthening of τi. In addition, the two strains for which the upper asymptotes differed the most (AK and C) displayed the shortest time constants. Having established large differences between increase rates (and asymptotes), the similarity of the decrease rates between strains becomes equally striking (Table 1). Finally, the difference inS0, the value used to initiate the iteration (Table 1), reflects differences in the sleep–wake distribution in baseline: C mice displayed the lowest value at light onset because their main sleep episode started ∼5 hr earlier (and thus SWS need had already dissipated), whereas in the remaining strains, the onset of the main sleep episode more or less coincided with the dark-to-light transition (Franken et al., 1999) (Figs.1e, 2).
A sleep deprivation dose–response curve
The simulation presented in experiment 1 demonstrated that the time constant for the accumulation of a need for SWS, but not for its decline during SWS, varied between genotypes. To confirm these predictions, we designed a dose–response experiment in which sleep was deprived for varying durations (i.e., dose) to verify that the delta power increase in subsequent SWS (i.e., response) varies with both genotype and SD duration. In this study, we compared AK and D2 mice for which the largest difference in increase rates was predicted (Table1).
Quantifying the increase rate of Process S during wakefulness
The SD doses did not differ between strains (Table2). Included in the actual SD duration is sleep latency, defined as the time between the end of the SD and sleep onset, which can also be taken as a measure of sleep need. In accordance with this, sleep latency was inversely related to SD duration (Table 2). Sleep latency did not differ significantly between the strains for any of the SDs, although it generally appeared to be shorter for D2 mice.
Table 2.
Duration of the sleep deprivation and of recovery sleep latency
| SD duration (hr) | Sleep latency (min) | |||
|---|---|---|---|---|
| AK | D2 | AK | D2 | |
| 35 min (day 9) | 0.96 (0.04) | 0.98 (0.08) | 12.2 (2.8) | 9.6 (3.5) |
| 35 min (day 4) | 1.16 (0.04) | 1.20 (0.11) | 19.7 (2.4) | 16.1 (4.4) |
| 70 min | 1.66 (0.06) | 1.61 (0.06) | 15.6 (4.2) | 7.2 (2.9) |
| 140 min | 2.74 (0.04) | 2.64 (0.06) | 8.5 (3.2) | 3.5 (1.6) |
| 6 hr | 6.27 (0.06) | 6.48 (0.22) | 2.1 (1.1) | 0.6 (0.5) |
| 9 hr | 9.32 (0.07) | 9.61 (0.16) | 0.3 (0.8) | −0.3 (0.1) |
| p | <0.0001 | <0.0001 | <0.0001 | 0.0008 |
The durations of the SDs in the dose–response study did not differ between strains [mean (SEM); two-way ANOVA factor strain: p = 0.3, factor dose: p < 0.0001, interaction: p = 0.4; Tukey's range test: 9 hr > 6 hr > 140 min > 70 min > 35 min (day 4) = 35 min (day 9); p < 0.05; n = 12 per dose except 6 hr SD; n = 14; data from both strains pooled for the Tukey test]. Sleep latency varied with SD dose and genotype [two-way ANOVA factor strain: p = 0.02; factor dose: p < 0.0001; interaction: p = 0.7; Tukey's range test: 9 hr = 6 hr = 140 min < 140 min = 35 min (day 9) = 70 min < 35 min (day 9) = 70 min = 35 min (day 4); p < 0.05; data from both strains pooled]; however, for none of the SDs did the two strains differ significantly (t tests; p > 0.1). Probability (p) values indicated in this table were obtained from the one-way ANOVA with factor SD dose.
The dose–response curve confirmed the outcome of experiment 1. Delta power increased as a function of SD length in both strains (Fig.3), with even the shortest SDs evoking a significant increase in delta power over the control values in baseline. Most importantly, the results also confirmed that the rate of increase is faster in AK than in D2 mice; for SDs >100 min, the increase in delta power was larger for AK mice (Fig. 3). On a smaller scale, similar observations were made for the relation between waking duration and subsequent delta power under undisturbed baseline conditions (Fig. 3). Also here, delta power was higher after longer waking bouts and, although not significant, seemed to increase at a faster rate for AK mice.
Fig. 3.

The relationship between waking duration and delta power. Left panel, The SD dose–response curve. The response in delta power (mean ± SEM; n = 6 per dose except 6 hr SD; n = 7) varied with SD dose and genotype [two-way ANOVA factors strain and dose:p < 0.0001; interaction: p = 0.0004; Tukey's range test: AK: 6 hr >9 hr >140 min >70 min = 35 min (day 4) = 35 min (day 9); D2: 6 hr = 9 hr >140 min = 35 min (day 4) = 70 min = 35 min (day 9);p < 0.05]. Delta power was higher in AK than in D2 mice for the 140 min (p = 0. 02) and 6 hr SD (p = 0.001; indicated by the gray stars). After the 9 hr SD, values tended to be higher (p = 0.09, t tests). The relationship between SD duration and delta power appeared linear for SDs of <9 hr (thinner lines; linear regression: AK: delta power = 24.6%/hr · SD duration + 92%; D2: delta power = 12.6%/hr · SD duration + 106%; p< 0.0001; r2 = 0.94 for both strains). Right panel, Relationship between the duration of spontaneous waking bouts and delta power in the last 4 hr of the three baseline light periods. The duration determines the level of delta power in subsequent SWS (two-way ANOVA factor strain:p = 0.7; factor “category”:p < 0.0001; interaction: p = 0.5; Tukey's range test for both AK and D2: <12 min = 12–25 min < 12–25 min = >24 min; p < 0.05,n = 6 per category per strain; see Materials and Methods for details). This relationship was quantified by linear regression (AK: delta power = 26.4%/hr · W duration + 93%;p = 0.003;r2 = 0.41; D2: delta power = 16.2%/hr · W duration + 95%; p = 0.0009;r2 = 0.53; n= 6 per category per strain). For both panels the solid black lines connect mean values for AK mice (black dots); dashed lines connect values for D2 mice (gray squares). Values represent the mean delta power in the first 225 (left panel) or 75–225 (right panel) 4 sec epochs scored as SWS after the end of wakefulness. Although scaling differed between panels, the delta power/waking time ratio is preserved, allowing slope comparisons.
For both spontaneous and enforced periods of wakefulness (disregarding the 9 hr SD), the relationship between waking duration and subsequent delta power appeared linear (Fig. 3) instead of the anticipated exponential saturating function. Especially for the shorter SDs (<6 hr) in AK, larger increases in delta power were anticipated. Enticed by these findings, we repeated the simulation of experiment 1 to assess whether Process S could be described better with a linear function. In addition, the assumption of a linear function eliminates the necessity of estimating an upper asymptote, which might have influenced the values of the time constants. The fit between simulated and empirical data did not further improve with the use of a linear function (Table3). However, this analysis again confirmed that the increase rate varied according to genotype. Given the different methods by which, and the data sets from which, the linear increase rates were obtained, they were remarkably similar [AK: 24.6–26.4–27.1%/hr; D2: 12.6–16.2–10.5%/hr for the SDs and spontaneous waking bouts in experiment 2 (Fig. 3) and the linear simulation of experiment 1 (Table 3), respectively].
Table 3.
Simulation of Process S assuming a linear buildup rate
| τi (%/hr) | τd(hr) | DIF2 | r | |
|---|---|---|---|---|
| AK | 27.1 (2.0) | 2.1 (0.2) | 386 (48) | 0.92 |
| C | 18.8 (2.2) | 1.8 (0.2) | 273 (57) | 0.90 |
| B6 | 15.0 (1.3) | 2.2 (0.1) | 221 (31) | 0.90 |
| Br | 21.1 (2.9) | 1.6 (0.2) | 412 (99) | 0.92 |
| D2 | 10.5 (0.9) | 2.1 (0.3) | 326 (97) | 0.86 |
| 129 | 18.9 (2.0) | 1.7 (0.1) | 336 (66) | 0.88 |
| p | <0.0001 | 0.2 | 0.5 | 0.5 |
The simulation of Process S was repeated with the assumption of a linear buildup. The decrease still followed an exponential decline with the same lower asymptotes (Table 1). Significant genotype differences were observed for the buildup rate [τi; mean (SEM); Tukey's range test: AK = Br > Br = 129 = C = B6 > C = B6 = D2; p < 0.05; n = 7 per strain]. The minimum square of the differences (DIF2) did not differ between the two approaches (two-way ANOVA with repeated measures: factor linear vs exponential: p = 0.3; factor strain: p = 0.3; interaction: p = 0.9; n = 7 per strain). The average difference in DIF2was 27 ± 22 %2 (linear − exponential; n = 42). p values underneath each column indicate results from a one-way ANOVA with factor strain.
Another unanticipated result was the lower than expected delta power values obtained after the 9 hr SD. For AK mice, the values obtained after the 9 hr SD were significantly lower than those obtained after the 6 hr SD (Fig. 3). Toward the end of the 9 hr SD, it became increasingly more difficult to keep the animals awake, and attempts to enter SWS doubled over the last 3 hr, resulting in an additional 30 min of SWS (Fig. 4). The length of these short (<10 sec on average) SWS episodes (i.e., the mean reaction time of the experimenters) did not change with time (analysis not shown). One explanation for the lower than expected delta power after the 9 hr SD is that the numerous short SWS episodes during the SD collectively are sufficient to counter a further accumulation of SWS need. In addition, because of the exponential nature of the decrease of Process S, per unit of SWS time, SWS need is more effectively reduced when the prevailing need for SWS is high (see below and Fig.5). To further explore this possibility, the time course of Process S during the 6 and 9 hr SD were simulated using the mean strain parameters obtained for the AK and D2 mice in experiment 1 (Table 1). The values of Process S reached at the end of the SDs were compared with the delta power at sleep onset. Given that the parameters were obtained in another set of mice and in a different experiment, the results of the simulation could predict remarkably well the delta power values reached after the 9 hr SD (Fig. 4). This again underscores the validity of the model and strongly suggests that it is indeed the amount of SWS in the last 3 hr of the SD that precludes a further buildup of delta power. Alternatively, differences in PS need or pressure accumulated over the SD, and which can affect the expression of delta power in SWS (Brunner et al., 1990; Endo et al., 1997), might have contributed to the lower than expected levels of delta power observed after the 9 hr SD. Inferring from the amount of PS expressed over the same period that the initial delta power was calculated, a higher PS pressure was present after the 9 hr SD (PS time in the first 15 min of SWS after SD; 6 vs 9 hr SD; AK: 1.5 ± 0.4/2.5 ± 0.7 min; D2: 0.3 ± 0.2/1.7 ± 0.5 min; two-way ANOVA factor strain: p = 0.04; factor dose:p = 0.02; interaction: p = 0.7). However, this difference was significant for D2 (p = 0.03), where delta power did not differ for the two SDs, and not for AK (p = 0.3;t tests), where delta power did significantly decrease.
Fig. 4.
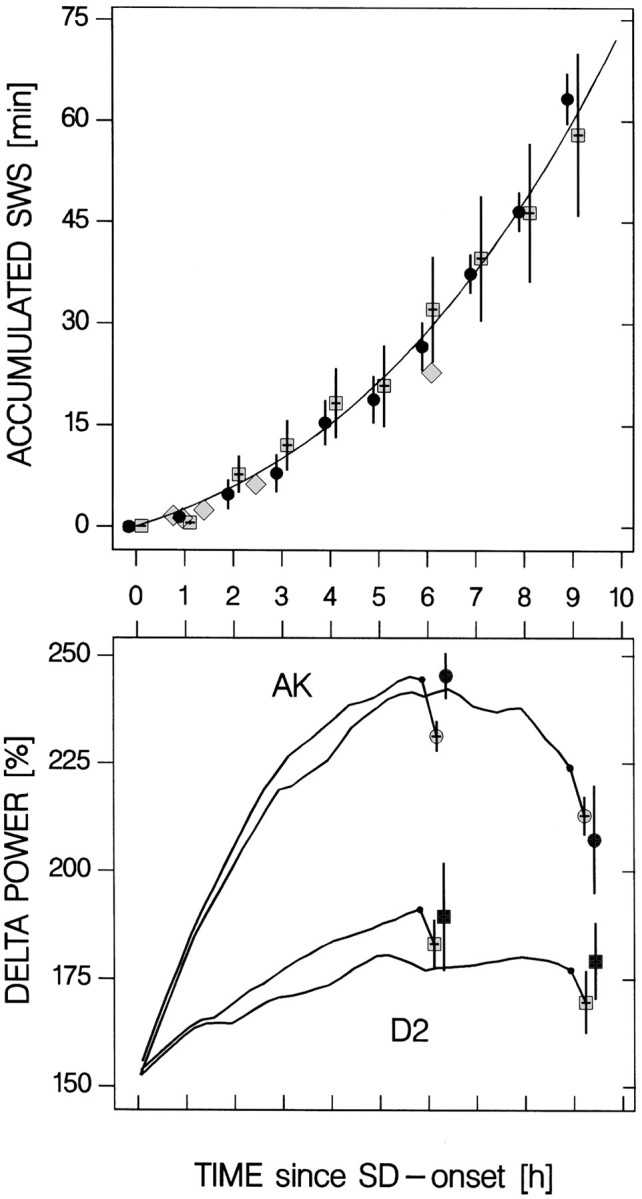
The amount of SWS and time course of Process S during the SDs. Top panel, Hourly values (mean ± SEM; n = 6 per hour per strain) for the accumulation of the amount of SWS during the 9 hr SD. Attempts to enter SWS increase as the SD progresses and result in a doubling in SWS time in the last 3 hr. SWS did not differ between strains at any of the time points (two-way ANOVA factor strain: p = 0.6; factor 1 hr interval: p < 0.0001; interaction:p = 1.0). Symbols are as in Figure 3. Thesix gray diamonds represent mean SWS values for both strains accumulated over the shorter SDs used in the dose–response curve. Genotype did not affect these values (two-way ANOVA factor strain: p = 0.9; factor dose: p< 0.0001; interaction: p = 0.9). Bottom panel, SWS expressed during the SD can explain the lower delta power values reached after the 9 hr as compared with the 6 hr SD. With the assumptions and the parameters of the simulation analyses (Table1), Process S can be followed through the 6 and 9 hr SDs. Delta power (filled black symbols:circles, AK, squares, D2; mean ± SEM) after the 6 (n = 7) and 9 hr (n = 6) SD can be predicted remarkably well (simulated values of S: open symbols; mean ± SEM). Delta power values are indicated at time + 0.1 hr to avoid overlap of error bars.
Quantifying the decrease rate of Process S during recovery sleep
According to the outcome of experiment 1, the rate at which Process S decreases during SWS does not vary with genotype. The exponential nature of the decrease during SWS is best illustrated during recovery sleep from a 6 hr SD during which SWS predominates and delta power is initially high (Fig. 6). It has to be kept in mind that the rate of decrease directly estimated from the data will differ from that determined with the simulation, because the presence of wakefulness (and PS) during recovery will counter the decrease, thus resulting in a slower overall decrease rate. On the other hand, for the same reasons, the lower asymptote estimated directly from the data will be higher, which as discussed above for interdependence between the upper asymptote and τi will in turn yield a faster τd.
In a first analysis, the delta power decrease over the first recording hour after the end of the respective SDs of the dose–response curve was expressed as a function of the delta power reached in the SWS immediately after the SD (Fig. 5). This decrease in delta power strongly depended on its initial level. According to the linear relationship between initial delta power and its subsequent decline, the hallmark of an exponential decreasing function, 88% of the variance in the decline could be explained by its initial value. This linear relationship translated into time constants, τd, that did not differ between AK and D2 (Fig.5). The amount of SWS expressed during the hour over which the decrease in delta power was calculated varied with dose (data not shown) and thus could have influenced the outcome of the previous analysis. However, the amount of SWS did not differ between AK and D2 for any of the SDs and did not vary in a dose-dependent manner.
In a second analysis, τd was estimated by a nonlinear regression analysis on the time course of delta power over the initial 6 hr after the 6 hr SD. Throughout this 6 hr recovery period, SWS amount was maintained at a high level in all strains [58% of total recording time (mean over six strains and 6 hr); range, 50% (C) to 65% (AK)]. The time course of delta power during recovery is illustrated for AK and D2 mice in Figure 6, and the mean results of the nonlinear regression analysis for all six inbred strains are shown in Table 4. Again, the decrease rates did not differ between genotypes despite marked differences in the estimated level of Process S at recovery sleep onset. For AK and D2, surprisingly similar values for τd were obtained in both the dose–response curve analysis (Fig. 5) (AK: 1.3 hr; D2: 1.1 hr) and the present analysis (Fig. 6) (1.2 hr in both). These two analyses confirm that (1) Process S decreases exponentially, and (2) the rate of decrease does not significantly vary between the genotypes.
Table 4.
Parameters describing the exponential decrease in delta power after 6 hr SD
| τd (hr) | S0(%) | LA (%) | |
|---|---|---|---|
| AK | 1.2 (0.1) | 272 (6) | 106 (3) |
| C | 1.3 (0.4) | 237 (15) | 112 (6) |
| B6 | 1.3 (0.2) | 223 (16) | 108 (7) |
| Br | 1.2 (0.2) | 261 (23) | 111 (4) |
| D2 | 1.2 (0.1) | 201 (15) | 109 (4) |
| 129 | 1.8 (0.6) | 206 (15) | 100 (7) |
| p | 0.8 | 0.008 | 0.7 |
The decrease in delta power in the first 6 hr after the 6 hr SD followed an exponential decline. The parameters defining this function are the time constant (τd), the level of S reached at the end of the SD (S0), and the lower asymptote (LA), which were determined by non-linear fitting (see Materials and Methods). Only S0 varied with genotype [mean (SEM); p values indicate the probability that the parameter does not vary with genotype; one-way ANOVA factor strain; S0: AK = Br = C = B6 > Br = C = B6 = 129 = D2; p < 0.05; Tukey's range test; n = 7 per strain]. Notice that here S0 indicates the value of S at the time of recovery sleep onset, whereas elsewhere the level of S at light onset of baseline is meant.
Experiment 3: QTL analysis of delta power at sleep onset after enforced and spontaneous periods of wakefulness
Because mice of a particular inbred strain can be considered genetically identical clones that differ from other inbred strains, the results of experiments 1 and 2 strongly suggest a genetic basis underlying the accumulation rate for SWS need. The segregation of this trait in recombinant offspring of strains for which this trait differed can be used for mapping. Here we present data from RI mice that were derived from a B6xD2 cross (BXD) and provide a preliminary mapping of genomic regions associated with the accumulation of SWS need.
QTLs associated with the rebound after 6 hr SD
The wake-dependent increase rates for SWS delta power (τi) between B6 and D2 differed when compared separately (Table 1) (p = 0.021; ttest; n = 7 per strain). In keeping with this, the level of delta power attained after the 6 hr SD differed between these two strains (Fig. 7) (p = 0.016; t tests;n = 7 per strain). Their RI offspring also varied for this quantitative trait (Fig. 7) (one-way ANOVA factor strain;p < 0.0001; Tukey's range test: BXD-5 > BXD-21, -28, -31, -20, -32; B6 > BXD-14, -12, -29, -30, -2, -21, -28, -31, -20, -32; p < 0.05), with additive genetic factors accounting for 37% of the total variance (i.e., heritability in inbred strains) (Hegmann and Possidente, 1981). Based on these findings, we pursued a QTL analysis by calculating nominal correlations between the SDP of the trait and the SDP of the genotype of each of the 788 MIT markers typed for these RI strains. For the delta power rebound after the 6 hr SD, the permutation test (see Materials and Methods) established a suggestive level for LOD scores of >1.77, corresponding to a p < 0.0043 level for a nominal correlation, and a significant level for LOD scores of >2.98, corresponding top < 0.00021. Using these criteria, two genomic regions were identified.
Fig. 7.

QTL analysis of the delta power rebound after 6 hr sleep deprivation in BXD-RI mice. Top panel, SDP of delta power in the first 15 min of SWS (mean ± SEM;n = 4–7 per strain) after the sleep deprivation.Black vertical bars mark progenitor strains B6 and D2.Zeros above the horizontal axis denote strains carrying the B6 allele (as opposed to a D2 allele) at the markers that gave the best LOD score on chromosome 13. Bottom panel, Point correlations between the SDP of the phenotype (i.e., delta power) and the genotype of the MIT markers on chromosome 13. p values of the correlations are converted into LOD scores. The genome-wide suggestive and significant levels are derived from an empirical probability distribution (see Materials and Methods). The positions of the markers are given in cM from centromere according to the MGI database (www.informatics.jax.org). The 38–53 cM range indicates the interval where the interpolated LOD scores > suggestive, roughly corresponding to a ±2 LOD score confidence interval.
A significant QTL was found on chromosome 13 with 37.8–53.4 cM as the region where LOD scores were above the suggestive threshold [D13Mit231, -254 (39.9/40.0 cM) LOD = 2.36; D13Mit126, -106, -193 (41.0/42.0/43.0 cM) LOD = 3.57; D13Mit107, -147, -145 (48.0/49.0/52.0 cM) LOD = 2.92]. The best LOD score (3.57) corresponds to a nominal p level of 0.00005 and a genome-wide p level of 0.01. We termed this QTLDps1 (delta power in SWS QTL 1). The LOD scores for all MIT markers typed in BXD RI strains for chromosome 13 are shown in Figure7. For only one RI strain (BXD-29), the phenotype clearly did not match its genotype (Fig. 7). A possible explanation might be the presence of a double cross-over between two neighboring markers genotyped as B6 alleles. Assuming a D2 genotype for BXD-29 would further increase the LOD score for Dps1 to 5.04. The only other suggestive QTL was found on chromosome 2 between 82.3 and 101.6 cM [D2Mit311, -343, -229, -456 (83.1/84.2/85.2/86.3 cM) LOD = 2.13; D2Mit147, -528 (87.0/87.4 cM) LOD = 2.52]. These findings were not related to or influenced by differences in PS or SWS time expressed during the period over which delta power was calculated (analyses not shown). The rate at which delta power decreases during recovery sleep after the SD was analyzed according to the procedure illustrated in Figure 5. As in the previous two experiments, no significant differences were present between strains and no QTLs were identified (analysis not shown).
QTLs associated with delta power at sleep onset in baseline
In an attempt to confirm the QTLs identified for delta power after enforced wakefulness, the same analysis was performed in undisturbed, baseline conditions for delta power at the onset of the main sleep period. However, in 19 of the 25 BXD-RI strains, an additional sleep period was apparent in the latter half of the dark period, best illustrated in the progenitor strain B6 (Fig.8). In 10 of these 19 BXD-RI strains, the delta power reached at the onset of these additional sleep periods was higher than at the onset of the main sleep period (Fig. 8). It can be argued whether this additional sleep period is still part of the active period and thus whether the higher of these two sleep onset values or the sleep onset value of the main sleep period best reflects the SWS need accumulated during the active period. Both possibilities were pursued, and the same four QTLs were detected by either analysis. Only the results of the latter analysis will be presented below.
Fig. 8.

Distribution of SWS in baseline and the time course of SWS delta power during the main sleep periods of the 25 BXD-RI strains tested. The SWS distribution (gray area) represents a 2 hr moving average of percentage recording time with a 15 min resolution (4–7 per strain). Delta power values (black dots connected with thick lines) represent means over consecutive 225 4 sec epochs scored as SWS. Only delta power values for SWS occurring within the main sleep period(s) are depicted. One to two major sleep periods were determined per mouse according to the SWS distribution (see Materials and Methods). The initial delta power values of these sleep periods were used in the QTL analysis. The number in the top right-hand corner of each panel indicates the BXD-RI strain ID. The progenitor strains, B6 and D2, are indicated on the bottom row. Data in the dark period are displayed twice to visualize the dark-to-light transition.
Although the main sleep period was always associated with the light period, both its onset and end varied with strain (sleep onset: −0.9 ± 0.2 hr; sleep end: 11.5 ± 0.1 hr; mean ± SEM;n = 128; times relative to lights on; one-way ANOVA with factor strain: p < 0.0001 for both variables;n = 4–7 per strain; 27 strains). Delta power at the onset of the main sleep period differed between the progenitor strains B6 and D2 (p = 0.051; t test,n = 7/strain) and varied among the BXD-RI strains (Fig.8) (one-way ANOVA factor strain: p = 0.0003; Tukey's range test: B6 > BXD-9, -28, -31; BXD-13 > BXD-31; BXD-19 > BXD-31; p < 0.05). For this trait, the permutation test established a suggestive threshold for QTLs with LOD scores of >1.83, corresponding to a p < 0.0037 level for a nominal correlation, and a significant level for QTLs with LOD scores of >3.14 or p < 0.00014. Four QTLs were identified with these criteria.
First and most important for our purposes is the suggestive QTL found on chromosome 13 in the range from 34.5 to 41.5 cM [D13Mit13 (35.0 cM) LOD = 2.30; D13Mit224 (37.5 cM) LOD = 2.44]. This QTL overlaps with the Dps1 QTL. The markers that gave the best correlation for Dps1 (D13Mit126, -106, -193) now have a LOD score of 1.61 (i.e., nominal p = 0.0065). This indicates that the two phenotypes (i.e., delta power after the SD and after the active period) are genetically correlated, which was supported by a significant correlation (r = 0.59;p = 0.0011; n = 27). Another suggestive QTL was found on chromosome 7 between 21.5 and 28.0 cM [D7Mit229 (23.0 cM) LOD = 2.22; D7Mit145 (26.5 cM) LOD = 2.18]. Two significant QTLs were found: on chromosome 12 between 52.4 and 58.2 cM [D12Mit280 (55.0 cM) LOD = 3.30; D12Mit18, -263, -8 (56.0 cM) LOD = 2.43; D12Nds2 (57.0 cM) LOD = 2.38; D12Mit150 (58.0 cM) LOD = 1.92] and on chromosome 17 between 16.6 and 22.9 cM [D17Mit16, -28, -233 (18.15/18.2/20.9 cM) LOD = 3.17; D17Mit11 (22.0 cM) LOD = 2.44]. These two QTL were termed Dps2and Dps3, respectively.
The QTLs found for SWS delta power at sleep onset in baseline that do not influence the rebound after sleep deprivation are more likely to be related to the distribution of SWS (that in turn drives delta power) rather than directly linked to the increase rate of a need for SWS. This could be demonstrated for the Dps3 QTL on chromosome 17. The distribution of SWS in BXD-RI mice was highly variable especially in the 6 hr preceding the main sleep period (Fig. 8). Changes in this period are likely to affect the expression of delta power at sleep onset. In an effort to quantify and capture these changes in one value, the sum of the difference between the maximum (usually related to the additional sleep period) and the subsequent minimum amount of SWS and the difference between that minimum and the subsequent SWS level reached in the main sleep period was calculated (values not shown). This value was largest for B6 mice because they exhibited a pronounced peak in the amount of SWS directly followed by an equally pronounced peak in the amount of wakefulness just before sleep onset (Fig. 8). Only one suggestive QTL was identified on chromosome 17 for this trait, which was identical to theDps3 QTL. With 1.83 and 3.17 as genome-wide suggestive and significant levels, this QTL was localized between 15.0 and 24.1 cM [D17Mit45, -135 (16.4/16.5 cM) LOD = 2.37; D17Mit16, -28, -233 (18.15/18.2/20.9 cM) LOD = 3.04; D17Mit11 (22.0 cM) LOD = 2.49; D17Mit136 (23.0 cM) LOD = 2.44; D17Mit49 (23.2 cM) LOD = 2.67].
DISCUSSION
The results of the experiments demonstrate that one of the parameters of the process underlying the homeostatic regulation of SWS need, i.e., its rate of increase in the absence of SWS, greatly varies between genotypes. In contrast, the exponential decrease of SWS need in the presence of SWS did not seem to vary. Preliminary mapping of this trait in recombinant inbred strains yielded one significant QTL on chromosome 13.
The distribution and duration of SWS and Process S
A different time course of delta power can be a consequence of different dynamics of Process S or a consequence of the distribution and amount of SWS. The simulation, which accounts for differences in the sleep–wake distribution, provides a tool to distinguish between these two alternatives. In the present analyses, the remarkable difference in the baseline time course of delta power between C and BR mice had to be attributed to differences in the SWS distribution because the estimates of the parameters of Process S did not differ. Indeed, the diurnal amplitude of the amount of sleep for C mice is strongly attenuated as compared with BR (Franken et al., 1999). A similar observation was made in gene-targeted mice of the 129 strain that lack albumin D-binding protein (DBP), a circadian transcription factor (Lopez-Molina et al., 1997; Ripperger et al., 2000). In these mice, a pronounced reduction of the diurnal amplitude of delta power was observed (Franken et al., 2000), which also could be attributed to differences in the distribution of SWS because the time constants describing Process S were unaffected [τi = 9.0 hr; τd = 1.7 hr; the 129 mice served as isogenic controls (Table 1)]. Dbp is localized within the chromosome 7 QTL, and a functional, different Dbp gene between B6 and D2 might affect the baseline sleep–wake distribution in BXD-RI strains and the delta power it drives.
The results of our experiments strongly reemphasize that delta power in SWS is a marker of a homeostatic sleep regulatory process, i.e., Process S in the Two-Process model of sleep regulation (Borbély, 1982; Daan et al., 1984). However, our data also add to the notion that SWS delta power and SWS duration are regulated differently (Dijk and Beersma, 1989; Dijk and Kronauer, 1999), which is inconsistent with the assumptions of the Two-Process model in which Process S, in interaction with a circadian process, determines sleep duration. In the present study, large strain differences in sleep duration could be observed without apparent differences in the dynamics of Process S. Aeschbach et al. (1996) arrived at the same conclusion when comparing delta power responses to an SD between human habitual long and short sleepers. Other support comes from observations in rats and mice in which during the course of recovery from SD, delta power can reach values below baseline (i.e., “negative rebound”), whereas the duration of SWS remains well above baseline (Franken et al., 1991a, 1999; Rechtschaffen et al., 1999). This negative rebound was shown to result from the SD-induced increase in SWS time (Franken et al., 1991b). Thus, although delta power in SWS is “driven” by the previous sleep–wake history, it does not seem to drive SWS duration.
This is the first study to demonstrate that the increase in SWS need, quantified in baseline and recovery from SD, varies within individuals of one species. One could argue that the strain differences in delta power do not relate to differences in the dynamics of Process S that it is thought to reflect but that the expression of delta power is merely affected. However, in all strains, regardless of differences in time constants, delta power did reliably reflect prior sleep duration. Furthermore, several non-EEG measures are also indicative of differences in SWS need. Thus, an increased SWS need in AK mice is also evidenced by a larger SWS amount (during baseline 2.8 hr more than in D2 mice) and their inability to maintain long waking bouts (Franken et al., 1999). Conversely, although D2 mice display the largest amount of wakefulness in the dark period, SWS fragmentation, a measure negatively correlated with SWS need (Franken et al., 1991a), was higher in subsequent sleep compared with other inbred strains (Franken et al., 1999).
The dynamics of Process S during wakefulness
The results of experiment 2 suggest that the relationships between spontaneous and enforced bouts of wakefulness and subsequent delta power do not differ. This illustrates that nonspecific factors that inevitably accompany the SD (e.g., increased stress and activity) do not seem to be a major contributor to the subsequent delta power rebound; if anything, the increase rate in delta power is higher for spontaneous waking bouts, possibly related to the absence of SWS during these bouts (see below).
The relationship between the time spent awake and EEG delta power seems linear. Performance, a non-EEG measure inversely related to SWS need, also changes linearly as a function of time spent awake (Kuo et al., 1998). Recovery of performance during sleep, on the other hand, shows an exponential saturating function with a time constant comparable to that of delta power (Jewett et al., 1999). The impression of an exponential saturating function for the increase of SWS need might result from the intrusion of short SWS episodes during longer SDs. In addition, as in rats and humans (Franken et al., 1991a, 1993;Aeschbach et al., 1999; Cajochen et al., 1999), delta power within wakefulness increased over the SDs in the six inbred strains of mice (data not shown). This might not only reflect an increased need for SWS, but at the same time might result in a slower buildup rate of Process S. The SWS during the SD, possibly in conjunction with a slowing of the increase rate, can readily account for a lack of a further increase or even a decrease in delta power observed after the 9 hr SD in the present study and after longer than 24 hr SDs in the rat (Rechtschaffen et al., 1999).
Our interpretation that SWS need increases at different rates might be related to strain differences in the “quality” of wakefulness. Differences in locomotor activity, metabolic rate, brain temperature, the response to the environment, and vigilance, among others, might affect subsequent sleep (Horne, 1988). For some of those variables, an effect on delta power has been suggested, but for the few variables that were compared between D2 and AK mice, no obvious differences seem present [Trullas and Skolnick, 1993; the Mouse Genome Informatics (MGI) database (www.informatics.jax.org)]. From the higher relative contribution of delta power to the waking EEG (Franken et al., 1998), one could infer that D2 mice in baseline spent a larger portion of their waking time in a more quiet or “drowsy” state. Implications of the expression of delta power during wakefulness on Process S remain to be established. Compared with wakefulness, SWS clearly represents the more homogeneous behavioral state. The similarity between strains of the decrease rate of SWS need during SWS might reflect this.
The genetics of Process S
Two genomic regions were identified that might contain genes that modify the rate at which SWS need accumulates. To propose candidate genes at this point is premature because these regions contain several hundreds of genes. Nevertheless, according to their position listed in the MGI database, several genes directly associated with the expression of delta power or mentioned in relation to a possible function of SWS are worth pointing out. The chromosome 2 QTL contains the gene encoding brain-glycogen phosphorylase, an enzyme that converts glycogen into glucose-1-phosphate during metabolic demand. Possibly, SWS serves to replenish glycogen stores that become depleted during wakefulness (Benington and Heller, 1995; Holden et al., 2000). Two other genes encode enzymes that regulate adenosine levels:S-adenosyl-homocysteine hydroxylase and adenosine deaminase. Adenosine has been implicated in mediating the EEG manifestation of SWS need; i.e., delta power (Benington and Heller, 1995;Porkka-Heiskanen et al., 2000). The genes for growth hormone releasing hormone (GHRH) and the somatostatin receptor are also localized in this region. GHRH and somatostatin regulate the pituitary release of growth hormone. All three hormones have been implicated in the regulation of SWS and the expression of delta power (Krueger and Obál, 1997;Van Cauter et al., 1998). Furthermore, the chromosome 2 QTL is homolog to the human chromosome 20q13.2 region that contains a gene that mediates a low-voltage EEG trait (Anokhin et al., 1992). TheDps1 QTL on chromosome 13 encompasses the gene encoding the neurotrophic tyrosine kinase-2 receptor on which brain-derived neurotrophic factor (BDNF) acts. BDNF mRNA expression parallels the expression of delta power and was found to increase with prolonged wakefulness and to decrease with recovery sleep (Peyron et al., 1998).
The basic assumption underlying the QTL analysis is that the QTLs that were found contain functionally polymorphic genes that affect the phenotype in the progenitor strains (Lander and Botstein, 1989; Lander and Schork, 1994). Because most quantitative traits are determined by several genes, any one of them is likely to explain only a certain percentage of the trait variance. The Dps1 QTL explained a large portion (49%) of the genetic variance in the rebound in delta power, suggesting the presence a major gene. We are currently refining and confirming the mapping of these QTLs in B6xD2 and AKxD2 intercross and backcross panels. Ultimately, this might lead to a resolution high enough to warrant either a candidate gene approach (if present) or, alternatively, positional cloning.
The efficiency of the QTL approach in identifying genes is being questioned, and, currently, forward genetics by mutagenesis is being favored (Nadeau and Frankel, 2000). Nevertheless, especially because the complete genomes of the B6 and D2 strains (and thus their polymorphisms) will be available soon (Marshall, 2000), QTL analysis will prove to be a powerful tool in identifying genes underlying complex traits such as sleep and its homeostatic regulation (Tafti et al., 1999) that do not easily lend themselves to the high-throughput screening necessary for mutagenesis.
Footnotes
This study was supported by the Swiss National Science Foundation (Grants 31.45751.95 and 3100-056000), the National Institutes of Health (Grant HL64148), and a Roche Research Foundation Fellowship. We thank Dr. Nihal Okaya for help with mathematical issues and Drs. Bruce O'Hara and Derk-Jan Dijk for helpful discussions and comments on this manuscript.
Correspondence should be addressed to Dr. Paul Franken, Department of Biological Sciences, Stanford University, Stanford, CA 94305-5020. E-mail: pfranken@stanford.edu.
REFERENCES
- 1.Achermann P, Dijk DJ, Brunner DP, Borbély AA. A model of human sleep homeostasis based on EEG slow-wave activity: quantitative comparison of data and simulations. Brain Res Bull. 1993;31:97–113. doi: 10.1016/0361-9230(93)90016-5. [DOI] [PubMed] [Google Scholar]
- 2.Aeschbach D, Cajochen C, Landolt H, Borbély AA. Homeostatic sleep regulation in habitual short sleepers and long sleepers. Am J Physiol. 1996;270:R41–53. doi: 10.1152/ajpregu.1996.270.1.R41. [DOI] [PubMed] [Google Scholar]
- 3.Aeschbach D, Matthews JR, Postolache TT, Jackson MA, Giesen HA, Wehr TA. Two circadian rhythms in the human electroencephalogram during wakefulness. Am J Physiol. 1999;277:R1771–1779. doi: 10.1152/ajpregu.1999.277.6.R1771. [DOI] [PubMed] [Google Scholar]
- 4.Anokhin A, Steinlein O, Fischer C, Mao Y, Vogt P, Schalt E, Vogel F. A genetic study of the human low-voltage electroencephalogram. Hum Genet. 1992;90:99–112. doi: 10.1007/BF00210751. [DOI] [PubMed] [Google Scholar]
- 5.Beijsterveldt CEM, Boomsma DI. Genetics of the human electroencephalogram (EEG) and event-related brain potentials (ERPs): a review. Hum Genet. 1994;94:319–330. doi: 10.1007/BF00201587. [DOI] [PubMed] [Google Scholar]
- 6.Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol. 1995;45:347–360. doi: 10.1016/0301-0082(94)00057-o. [DOI] [PubMed] [Google Scholar]
- 7.Borbély AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- 8.Brunner DP, Dijk DJ, Tobler I, Borbély AA. Effect of partial sleep deprivation on sleep stages and EEG power spectra: evidence for non-REM and REM sleep homeostasis. Electroencephalogr Clin Neurophysiol. 1990;75:492–499. doi: 10.1016/0013-4694(90)90136-8. [DOI] [PubMed] [Google Scholar]
- 9.Cajochen C, Khalsa SBS, Wyatt JK, Czeisler CA, Dijk DJ. EEG and ocular correlates of circadian melatonin phase and human performance decrements during sleep loss. Am J Physiol. 1999;277:R640–649. doi: 10.1152/ajpregu.1999.277.3.r640. [DOI] [PubMed] [Google Scholar]
- 10.Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daan S, Beersma DGM, Borbély AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol. 1984;246:R161–183. doi: 10.1152/ajpregu.1984.246.2.R161. [DOI] [PubMed] [Google Scholar]
- 12.Dijk DJ, Beersma DG. Effects of SWS deprivation on subsequent EEG power density and spontaneous sleep duration. Electroencephalogr Clin Neurophysiol. 1989;72:312–320. doi: 10.1016/0013-4694(89)90067-9. [DOI] [PubMed] [Google Scholar]
- 13.Dijk DJ, Czeisler CA. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J Neurosci. 1995;15:3526–3538. doi: 10.1523/JNEUROSCI.15-05-03526.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dijk DJ, Kronauer RE. Commentary: models of sleep regulation: successes and continuing challenges. J Biol Rhythms. 1999;14:569–573. doi: 10.1177/074873099129000902. [DOI] [PubMed] [Google Scholar]
- 15.Dijk DJ, Beersma DG, Daan S. EEG power density during nap sleep: reflection of an hourglass measuring the duration of prior wakefulness. J Biol Rhythms. 1987;2:207–219. doi: 10.1177/074873048700200304. [DOI] [PubMed] [Google Scholar]
- 16.Endo T, Schwierin B, Borbély AA, Tobler I. Selective and total sleep deprivation: effect on the sleep EEG in the rat. Psychiatry Res. 1997;66:97–110. doi: 10.1016/s0165-1781(96)03029-6. [DOI] [PubMed] [Google Scholar]
- 17.Franken P, Dijk DJ, Tobler I, Borbély AA. Sleep deprivation in the rat: effects on electroencephalogram power spectra, vigilance states, and cortical temperature. Am J Physiol. 1991a;261:R198–208. doi: 10.1152/ajpregu.1991.261.1.R198. [DOI] [PubMed] [Google Scholar]
- 18.Franken P, Tobler I, Borbély AA. Sleep homeostasis in the rat: simulation of the time course of EEG slow-wave activity. Neurosc Lett. 1991b;130:141–144. doi: 10.1016/0304-3940(91)90382-4. [DOI] [PubMed] [Google Scholar]
- 19.Franken P, Tobler I, Borbély AA. Effects of 12-h sleep deprivation and of 12-h cold exposure on sleep regulation and cortical temperature in the rat. Physiol Behav. 1993;54:885–894. doi: 10.1016/0031-9384(93)90297-s. [DOI] [PubMed] [Google Scholar]
- 20.Franken P, Tobler I, Borbély AA. Varying photoperiod in the laboratory rat: profound effect on 24-h sleep pattern but no effect on sleep homeostasis. Am J Physiol. 1995;269:R691–701. doi: 10.1152/ajpregu.1995.269.3.R691. [DOI] [PubMed] [Google Scholar]
- 21.Franken P, Malafosse A, Tafti M. Genetic variation in EEG activity during sleep in inbred mice. Am J Physiol. 1998;275:R1127–1137. doi: 10.1152/ajpregu.1998.275.4.R1127. [DOI] [PubMed] [Google Scholar]
- 22.Franken P, Malafosse A, Tafti M. Genetic determinants of sleep regulation in inbred mice. Sleep. 1999;22:155–169. [PubMed] [Google Scholar]
- 23.Franken P, Lopez-Molina L, Marcacci L, Schibler U, Tafti M. The transcription factor DBP affects circadian sleep consolidation and rhythmic EEG activity. J Neurosci. 2000;20:617–625. doi: 10.1523/JNEUROSCI.20-02-00617.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hegmann JP, Possidente B. Estimating genetic correlations from inbred strains. Behav Genetics. 1981;11:103–114. doi: 10.1007/BF01065621. [DOI] [PubMed] [Google Scholar]
- 25.Holden CP, Shepel PN, Mackiewicz M, Pack AI, Geiger JD. Brain glycogen levels are decreased with increased wakefulness and may represent a homeostatic drive for sleep. Soc Neurosci Abstr. 2000;26:1755. doi: 10.1523/JNEUROSCI.22-13-05581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horne JA. Why we sleep: the functions of sleep in humans and other mammals. Oxford UP; New York: 1988. [Google Scholar]
- 27.Jewett ME, Dijk DJ, Kronauer RE, Dinges DF. Dose-response relationship between sleep duration and human psychomotor vigilance and subjective alertness. Sleep. 1999;22:171–179. doi: 10.1093/sleep/22.2.171. [DOI] [PubMed] [Google Scholar]
- 28.Krueger JM, Obál F. Sleep regulatory substances. In: Schwartz WJ Jr, editor. Sleep science: integrating basic research and clinical practice. Karger; Basel: 1997. pp. 175–194. [Google Scholar]
- 29.Kuo A, Carlin MM, Powell JW, Dinges DF. Chronic restriction of sleep to 4 hours per night for 14 nights changes performance linearly but not subjective sleepiness. Sleep. 1998;21[Suppl]:241. [Google Scholar]
- 30.Lander ES, Botstein D. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics. 1989;121:185–199. doi: 10.1093/genetics/121.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lander ES, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 32.Lander ES, Schork NJ. Genetic dissection of complex traits. Science. 1994;265:2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- 33.Lopez-Molina L, Conquet F, Dubois-Dauphin M, Schibler U. The DBP gene is expressed according to a circadian rhythm in the SCN and influences circadian behavior. EMBO J. 1997;16:6762–6771. doi: 10.1093/emboj/16.22.6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marshall E. Public-private project to deliver mouse genome in 6 months. Science. 2000;290:242–243. doi: 10.1126/science.290.5490.242a. [DOI] [PubMed] [Google Scholar]
- 35.McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- 36.Nadeau JH, Frankel WN. The roads from phenotypic variation to gene discovery: mutagenesis versus QTLs. Nat Genet. 2000;25:381–384. doi: 10.1038/78051. [DOI] [PubMed] [Google Scholar]
- 37.Neckelmann D, Ursin R. Sleep stages and EEG power spectrum in relation to acoustical stimulus arousal threshold in the rat. Sleep. 1993;16:467–477. [PubMed] [Google Scholar]
- 38.Peyron C, Wurts SW, Srere HK, Heller HC, Edgar DM, Kilduff TS. mRNA level of brain-derived neurotrophic factor increases in several brain regions after sleep deprivation. Soc Neurosci Abstr. 1998;24:1430. [Google Scholar]
- 39.Porkka-Heiskanen T, Strecker RE, McCarley RW. Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience. 2000;99:507–517. doi: 10.1016/s0306-4522(00)00220-7. [DOI] [PubMed] [Google Scholar]
- 40.Rechtschaffen A, Bergmann BM, Gilliland MA, Bauer K. Effects of method, duration, and sleep stage on rebounds from sleep deprivation in the rat. Sleep. 1999;22:11–31. doi: 10.1093/sleep/22.1.11. [DOI] [PubMed] [Google Scholar]
- 41.Ripperger JA, Shearman LP, Reppert SM, Schibler U. CLOCK, an essential pacemaker component, controls expression of the circadian transcription factor DBP. Genes Dev. 2000;14:679–689. [PMC free article] [PubMed] [Google Scholar]
- 42.Steriade M. Coherent oscillations and short-term plasticity in corticothalamic networks. Trends Neurosci. 1999;22:337–345. doi: 10.1016/s0166-2236(99)01407-1. [DOI] [PubMed] [Google Scholar]
- 43.Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- 44.Tafti M, Malafosse A, Franken P. Genetic Determinants of theta rhythm in mice. J Sleep Res. 1998;7[Suppl 2]:269. [Google Scholar]
- 45.Tafti M, Chollet D, Valatx JL, Franken P. Quantitative trait loci approach to the genetics of sleep in recombinant inbred mice. J Sleep Res. 1999;8[Suppl 1]:37–43. doi: 10.1046/j.1365-2869.1999.00007.x. [DOI] [PubMed] [Google Scholar]
- 46.Tobler I, Borbély AA. Sleep EEG in the rat as a function of prior waking. Electroencephalogr Clin Neurophysiol. 1986;64:74–76. doi: 10.1016/0013-4694(86)90044-1. [DOI] [PubMed] [Google Scholar]
- 47.Trullas R, Skolnick P. Differences in fear motivated behaviors among inbred mouse strains. Psychopharmacology. 1993;111:323–331. doi: 10.1007/BF02244948. [DOI] [PubMed] [Google Scholar]
- 48.Van Cauter E, Plat L, Copinschi G. Interrelations between sleep and the somatotropic axis. Sleep. 1998;21:553–566. [PubMed] [Google Scholar]
- 49.Werth E, Dijk DJ, Achermann P, Borbély AA. Dynamics of the sleep EEG after an early evening nap: experimental data and simulations. Am J Physiol. 1996;271:R501–510. doi: 10.1152/ajpregu.1996.271.3.R501. [DOI] [PubMed] [Google Scholar]
- 50.Williams RW, Airey DC, Kulkarni A, Zhou G, Lu L (2001) Genetic dissection of the olfactory bulbs of mice: QTLs on chromosomes 4, 6, 11, and 17 modulate bulb size. Behav Genet, in press. [DOI] [PubMed]