

Abstract
Alzheimer's disease (AD) is a devastating dementia of late life that is correlated with a region-specific neuronal cell loss. Despite progress in uncovering many of the factors that contribute to the etiology of the disease, the cause of the nerve cell death remains unknown. One promising theory is that the neurons degenerate because they reenter a lethal cell cycle. This theory receives support from immunocytochemical evidence for the reexpression of several cell cycle-related proteins. Direct proof for DNA replication, however, has been lacking. We report here the use of fluorescent in situ hybridization to examine the chromosomal complement of interphase neuronal nuclei in the adult human brain. We demonstrate that a significant fraction of the hippocampal pyramidal and basal forebrain neurons in AD have fully or partially replicated four separate genetic loci on three different chromosomes. Cells in unaffected regions of the AD brain or in the hippocampus of nondemented age-matched controls show no such anomalies. We conclude that the AD neurons complete a nearly full S phase, but because mitosis is not initiated, the cells remain tetraploid. Quantitative analysis indicates that the genetic imbalance persists for many months before the cells die, and we propose that this imbalance is the direct cause of the neuronal loss in Alzheimer's disease.
Keywords: cell cycle, PCNA, cyclin B, hippocampus, nucleus basalis, FISH (fluorescent in situ hybridization), neurodegeneration
The death of nerve cells during early development is a constructive part of pruning and shaping the emerging nervous system. The loss of neurons during adult life, however, is a pathological process that produces behavioral disorders and potentially the death of the organism. Recently, several laboratories have offered evidence that an attempt by the cell to reenter a mitotic cycle precedes many instances of neuronal death (Smith and Lippa, 1995; Arendt et al., 1996, 1998a,b; Vincent et al., 1996, 1997, 1998; McShea et al., 1997, 1999; Nagy et al., 1997a, 1998;Busser et al., 1998; Smith et al., 1999). Together, these studies suggest that the paradoxical association of the generative process of cell division with the degenerative process of cell death is found at all stages of the existence of a neuron. The prohibition against cell division begins at the earliest stages of maturation of a neuron. Hindbrain neurons in mice lacking the retinoblastoma tumor suppressor gene are unable to avoid reentering the cell cycle and die by apoptosis immediately after their emigration from the ventricular zone (Lee et al., 1994). Neuronal cell death later in development has also been linked to ectopic cell cycling. Using mutant models of target-related cell death, Herrup and Busser (1995) showed that target-deprived neurons initiated the synthesis of cell cycle enzymes [cyclin D and proliferating cell nuclear antigen (PCNA)] and incorporated bromodeoxyuridine (BrdU) into high molecular weight DNA just before dying. During this same developmental period, if cell cycle reentrance is forced by ectopically driving an oncogene with a neuronal-specific promotor, the targeted neurons will die rather than divide (al-Ubaidi et al., 1992; Feddersen et al., 1992). Severalin vitro cell death models have illustrated the same correlation (Park et al., 1997, 2000; Copani et al., 1999; Giovanni et al., 1999, 2000; Wu et al., 2000).
Of significant clinical relevance is the increasing evidence that neuronal degeneration in the adult organism is also accompanied by apparent cell cycle involvement. In both Alzheimer's disease (Smith and Lippa, 1995; Arendt et al., 1996, 1998a,b; Vincent et al., 1996, 1997, 1998; McShea et al., 1997, 1999; Nagy et al., 1997a, 1998;Busser et al., 1998; Smith et al., 1999) and stroke (Hayashi et al., 2000), cell cycle-related proteins appear in neurons at risk for death. These observations suggest that the same prohibitions against cell division that followed a neuron from the ventricular zone through the developmental process remain in effect until the end of life. A persistent question that accompanies this conclusion, however, is whether the immunocytochemical appearance of cell cycle enzymes is indicative of a true cell cycle or only the dysregulation of enzyme synthesis. Direct proof of DNA replication has been missing in the Alzheimer's disease brain. We report here the use of fluorescentin situ hybridization (FISH) to examine human hippocampal neurons in autopsy material from Alzheimer's disease patients and controls. We find direct evidence for attempted cell cycling in the Alzheimer's disease neurons and propose that the ultimate death of the nerve cells is attributable to the genetic imbalance caused by the tetraploid status of their genome.
This work describes original experiments that have not been published previously in any venue except in abstract form (Yang et al., 2000).
MATERIALS AND METHODS
FISH probes. Chromosome 11 probes were made from two overlapping bacterial artificial chromosomes (BACs) obtained from RPCI11 library (reference numbers 794I11 and 407N16). These probes come from the region of human chromosome 11 that encodes the β-site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1), located at 11 (q23.3-q24.1). The centromeric probes for chromosomes 18 and 21 were obtained from Oncor (Gaithersburg, MD). The chromosome 18 probe was supplied with biotin-substituted uridine residues, whereas the chromosome 21 probe was supplied with digoxygenin-substituted residues. A second set of probes for chromosome 21 was made from two overlappingBACs (RPCI11 library, reference numbers 35J21 and 575G22) derived from the BACE2 locus (at 21q22.3). BAC sequences were labeled by standard nick-translation protocols using digoxygenin-labeled dUTP. After digoxygenin labeling, probes were concentrated with human Cot-1 (Life Technologies) for at least 2 hr to block repeat human sequences. The chromosomal location of the various probes is illustrated in Figure 2.
Fig. 2.

Chromosomal location of the probes used in this study. The arrows indicate the approximate location of the probes displayed on chromosomal drawings copied from the NCBI web site (http://www.ncbi. nlm.nih.gov/LocusLink/).
Histology. Formalin-fixed brain autopsy tissue was obtained from the Neuropathology Core of the University Hospitals/Case Western Reserve University Alzheimer's Disease Research Center. After being embedded in paraffin, 10 μm sections were cut and mounted on Superfrost+ glass slides. The specifics of each of the cases examined are detailed in Table 1. Cultured human lymphocytes (line GM07038A) were received from the Human Genetics service of University Hospitals/Case Western Reserve University. Cells were grown in suspension for 70 hr, treated with colcemid (Life Technologies; 10 μg/ml), grown for an additional 2 hr, then swelled and lysed in hypotonic solution, fixed in methanol/acetic acid (3:1), smeared onto glass slides, and dried.
Table 1.
Summary of human material used
| Case # | PMI (hr) | Age (years) | Sex | |
|---|---|---|---|---|
| Control cases | 1 | 13 | 69 | F |
| 2 | 6 | 74 | F | |
| 3 | 16 | 72 | F | |
| 4 | 19 | 87 | F | |
| Average | 13.5 | 75.5 | ||
| Alzheimer's disease cases | 5 | 3 | 80 | M |
| 6 | 34 | 85 | M | |
| 7 | 24 | 83 | M | |
| 8 | 13.5 | 96 | ||
| 9 | 8 | 69 | M | |
| 10 | 23 | 77 | F | |
| 11 | 6 | 68 | F | |
| Average | 15.9 | 79.7 |
All material was from the University Hospitals of Cleveland/Case Western Reserve University Alzheimer's Disease Research Center Neuropathology Core. PMI, Postmortem interval (time in hours between death and autopsy); age, age at death.
For lymphocytes, the dried slides were transferred to 2× SSC for 60 min at 37°C, then dehydrated and air dried. The bound DNA was then denatured in 70% formamide/2× SSC at 73°C for 2 min, after which the sections were exposed to digoxygenin-substitutedBACE1 probe and incubated overnight at 37°C. The probe had been denatured previously at 73°C for 10 min and reannealed in 50% formamide/2× SSC hybridization buffer for at least 2 hr. Sites of hybridization were revealed with mouse anti-digoxygenin antibody (Boehringer Mannheim, Indianapolis, IN) and an Alexa-488-substituted goat anti-mouse IgG (Molecular Probes, Eugene, OR).
For the CNS tissue, sections were deparaffinized, pretreated with 30% Pretreatment Powder (Oncor) for 15 min at 45°C, and then digested with protease (Oncor; 0.25 mg/ml) for 25 min at 45°C. After rinsing, slides were dehydrated through alcohols and allowed to air dry. Probe was applied to the section, and a glass coverslip was overlaid to prevent evaporation. Slides were heated to 90°C for 12 min to denature the DNA and then incubated overnight at 37°C. Slides were soaked in 50% formamide/2× SSC for 5 min to remove the coverslip; then they were rinsed twice in the same buffer for 15 min at 37°C and once in 0.1× SSC for 30 min and transferred to phosphate buffer at room temperature. Primary antibody was applied at a 1:100 dilution (in PBS with 1% normal goat serum) for 20 min at 37°C; then the sections were rinsed three times in phosphate buffer. Secondary antibody was applied at a 1:150 dilution, and the sections were incubated for 20 min at 37°C. After rinsing, the sections were counterstained with propidium iodide (1:40).
Quantitation. Sections were labeled as described above. For each control and Alzheimer's disease case, 100 hippocampal cells were examined in each of the four CA fields. A cell was counted only if the equator of the nucleus was visible in the section. The number of spots was determined at 1000× under fluorescent illumination. Counts of two independent investigators (Y.Y. and K.H.) were comparable. Four control and six Alzheimer's disease cases were counted, and the frequency of cells with zero through four spots was tallied.
RESULTS
Examination of cresyl violet-stained sections of hippocampus and basal forebrain from the Alzheimer's disease cases listed in Table1 showed tissue atrophy and significant cell loss (Fig. 1A–D). As reported previously by several laboratories, including our own, immunocytochemical staining for cell cycle enzymes revealed the unexpected presence of cyclin B and PCNA in hippocampal neurons. Our laboratory has also reported evidence of ectopic cell cycle proteins in two other sites of known cell loss, the dorsal raphe and the locus coeruleus (Busser et al., 1998). Another subcortical structure that is recognized as suffering significant cell loss in Alzheimer's disease is the basal nucleus of Meynert (Whitehouse et al., 1982), but this region has not yet been examined for the presence of cell cycle enzymes. Thus, as part of the present study, we have extended our analysis to the at-risk neurons in this location. We found clear evidence for the ectopic expression of the cell cycle proteins PCNA and cyclin B in the large neurons of this structure (Fig.1C,D). As described previously for hippocampus, dorsal raphe, and locus coeruleus, the fraction of the total neurons that were stained was small. This has been interpreted as a reflection that not all neurons are in the process of cell death at any one time. Rather, only the dying neurons will express PCNA and cyclin B. The location of the antigens was predominantly cytoplasmic, which is also consistent with previous findings. During a normal cell cycle, both PCNA and cyclin B are found in the nucleus, although both are known to shuttle in and out of the cytoplasm at other times. The reason for their predominantly cytoplasmic location in the Alzheimer's disease brain is unknown.
Fig. 1.

The appearance of the hippocampus and basal nucleus in Alzheimer's disease. A, Loss of hippocampal pyramidal cells is evident in this cresyl violet-stained field from AD case 3 when compared with control (B). A similar reduction in cell density is visible in the large neurons of the basal nucleus of Meynert (C), compared with control (D). Immunocytochemistry reveals presence of various cell cycle-related proteins, including the DNA polymerase subunit, PCNA (E), and the G2 cyclin, cyclin B (F).
The expression of the cell cycle proteins is not, in and of itself, direct evidence that a neuron has entered a formal cell cycle process. Indeed, the unusual cytoplasmic location of the PCNA and cyclin B proteins might be taken as evidence that some less organized derepression of protein synthesis is at work. If this were an animal model, we could approach the problem by supplying an exogenous DNA precursor such as BrdU several hours before death (Herrup and Busser, 1995). Even with this technique, however, questions could be raised about the contribution of DNA repair to the observed labeling. In any event, such an experimental approach in human patients with Alzheimer's disease is precluded. We decided instead to look for physical evidence of DNA replication by assessing the ploidy of the neuronal genome at different chromosomal locations through the use of FISH.
The human genome consists of 22 pairs of autosomes plus two sex chromosomes. Any unique DNA sequence should thus be represented twice, but only twice, in every cell nucleus (except in known polyploid cells such as liver hepatocytes). To approach the problem of DNA content in brain neurons, we selected four large DNA sequences as probes. The first probe consisted of overlapping BAC clones encompassing theBACE1 locus on human chromosome 11 (Fig.2). To assess the fidelity of our technique, we hybridized this probe to a population of colcemid-arrested human lymphocytes. After being derivatized with digoxygenin-substituted nucleotides, the BACE1 probe was applied to the cells, and its presence was revealed with an anti-digoxygenin antibody. The results are shown in Figure3. Most of the cells in our sample were in interphase, and two spots of fluorescence were seen easily in the nuclei of most of these cells (Fig. 3A). Mitotic figures were also present, and because the BACE1 locus is near the telomere of the long arm of chromosome 11 (q23.3-q24.1), labeling near the end of a mid-sized chromosome was consistently found in metaphase nuclei (Fig. 3B, arrows).
Fig. 3.

Localization of FISH probes in cultured cells. Pictured here is a metaphase spread of colcemid-blocked human lymphocytes hybridized with the BACE1 BACs.A, There are two spots (arrows) of hybridization in most interphase cells. B, Note the location of the specific hybridization (arrows) to the end of a condensed chromosome 11 in metaphase cells. Scale bar, 10 μm.
We then adapted the FISH techniques to formalin-fixed paraffin-embedded brain tissue. Distinct hybridization signals were found in all cell types, including neurons, astrocytes, and microglial cells. Specific hybridization was lost when excess unlabeled probe was added to the hybridization mixture (data not shown). As expected, when we examined control tissue sections from newborn cerebellum (Fig.4A), 40-year-old hippocampus (Fig. 4B), or age-matched controls for the Alzheimer's disease cases (Figs. 4C), no neuronal nucleus contained more than two spots of hybridization. Note that in this and all subsequent Figures, small specks of background fluorescence are visible. These are easily distinguished from the specific locus of hybridization by their small size and reduced luminosity. Tissue from Alzheimer's disease brain, however, produced a significantly different picture. Hippocampal pyramidal cells from seven pathologically verified cases of Alzheimer's disease were examined (Table 1). Hybridization of the chromosome 11 probe in all seven cases consistently revealed cells with three and four spots of hybridization (Fig.5A,B). Such polyploid cells were found only in the Alzheimer's disease brain and only in regions in which nerve cell death had been reported previously. Samples from the same Alzheimer's brains, taken from regions unaffected by disease pathology, showed no such anomalies. Neurons in primary motor cortex (Fig. 5C) or cerebellum (Fig. 5D) showed two or fewer spots of bright fluorescence per nucleus. Thus, a fraction of the cells in Ammon's horn of patients with Alzheimer's disease have replicated their genome in the region around the end of the long arm of chromosome 11.
Fig. 4.

Normal diploid chromosomal complement is found in most neurons at most ages. In each panel, arrowsindicate specific spots of hybridization. A,BACE1 BAC hybridization to cerebellar granule cells of newborn human cerebellum (propidium iodide counterstain). The high density of cells in the internal granule cell layer results in the apparent overlap of cells in these 10 μm paraffin sections.B, BACE1 hybridization to hippocampal pyramidal cells of a nondemented 40-year-old female. C, Hippocampal neuron from area CA4 of a control case hybridized with theBACE1 probe. Two or fewer spots of hybridization are seen. The bright yellow/orange fluorescent material in the cell cytoplasm is lipofuscin, which autofluoresces at the wavelengths used. Note that in each of these control situations, we find two or fewer spots of hybridization per cell. Scale bars, 10 μm.
Fig. 5.

FISH applied to neurons in affected areas of Alzheimer's disease brain. A, Confocal image of a hippocampal neuron from area CA3 of an Alzheimer's disease brain (case 9). Note the presence of four bright spots of fluorescence (arrows) in the nucleus, suggesting a doubling of the number of chromosomes 11 in this cell. B, Hippocampal neuron from area CA4 of the same case (9). Three spots of bright hybridization are found in this single nucleus, revealing aneuploidy for this portion of chromosome 11. C, Neuron from the motor cortex of the same Alzheimer's disease case (9) hybridized with the same BACE1 probe. D, Granule cells from the cerebellum of an Alzheimer's disease brain. In these neurons, as in all others that we examined from this region, two or fewer spots of hybridization (arrows) were found. A was taken on a Zeiss confocal microscope.B–D were taken at 1000× on a Leitz fluorescence microscope. Scale bars: A, D, 10 μm.
To determine whether this apparent DNA replication was unique to chromosome 11, we used a second set of overlapping BACs from theBACE2 locus. Using identical protocols, we hybridized this probe to hippocampal sections from the same Alzheimer's disease cases. As with the chromosome 11 probes, three and four spots of FISH hybridization were found with this second probe in Alzheimer's disease cases (Fig. 6A). TheBACE2 locus is near the end of the long arm of human chromosome 21 (q22.3) (Fig. 2), and thus, at least two chromosomal loci have replicated their DNA during the progression of the disease in these individuals.
Fig. 6.

Different probes from different chromosomes all reveal aneuploidy in hippocampal neurons in Alzheimer's disease cases.A, Hippocampal pyramidal cell from area CA4 of Alzheimer's disease case 9 probed with the BACE2 probe. Thearrows indicate four spots of specific fluorescence found in the nucleus. B, A cell from the same region of the same case hybridized with the chromosome 21 centromeric probe. Three spots of hybridization are clearly visible in the nucleus.C, A CA3 hippocampal pyramidal neuron from control brain. Two spots of hybridization (arrows) were found when the same chromosome 21 centromeric probe was used. No cell with more than two spots was found in this or any other control case.D, A confocal image of a cell from Alzheimer's disease case 9 hybridized with the probe to the centromere of chromosome 18. Note the three hybridization spots in the nucleus of this cell. Scale bar, 10 μm.
During the cell cycle, DNA replication begins at origins of replication in the arms of the chromosomes and ultimately involves the centromeric regions (Nasmyth et al., 2000). To gain additional insight into whether the entire genome had replicated, we used a probe specific to centromeric satellite DNA of chromosome 21 (Fig. 2). This third probe also revealed three and four spots in hippocampal neurons of Alzheimer's disease tissue (Fig. 6B), but not in age-matched control neurons (Fig. 6C). We obtained identical results with a centromeric probe from chromosome 18; this probe too revealed euploidy in control neurons and polyploidy in Alzheimer's disease tissue (Fig. 6D).
The qualitative discovery of polyploidy in the at-risk neurons in Alzheimer's disease is strong evidence that the cells have entered a cell cycle that has proceeded through S phase. Because no neuron has ever been observed in M phase, however, the cells must remain stalled in a state resembling G2. To add quantitative detail to the qualitative picture, we performed cell counts of the FISH preparations. Six of the Alzheimer's disease cases as well as four controls were examined. A minimum of 400 hippocampal pyramidal neurons were counted per case. In each of the Alzheimer's disease brains, but in none of the controls, we found cells with three or four spots of hybridization. When the results from counts using all four probes were combined, the frequencies ranged from 0.005 to 0.100 of the total number of cells, with a mean frequency of 0.037 ± 0.028 (SD) (Table 2). As discussed below, these values suggest that the cells remain in a tetraploid state for some time before dying.
Table 2.
Genomic content of hippocampal pyramidal cells
| Spots per nucleus | |||
|---|---|---|---|
| 0 | 1–2 | 3–4 | |
| Control (n = 4) | 36.4% ± 6.2 | 63.6% ± 6.2 | 0 |
| Alzheimer's disease (n = 6) | 25.1% ± 3.9 | 71.1% ± 3.3 | 3.7% ± 0.9 |
Cells were scored for the presence of fluorescent spots indicating the presence of one of four specific gene loci. Four hundred hippocampal cells were counted for each case, 100 cells from each of the four CA fields. Values represent the pooled findings from all cases and all probes expressed as the percentage of the total cells counted. Note that the frequency of cells with zero spots or with one to two spots is not significantly different and suggests no major technical difference in hybridization efficiency between Alzheimer's disease and control cells.
The observation of DNA replication is not unique to hippocampus. We examined the large neurons of the basal nucleus of Meynert using both the chromosome 11 and 21 BAC probes (Fig.7). The results were nearly identical to those from hippocampus. In basal forebrain neurons from patients with Alzheimer's disease, we found consistent evidence for polyploidy, whereas in control cases we were able to demonstrate more than two spots of hybridization in only one case. The nucleus basalis has been reported by others to show significant regional variation in atrophy, and our cell cycle findings are consistent with these earlier results. In every case, however, when immunocytochemistry revealed the presence of ectopic cell cycle enzymes, FISH applied to adjacent sections revealed evidence for polyploidy in a fraction of the neuronal nuclei. One feature of the hybridization that differed significantly between the hippocampal and basal nuclear neurons was the distribution of the fluorescent spots. In hippocampus, the multiple spots tended to distribute in an apparently random fashion throughout the nucleus (Figs. 5A, 6A). In the basal nucleus, however, the four spots were most commonly seen as two doublets (Fig.7, insets).
Fig. 7.
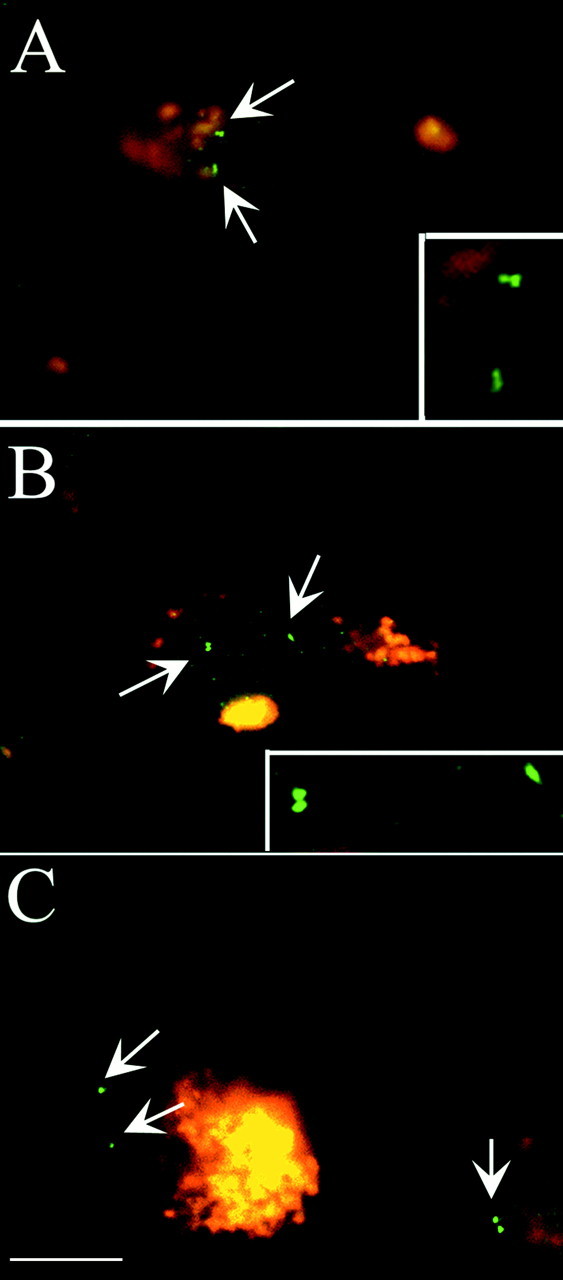
Aneuploidy in the large neurons of the basal nucleus of Meynert. A, B, Neurons illustrated here were probed with either BACE1(A) or BACE2 (B) probes. In each case, examples of polyploid neurons were found in Alzheimer's disease but not control brains. C, A control brain probed with the BACE1 probe from chromosome 11. This field is typical of the results in the control cases in which two or fewer spots of FISH hybridization (arrows) were found. Scale bar, 10 μm.
DISCUSSION
We report here direct evidence in support of the hypothesis that the neurons in the Alzheimer's disease brain reenter the cell cycle before they die. The methodology we used to determine this, FISH, is uniquely suited to assess cell cycle progression because it clearly distinguishes DNA replication from other synthetic events such as DNA repair. The observation that loci on three different chromosomes are present in four copies rather than two suggests that in Alzheimer's disease the entire genome of the neuron is duplicated before its death. Our data show that the association of polyploid cells with the neurons of an at-risk population is not restricted to hippocampal pyramidal cells. The large catecholaminergic neurons of the basal nucleus of Meynert have also undergone DNA replication. Although we have not yet applied FISH to the locus coeruleus or dorsal raphe, we have every reason to believe that earlier immunocytochemical findings in these regions are also indicative of true cell cycle anomalies. Thus, entry into an unscheduled cell cycle appears to precede the neuronal loss in Alzheimer's disease, consistent with earlier immunocytochemical studies (Vincent et al., 1996; McShea et al., 1997;Nagy et al., 1997a; Arendt et al., 1998b; Busser et al., 1998). Note, however, that immunocytochemical demonstration of cell cycle enzyme expression does not provide such unambiguous evidence for a concerted cell division process in a normally postmitotic neuron.
Full DNA replication means that both the G1 and S phases have been completed. The presence of cyclin B (a G2 cyclin) suggests that the cycle has proceeded into G2 and that there is a block in the cell cycle at some point before M phase. The nature of this block is unknown, but it has been shown that the cdc2/cyclin B complex is present and active in Alzheimer's disease brain tissue (Vincent et al., 1996, 1997). Active cdc2 is a mitosis-promoting factor capable of driving cells in culture into the series of elaborate chromosomal movements required for M phase of the cell cycle. The presence of this activity, along with evidence for a completed S phase, raises the question of what factor or factors are missing in the adult neurons that block their resolution of the cell cycle process.
This question is of particular interest, given the spatial distribution of the spots of hybridization in our samples. In hippocampus, the spots are scattered throughout the cell, as illustrated in Figures 5 and 6. This distribution suggests that proteins such as the cohesin complexes that normally hold replicated daughter chromosomes together (for review, see Nasmyth et al., 2000) must either be degraded or perhaps never synthesized. By contrast, in the basal nucleus, the four hybridization signals almost always appear as two pairs of spots (Fig.7). This picture is more consistent with a cell in which a well regulated division process is halted at a normal G2 cell cycle checkpoint. The genome content is 4n rather than 2n, yet the cell has maintained the pairing of the newly replicated sister chromatids in anticipation of chromosome condensation. Indeed, the picture in hippocampus closely resembles that found in a cell in which the cycle has processed to anaphase when the SCC1 cohesin subunits are cleaved, allowing centromere separation (Waizenegger et al., 2000). There has been no chromosome condensation, however, and the suggestion is that the genetic material of the pyramidal cells is in substantial disarray. The reason for this regional difference is unclear, as are its consequences.
The details of our findings have important implications for the mechanics of the neuronal cell death process in Alzheimer's disease. Our quantitative analysis suggests that nearly 4% of the at-risk neurons are in a polyploid state in the hippocampus of affected individuals. Because our FISH sampling represents a “snapshot” of a single moment in time, this value is unexpectedly large. Indeed, 4% is likely to be an underestimate of the true number of polyploid neurons because, as indicated by the 0 spot column in Table 2, the efficiency of hybridization is not 100%. A simple calculation demonstrates the significance of this value. In the developing mouse cerebellum, most neurons die within 20 hr after entering the cell cycle (Herrup and Busser, 1995) and thus would be visibly labeled with cell cycle markers for only that short amount of time. Given that the time course of Alzheimer's disease is roughly 10 years from diagnosis to death, and if the same rate of cell death were applied, then the fraction of neurons visibly present in a cell cycle would be (the number of hours in 10 years) or . On the basis of these simple assumptions, our observed rates suggest that the polyploid neurons must survive for hundreds of days after their genome has replicated. We therefore propose that the tetraploidy of the cells is itself lethal and represents the fundamental cause of the demise of the cells. The slow loss of synapses and atrophy of neuritic processes is viewed in this model as a consequence of the persistent abnormal gene dose. Recently Stadelmann et al. (1999) reported finding evidence for the apoptotic demise of the Alzheimer's disease neurons. Their observed fraction of caspase-3-positive cells ( to ) is close to the rate calculated above and suggests that this process represents the final rapid demise of the polyploid neurons.
This is only the second reported application of FISH techniques to cells of the human adult CNS and the first using paraffin-embedded histological material. One previous report has appeared (Sendera et al., 2000) in which FISH was used on fresh frozen cerebellar tissue to demonstrate trisomy for chromosome 21 in a suspected case of Down's syndrome. From a technicalstandpoint, it is noteworthy that our material was obtained from conventional brain autopsy tissue that had been formalin-fixed in the standard manner and received no other special treatment. Thus, the approach we have adopted will be widely useful in exploring somatic mosaicism of genetic content. Aneuploidies such as trisomy 21 are devastating conditions leading to severe mental retardation. When the nondisjunction event occurs after gametogenesis, however, the condition can be much milder. Through the application of FISH, the extent of mosaicism can be retrospectively determined on a region by region basis using normally acquired pathology samples of brain tissue. The correlation of the degree of mosaicism in different areas with the clinical description of mental function may help provide insight into the mechanisms of action of different chromosomal aneuploidies. This analysis would proceed in much the same the way that chimeras in chick and mouse have been used to discover the sites of action of various genetic mutations.
It should be noted that the existence of chromosomal imbalance in Alzheimer's disease has been proposed previously (Potter, 1991) on the basis of the location of the βAPP gene on chromosome 21 (Kang et al., 1987; Tanzi et al., 1987) and the early onset of Alzheimer's disease-like pathology in the brains of patients with Down's syndrome (trisomy 21). In support of this hypothesis, abnormal segregation of chromosomes has been reported in fibroblast cells bearing certain presenilin-1 mutations (Li et al., 1997; Geller and Potter, 1999). Our results suggest that the extent of chromosomal imbalance in Alzheimer's disease does exist, but it represents full DNA replication rather than a specific nondisjunction-induced trisomy of chromosome 21.
In Alzheimer's disease as well as in experimental animals, attempting cell division in a mature neuron appears to be a lethal effort (al-Ubaidi et al., 1992; Feddersen et al., 1992; Jacks et al., 1992;Lee et al., 1992; Herrup and Busser, 1995). The source of the signal that drives the neurons in Alzheimer's disease to begin the process thus becomes a question of utmost importance. The β-amyloid peptide itself has been shown to be capable of driving cell division and cell death in cultured neurons (Copani et al., 1999); in a similar assay, conditioned medium from β-amyloid-stimulated microglial cells can also trigger neuronal cell division leading to cell death (Giovanni et al., 1999; Wu et al., 2000). These latter data are particularly intriguing given the epidemiological findings of a protective effect of high doses of nonsteroidal anti-inflammatory drugs (P. McGeer and McGeer, 1995; E. G. McGeer and McGeer, 1997). Thus, the inflammatory process may be one source of the mitotic “pressure” in Alzheimer's disease brain. Whatever the cause, however, the finding that unscheduled reentrance into the cell cycle is the likely cause of the neuronal degeneration in Alzheimer's disease opens various new therapeutic avenues.
Footnotes
This work was supported by the National Institute on Aging through Alzheimer's Disease Research Center Grant AG08120 and by National Institutes of Neurological Diseases and Stroke Grant NS20591 to K.H. It was also made possible by a generous grant from the Blanchette Hooker Rockefeller Foundation.
Correspondence should be addressed to Karl Herrup, Alzheimer Research Laboratory, Department of Neuroscience, Case Western Reserve University, School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106. E-mail: kxh26@po.cwru.edu.
REFERENCES
- 1.al-Ubaidi MR, Hollyfield JG, Overbeek PA, Baehr W. Photoreceptor degeneration induced by the expression of simian virus 40 large tumor antigen in the retina of transgenic mice. Proc Natl Acad Sci USA. 1992;89:1194–1198. doi: 10.1073/pnas.89.4.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arendt T, Rodel L, Gartner U, Holzer M. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer's disease. NeuroReport. 1996;7:3047–3049. doi: 10.1097/00001756-199611250-00050. [DOI] [PubMed] [Google Scholar]
- 3.Arendt T, Holzer M, Gartner U. Neuronal expression of cyclin dependent kinase inhibitors of the INK4 family in Alzheimer's disease. J Neural Transm. 1998a;105:949–960. doi: 10.1007/s007020050104. [DOI] [PubMed] [Google Scholar]
- 4.Arendt T, Holzer M, Gartner U, Bruckner MK. Aberrancies in signal transduction and cell cycle related events in Alzheimer's disease. J Neural Transm [Suppl] 1998b;54:147–158. doi: 10.1007/978-3-7091-7508-8_14. [DOI] [PubMed] [Google Scholar]
- 5.Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci. 1998;18:2801–2807. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Copani A, Condorelli F, Caruso A, Vancheri C, Sala A, Giuffrida Stella AM, Canonico PL, Nicoletti F, Sortino MA. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 1999;13:2225–2234. [PubMed] [Google Scholar]
- 7.Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT. Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron. 1992;9:955–966. doi: 10.1016/0896-6273(92)90247-b. [DOI] [PubMed] [Google Scholar]
- 8.Geller LN, Potter H. Chromosome missegregation and trisomy 21 mosaicism in Alzheimer's disease. Neurobiol Dis. 1999;6:167–179. doi: 10.1006/nbdi.1999.0236. [DOI] [PubMed] [Google Scholar]
- 9.Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F x DP, in B-amyloid-induced neuronal death. J Biol Chem. 1999;274:19011–19016. doi: 10.1074/jbc.274.27.19011. [DOI] [PubMed] [Google Scholar]
- 10.Giovanni A, Keramaris E, Morris EJ, Hou ST, O'Hare M, Dyson N, Robertson GS, Slack RS, Park DS. E2F1 mediates death of β-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J Biol Chem. 2000;275:11553–11560. doi: 10.1074/jbc.275.16.11553. [DOI] [PubMed] [Google Scholar]
- 11.Hayashi T, Sakai K, Sasaki C, Zhang WR, Abe K. Phosphorylation of retinoblastoma protein in rat brain after transient middle cerebral artery occlusion. Neuropathol Appl Neurobiol. 2000;26:390–397. doi: 10.1046/j.1365-2990.2000.00264.x. [DOI] [PubMed] [Google Scholar]
- 12.Herrup K, Busser JC. The induction of multiple cell cycle events precedes target-related neuronal death. Development. 1995;121:2385–2395. doi: 10.1242/dev.121.8.2385. [DOI] [PubMed] [Google Scholar]
- 13.Jacks T, Fazeli A, Schmitt E, Bronson R, Goodell M, Weinberg R. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 14.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 15.Lee E-H, Hu N, Yuan SS, Cox LA, Bradley A, Lee WH, Herrup K. Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev. 1994;8:2008–2021. doi: 10.1101/gad.8.17.2008. [DOI] [PubMed] [Google Scholar]
- 16.Lee EY-HP, Chang C-Y, Hu N, Wang Y-CJ, Lai C-C, Herrup K, Lee W-H, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Xu M, Zhou H, Ma J, Potter H. Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell. 1997;90:917–927. doi: 10.1016/s0092-8674(00)80356-6. [DOI] [PubMed] [Google Scholar]
- 18.McGeer EG, McGeer PL. The role of the immune system in neurodegenerative disorders. Mov Disord. 1997;12:855–858. doi: 10.1002/mds.870120604. [DOI] [PubMed] [Google Scholar]
- 19.McGeer P, McGeer E. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Rev. 1995;21:195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 20.McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- 21.McShea A, Wahl AF, Smith MA. Re-entry into the cell cycle: a mechanism for neurodegeneration in Alzheimer disease. Med Hypotheses. 1999;52:525–527. doi: 10.1054/mehy.1997.0680. [DOI] [PubMed] [Google Scholar]
- 22.Nagy Z, Esiri MM, Smith AD. Expression of cell division markers in the hippocampus in Alzheimer's disease and other neurodegenerative conditions. Acta Neuropathol (Berl) 1997a;93:294–300. doi: 10.1007/s004010050617. [DOI] [PubMed] [Google Scholar]
- 23.Nagy Z, Esiri MM, Cato AM, Smith AD. Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol (Berl) 1997b;94:6–15. doi: 10.1007/s004010050665. [DOI] [PubMed] [Google Scholar]
- 24.Nagy Z, Esiri MM, Smith AD. The cell division cycle and the pathophysiology of Alzheimer's disease. Neuroscience. 1998;87:731–739. doi: 10.1016/s0306-4522(98)00293-0. [DOI] [PubMed] [Google Scholar]
- 25.Nasmyth K, Peters J-M, Uhlmann F. Splitting the chromosome: cutting the ties that bind sister chromatids. Science. 2000;288:1379–1384. doi: 10.1126/science.288.5470.1379. [DOI] [PubMed] [Google Scholar]
- 26.Park DS, Morris EJ, Greene LA, Geller HM. G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J Neurosci. 1997;17:1256–1270. doi: 10.1523/JNEUROSCI.17-04-01256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park DS, Morris EJ, Bremner R, Keramaris E, Padmanabhan J, Rosenbaum M, Shelanski ML, Geller HM, Greene LA. Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J Neurosci. 2000;20:3104–3114. doi: 10.1523/JNEUROSCI.20-09-03104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Potter H. Review and hypothesis: Alzheimer disease and Down syndrome–chromosome 21 nondisjunction may underlie both disorders. Am J Hum Genet. 1991;48:1192–1200. [PMC free article] [PubMed] [Google Scholar]
- 29.Sendera TJ, Ma SY, Jaffar S, Kozlowski PB, Kordower JH, Mawal Y, Saragovi HU, Mufson EJ. Reduction in TrkA-immunoreactive neurons is not associated with an overexpression of galaninergic fibers within the nucleus basalis in Down's syndrome. J Neurochem. 2000;74:1185–1196. doi: 10.1046/j.1471-4159.2000.741185.x. [DOI] [PubMed] [Google Scholar]
- 30.Smith MZ, Nagy Z, Esiri MM. Cell cycle-related protein expression in vascular dementia and Alzheimer's disease. Neurosci Lett. 1999;271:45–48. doi: 10.1016/s0304-3940(99)00509-1. [DOI] [PubMed] [Google Scholar]
- 31.Smith TW, Lippa CF. Ki-67 immunoreactivity in Alzheimer's disease and other neurodegenerative disorders. J Neuropathol Exp Neurol. 1995;54:297–303. doi: 10.1097/00005072-199505000-00002. [DOI] [PubMed] [Google Scholar]
- 32.Stadelmann C, Deckwerth T, Srinivasan A, Bancher C, Brück W, Jellinger K, Lassmann H. Activation of caspase-3 in single neurons and autophagic granules of granulovacular degeneration in Alzheimer's disease: evidence for apoptotic cell death. Am J Pathol. 1999;155:1459–1466. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St. George-Hyslop P, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235:880–884. doi: 10.1126/science.2949367. [DOI] [PubMed] [Google Scholar]
- 34.Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer's disease? J Cell Biol. 1996;132:413–425. doi: 10.1083/jcb.132.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vincent I, Jicha G, Rosado M, Dickson D. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vincent I, Zheng JH, Dickson DW, Kress Y, Davies P. Mitotic phosphoepitopes precede paired helical filaments in Alzheimer's disease. Neurobiol Aging. 1998;19:287–296. doi: 10.1016/s0197-4580(98)00071-2. [DOI] [PubMed] [Google Scholar]
- 37.Waizenegger IC, Hauf S, Meinke A, Peters JM. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell. 2000;103:399–410. doi: 10.1016/s0092-8674(00)00132-x. [DOI] [PubMed] [Google Scholar]
- 38.Whitehouse P, Price D, Struble R, Clark A, Coyle J, DeLong M. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 39.Wu Q, Combs C, Cannady S, Geldmacher D, Herrup K. Beta-amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol Aging. 2000;21:797–806. doi: 10.1016/s0197-4580(00)00219-0. [DOI] [PubMed] [Google Scholar]
- 40.Yang Y, Cicero SA, Geldmacher DS, Herrup K. DNA replication occurs in at-risk neurons in the Alzheimer's disease brain. Soc Neurosci Abstr. 2000;26:1550. [Google Scholar]