

Abstract
Minocycline, a semisynthetic tetracycline derivative, protects brain against global and focal ischemia in rodents. We examined whether minocycline reduces excitotoxicity in primary neuronal cultures. Minocycline (0.02 μm) significantly increased neuronal survival in mixed spinal cord (SC) cultures treated with 500 μm glutamate or 100 μm kainate for 24 hr. Treatment with these excitotoxins induced a dose-dependent proliferation of microglia that was associated with increased release of interleukin-1β (IL-1β) and was followed by increased lactate dehydrogenase (LDH) release. The excitotoxicity was enhanced when microglial cells were cultured on top of SC cultures. Minocycline prevented excitotoxin-induced microglial proliferation and the increased release of nitric oxide (NO) metabolites and IL-1β. Excitotoxins induced microglial proliferation and increased the release of NO metabolites and IL-1β also in pure microglia cultures, and these responses were inhibited by minocycline. In both SC and pure microglia cultures, excitotoxins activated p38 mitogen-activated protein kinase (p38 MAPK) exclusively in microglia. Minocycline inhibited p38 MAPK activation in SC cultures, and treatment with SB203580, a p38 MAPK inhibitor, but not with PD98059, a p44/42 MAPK inhibitor, increased neuronal survival. In pure microglia cultures, glutamate induced transient activation of p38 MAPK, and this was inhibited by minocycline. These findings indicate that the proliferation and activation of microglia contributes to excitotoxicity, which is inhibited by minocycline, an antibiotic used in severe human infections.
Keywords: ischemia, Alzheimer's disease, inflammation, glutamate, mitogen-activated protein kinase, MAPK, cell culture
Neurotoxicity of excitatory glutamate is a contributing factor in acute neuronal damage, including traumatic brain injury and stroke, and in most of the chronic neurodegenerative diseases, such as Alzheimer's disease and multiple sclerosis (Beal, 1995; Dirnagl et al., 1999; Lee et al., 1999; Smith et al., 2000). Overstimulation of the NMDA and AMPA/kainate glutamate receptors (GluR) is a key event in excitotoxicity (Choi, 1992; Dugan and Choi, 1994; Lee et al., 1999). However, identification of indirect mechanisms involving non-neuronal cells in this cascade is important, because inhibition of NMDA or AMPA/kainate receptors has been associated with toxic side effects and because activation of these receptors may occur too early for the clinically relevant time frame of therapeutic intervention (Barone and Feuerstein, 1999; DeGraba and Pettigrew, 2000).
Inflammation has an important role in pathogenesis of brain diseases (Beal, 1995; Rothwell et al., 1996; Barone and Feuerstein, 1999; Dirnagl et al., 1999; Lee et al., 1999; McGeer and McGeer, 1999;Touzani et al., 1999; Cooper et al., 2000). Inflammation is regarded as an attractive pharmacological target, because it progresses over several days after injury and because intervention with inflammatory mechanisms, which are not fundamental for physiological brain functions, may not result in intolerable side effects (Barone and Feuerstein, 1999), as the wide clinical use of nonsteroidal anti-inflammatory drugs demonstrates.
The fundamental observation suggesting a role of glia in glutamate-induced neuronal death is that excitotoxicity is associated with activation of astrocytes and microglia in vivo(Kreutzberg, 1996; Schousboe et al., 1997). Moreover, induction of interleukin-1β (IL-1β) and tumor necrosis factor-α (TNFα), or inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), occurs in response to brain injury (Munoz-Fernandez and Fresno, 1998;Barone and Feuerstein, 1999; Dirnagl et al., 1999; Venters et al., 2000) Furthermore, inhibition of IL-1β reduces neuronal damage caused by excitotoxin injections, traumatic brain injury, or ischemia (Rothwell et al., 1996; Sanderson et al., 1999; Touzani et al., 1999). Finally, Tsirka and coworkers (Tsirka et al., 1996; Rogove and Tsirka 1998; Wang et al., 1998) showed, using tissue plasminogen activator and plasminogen-deficient mice, that microglia activation is necessary, but not sufficient, to trigger neuronal degeneration after excitotoxic insult. Altogether, compounds with anti-inflammatory properties or with an ability to inhibit microglial activation may represent potential therapies against excitotoxic brain diseases.
We have shown previously that minocycline, a tetracycline derivative with anti-inflammatory effects unrelated to its antimicrobial action, protects against brain ischemia (Yrjänheikki et al., 1998, 1999). The neuroprotection was associated with reduced activation of microglia, but not astrocytes, and with inhibition of induction of IL-1β-converting enzyme (ICE) mRNA, suggesting that minocycline may function by reducing cytotoxic properties of microglia, triggered either by ischemia or secondarily by excitotoxicity. In a transgenic animal model of Huntington's disease, minocycline inhibits caspase expression and delays mortality (Chen et al., 2000). Herein, we show that stimulation of glutamate receptors induces an activation and robust proliferation of microglia, which subsequently releases IL-1β and NO. This leads to neuronal cell death. We also demonstrate that glutamate-induced microglial activation occurs through the p38 mitogen-activated protein kinase (p38 MAPK) pathway and that minocycline inhibits p38 MAPK activation in microglia, reduces microglial activation, and provides neuroprotection against excitotoxicity.
MATERIALS AND METHODS
Primary spinal cord cultures. All cell culture protocols used in this study were approved by the Animal Care and Use Committee of Kuopio University and follow the NIH guidelines for humane care of animals. Spinal cords (SCs) were dissected out from 14-d-old rat embryos (Wistar, University of Kuopio, Finland), and the meninges and dorsal root ganglia were removed. Tissues were minced and trypsinized (0.25% trypsin-EDTA in 0.1 mphosphate buffer; Life Technologies, Roskilde, Denmark) for 15 min at 37°C. After centrifugation for 5 min at 800 rpm, the tissues were resuspended into high-glucose DMEM (Life Technologies) containing 10% fetal bovine serum (FBS) and 10% heat-inactivated horse serum (HS-HI) and triturated with a fire-polished Pasteur pipette. The single-cell suspension was collected, and its cell density was counted with a Burker hemocytometer. Cells were cultured on poly-l-lysine-coated 96-well plates (1 × 105 cells per well) or 24-well plates (2.5 × 105 cells per well) at 37°C in a 7.5% CO2 incubator. The medium was changed on the following day to DMEM containing 5% FBS and 5% HS-HI. After 4 d in vitro, 5 μm cytosine β-d-arabinofuranoside (Sigma, St. Louis, MO) was added for 24 hr to inhibit the growth of non-neuronal cells. This procedure results in mixed SC cultures, consisting of all neuronal populations (70%) present in the spinal cord, astrocytes (25%), and a few other non-neuronal cell types, including microglia (5%). We have shown previously that these neurons express functional glutamate receptors of different types and both phosphorylated and nonphosphorylated neurofilaments (Vartiainen et al., 1999).
Primary mixed glial and microglia cultures. Cortices and midbrains were dissected from newborn rat pup (Wistar), and the meninges and blood vessels were removed. Tissues were collected into 0.1 m PBS, washed four times with cold 0.1m PBS, homogenized mechanically without enzymes, and filtered through a 70 μm nylon cell strainer (Falcon; Fisher Scientific, Pittsburgh, PA). PBS was replaced in two centrifugation steps (1000 × g, for 10 min at 4°C) with low-glucose DMEM (Life Technologies) plus 10% FBS. The cells were suspended into culture medium and plated into cell culture plates at 6 × 105cells/mm2 and maintained in a humidified incubator at 37°C and 5% CO2. The medium was changed at the second day in vitro and once per week thereafter. This procedure results in mixed glial cultures, consisting of dividing astrocytes and microglial cells. After 2 weeks in vitro, microglia were harvested once per week by carefully collecting the medium until the mixed cultures were 2–3 months old. The purity of microglial cultures is ∼98%.
Cell exposure experiments. SC cultures were exposed at the seventh day in vitro to the following excitotoxic compounds (from Research Biochemicals, Natick, MA): 500 μm glutamate for 24 hr and 100 μm kainate for 24 hr. In some experiments, 0.02 μm minocycline (Sigma), 10 μm PD98059 (Tocris Cookson, Bristol, UK), a specific p44/42 MAPK inhibitor, or 10 μmSB203580 (Tocris Cookson), a specific p38 MAPK inhibitor, was added 30 min before administering excitotoxic compounds and were present through the exposure. In addition, for a dose–response curve of minocycline, the cultures exposed to 500 μm glutamate were treated with 2 μm, 200 nm, 20 nm, 10 nm, 1 nm, 0.1 nm, and 0.01 nmminocycline. All compounds were dissolved in the culture medium supplemented with 5% HS-HI that was used alone as a 0 control. The cultures were exposed for 24 hr, unless mentioned otherwise. In a set of experiments, microglial cells were cultured on top of 5-d-old SC cultures and exposed at day 7 as described above.
Mixed glial cultures were exposed to excitotoxins (500 μm) after 2 weeks in vitro and pure microglial cultures on the third day in vitro. Minocycline (0.2 μm) was added in some cultures 30 min before exposure. All compounds were dissolved in the cell culture medium supplemented with 10% FBS that was used alone as a 0 control.
Lactate dehydrogenase assay. The release of lactate dehydrogenase (LDH) was measured from culture medium using a Sigma Kinetic LDH kit. The culture medium samples were collected 24 hr after the onset of excitotoxic exposures, prepared cell-free by centrifugation, and measured immediately with a Multiskan MS ELISA reader (Labsystems, Helsinki, Finland), taking absorbance measurements at 340 nm every 30 sec for 3 min.
NO and IL-1β assays. The production of NO was quantified by measuring the released NO metabolites (nitrates and nitrites) with Griess reagent (Sigma). After a 24 hr exposure, the culture medium samples were collected and prepared cell-free by centrifugation. The medium (50 μl) was incubated with the same volume of Griess reagent at room temperature (RT) for 15 min before measuring absorbance at 540 nm in a Multiskan ELISA reader (Labsystems) with appropriate standards. Interleukin-1β samples were prepared similar to NO samples and determined using a rat interleukin-1β ELISA kit (Endogen, Woburn, MA) according to the manufacturer's instructions and a Multiscan MS ELISA reader (Labsystems).
Immunocytochemistry. The cultures were fixed with 4% paraformaldehyde in 0.1 m PBS for 20–30 min and rinsed in 0.1 m PBS. The nonspecific binding was blocked with 1% bovine serum albumin and 0.3% Triton X-100 in 0.1m PBS for 30 min at RT. Subsequently, cultures were incubated with primary antibodies to microglia (mouse monoclonal antibody, OX-42, against CD 11b surface antigen; 1:1500 dilution; Serotec, Oxford, UK), to neuronal nuclei (NeuN) (mouse monoclonal antibody; 1:100; Chemicon, Temecula, CA), or to phospho-p38 or -p42/44 MAPK (rabbit polyclonal antibody; 1:1000 and 1:250 dilution; New England Biolabs, Beverly, MA) in the blocking buffer for 48 hr at 4°C. The cultures were rinsed with 0.1 m PBS, incubated with the secondary antibody biotinylated anti-mouse IgG (1:200 dilution; Amersham Pharmacia Biotech, Berkshire, UK) or biotinylated anti-rabbit IgG (1:200 dilution; Amersham Pharmacia Biotech) for 2 hr at RT, and reacted with alkaline phosphatase– avidin–biotin complex (1:200 dilution; Vector Laboratories, Burlingame, CA) for 2 hr at room temperature and rinsed. The color was obtained from an alkaline phosphatase substrate kit (Vector Laboratories). In one set of experiments, phospho-p38 MAPK-immunostained cultures were double-stained with OX-42 antibody (1:800 dilution) using anti-mouse IgG fluorescein (1:70 dilution; Jackson ImmunoResearch, West Grove, PA) as a secondary antibody.
Immunoreactive cells were counted in a blinded manner from 6–10 random fields of 4 × 10−3mm2 area per well and from three to five wells per treatment.
Proliferation assay. Thymidine analog 5′-bromo-2′deoxyuridine-5′-monophosphate (BrdU) (Sigma) was added to the medium at 5 μm concentration. After 24 hr, the cultures were fixed with 4% paraformaldehyde in 0.1m PBS for 20–30 min and rinsed in 0.1m PBS. The DNA was denatured by incubating the cultures first with 50% formamide in 2× SSC for 2 hr at 65°C, followed by 2N HCl treatment for 30 min at 37°C. After neutralization with 0.1 m boric acid, pH 8.5, for 10 min at RT, the cultures were rinsed in 0.1 m PBS and incubated with a mouse monoclonal anti-BrdU (0.25 μg/ml; Boehringer Mannheim, Mannheim, Germany) in the blocking buffer at RT for 24 hr. Otherwise, the procedure was identical to the immunocytochemistry described above. The immunoreactive cells were counted in a ratio of total cell number from 6–10 random fields of 4 × 10−3 mm2area per well and from three to five wells per treatment in a blinded manner.
Detection of apoptosis. Apoptotic cell death was demonstrated by nuclear bis-benzimide (Hoechst 33342; 5 μg/ml for 5 min; Sigma) staining of 4% paraformaldehyde-fixed cultures and by DNA ladder formation. The bis-benzimide-stained cultures were examined in a Nikon (Tokyo, Japan) Diaphot 300 inverted fluorescence microscope equipped with appropriate filter sets.
For detection of DNA ladders, the cells were plated on the poly-l-lysine-coated six-well plates. After an exposure of 24 hr, the cells were gently rinsed with 0.1 m PBS and harvested with a cell scraper into 0.3 ml of ice-cold 0.1 mPBS. Genomic DNA was isolated and used as a template in a ligation-mediated PCR (LM-PCR) (ApoAlert LM-PCR Ladder Assay kit; Clontech, Palo Alto, CA). Briefly, the 5′-phosphorylated blunt ends of the DNA fragments generated by apoptosis were first ligated (T4 DNA ligase; 16°C, 12–16 hr) with dephosphorylated adapters composed of a 12-mer and a 24-mer. Because of the adapter design, only the 24-mer was ligated and the 12-mer was released in the subsequent heating step. The protruding ends were first filled in using a thermostable DNA polymerase (72°C, 8 min) and next subjected to PCR with the 24-mers serving as primers. Thermal cycling was composed of a denaturation step (94°C, 1 min) and combined with an annealing and polymerization step (72°C, 3 min). Samples of 5 μl were taken after completing of 23, 26, 29, 32, and 35 cycles and analyzed by electrophoresis on a 1.2% agarose gel. The DNA ladders were visualized after ethidium bromide staining using UV light, and the intensity of DNA fragments was analyzed by ImageQuant software. The LM-PCR experiment was run twice with two different set of samples.
Western blot analysis of mitogen-activated protein kinases.Cells were lysed in the lysis buffer (20 mmTris-HCl, 5 mm EGTA, 1% Triton X-100, 1 mm Na3V4O3, and 1 mm PMSF). After protein concentrations were analyzed with Bio-Rad (Hercules, CA) Protein Assay reagent, the samples were incubated with Laemli's buffer for 5 min at 100°C. Protein (10 μg) was loaded per lane, and the proteins were separated by electrophoresis in 10% SDS-PAGE gel, using a MiniProtean apparatus (Bio-Rad) and transferred onto Hybond-P membrane by MiniTransBlot (Bio-Rad) wet blotting apparatus according to the manufacturer's instructions. The membrane was blocked by 5% skimmed milk solution in 0.1 mm PBS for 1 hr at RT. Activated forms of p38 and p42/44 MAPKs were detected by incubating membranes with rabbit polyclonal phospho-p38 and -p42/42 MAPK antibodies (1:1000 dilution; New England Biolabs) for 2 hr at RT. Similarly, GluR4 AMPA/kainate receptor was detected by incubating membranes with rabbit polyclonal GluR4 antibody (1:200 dilution; Chemicon) for 2 hr at RT. Secondary HRP-labeled anti-rabbit IgG antibodies (1:2000; Amersham Pharmacia Biotech) were added for 2 hr at RT. The protein bands were visualized using ECL Plus kit (Amersham Pharmacia Biotech), scanned on STORM fluoroimager (Molecular Dynamics, Sunnyvale, CA), and quantified using ImageQuant software (Molecular Dynamics).
Statistical analysis. Data are presented as the mean ± SD. Statistical comparisons were made by single-factor ANOVA ,followed by Tukey's post hoc test controlling Type I error.p values < 0.05 were considered significant.
RESULTS
Minocycline prevents excitotoxic cell death in mixed SC cultures
Our preliminary experiments demonstrated that a 24 hr exposure to 500 μm glutamate or 100 μm kainate resulted in death of 60–40% of the neurons when compared with a 24 hr exposure to 1 mm glutamate or 500 μm kainate, which resulted in 100% neuronal death (data not shown). Neuron counts from the NeuN-immunostained cultures confirmed that 40–50% of the neurons survived the exposure to 500 μm glutamate and 100 μm kainate (Fig.1c). Administration of minocycline 30 min before the exposure to 500 μm glutamate reduced the LDH release dose-dependently (Fig. 1a). The IC50for minocycline was 10 nm, but the maximal and most consistent (small variation) protection (∼40%) was achieved by the treatment with 20 nm to 2 μm minocycline. The higher doses did not increase the neuronal survival (data not shown). Therefore, 20 nm minocycline was used for the next experiments. Figure 1b demonstrates that, in separate experiments, administration of 20 nm minocycline 30 min before the exposure decreased glutamate-induced LDH release by 58% (p < 0.01; single-factor ANOVA) and kainate-induced LDH release by 41% (p < 0.01; single-factor ANOVA) (Fig. 1b). The reduced loss of neurons in minocycline-treated cultures was also found in NeuN-stained cultures (Fig. 1c). Bis-benzimide revealed neuronal nuclei with fragmented DNA in glutamate and kainate-exposed cultures (Fig.1d–f). The DNA fragmentation, an indication of apoptosis, was confirmed by two separate semiquantitative ligation-mediated PCR assay experiments, which demonstrated that minocycline treatment reduced glutamate-induced DNA fragmentation (Fig.1g) by ∼86% according to quantitations of the DNA laddering (data not shown).
Fig. 1.
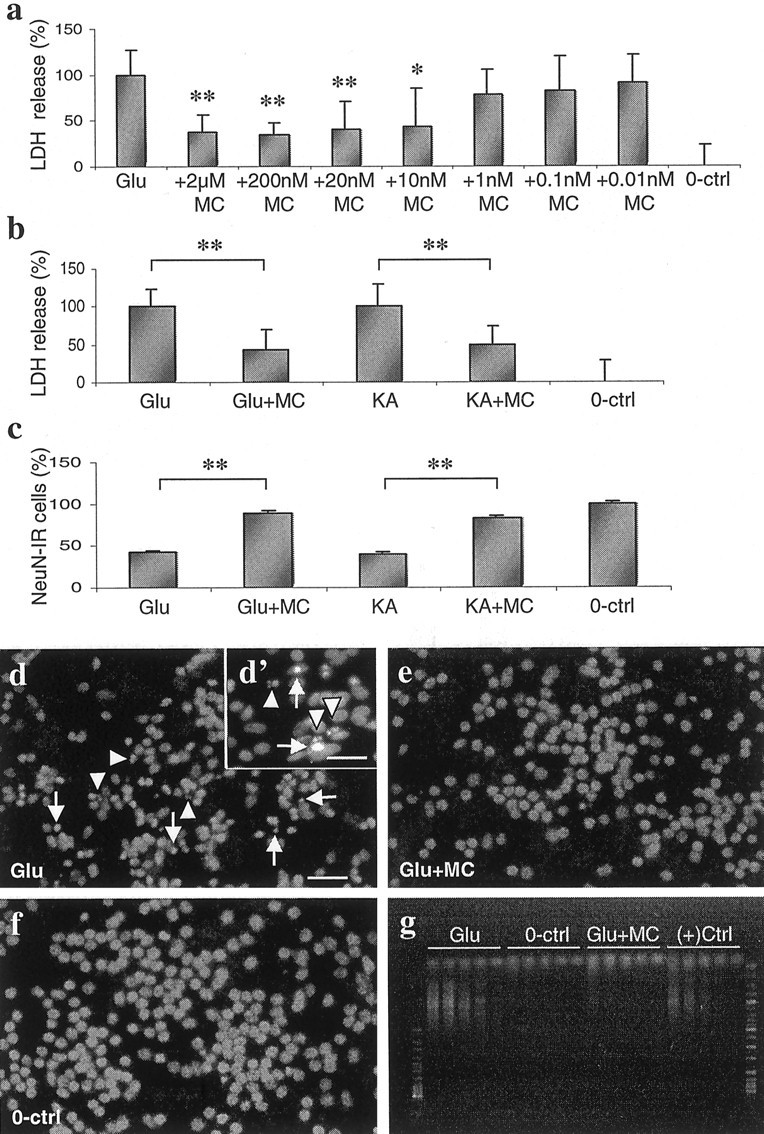
Excitotoxin-induced neuronal death is inhibited by minocycline treatment. a, Dose-dependent neuroprotection of minocycline detected by measuring LDH release from SC cell culture medium after 24 hr exposure to 500 μm glutamate with and without 2 μm to 0.01 nm minocycline treatment. Ten nanomolar minocycline was the lowest dose to provide significant neuroprotection, but the most consistent (with the least variation) and efficient protection was seen at 20 nm to 2 μm concentrations. Data are presented as the mean ± SD pooled from two independent experiments (n = 6). *p < 0.05; **p < 0.01 versus glutamate; single-factor ANOVA. 100% LDH release refers to the LDH release observed 24 hr after adding 500 mm glutamate alone.b, LDH release into SC cell culture medium, measured after a 24 hr exposure to 500 μm glutamate and 100 μm kainate (KA), is significantly reduced by 0.02 μm minocycline treatment. Data are presented as the mean ± SD pooled from three independent experiments (n = 12). **p < 0.01; single-factor ANOVA. 100% LDH release refers to the LDH release observed 24 hr after adding 500 μm glutamate or 100 μm kainate alone. c, The neuronal cell loss in SC cell cultures caused by 24 hr exposure to 500 μm glutamate and 100 μm kainate was decreased by 0.02 μm minocycline treatment. NeuN-immunoreactive cells were counted in a blind manner. Data are presented as the mean ± SD pooled from two independent experiments (n = 6). **p < 0.01; single-factor ANOVA. d–f, Representative fluorescence micrographs of bis-benzimide chromatin staining in SC cell cultures exposed to 500 μm glutamate for 24 hr with and without 0.02 μm minocycline treatment. Healthy surviving neurons have a round and large nucleus, whereas in preapoptotic neurons the nuclei are condensed (arrows) and in apoptotic neurons chromatin has fragmented (arrowheads). Preapoptotic and apoptotic neurons can been seen in cultures exposed to glutamate (d). In minocycline-pretreated (e) and control (f) cultures, preapoptotic and apoptotic neurons are not observed. Scale bars: d–g, 200 μm; d′, 100 μm.g, The apoptotic neuronal death and its inhibition by minocycline was also studied by semiquantitative ligation-mediated PCR assay. The number of DNA fragments generated in 500 μmglutamate-exposed (Glu), unexposed (0-ctrl), and 0.02 μmminocycline-treated glutamate-exposed (Glu+MC) cultures was amplified by 23, 26, 29, 32, and 35 (the five lanesfrom right to left in the gel, representing each kind of sample) thermal cycles and compared with a positive control sample [(+)Ctrl, provided by the manufacturer of the kit], which was run in parallel with other samples. DNA ladder became clearly visible only in the samples from glutamate-treated cells. The experiment was repeated twice with similar results.
Minocycline inhibits microglial proliferation, which precedes increased LDH release in mixed SC cultures
After a 24 hr exposure to 500 μm glutamate or 100 μm kainate, a twofold to threefold increase in the number of microglial cells was found (Fig.2a,b). Minocycline treatment completely prevented the increase of OX-42-positive cells in glutamate- or kainate-exposed cultures. Minocycline slightly reduced the number of microglial cells even in control cultures, which were not challenged with excitotoxins (Fig. 2a). When comparing the time course of the changes in OX-42-positive cells and LDH release, 500 μm glutamate increased the number of OX-42-positive cells from 65 ± 14 to 132 ± 13 per 4 × 10−3 mm2(mean ± SD; p < 0.01; single-factor ANOVA) at 12 hr and to 165 ± 16 per 4 × 10−3 mm2(p < 0.05; single-factor ANOVA) at 24 hr, whereas the increase in LDH was not significantly changed at 12 hr (p = 0.5; single-factor ANOVA) but became significant only at 24 hr after administration of glutamate (p < 0.01; single-factor ANOVA). In addition, sublethal concentrations of excitotoxins, 20 μmkainate or 50 μm glutamate, increased the number of OX-42-positive cells twofold to threefold, and this increase was inhibited by 0.02 μm minocycline (data not shown). Also, neuron-free cultures from newborn rat midbrain and forebrain responded to excitotoxins with a twofold to threefold increase in the number of microglia in a minocycline-sensitive manner (Fig. 2c). This was the only comparison between the brain and SC cultures, and the results were in agreement. Finally, because activated microglia have been reported to increase the release of IL-1β (Rothwell et al., 1996; Pearson et al., 1999; Streit et al., 1999), we studied changes in IL-1β release in mixed SC cultures. Toxic concentrations of glutamate or kainate increased the release of IL-1β (Fig. 2d), and this effect was reduced by minocycline treatment at the neuroprotective concentration of 0.02 μm. Altogether, these results indicated that excitotoxins induce microglial proliferation and activation independent of neuronal injury. If the hypothesis that microglia proliferation contributes to excitotoxicity is correct, then increasing the number of microglial cells present in the mixed cultures should result in enhanced excitotoxic cell death. We therefore cultured pure microglia cells on top of SC cultures, and this increased the proportion of microglia cells from 5 to 15% (threefold). Both basal LDH release as well as glutamate- and kainate-induced LDH releases were significantly enhanced 24 hr later in these microglia-rich cultures compared with normal SC cultures, and minocycline was still able to provide significant (p < 0.05; single-factor ANOVA) neuroprotection (Fig. 2e).
Fig. 2.
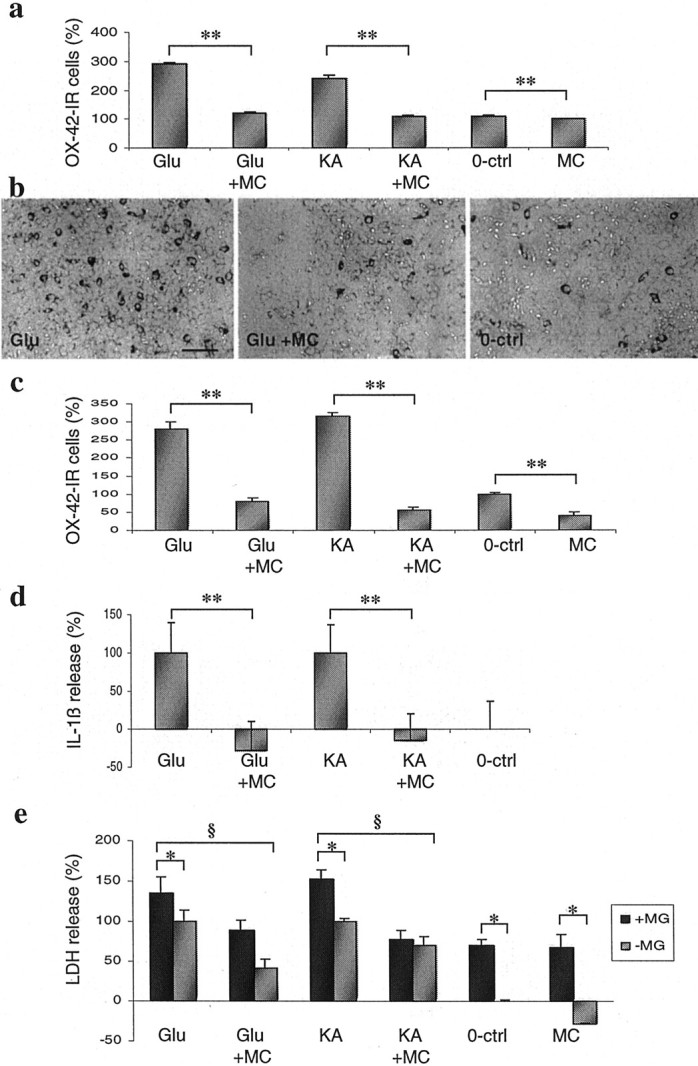
Minocycline (MC) inhibits excitotoxin-induced proliferation of microglial cells independently of neuronal cell death. a, In SC cultures, the number of OX-42-immunoreactive (OX-42-IR) microglial cells is significantly increased after a 24 hr exposure to 500 μmglutamate (Glu) and 100 μm kainate (KA). Minocycline (0.02 μm) treatment completely prevents the microglia proliferation and decreases the microglia cell number even in unexposed control cultures (0-ctrl). Data are presented as the mean ± SD counted in a blind manner and pooled from two independent experiments (n = 8). **p < 0.01; single-factor ANOVA. b, Representative photomicrographs of the effect of glutamate and minocycline on OX-42-immunoreactive cells. Scale bar, 250 μm. c, A 24 hr exposure of neuron-free glial cultures to 500 μmglutamate and 500 μm kainate results in microglia proliferation, which is inhibited by 0.2 μm minocycline treatment. Minocycline also inhibits the spontaneous microglia proliferation. Data are presented as the mean ± SD counted in a blind manner (n = 3). **p < 0.05; single-factor ANOVA. d, Exposure of SC cultures to excitotoxins causes increased IL-1β release, which is reduced by minocycline treatment. The exposure and treatment of these cultures were done as described in Figure 1a. Data are presented as the mean ± SD pooled from two independent experiments (n = 6). **p < 0.01; single-factor ANOVA. e, Increased number of microglia in SC cell cultures enhances the excitotoxicity, which was inhibited by minocycline. Adding microglia onto SC cell cultures (+MG), excitotoxicity of 500 μm glutamate and 100 μm kainate is increased significantly (*p < 0.05; single-factor ANOVA) when compared with the normal mixed SC culture (−MG). The basal LDH release is also enhanced significantly (*p < 0.05; single-factor ANOVA) in microglia-rich cultures. Minocycline (0.02 μm) treatment 30 min before excitotoxin exposures was able to provide significant (§p < 0.05; single-factor ANOVA) neuroprotection. Data were presented as the mean ± SD pooled from two independent experiments (n = 6).
Excitotoxin-induced activation of p38 MAPK in microglia is prevented by minocycline, and inhibition of p38 MAPK is neuroprotective in mixed SC cultures
p38 and p44/42 MAPK are activated within 5–30 min after external stimulation of the brain (Murray et al., 1998; Alessandrini et al., 1999; Sugino et al., 2000) or various in vitro cell preparations (Bhat et al., 1998; Chen and Wang, 1999; Mukherjee et al., 1999). In our cell culture model, MAPK activation peaks at 5–10 min, starting to decline after 30 min (N. Vartianen, G. Goldsteins, and J. Koistinaho, unpublished observations). Treatment with 500 μm glutamate or 100 μmkainate for 10 min increased immunoreactivity for active, phospho-p38 MAPK but not phospho-p44/42 MAPK in mixed SC cultures (Fig.3a–e). Phospho-p38 MAPK was exclusively localized in microglial cells (Fig. 3b–e). Phospho-p44/42 MAPK was found in astrocytes, several neurons, and in some solitary microglial cells (data not shown). No MAPK response was seen in astrocytes, even when pure astrocyte cultures were studied with immunoblotting (data not shown). Treatment with 0.02 μm minocycline inhibited the excitotoxin-induced immunoreactivity for phospho-p38 (Fig.3a) and had no clear effect on phospho-p44/42 MAPK (data not shown). To find out whether p38 MAPK or p44/42 MAPK activation is required for excitotoxic neuronal death, the effect of SB203580, a p38 MAPK inhibitor, and PD98059, a p44/42 MAPK inhibitor, on glutamate- and kainate-induced cell death was tested. SB203580 (10 μm), but not 10 μmPD98059, provided neuroprotection against 24 hr exposure to 500 μm glutamate and 100 μmkainate (Fig. 3f). This neuroprotection was also associated with reduced IL-1β release (data not shown).
Fig. 3.
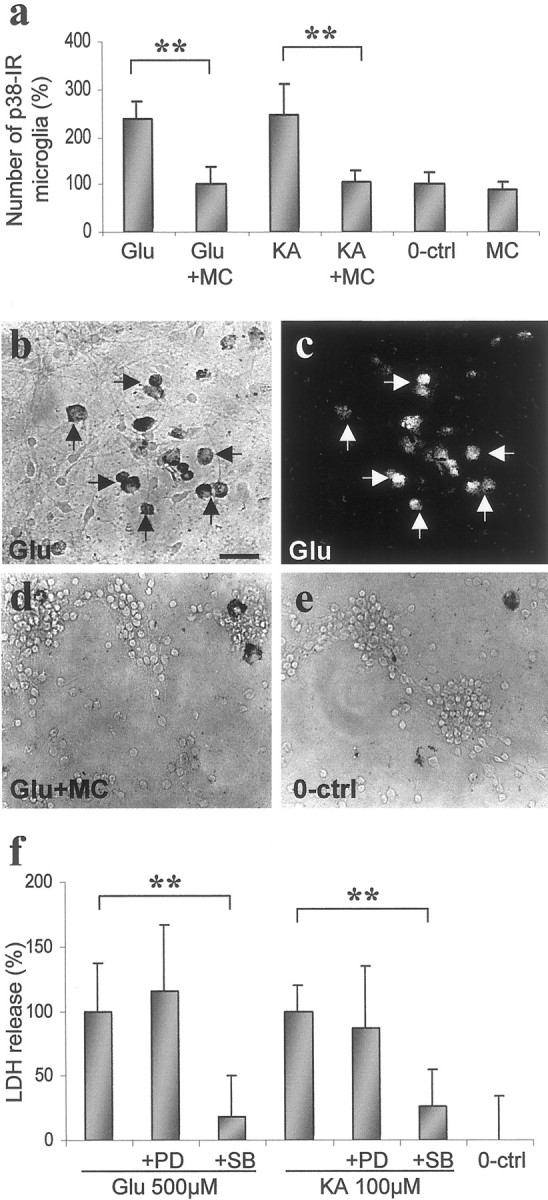
Minocycline (MC) treatment inhibits activation of p38 MAPK in microglia at neuroprotective doses, and inhibition of p38 MAPK is neuroprotective in SC cell cultures.a, Quantitation of phospho-p38 MAPK-immunoreactive microglia in SC cell cultures after 10 min stimulation with 500 μm glutamate (Glu) and 100 μm kainate (KA) with and without 0.02 μm minocycline treatment. Both glutamate and kainate increased the number of immunoreactive cells within 10 min. Minocycline decreased the induced p38 MAPK activity in microglial cells. Data are presented as the mean ± SD counted in a blind manner from three independent experiments (n = 9). **p < 0.01; single-factor ANOVA.b–e, Representative photomicrographs of the effect of glutamate and minocycline on phospho-p38-immunoreactive (p38-IR) cells of SC cell cultures. Double-staining with phospho-p38 (b) and OX-42 (c) antibodies indicates that p38 MAPK activity is increased by glutamate stimulation only in microglia. Scale bar, 200 μm. f, LDH release induced by 500 μm glutamate and 100 μm kainate (24 hr) was reduced by 10 μmSB203580 (SB), a specific p38 MAPK inhibitor, but not by 10 μm PD98059 (PD), a specific p44/42 MAPK inhibitor. Data are presented as the mean ± SD from three independent experiments (n = 10–13). **p < 0.01; single-factor ANOVA.0-ctrl, Unexposed 0 control.
Excitotoxins induce p38 MAPK-mediated proliferation and activation of microglial cells independently of neurons and astrocytes: inhibition by minocycline
Previous studies have demonstrated the presence of metabotropic, NMDA, and AMPA/kainate receptors in activated microglial cells (Gottlieb and Matute, 1997; Biber et al., 1999; Noda et al., 2000). We confirmed the presence of GluR4 AMPA/kainate receptors in pure microglia cultures by immunoblotting (Fig.4a) and immunocytochemistry (Fiebich et al., 2000). Treatment with glutamate and kainate for 24 hr induced proliferation of pure microglia twofold to threefold compared with spontaneous proliferation of the control cultures (Fig.4b). The excitotoxin-induced proliferation was associated with increased release of NO metabolites and IL-1β (Fig.4c,d). In our microglia cell cultures, induction of phospho-MAPKs peaks at 5–15 min after stimulation and decreases back to basal levels within 60 min (data not shown). Phospho-p38 MAPK and phospho-p42/44 MAPK were detectable in untreated microglia, but glutamate treatment increased the amount of phospho-p38 MAPK further by 45% transiently, i.e., at 5 min (Fig.5a), whereas no change was seen in phospho-p44/42 MAPK (Fig. 5b). Treatment of pure microglia with 0.2 μm minocycline or with 10 μm SB203580 prevented the excitotoxin-induced proliferation of microglia (Fig. 4b). In addition, minocycline treatment inhibited the IL-1β and NO metabolites release triggered by glutamate or kainate (Fig. 4c,d). Finally, minocycline treatment reduced the basal amount of phospho-p38 MAPK by 50% and completely blocked the glutamate-induced increase of phospho-p38MAPK (p < 0.05; single-factor ANOVA), (Fig. 5a) when administered 30 min before 5 min glutamate exposure. Minocycline also decreased the levels of phospho-p44/42 MAPK (Fig. 5b).
Fig. 4.
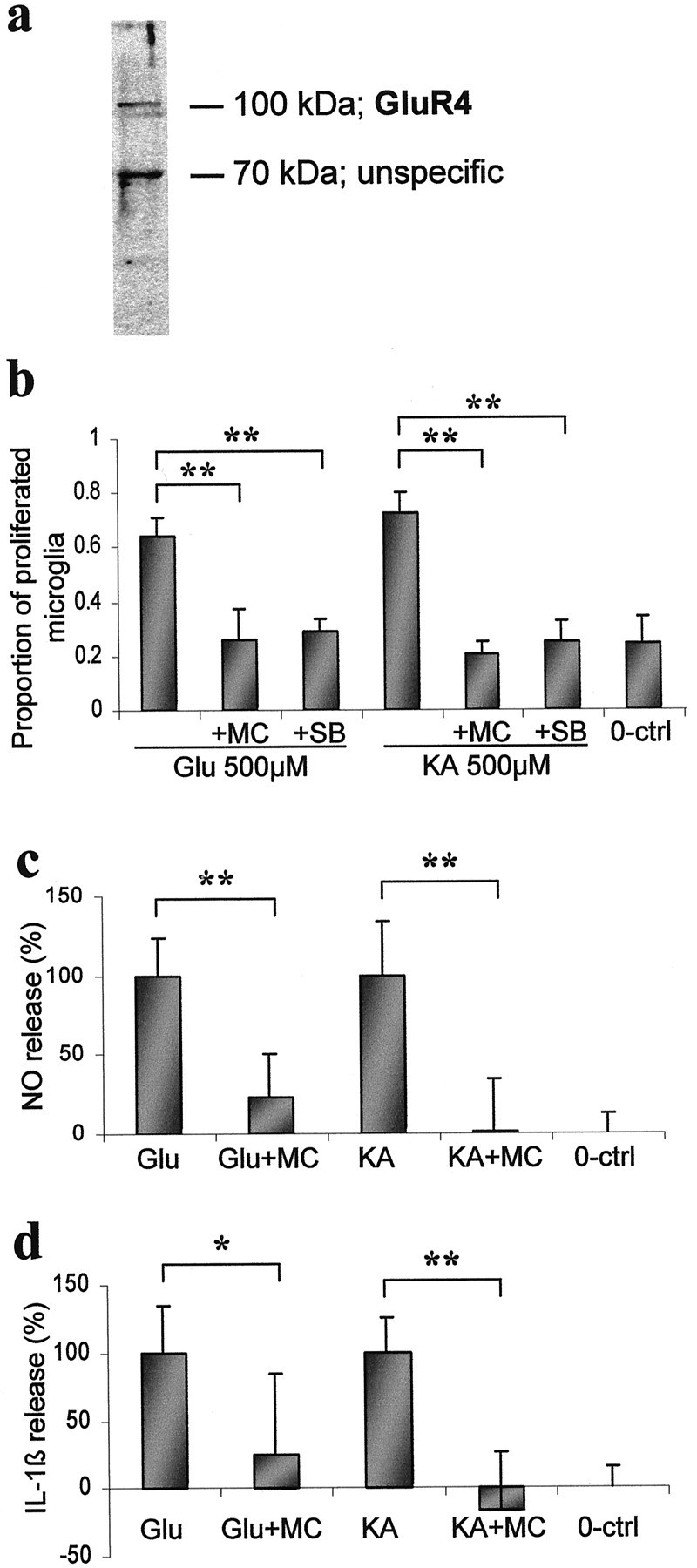
Stimulation of glutamate receptors causes microglial proliferation and activation, which are decreased by minocycline (MC). a, Immunoblot analysis of AMPA/kainate receptor GluR4 in pure microglia cultures. A specific immunoreactive band at appropriate size is detected. b, Quantitation of BrdU-positive cells in pure microglial culture 24 hr after stimulation with 500 μm glutamate (Glu) and 100 μm kainate (KA). Both 0.2 μm minocycline and 10 μm SB203580 (SB), a specific p38 MAPK inhibitor, treatments reduce the excitotoxin-induced microglial proliferation. Data are present as the mean ± SD pooled from two independent experiments (n = 9–11). **p < 0,01; single-factor ANOVA. A 24 hr stimulation with excitotoxins also causes increased NO (c) and IL-1β (d) release by a mechanism, which is reduced by 0.02 μm minocycline treatment. Data are presented as the mean ± SD pooled from two independent experiments (n = 8). *p < 0.05; **p < 0.01; single-factor ANOVA. 0-ctrl, Unexposed 0 control.
Fig. 5.
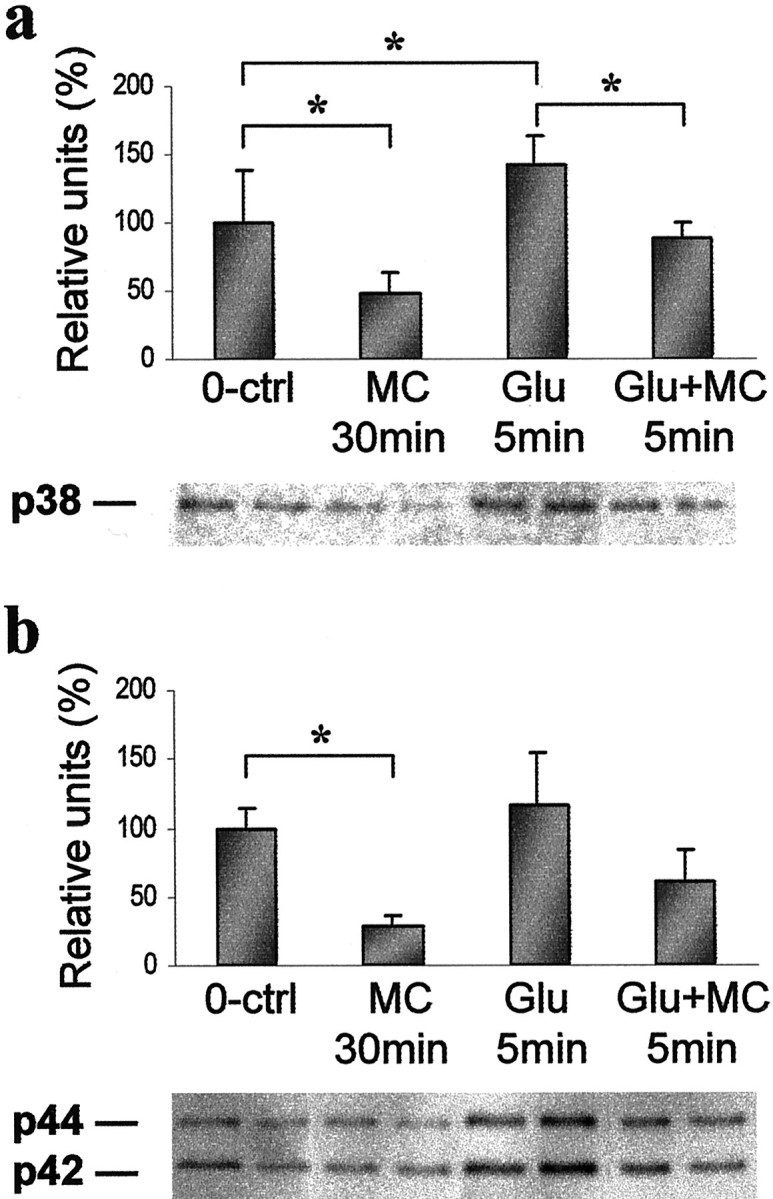
Glutamate-stimulated pure microglial cultures show an increased amount of phospho-p38 MAPK, which is reduced by minocycline. Quantitative immunoblot analysis of phospho-p38 (a) and p44/42 MAPK (b) after 5 min stimulation with 500 μm glutamate alone (Glu 5min), 5 min stimulation with 500 μmglutamate with minocycline administered 30 min before glutamate (Glu+MC 5min), 0.2 μm minocycline alone (MC 30min), and untreated cultures (0-ctrl). Below the graphs, representative immunoblots are shown. The experiments were repeated twice with similar results (*p < 0.05).
DISCUSSION
Inflammation and excitotoxicity are two major components of brain injury and diseases, including stroke and Alzheimer's disease (Beal, 1995; Dirnagl et al., 1999; Lee et al., 1999; Smith et al., 2000). Our results demonstrate for the first time that excitotoxins induce activation and proliferation of microglial cells and thereby enhance the release of microglial toxins, including IL-1β and NO, resulting in increased neuronal death. This conclusion is in line with previousin vivo and in vitro studies, which have suggested that activation of microglia contributes to ischemic and excitotoxic neuronal death (Giulian and Corpuz, 1993; Giulian and Vaca, 1993; Kim and Ko, 1998; Rogove and Tsirka 1998; Yrjänheikki et al., 1998, 1999). Because minocycline inhibits this interplay between excitotoxicity and inflammation by inhibiting the p38 MAPK pathway in microglia cells, p38 MAPK, and possibly its upstream kinases in these cells represent potential targets for pharmacotherapy of brain diseases.
Activated microglial cells accumulate around the injured area in the human and rodent brain (Kreutzberg, 1996; Schousboe et al., 1997). This microglial reaction can be detected as early as 20 min after ischemia and is maximal 4–6 d after the insult (Morioka et al., 1991). In Alzheimer's disease, which represents a chronic neurodegenerative disease, activated microglia are regularly associated with amyloid plaques (Beal, 1995; McGeer and McGeer, 1999). Although it has been well demonstrated that microglial cells synthesize and release cytotoxic molecules, including cytokines and proteases (Giulian and Corpuz, 1993; Giulian and Vaca, 1993; Rothwell et al., 1996; Tsirka et al., 1996; Kim and Ko, 1998; Rogove and Tsirka, 1998; Flavin et al., 2000), it is difficult to determine with in vivo models whether or not microglial cells play a role in neuronal cell death. Strong depolarization by inducing spreading depression in the rat brain upregulates the number of activated microglial cells in 3 d without association with neuronal death (Caggiano and Kraig, 1996), indicating that microglial activation alone is not necessarily a sufficient stimulus to cause acute neuronal death. On the other hand,in vitro studies have suggested that activated microglial cells do play a role in neuronal death. Activation of these cells may exert detrimental effects by release of inflammatory molecules (such as IL-1β, TNFα and NO) (Bhat et al., 1998; Fiebich et al., 1998), extracellular proteases (Tsirka et al., 1996; Rogove and Tsirka, 1998;Flavin et al., 2000), or presently uncharacterized factors that enhance excitotoxicity by acting through NMDA receptors (Giulian and Vaca, 1993; Kim and Ko, 1998). We did not attempt to define the microglial factor(s) responsible for enhanced excitotoxicity but demonstrated that microglial cells contribute to excitotoxic cell death. This conclusion is based on the following findings: (1) increase of activated microglial cells precedes excitotoxic cell death, (2) excitotoxicity can be enhanced by adding microglial cells to mixed neuronal cultures, and (3) inhibition of a kinase (p38 MAPK), which is found in its active form only in microglia, blocks microglial proliferation and activation and is simultaneously protective against excitotoxicity. Although the evidence from cell culture studies strongly supports the neurotoxic role of microglia, the role of these cells in the brain may be different and may depend on the injury stimulus and functional state of other surrounding cells.
Although microglial cells seem to express fewer glutamate receptors than neurons, they express a subset of NMDA and AMPA/kainate receptors after global brain ischemia (Gottlieb and Matute, 1997), and in vitro studies have demonstrated the presence of at least AMPA/kainate (Noda et al., 2000) and group I metabotropic (Biber et al., 1999) glutamate receptors in unstimulated microglia cells. We confirmed the presence of GluR4 AMPA/kainate receptor in our microglia preparation by immunoblotting and showed a rapid activation and proliferation of microglial cells after stimulation with 500 μm glutamate or 100 μmkainate. Because both kainate and glutamate were efficient in activating microglia, it is very likely that the effect was mediated substantially through AMPA/kainate type glutamate receptors, although the effect of glutamate could also be partially mediated through group I metabotropic receptors. Our preliminary studies with specific agonists suggest, however, that metabotropic receptors play no role in microglial activation (T. Tikka, B. Fiebich, and J. Koistinaho, unpublished observations).
Walton et al. (1998) have demonstrated that global brain ischemia induces activation of p38 MAPK exclusively in microglial cells, and similar results have been obtained in focal brain ischemia experiments on rats (Yrjänheikki and Koistinaho, 1999) and mice (Koistinaho et al., 2000). In recent studies, transient activation of p38 MAPK was found in neurons after global brain ischemia in gerbils (Sugino et al., 2000) and focal ischemia of the rat (Tian et al., 2000), but also in these studies microglial cells showed a prominent activation of p38 MAPK. The activation of MAP kinases, including p38 MAPK, clearly depends on the model used and the time point when tissues are analyzed after stimulation. We found that glutamate and kainate induce transient activation of p38 MAPK exclusively in microglial cells, partially in line with the previous studies. Because p38 MAPK is thought to mediate inflammatory responses in various cell types (Lee et al., 1994; Chen and Wang, 1999), including microglia (Bhat et al., 1998), possibly by activating transcription factors that positively regulate inductions of inflammatory genes (Bhat et al., 1998), inhibition of p38 MAPK can be expected to be beneficial in injuries involving microglial activation and inflammation. Specific inhibitors of p38 MAPK have been proven to reduce peripheral inflammation (Underwood et al., 2000; Warny et al., 2000), slow down microglial activation (Bhat et al., 1998), and protect brain against ischemic insults (Sugino et al., 2000). Our results confirm the role of p38 MAPK in both microglial activation and excitotoxic cell death, when in vitro models are applied. It should be noted that, although p44/42 MAPK is thought to be a survival kinase (Grewal et al., 1999) and inhibition of p44/42 MAPK in our culture model and in some previous in vivo studies was without effect (Sugino et al., 2000), p44/42 MAPK has been shown to contribute to ischemic neuronal death in vivo (Alessandrini et al., 1999) and to seizure-induced neuronal damage in vitro (Murray et al., 1998). On the other hand, some studies have reported no activation of p44/42 MAPK after focal brain ischemia (Tian et al., 2000). We did not detect activation of p44/42 MAPK in our culture system but observed a considerable basal expression of p44/42 MAPK, which might mask the possible activation by excitotoxicity. Altogether, it is evident that the role of MAPK signaling in brain injury depends on several factors, including the model used and the time or the route of inhibitor administration.
Minocycline and doxycycline are semisynthetic tetracycline derivatives, which exert several anti-inflammatory effects independent of their anti-microbial action. These tetracyclines and their non-antibiotic derivatives (chemically modified tetracyclines) inhibit matrix metalloproteases, nitric oxide synthases, cyclooxygenase-2, and phospholipase A2 (Amin et al., 1996; Golub et al., 1998; Patel et al., 1999). In addition, they have been associated with reduced free-radical formation and inhibition of interleukin-1β synthesis (Gabler and Creamer, 1991; Yrjänheikki et al., 1998). We have shown previously that minocycline provides significant protection against global and focal brain ischemia that is associated with reduced microglial activation, reduced expression of iNOS and ICE, and diminished activation of COX-2 (Yrjänheikki et al., 1998, 1999). In addition, minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in an animal model of Huntington's disease (Chen et al., 2000). Our present results support the previous findings and suggest that minocycline provides neuroprotection by inhibition of microglial activation and proliferation through inhibition of the p38 MAPK pathway. Because activation of the p38 MAPK pathway is involved in induction of several proinflammatory genes, inhibition of p38 MAPK may be a mechanism by which minocycline exerts a wide range of anti-inflammatory effects in the brain and peripheral system.
Altogether, compounds that inhibit p38 MAPK activation in microglia represent potential therapies against excitotoxic brain insults. We found doses of 0.02–2 μm, corresponding to 1–0.01 μg/ml concentrations, to be efficient. Because minocycline concentration up to 1 μg/ml can be achieved in the dog CSF and brain (Barza et al., 1975), the doses used in the present study may have clinical relevance. Considering that the identification of minocycline as such an anti-inflammatory compound indicates that semisynthetic tetracycline derivatives, which are clinically well tolerated, are good candidates for treatment of human brain diseases.
Footnotes
The study was supported by the Sigrid Juselius Foundation (J.K., T.T.), the Saastamoinen Foundation (T.T.), and the Finnish Cultural Foundation of Northern Savo (T.T.).
Correspondence should be addressed to Jari Koistinaho, A. I. Virtanen Institute for Molecular Sciences, University of Kuopio, P.O. Box 1627, 70211 Kuopio, Finland. E-mail: jari.koistinaho@uku.fi.
REFERENCES
- 1.Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amin AR, Atttur MG, Thakker GD, Patel PD, Vyas PR, Patel RN, Patel IR, Abramson SB. A novel mechanism of action of tetracyclines: effects on nitric oxide synthases. Proc Natl Acad Sci USA. 1996;93:14014–14019. doi: 10.1073/pnas.93.24.14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Barza M, Brown RB, Shanks C, Gamble C, Weinstein L. Relation between lipophilicity and pharmacological behavior of minocycline, doxycycline, tetracycline, and oxytetracycline in dogs. Antimicrob Agents Chemother. 1975;8:713–720. doi: 10.1128/aac.8.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 6.Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-α gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biber K, Laurie DJ, Berthele A, Sommer B, Tölle TR, Gebicke-Härter PJ, van Calker D, Boddeke HWGM. Expression and signaling of group I metabotropic glutamate receptors in astrocytes and microglia. J Neurochem. 1999;72:1671–1680. doi: 10.1046/j.1471-4159.1999.721671.x. [DOI] [PubMed] [Google Scholar]
- 8.Caggiano AO, Kraig RP. Eicosanoids and nitric oxide influence induction of reactive gliosis from spreading depression in microglia but not astrocytes. J Comp Neurol. 1996;369:93–108. doi: 10.1002/(SICI)1096-9861(19960520)369:1<93::AID-CNE7>3.0.CO;2-F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen CC, Wang JK. p38 but not p44/42 mitogen-activated protein kinase is required for nitric oxide synthase induction mediated by lipopolysaccharide in RAW 264.7 macrophages. Mol Pharmacol. 1999;55:481–488. [PubMed] [Google Scholar]
- 10.Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6:797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- 11.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 12.Cooper NR, Kalaria RN, McGeer PL, Rogers J. Key issues in Alzheimer's disease inflammation. Neurobiol Aging. 2000;21:451–453. doi: 10.1016/s0197-4580(00)00148-2. [DOI] [PubMed] [Google Scholar]
- 13.DeGraba TJ, Pettigrew LC. Why do neuroprotective drugs work in animals but not humans? Neurol Clin. 2000;18:475–493. doi: 10.1016/s0733-8619(05)70203-6. [DOI] [PubMed] [Google Scholar]
- 14.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 15.Dugan LL, Choi DW. Excitotoxicity, free radicals, and cell membrane changes. Ann Neurol [Suppl] 1994;35:S17–S21. doi: 10.1002/ana.410350707. [DOI] [PubMed] [Google Scholar]
- 16.Fiebich BL, Butcher RD, Gebicke-Haerter PJ. Protein kinase C-mediated regulation of inducible nitric oxide synthase expression in cultured microglial cells. J Neuroimmunol. 1998;92:170–178. doi: 10.1016/s0165-5728(98)00201-x. [DOI] [PubMed] [Google Scholar]
- 17.Fiebich BL, Mueksch B, Boehringer M, Hull M. Interleukin-1beta induces cyclooxygenase-2 and prostaglandin E(2) synthesis in human neuroblastoma cells: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappa B. J Neurochem. 2000;75:2020–2028. doi: 10.1046/j.1471-4159.2000.0752020.x. [DOI] [PubMed] [Google Scholar]
- 18.Flavin MP, Zhao G, Ho LT. Microglial tissue plasminogen activator (tPA) triggers neuronal apoptosis in vitro. Glia. 2000;29:347–354. [PubMed] [Google Scholar]
- 19.Gabler WL, Creamer HR. Suppression of human neutrophil functions by tetracyclines. J Periodontal Res. 1991;26:52–58. doi: 10.1111/j.1600-0765.1991.tb01626.x. [DOI] [PubMed] [Google Scholar]
- 20.Giulian D, Corpuz M. Microglial secretion products and their impact on the nervous system. Adv Neurol. 1993;59:315–320. [PubMed] [Google Scholar]
- 21.Giulian D, Vaca K. Inflammatory glia mediate delayed neuronal damage after ischemia in the central nervous system. Stroke. 1993;[Suppl] 12:I84–I90. [PubMed] [Google Scholar]
- 22.Golub LM, Lee HM, Ryan ME, Giannobile WV, Payne J, Sorsa T. Tetracyclines inhibit connective tissue breakdown by multiple non-antimicrobial mechanisms. Adv Dent Res. 1998;12:12–26. doi: 10.1177/08959374980120010501. [DOI] [PubMed] [Google Scholar]
- 23.Gottlieb M, Matute C. Expression of ionotropic glutamate receptor subunits in glial cells of the hippocampal CA1 area following transient forebrain ischemia. J Cereb Blood Flow Metab. 1997;17:290–300. doi: 10.1097/00004647-199703000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Grewal SS, York RD, Stork PJ. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- 25.Kim WK, Ko KH. Potentiation of N-methyl-d-aspartate-mediated neurotoxicity by immunostimulated murine microglia. J Neurosci Res. 1998;54:17–26. doi: 10.1002/(SICI)1097-4547(19981001)54:1<17::AID-JNR3>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 26.Koistinaho J, Koistinaho M, Kettunen MI, Cordell B, Kauppinen RA, Higgins LS. Increased vulnerability to focal brain ischemia in APP751 transgenic mice. Eur J Neurosci. 2000;[Suppl 11] 12:120. [Google Scholar]
- 27.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 28.Lee JC, Laydon J T, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 29.Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;[Suppl 6738] 399:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- 30.McGeer EG, McGeer PL. Brain inflammation in Alzheimer disease and the therapeutic implications. Curr Pharm Des. 1999;5:821–836. [PubMed] [Google Scholar]
- 31.Morioka T, Kalehua AN, Streit WJ. The microglial reaction in the rat dorsal hippocampus following transient forebrain ischemia. J Cereb Blood Flow Metab. 1991;11:966–973. doi: 10.1038/jcbfm.1991.162. [DOI] [PubMed] [Google Scholar]
- 32.Mukherjee PK, DeCoster MA, Campbell FZ, Davis RJ, Bazan NG. Glutamate receptor signaling interplay modulates stress-sensitive mitogen-activated protein kinases and neuronal cell death. J Biol Chem. 1999;274:6493–6498. doi: 10.1074/jbc.274.10.6493. [DOI] [PubMed] [Google Scholar]
- 33.Munoz-Fernandez MA, Fresno M. The role of tumour necrosis factor, interleukin 6, interferon-gamma and inducible nitric oxide synthase in the development and pathology of the nervous system. Prog Neurobiol. 1998;56:307–340. doi: 10.1016/s0301-0082(98)00045-8. [DOI] [PubMed] [Google Scholar]
- 34.Murray B, Alessandrini A, Cole AJ, Yee AG, Furshpan EJ. Inhibition of the p44/42 MAP kinase pathway protects hippocampal neuronas in a cell-culture model of seizure activity. Prog Natl Acad Sci USA. 1998;95:11975–11980. doi: 10.1073/pnas.95.20.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noda M, Nakanishi H, Nabekura J, Akaike N. AMPA-kainate subtypes of glutamate receptor in rat cerebral microglia. J Neurosci. 2000;20:251–258. doi: 10.1523/JNEUROSCI.20-01-00251.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel RN, Attur MG, Dave MN, Patel IV, Stuchin SA, Abramson SB, Amin AR. A novel mechanism of action of chemically modified tetracyclines: inhibition of COX-2-mediated prostaglandin E2 production. J Immunol. 1999;163:3459–3467. [PubMed] [Google Scholar]
- 37.Pearson VL, Rothwell NJ, Toulmond S. Excitotoxic brain damage in the rat induces interleukin-1beta protein in microglia and astrocytes: correlation with the progression of cell death. Glia. 1999;25:311–323. [PubMed] [Google Scholar]
- 38.Rogove AD, Tsirka SE. Neurotoxic responses by microglia elicited by excitotoxic injury in the mouse hippocampus. Curr Biol. 1998;8:19–25. doi: 10.1016/s0960-9822(98)70016-8. [DOI] [PubMed] [Google Scholar]
- 39.Rothwell NJ, Luheshi G, Toulmond S. Cytokines and their receptors in the central nervous system: physiology, pharmacology, and pathology. Pharmacol Ther. 1996;69:85–95. doi: 10.1016/0163-7258(95)02033-0. [DOI] [PubMed] [Google Scholar]
- 40.Sanderson KL, Raghupathi R, Saatman KE, Martin D, Miller G, McIntosh TK. Interleukin-1 receptor antagonist attenuates regional neuronal cell death and cognitive dysfunction after experimental brain injury. J Cereb Blood Flow Metab. 1999;19:1118–1125. doi: 10.1097/00004647-199910000-00008. [DOI] [PubMed] [Google Scholar]
- 41.Schousboe A, Sonnewald U, Civenne G, Gegelashvili G. Role of astrocytes in glutamate homeostasis. Implications for excitotoxicity. Adv Exp Med Biol. 1997;429:195–206. doi: 10.1007/978-1-4757-9551-6_14. [DOI] [PubMed] [Google Scholar]
- 42.Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6:62–66. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- 43.Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 44.Sugino T, Nozaki K, Takagi Y, Hattori I, Hashimoto N, Moriguchi T, Nishida E. Activation of mitogen-activated protein kinases after transient forebrain ischemia in gerbil hippocampus. J Neurosci. 2000;20:4506–4514. doi: 10.1523/JNEUROSCI.20-12-04506.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tian D, Litvak V, Lev S. Cerebral ischemia and seizures induce tyrosine phosphorylation of PYK2 in neurons and microglial cells. J Neurosci. 2000;20:6478–6487. doi: 10.1523/JNEUROSCI.20-17-06478.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Touzani O, Boutin H, Chuquet J, Rothwell N. Potential mechanisms of interleukin-1 involvement in cerebral ischaemia. J Neuroimmunol. 1999;100:203–215. doi: 10.1016/s0165-5728(99)00202-7. [DOI] [PubMed] [Google Scholar]
- 47.Tsirka SE, Rogove AD, Strickland S. Neuronal cell death and tPA. Nature. 1996;384:123–124. doi: 10.1038/384123b0. [DOI] [PubMed] [Google Scholar]
- 48.Underwood DC, Osborn RR, Kotzer CJ, Adams JL, Lee JC, Webb EF, Carpenter DC, Bochnowicz S, Thomas HC, Hay DW, Griswold DE. SB 239063, a potent p38 MAP kinase inhibitor, reduces inflammatory cytokine production, airways eosinophil infiltration, and persistence. J Pharmacol Exp Ther. 2000;293:281–288. [PubMed] [Google Scholar]
- 49.Vartiainen N, Tikka T, Keinänen R, Chan PH, Koistinaho J. Glutamatergic receptors regulate expression, phosphorylation and accumulation of neurofilaments in spinal cord neurons. Neuroscience. 1999;93:1123–1133. doi: 10.1016/s0306-4522(99)00200-6. [DOI] [PubMed] [Google Scholar]
- 50.Venters HD, Dantzer R, Kelley KW. A new concept in neurodegeneration: TNFalpha is a silencer of survival signals. Trends Neurosci. 2000;23:175–180. doi: 10.1016/s0166-2236(99)01533-7. [DOI] [PubMed] [Google Scholar]
- 51.Walton KM, Dirocco R, Bartlett BA, Koury E, Marcy VR, Jarvis B, Schaefer EM, Bhat RV. Activation of p38MAPK in microglia after ischemia. J Neurochem. 1998;70:1764–1767. doi: 10.1046/j.1471-4159.1998.70041764.x. [DOI] [PubMed] [Google Scholar]
- 52.Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 53.Warny M, Keates AC, Keates S, Castagliuolo I, Zacks JK, Aboudola S, Qamar A, Pothoulakis C, LaMont JT, Kelly CP. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J Clin Invest. 2000;105:1147–1156. doi: 10.1172/JCI7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yrjänheikki J, Koistinaho J (1999) Mitogen-activated protein kinase p38 is activated in focal brain ischemia. Presented at the 1st Biennial Kuopio Symposium, Kuopio, Finland, June [Abstr 82].
- 55.Yrjänheikki J, Keinänen R, Pellikka M, Hökfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yrjänheikki J, Tikka T, Keinänen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]