

Abstract
Perispinal (intrathecal) injection of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein gp120 creates exaggerated pain states. Decreases in response thresholds to both heat stimuli (thermal hyperalgesia) and light tactile stimuli (mechanical allodynia) are rapidly induced after gp120 administration. gp120 is the portion of HIV-1 that binds to and activates microglia and astrocytes. These glial cells have been proposed to be key mediators of gp120-induced hyperalgesia and allodynia because these pain changes are blocked by drugs thought to affect glial function preferentially. The aim of the present series of studies was to determine whether gp120-induced pain changes involve proinflammatory cytokines [interleukin-1β (IL-1) and tumor necrosis factor-α (TNF-α)], substances released from activated glia. IL-1 and TNF antagonists each prevented gp120-induced pain changes. Intrathecal gp120 produced time-dependent, site-specific increases in TNF and IL-1 protein release into lumbosacral CSF; parallel cytokine increases in lumbar dorsal spinal cord were also observed. Intrathecal administration of fluorocitrate (a glial metabolic inhibitor), TNF antagonist, and IL-1 antagonist each blocked gp120-induced increases in spinal IL-1 protein. These results support the concept that activated glia in dorsal spinal cord can create exaggerated pain states via the release of proinflammatory cytokines.
Keywords: Hargreaves test, von Frey test, microglia, astrocytes, interleukin-1, tumor necrosis factor, rats, thermal hyperalgesia, mechanical allodynia
Sensitization of spinal cord dorsal horn neurons by neuroactive substances such as substance P, glutamate, and nitric oxide (NO) leads to enhanced pain states (Haley and Wilcox, 1992). Although classic views of pain facilitation have only focused on neurons, these neuroactive substances also activate glia (Hartung et al., 1988; Marriott et al., 1991; Murphy, 1993; Kreutzberg, 1996). As a result, microglia and astrocytes release a wide variety of neuroactive substances, including several known to activate spinal cord pain transmission neurons (Hartung et al., 1988; Marriott et al., 1991;Dutton, 1993; Kreutzberg, 1996; Murphy and Grzybicki, 1996).
A growing literature supports the idea that some forms of exaggerated pain may involve glial activation. Subcutaneous formalin, subcutaneous zymosan (yeast cell walls), and peripheral nerve injury produce (1) decreased thresholds to mechanical stimuli (mechanical allodynia) and heat stimuli (thermal hyperalgesia) and (2) activated microglia and astrocytes within spinal cord dorsal horn (Watkins et al., 1995a; Coyle, 1998; DeLeo and Colburn, 1999; Fu et al., 1999). Indeed, glial activation correlates with pain behaviors (Garrison et al., 1991; Coyle, 1998). Moreover, functional disruption of spinal glia blocks both thermal hyperalgesia and mechanical allodynia produced by these procedures (Meller et al., 1994; Watkins et al., 1997) and by intraperitoneal bacteria (Watkins et al., 1995a,b), nerve inflammation (Hammack et al., 1999; Chacur et al., 2000), and perispinal (intrathecal) injection of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein gp120 (Milligan et al., 2000).
Direct in vitro antigen stimulation of glia by substances such as bacterial cell walls [lipopolysaccharide (LPS)] and viral envelope proteins (gp120) activates these cells, causing release of glutamate and NO, as well as release of proinflammatory cytokines including interleukin-1β (IL-1β) (Murphy, 1993; Kettenmann and Ransom, 1995; Kreutzberg, 1996; Murphy and Grzybicki, 1996). Although glutamate and NO have long been known to facilitate pain (Meller et al., 1992a), spinal IL-1 has only been recently recognized as exerting such effects. Indeed, intrathecal IL-1 induces nociception (Tadano et al., 1999) and mechanical and thermal hyperalgesia (Meller et al., 1994). Endogenous spinal IL-1 mediates exaggerated pain states produced by subcutaneous inflammation (Watkins et al., 1997), intraperitoneal LPS (Watkins et al., 1994), and nerve inflammation (Hammack et al., 1999; Chacur et al., 2000), because intrathecal IL-1 receptor antagonists block these pain states.
Because spinal IL-1 can exaggerate pain and in vitro immune glial activation releases IL-1, the purpose of the present studies was to determine whether in vivo spinal immune challenge creates IL-1-mediated exaggerated pain states. Because many viruses and bacteria “home” to the spinal cord of humans, such a result would potentially have striking implications for pathological pain associated with such clinical conditions. Spinal immune activation was induced by intrathecal administration of HIV-1 gp120, a procedure that we have shown previously to produce both thermal hyperalgesia and mechanical allodynia (Milligan et al., 2000). A combination of behavioral assessments, cytokine protein assays, and immunohistochemistry was used to assess potential mediation of these gp120-induced pain phenomena by endogenously released spinal IL-1.
MATERIALS AND METHODS
Subjects
Pathogen-free adult male Sprague Dawley rats (300–450 gm; Harlan Labs, Madison, WI) were used in all experiments. Rats were housed in temperature-controlled (23 ± 3°C) and light-controlled (12/12 hr light/dark cycle; lights on at 0700 hr) rooms with standard rodent chow and water available ad libitum. Behavioral testing was performed between 0700 and 1200 hr. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Colorado at Boulder.
Drugs
Frozen solutions of recombinant gp120 (product 1021; lot numbers 8JE28O7, 8JE28D14, and 8D159M2; ImmunoDiagnostics, Bedford, MA) were thawed, aliquoted at 1 μg/μl, and stored at −75°C. Vehicle, composed of 0.2 μm pore-filtered 0.1% bovine serum albumen (Sigma-Aldrich, St. Louis, MO) in sterile PBS, pH 7.4 (Life Technologies, Gaithersburg, MD), was aliquoted and stored at −75°C. Frozen aliquots of gp120 and vehicle were thawed immediately before administration, and gp120 was diluted to 0.5 μg/μl in all experiments. Aliquots were kept on ice during use and discarded within 1 hr.
Behavioral measures
Hargreaves test for thermal hyperalgesia. The Hargreaves test, which measures response latencies to hindpaw thermal stimulation (Hargreaves et al., 1988), was performed as described previously (Milligan et al., 2000). Briefly, rats were habituated to the experimental context (room and apparatus) before surgery for 3–4 consecutive days for 1 hr/d. After intrathecal surgery (see below), rats were placed in the experimental context for 20 min followed by predrug baseline (BL) paw withdrawal assessment. The BL was calculated from an average of three consecutive withdrawal latencies of both the left and right hindpaws measured at 15 min intervals. Voltage to the light source was adjusted to yield baseline latencies ranging from 10 to 13 sec. This procedure was followed by intraperitoneal and intrathecal injections, as described below. The order of paw testing varied randomly. Because there were no left versus right hindpaw differences throughout testing, the values for the left and right hindpaw withdrawal latencies were averaged. A cutoff time of 20 sec was imposed to avoid tissue damage.
Von Frey test for mechanical allodynia. The von Frey test measures paw withdrawal responses to a range of calibrated low-threshold mechanical stimuli. This test was performed as described previously (Milligan et al., 2000). Briefly, rats were habituated to the experimental context (room and apparatus) before surgery on 4 consecutive days for 1 hr/d. After intrathecal surgery (see below), rats were placed in the experimental context for 20–30 min followed by predrug BL assessment. The BL was calculated from an average of three consecutive withdrawal responses of both the left and right hindpaws measured at 15–20 min intervals. A logarithmic series of 10 calibrated Semmes-Weinstein monofilaments (von Frey hairs; Stoelting, Wood Dale, IL) was applied randomly to the left and right hindpaws to determine the threshold stiffness required for a paw withdrawal response. Log stiffness of the hairs is defined as log10(grams × 10,000). The 10 stimuli had the following log-stiffness values (the value in grams is given in parentheses): 3.61 (0.407 gm), 3.84 (0.692 gm), 4.08 (1.202 gm), 4.17 (1.479 gm), 4.31 (2.041 gm), 4.56 (3.630 gm), 4.74 (5.495 gm), 4.93 (8.511 gm), 5.07 (11.749 gm), and 5.18 (15.136 gm). The range of monofilaments used in these experiments (0.407–15.136 gm) has been shown previously to produce a logarithmically graded slope when interpolating a 50% response threshold of stimulus intensity [calculated as log10 (milligrams × 10)] (Chaplan et al., 1994).
The monofilament was applied perpendicularly to the midplantar or heel of the hindpaw for 8 sec. Threshold assessment was conducted as described previously (Milligan et al., 2000). Paw withdrawal responses were measured for 120 min, at 20 min intervals, for both the left and right hindpaw, beginning 20 min after intrathecal injection. In instances in which rats failed to respond to the strongest stimulus (15.136 gm), the upper cutoff value was assigned. Monofilaments with greater stimulus intensities lifted the paw during stimulus presentation and were deemed unreliable. Responses that occurred to the weakest stimulus (0.407 gm) were assigned the lower cutoff value for that time point. Because there were no differences in mechanical thresholds between left and right hindpaws throughout testing, data obtained from the left and right hindpaws were averaged.
The log stiffness that would have resulted in the 50% paw withdrawal rate was computed as described previously (Milligan et al., 2000). Briefly, thresholds were estimated by fitting a Gaussian integral psychometric function to the observed withdrawal rates, for each of the tested von Frey hairs, using a maximum-likelihood fitting method (Harvey, 1986; Treutwein and Strasburger, 1999). This method is a streamlined, functionally equivalent version of methods used previously (Dixon, 1980; Chaplan et al., 1994) and yields exactly the same results for the same set of data (Dixon, 1980; Chaplan et al., 1994; Milligan et al., 2000). Estimated thresholds derived from a Gaussian integral function yield a mathematical continuum and thus are appropriate for parametric statistical analyses (Harvey, 1986; Treutwein and Strasburger, 1999; Milligan et al., 2000). The computer program PsychoFit may be downloaded from L. O. Harvey's website (http://psych.colorado.edu/∼lharvey). These computed log-stiffness threshold values were then used for subsequent statistical analyses.
Intrathecal surgery and injections
Chronic lumbosacral indwelling catheters were constructed and implanted as described previously (Milligan et al., 1999b). The indwelling catheters were used to microinject drugs over the lumbosacral spinal cord only on the test day. This occurred between 4 and 10 d after surgery. All microinjections were conducted as described previously (Milligan et al., 1999b).
Verification of catheter placement was conducted immediately before spinal tissue dissection by visualization of the catheter tip at the level of the lumbosacral spinal cord. Approximately 3% of all animals that were catheterized failed verification for subdural catheter placement and were not included in the data analyses.
Cytokine measures: ELISA
Tissue and CSF collection. In preparation for spinal cytokine assays, intraperitoneal sodium pentobarbital (50 mg/kg; Abbott Labs, North Chicago, IL) was injected immediately after behavioral testing. This was supplemented with methoxyflurane (Pitman-Moore, Mundelein, IL) as required to maintain a surgical plane of anesthesia. Anesthesia was followed by exposure of the cervical and/or lumbosacral enlargement by laminectomy. A nick was made in the lumbar dura, and polyethylene-10 tubing (PE-10 Intramedic Tubing; Becton Dickinson Primary Care Diagnostics, Sparks, MD), attached at one end to a syringe, was inserted caudally into CSF. Approximately 10 μl of CSF was withdrawn and immediately flash frozen in liquid nitrogen. CSF was then withdrawn from the cisterna magna using a syringe attached to a 30 ga needle and immediately flash frozen in liquid nitrogen. After verifying the intrathecal catheter placement, the cervical and/or lumbosacral spinal cords were then dissected free and placed on an ice-chilled glass plate. The dorsal aspects of these tissues were then flash frozen in liquid nitrogen. Animals remained at the surgical plane of anesthesia throughout this procedure to minimize degradation of the samples during the collection procedure. Catheter verification plus sample collections require a maximum of 20–25 min; animals were then immediately killed by cervical dislocation. All samples were stored at −75°C until the time of assay (see below).
Sample preparation and assay. Procedures for tissue processing and ELISAs were identical to those described in detail previously (Hansen et al., 2000a,b). CSF samples were prepared for assay by being slowly thawed and then quick-spun (Quick Spin Mini-Centrifuge; National Labnet Company, Woodbridge, NY). Supernatants were removed for immediate use in ELISAs. For spinal tissues, protein was mechanically dissociated by sonication, and the total protein concentration was determined by the Bradford protein assay. Sonicated samples were centrifuged (Quick Spin Mini-Centrifuge; National Labnet Company), and supernatants were removed and stored at 4°C until the time ELISAs were performed.
IL-1β and tumor necrosis factor-α (TNF-α) protein were assayed using commercially available rat-specific ELISA kits (R&D Systems, Minneapolis, MN), in accordance with the manufacturer's instructions. The sensitivities of the IL-1 and TNF assays were each 0.5 pg/100 μl. The manufacturer's specifications state that the IL-1 assay shows no cross-reactivity for recombinant (r)-human IL-1 receptor antagonist, IL-1 receptor type I, or IL-1 receptor type II or r-rat glial cell line-derived neurotrophic factor (GDNF), IL-1α, IL-2, IL-4, interferon-γ, β-nerve growth factor, or TNF-α. The manufacturer's specifications state that the TNF assay shows no cross-reactivity for r-human TNF-α, TNF-β, TNF soluble receptor type I, or TNF soluble receptor type II; r-rat IL-1β, IL-2, IL-4, GDNF, interferon-γ, or β-nerve growth factor; or r-mouse TNF soluble receptor type I or II. TNF protein tissue content was not analyzed because of the unavailability of reliable TNF ELISA procedures for CNS tissue.
Data analysis
All statistical comparisons were computed using Statview 5.0.1 for the Macintosh. Data from the Hargreaves test were analyzed as the withdrawal latency in seconds, and data from the von Frey test were analyzed as the interpolated 50% threshold in log10 of stimulus intensity (monofilament stiffness in milligrams × 104). Predrug BL measures were analyzed by one-way ANOVA for Hargreaves and von Frey tests. Postdrug measures were analyzed by repeated measures ANOVA for Hargreaves and von Frey tests. Statistical analyses conducted for all tissue and CSF ELISAs and for image analysis of glial activation markers were by ANOVA. Where appropriate, ANOVAs were followed by Fisher's protected least significant difference post hoc analysis. Serum levels of IL-1 were analyzed by a two-tailed Student's t test.
Experiment 1: effect of intrathecal IL-1 receptor antagonist on intrathecal gp120-induced enhanced pain states and lumbar dorsal spinal cord IL-1 protein
Separate groups of rats were used for the mechanical allodynia and thermal hyperalgesia measures. Endotoxin-free solutions of recombinant met-human IL-1 receptor antagonist (IL-1ra; 100 μg/μl; lot number 2010316L6; Amgen, Thousand Oaks, CA) were stored at 4°C. Intrathecal IL-1ra (100 μg/μl) was injected after BL behavioral assessments, that is, 35 min before intrathecal gp120 or vehicle (n = 5–6/group). Control groups received equivolume intrathecal IL-1ra vehicle (lot number 0210306L6; Amgen) (n = 5–6 per group). Behavioral assessments for mechanical allodynia and thermal hyperalgesia were conducted every 20 min from 20 to 120 min after intrathecal gp120, as described above. Rats were anesthetized immediately after the 120 min behavioral assessment. Lumbar dorsal spinal cord tissue was collected and assayed for IL-1 protein.
Experiment 2: effect of intrathecal gp120 on systemic blood levels of IL-1
Intrathecal implanted rats were injected with either gp120 (n = 5) or equivolume vehicle (n = 6) in a manner identical to that used in Experiment 1. Rats were anesthetized 120 min later, and trunk blood was collected by decapitation. Serum was collected from the samples after centrifugation and stored at −70°C until assayed.
Experiment 3: time course of intrathecal gp120 effects on lumbosacral CSF and lumbar dorsal spinal cord levels of IL-1 and TNF protein
Rats were injected intrathecally with either gp120 or vehicle (n = 5–8/group). Rats were anesthetized 20, 40, 60, 90, or 120 min later. Lumbar dorsal spinal cord and lumbosacral CSF were collected to define the time course of cytokine changes in the spinal region where the gp120 injection occurred. Cervical dorsal spinal cord and cervical CSF were collected to determine the site specificity of gp120 effects.
Experiment 4: effect of intrathecal TNF antagonist on intrathecal gp120-induced enhanced pain states and intrathecal gp120-induced elevations of IL-1 protein in lumbosacral CSF and lumbar dorsal spinal cord
Lyophilized TNF binding protein (TNFbp; endotoxin-free polyethylene glycol recombinant–met-human soluble TNF receptor type 1; lot number 36000D8; Amgen) was reconstituted at 30 μg/μl in sterile distilled water, aliquoted on ice, and stored at −75°C. Intrathecal TNFbp (300 μg/10 μl) was injected after BL von Frey assessments, that is, 35 min before intrathecal gp120 or vehicle (n = 5–7/group). Control groups received intrathecal equivolume vehicle (lot number 1105208E8; Amgen). The behavioral assessment for mechanical allodynia was conducted as described above. Rats were anesthetized immediately after the 120 min behavioral assessment, and lumbosacral CSF and lumbar dorsal spinal cord were collected and assayed as described above.
Experiment 5: effect of intrathecal fluorocitrate on gp120-induced increases in lumbosacral CSF and lumbar dorsal spinal cord IL-1 protein
Fluorocitrate was dissolved in 10 μl of 2 m HCl (0.3% of final volume) and diluted in PBS to a final concentration of 1 nmol/μl, pH 6.0. Intrathecal fluorocitrate (1 nmol/μl; Sigma-Aldrich) or vehicle was injected after BL assessment, that is, 35 min before intrathecal gp120 or equivolume vehicle (0.3% 2m HCl in sterile PBS, pH 6.0) (n = 5–7/group). Behavioral assessments for thermal hyperalgesia and mechanical allodynia were conducted in separate groups of rats; these data have been published previously (Milligan et al., 2000). Rats were anesthetized immediately after the 120 min behavioral measure. Lumbosacral CSF and lumbar dorsal spinal cord were collected and assayed for IL-1 protein.
Experiment 6: effect of intrathecal gp120 on microglia and astrocyte activation marker expression in lumbar dorsal spinal cord
Single-label light microscopy. Rats used for immunohistochemistry procedures (n = 2–6/group) were transcardially perfused first with isotonic saline (250 ml) and then with 4% paraformaldehyde/0.1 m phosphate buffer (4% PFA/PB, pH 7.4; 250 ml). After dissection and immersion post-fixation in 4% PFA/PB for an additional 2 hr, the lumbar spinal cords were stored overnight in PBS, pH 7.4, with 0.1% azide at 4°C. Spinal cords were embedded in a single gelatin block (Sigma-Aldrich). This block was then fixed in 4% PFA/PB, cryoprotected overnight in 22% sucrose/PB, sectioned on a cryostat at 20 μm, and thaw-mounted on electrically charged glass slides (Fisherbrand Superfrost Plus Slides; Fisher Scientific, Pittsburgh, PA). Only caudal sections of lumbar spinal cord were collected because hindpaw afferent information projects to this region (Tabo et al., 1999). The thaw-mounted cryostat sections (three sections per spinal level per animal) were processed for immunoreactivity.
For immunohistochemistry, sections were treated to suppress endogenous peroxidase and to prevent staining of endogenous biotin. The sections were incubated in either primary mouse monoclonal anti-rat GFAP [1:500; to visualize astrocyte activation (Garrison et al., 1994); ICN Biomedicals, Costa Mesa, CA] or primary mouse monoclonal anti-rat OX-42 [1:500; to visualize microglial activation (Kaltschmidt et al., 1994); BioSource, Camarillo, CA] for 48 hr at 4°C. The slides were then incubated in secondary biotinylated goat anti-mouse IgG antibody (1:500; Jackson ImmunoResearch, West Grove, PA) overnight at 4°C. Finally, sections were reacted using the avidin–biotin complex procedure (ABC, Vector Elite kit; 1:100 in PBS-Triton; 2 hr; Vector Laboratories, Burlingame, CA) and 3′,3-diaminobenzidine (DAB; Sigma-Aldrich). Glucose oxidase (Sigma-Aldrich; type V-s; 0.02%) and β-d-glucose (0.1%) were used to generate hydrogen peroxide. Nickelous ammonium sulfate was added to the DAB solution (0.025%, w/v) to intensify the reaction product. One series of sections was processed as described above except that the primary antibody was omitted from the incubation buffer (omission controls). The slides were dried overnight, cleared, and coverslipped.
Slides were viewed with an Olympus Vannox II bright-field microscope. Images were collected with a Cohu CCD camera coupled to an Apple PowerMac 7200 equipped with NIH Image software (version 1.60) and stored in a ZIP disk. The obtained photomicrographs were exported to and labeled using Adobe Photoshop version 5.0. Brightness and contrast were kept constant with the images not altered.
Quantification of computer-assisted image analysis. To analyze glial activation, a new procedure was developed. Because of the complex cell morphologies of astrocytes and microglia, cell counts do not provide sufficient power to quantify activation, especially at relatively short postdrug times. Early glial activation is typified by hypertrophy and thus can be assessed by calculation of the percent of the field occupied by the stained cells. NIH Image simplifies this process, because there are macros that function specifically to calculate the percent of the field that is black. This analysis routine changes the light microscopy image into a digitized black-and-white (no gray) image, based on thresholds set by the investigator. Because the image analysis system has 254 levels of intensity, the upper limit threshold is 254. The lower limit threshold is varied to determine how much “signal” is accepted for analysis. Setting the lower threshold at 0% (at gray scale = 0; no intensity level excluded) results in 100% of the field being digitized as black. In contrast, setting the lower threshold at 100% (at gray scale = 254; all intensity levels are excluded) results in 0% of the field digitized as black. When the lower cutoff threshold is raised from 0 to 100% by 10% increments, a function of how “the percent of field black” changes with increasing threshold is derived. The dimensions of the selected field are set constant for all analyses. The intensity of the light source is calibrated each day. NIH Image calculates the percent-of-field-black functions (0–100%) that were analyzed for the dorsal horns of each rat, with equal representation of all experimental conditions on all slides. The data were then expressed as the percent of field black. On the basis of the percent-of-field-black functions derived, the midpoint was selected for statistical analysis for all subjects. Because control groups for 4, 8, and 18 hr showed no differences, the data from these animals were pooled to form a single control group.
RESULTS
Experiment 1: effect of intrathecal IL-1 receptor antagonist on intrathecal gp120-induced enhanced pain states and lumbar dorsal spinal cord IL-1 protein
We reported previously that drugs that preferentially disrupt glial function block gp120-induced enhanced pain states (Milligan et al., 2000). This predicts that disrupting the actions of specific substances released by gp120-activated glia should also block the concomitant pain changes. This experiment tested whether IL-1 is a key mediator. The effects of IL-1ra were tested on gp120-induced mechanical allodynia and thermal hyperalgesia. IL-1 protein levels were also assayed from the lumbar dorsal spinal cord of these same animals.
Mechanical allodynia
Before intrathecal injections, all groups exhibited comparable baseline thresholds (Fig. 1) [ANOVA,F(1,18) = 1.317; p > 0.2]. As in our previous study (Milligan et al., 2000), intrathecal gp120 produced a robust mechanical allodynia (Fig. 1, Vehicle + gp120 group). Pretreatment with intrathecal IL-1ra abolished this gp120-induced allodynia (Fig. 1, IL1ra + gp120 group). These differences resulted in reliable main effects of intrathecal IL-1ra [F(1,18) = 67.597; p < 0.0001] and intrathecal gp120 [F(1,18) = 73.608; p< 0.0001] and a reliable interaction of intrathecal IL-1ra and intrathecal gp120 [F(1,18) = 89.605;p < 0.0001]. Post hoc means comparisons revealed that gp120 (Fig. 1, Vehicle + gp120 group) produced a decrease in hindpaw low-mechanical response thresholds compared with all other treatment groups (Fig. 1, Vehicle + Vehicle,IL1ra + Vehicle, and IL1ra + gp120 groups;p < 0.0001 for all comparisons).
Fig. 1.

Blockade of intrathecal gp120-induced mechanical allodynia by intrathecal IL-1ra. Rats were assessed for low-threshold mechanical sensitivity (von Frey test) both before (BL) and 20–120 min after completion of intrathecal drug administration. Replicating our previous study (Milligan et al., 2000), intrathecal gp120 produced low-threshold mechanical allodynia in rats pretreated with the vehicle of IL-1ra (Vehicle + gp120;black squares), compared with controls (Vehicle + Vehicle; white squares). Although IL-1ra had no effect in the absence of gp120 (IL1ra + Vehicle;white circles), IL-1ra blocked mechanical allodynia induced by gp120 (IL1ra + gp120; black circles). i.t., Intrathecal.
Thermal hyperalgesia
All groups exhibited comparable baseline thresholds before intrathecal injections [F(1,17) < 1] (Fig. 2). As in our previous study (Milligan et al., 2000), intrathecal gp120 produced a robust thermal hyperalgesia (Fig. 2, Vehicle + gp120 group). Pretreatment with intrathecal IL-1ra greatly attenuated this gp120-induced hyperalgesia (Fig. 2, IL1ra + gp120 group). These differences resulted in reliable main effects of IL-1ra [F(1,17) = 10.517; p< 0.005] and of intrathecal gp120 [F(1,17) = 44.268; p< 0.0001] and a reliable interaction of IL-1ra and gp120 [F(1,17) = 17.861; p< 0.001]. Post hoc means comparisons revealed that gp120 (Fig. 2, Vehicle + gp120 group) produced a decrease in hindpaw withdrawal latencies compared with all other treatment groups (Fig. 2, Vehicle + Vehicle, IL1ra + Vehicle, andIL1ra + gp120 groups; p < 0.0001 for all comparisons). Additionally, rats pretreated with intrathecal IL-1ra followed by intrathecal gp120 showed a small but significant decrease in hindpaw threshold responses compared with the Vehicle + Vehicle and the IL1ra + Vehicle groups (Fig. 2;p < 0.05 for each comparison).
Fig. 2.

Blockade of intrathecal gp120-induced thermal hyperalgesia by intrathecal IL-1ra. Rats were assessed for heat sensitivity (Hargreaves test) both before (BL) and 20–120 min after completion of intrathecal drug administration. Replicating our previous study (Milligan et al., 2000), intrathecal gp120 produced thermal hyperalgesia in rats pretreated with the vehicle of IL-1ra (Vehicle + gp120; black squares), compared with controls (Vehicle + Vehicle; white squares). Although IL-1ra had no effect in the absence of gp120 (IL1ra + Vehicle; white circles), IL-1ra blocked thermal hyperalgesia induced by gp120 (IL1ra + gp120; black circles).
Lumbar dorsal spinal cord IL-1 protein
Intrathecal gp120 caused a large elevation in the lumbar dorsal spinal cord content of IL-1 protein (Fig.3, Vehicle + gp120 group). This result complements the finding that IL-1ra blocks gp120-induced pain states (Figs. 1, 2). Intrathecal IL-1ra blocked increases in the lumbar dorsal spinal cord IL-1 protein produced by intrathecal gp120 (Fig. 3, IL1ra + gp120 group). These differences resulted in a reliable main effect of gp120 [F(1,34) = 8.936; p< 0.01] and a reliable interaction of IL-1ra and gp120 [F(1,34) = 6.114; p< 0.025]. Post hoc means comparisons revealed that gp120 (Fig. 3, Vehicle + gp120 group) produced a reliable increase in IL-1 protein compared with all other treatment groups (Fig. 3,Vehicle + Vehicle, IL1ra + Vehicle, andIL1ra + gp120 groups; p < 0.0001 for all comparisons).
Fig. 3.

Elevations of lumbar dorsal spinal cord IL-1 produced by intrathecal gp120 are blocked by pretreatment with IL-1ra. After completion of behavioral testing (see Figs. 1, 2), lumbar dorsal spinal cord was collected and assayed by ELISA for IL-1 protein. Relative to controls (Vehicle + Vehicle; white bar), intrathecal gp120 increased lumbar dorsal spinal cord IL-1 protein (Vehicle + gp120; black bar). Although IL-1ra had no effect in the absence of gp120 (IL1ra + Vehicle; herringbone bar), IL-1ra blocked the gp120-induced increase of IL-1 at this time (IL1ra + gp120; striped bar).
Experiment 2: effect of intrathecal gp120 on systemic blood levels of IL-1
Because the animals in Experiment 1 were not transcardially perfused before lumbar tissue and CSF collection, it was possible that gp120-induced elevations in IL-1 actually represented induction of IL-1 in blood rather than in spinal tissues. This experiment tested whether intrathecal gp120 elevates blood-borne IL-1 levels. The results demonstrate that elevations in CSF and tissue levels of IL-1 after intrathecal gp120 cannot be accounted for by elevations in systemic blood IL-1. Intrathecal vehicle-treated animals had serum IL-1 levels of 81.163 ± 23.197 pg/ml, whereas intrathecal gp120-treated animals had serum levels of 40.196 ± 11.134 pg/ml (p > 0.05).
Experiment 3: time course of intrathecal gp120 effects on lumbosacral CSF and lumbar dorsal spinal cord levels of IL-1 and TNF protein
The results of Experiment 1 demonstrated that (1) intrathecal gp120-induced exaggerated pain states can be blocked by intrathecal IL-1ra, (2) intrathecal gp120 reliably increases lumbar dorsal spinal cord IL-1 protein levels at the single time point examined (∼135 min after intrathecal gp120), and (3) IL-1ra not only blocked behavior but also blocked gp120-induced increases in IL-1 in lumbar dorsal spinal cord at this 135 min time point. It is not surprising that IL-1 protein is increased in tissue by 2 hr after glial activation. However, counter to general ideas of CNS cytokine regulation, the IL-1ra data of Experiment 1 suggest that IL-1 protein must be rapidly released to account for behavioral changes observed as early as 20 min after intrathecal gp120. To determine whether IL-1 protein release occurs early enough to account for the behavioral changes observed, this experiment examined a time course of gp120-induced IL-1 changes in lumbar dorsal spinal cord and lumbosacral CSF. It should be noted that proinflammatory cytokines can be produced intracellularly but then not released (Watkins et al., 1999; Vitkovic et al., 2000). Thus CSF assays were added to assess whether release occurs. Furthermore, because proinflammatory cytokines rarely exert their effects alone, CSF levels of TNF were assayed as well. TNF was chosen for assay because TNF often precedes IL-1 release (Fong et al., 1989) and synergizes with IL-1 actions (Benveniste, 1997).
Cervical and lumbar IL-1 protein levels in dorsal spinal cord
A site-specific action of lumbosacral intrathecal gp120 was found because cervical dorsal spinal cord tissue levels of IL-1 protein were barely detectable throughout the time course (Fig.4A,B). Comparison of lumbar and cervical dorsal spinal cord levels revealed that lumbosacral intrathecal gp120 injection produced very large increases in IL-1 in the lumbar dorsal spinal cord region compared with the cervical dorsal region (Fig. 4A,B). These differences resulted in reliable main effects of gp120 [F(1,116) = 14.085; p< 0.0005], tissue region [F(1,116)= 35.216; p < 0.0001], and time [F(1,55) = 12.286; p< 0.0001] and interactions of gp120 and tissue region [F(1,116) = 4.901; p< 0.05], gp120 and time [F(1,116) = 5.529; p < 0.0005], and tissue region and time [F(4,116) = 4.327; p< 0.005]. Post hoc means comparisons revealed that IL-1 protein levels in the lumbosacral region were reliably higher compared with the cervical region (p < 0.0001).
Fig. 4.

Time course of IL-1 protein changes in the dorsal spinal cord after lumbosacral intrathecal gp120 administration. Rats were administered intrathecal gp120 (black squares) or intrathecal vehicle (black-and-white squares) either 20, 40, 60, 90, or 120 min before tissue collection. A, gp120 increased IL-1 protein in the dorsal lumbar spinal cord, relative to vehicle controls. B, Site specificity of this effect was found based on the fact that increases in IL-1 protein in the cervical spinal cord were both much lower in magnitude and slower to occur. Note that the assay units in this figure are in picograms (see Fig. 5, units in nanograms).
Lumbosacral intrathecal gp120 produced time-dependent increases in lumbar dorsal spinal cord IL-1 protein (Fig. 4A,black squares). Smaller elevations in IL-1 protein were observed in vehicle controls (Fig. 4A,black-and-white squares). These differences resulted in reliable main effects of gp120 [F(1,55) = 12.235; p< 0.001] and time [F(4,55) = 9.942;p < 0.0001] and a reliable interaction of gp120 and time [F(1,55) = 2.625;p < 0.05]. Post hoc means comparisons revealed that at 120 min after injection, gp120 produced a reliable increase (p < 0.05) of IL-1 protein levels compared with vehicle controls.
Cervical and lumbosacral IL-1 protein levels in CSF
A site-specific action of lumbosacral intrathecal gp120 was found because cervical CSF levels of IL-1 were virtually nonexistent throughout the time course (Fig.5A,B). Lumbosacral intrathecal gp120 produced time-dependent increases of IL-1 protein in lumbosacral CSF (Fig. 5A, black squares). Small elevations in IL-1 protein were observed in vehicle controls (Fig. 5A,black-and-white squares). These differences resulted in reliable main effects of gp120 [F(1,58) = 20.279; p< 0.0001] and time [F(4,58) = 3.879; p < 0.01]. No reliable interactions were found. Post hoc means comparisons revealed that at 60, 90, and 120 min after injection, gp120 produced a reliable increase in IL-1 in lumbosacral CSF compared with vehicle controls at these same time points (p < 0.05 for each comparison).
Fig. 5.

Time course of IL-1 protein changes in lumbosacral CSF after lumbosacral intrathecal gp120 administration. Rats were administered intrathecal gp120 (black squares) or intrathecal vehicle (black-and-white square) either 20, 40, 60, 90, or 120 min before tissue collection. A, gp120 increased IL-1 protein in lumbosacral CSF, relative to vehicle controls. B, Site specificity of this effect was found based on the fact that increases in IL-1 protein in the cervical CSF were both much lower in magnitude and slower to occur. Note that the assay units in this figure are in nanograms versus picograms in Figure4.
Cervical and lumbosacral TNF protein levels in CSF
Lumbosacral intrathecal gp120 produced time-dependent increases in TNF protein in lumbosacral CSF (Fig.6A, black squares). Smaller changes in TNF protein were observed in vehicle controls (Fig. 6A, black-and-white squares). These differences resulted in reliable main effects of gp120 [F(1,55) = 32.252;p < 0.0001] and time [F(4,55) = 4.633; p< 0.01] and a reliable interaction of gp120 and time [F(4,55) = 4.065; p< 0.01]. Post hoc means comparisons revealed that gp120 produced reliable increases in TNF protein levels 40, 60, and 90 min after injection compared with vehicle controls at these same time points (p < 0.05 for each comparison). Site-specific actions of lumbosacral intrathecal gp120 were again supported because cervical CSF levels of TNF protein were virtually undetectable throughout the time course (Fig.6B).
Fig. 6.

Time course of TNF protein changes in lumbosacral CSF after lumbosacral intrathecal gp120 administration. Rats were administered intrathecal gp120 (black squares) or intrathecal vehicle (black-and-white squares) either 20, 40, 60, 90, or 120 min before tissue collection. A, gp120 increased TNF protein in lumbosacral CSF, relative to vehicle controls. B, Site specificity of this effect was found based on the fact that increases in TNF protein in cervical CSF were both much lower in magnitude and slower to occur.
Experiment 4: effect of intrathecal TNF antagonist on intrathecal gp120-induced enhanced pain states and intrathecal gp120-induced elevations of IL-1 protein in lumbosacral CSF and lumbar dorsal spinal cord
Experiment 3 demonstrated that lumbosacral intrathecal gp120 led to the rapid release of TNF and IL-1 from this spinal region. TNF release at the site of gp120 injection was at least as rapid as IL-1 release. Because cytokines often act in cascades, with TNF both inducing IL-1 and synergizing with IL-1 actions (Fong et al., 1989;Benveniste, 1997), TNF may also be required for gp120-induced pain changes to occur. Thus, this experiment examined the effects of intrathecal pretreatment with the TNF functional antagonist TNFbp (also known as soluble TNF receptor). Assessment of mechanical allodynia was chosen because it is a robust test for enhanced pain and our previous studies have not shown allodynia in the absence of hyperalgesia (this paper) (Milligan et al., 2000). Additionally, the effects of TNFbp pretreatment on gp120-induced IL-1 protein changes in lumbosacral dorsal spinal cord and CSF were assessed in these same animals. Because lumbosacral CSF samples are too small to allow more than one cytokine to be measured, CSF TNF levels were not assayed.
gp120-induced allodynia is attenuated by TNFbp
All groups exhibited comparable baseline thresholds before intrathecal injections [F(3,32) = 2.844; p > 0.05] (Fig.7). Lumbosacral intrathecal gp120 again rapidly produced mechanical allodynia (Fig. 7, Vehicle + gp120 group). TNFbp reduced allodynia that was produced by intrathecal gp120 (Fig. 7, TNFbp + gp120 group). These differences resulted in a reliable main effect of gp120 [F(1,22) = 57.384; p< 0.0001] and a reliable interaction of TNFbp and gp120 [F(1,22) = 5.212; p< 0.05]. Post hoc means comparisons revealed that TNFbp + gp120 produced less allodynia than did gp120 alone (p < 0.0001). However, blockade of allodynia was only partial, because von Frey thresholds were decreased in theTNFbp + gp120 group compared with either the Vehicle + Vehicle group or the TNFbp + vehicle group (Fig. 7;p < 0.0001 for each comparison). Intrathecal gp120 decreased paw withdrawal thresholds compared with all other treatment groups (p < 0.0001 for each comparison).
Fig. 7.

Blockade of intrathecal gp120-induced mechanical allodynia by intrathecal TNFbp. Rats were assessed for low-threshold mechanical sensitivity (von Frey test) both before (BL) and 20–120 min after completion of intrathecal drug administration. Rats were administered either intrathecal TNFbp or vehicle before either intrathecal gp120 or vehicle. Replicating our previous work (Milligan et al., 2000) and Experiment 1, intrathecal gp120 produced mechanical allodynia in the absence of TNFbp (Vehicle + gp120;black squares), compared with controls (Vehicle + Vehicle; white squares). Although TNFbp had no effect in the absence of gp120 (TNFbp + vehicle;white circles), TNFbp partially blocked gp120-induced mechanical allodynia (TNFbp + gp120; black circles). ELISA results from these same animals are below (see Fig. 8).
TNFbp blocks gp120-induced elevations of IL-1 in lumbar dorsal spinal cord
Lumbosacral intrathecal gp120 again produced large increases in IL-1 protein in lumbar dorsal spinal cord (Fig.8A, Vehicle + gp120 group). Intrathecal TNFbp abolished the increases in IL-1 in lumbar dorsal spinal cord produced by intrathecal gp120 (Fig.8A, TNF-bp + gp120 group). These differences resulted in a reliable main effect of intrathecal gp120 [F(1,17) = 8.794; p< 0.01] and a reliable interaction of TNFbp and gp120 [F(1,17) = 6.045; p = 0.025]. Post hoc means comparisons revealed that gp120 (Fig. 8A, Vehicle + gp120 group) produced a reliable increase in IL-1 protein levels compared with all other treatment groups (Fig. 8A, Vehicle + Vehicle, TNF-bp + Vehicle, and TNF-bp + gp120 groups; p < 0.0001 for each comparison).
Fig. 8.

Elevations of dorsal lumbar spinal cord IL-1 and lumbosacral CSF IL-1 produced by intrathecal gp120 are both blocked by pretreatment with intrathecal TNFbp. After completion of behavioral testing (see Fig. 7), dorsal lumbar spinal cord (A) and lumbosacral CSF (B) were collected and assayed by ELISA for IL-1 protein. Relative to controls (Vehicle + Vehicle; white bar), intrathecal gp120 increased dorsal lumbar spinal cord IL-1 protein content (Vehicle + gp120; black bar). Although TNFbp had no effect in the absence of gp120 (TNF-bp + Vehicle; herringbone bar), TNFbp blocked the gp120-induced increase of IL-1 at this time (TNF-bp + gp120; striped bar).
TNFbp blocks gp120-induced elevations of IL-1 in lumbosacral CSF
Lumbosacral intrathecal gp120 again produced large increases in IL-1 protein in lumbosacral CSF (Fig. 8B,Vehicle + gp120 group). TNFbp abolished the increases in IL-1 in lumbosacral CSF produced by intrathecal gp120 (Fig.8B, TNF-bp + gp120 group). These differences resulted in reliable main effects of TNFbp [F(1,34) = 4.858; p< 0.05] and gp120 [F(1,34) = 8.501;p < 0.01] and a reliable interaction of TNFbp and gp120 [F(1,34) = 4.756;p < 0.05]. Post hoc means comparisons revealed that gp120 (Fig. 8B, Vehicle + gp120 group) produced a reliable increase in IL-1 protein levels in CSF compared with all other treatment groups (Fig.8B, Vehicle + Vehicle, TNF-bp + Vehicle, and TNF-bp + gp120 groups; p< 0.0001 for each comparison).
Experiment 5: effect of intrathecal fluorocitrate on gp120-induced increases in lumbosacral CSF and lumbar dorsal spinal cord IL-1 protein
We have reported previously that fluorocitrate, a glial metabolic inhibitor (Hassel et al., 1992), blocks gp120-induced exaggerated pain states (Milligan et al., 2000). Although not reported previously, lumbar dorsal spinal cord and lumbosacral CSF were collected from all animals in that study after completion of the 120 min behavioral assessments. Results from Experiments 1–4 demonstrate that IL-1 may be a proximate mediator of gp120-induced exaggerated pain states. This suggests that fluorocitrate may disrupt gp120-induced increases in both lumbosacral CSF and lumbar dorsal spinal cord IL-1 protein.
Fluorocitrate attenuates gp120-induced IL-1 increases in lumbar dorsal spinal cord
Lumbosacral intrathecal gp120 again greatly increased IL-1 protein in lumbar dorsal spinal cord (Fig.9A, Vehicle + gp120group). Intrathecal fluorocitrate greatly reduced this gp120-induced increase in IL-1 (Fig. 9A, Fluorocitrate + gp120group). These differences resulted in reliable main effects of intrathecal fluorocitrate [F(1,1) = 4.884; p = 0.03] and gp120 [F(1,1) = 24.800; p< 0.0001] and an interaction of fluorocitrate pretreatment and gp120 treatment [F(1,19) = 4.624;p = 0.04]. Post hoc means comparisons revealed that gp120 (Fig. 9A, Vehicle + gp120group) produced a reliable increase in IL-1 protein levels compared with all other treatment groups (Fig. 9A, Vehicle + Vehicle, Fluorocitrate + Vehicle, andFluorocitrate + gp120 groups; p < 0.01 for each comparison).
Fig. 9.

Elevations of dorsal lumbar spinal cord IL-1 and lumbosacral CSF IL-1 produced by intrathecal gp120 are both attenuated by pretreatment with intrathecal fluorocitrate. The behavioral data from these animals, which demonstrate that intrathecal fluorocitrate blocks gp120-induced pain states, have been published previously (Milligan et al., 2000). After completion of behavioral testing (Milligan et al., 2000), dorsal lumbar spinal cord (A) and lumbosacral CSF (B) were collected and assayed by ELISA for IL-1 protein. Relative to controls (Vehicle + Vehicle; white bar), intrathecal gp120 increased dorsal lumbar spinal cord IL-1 protein content (Vehicle + gp120; black bar). Although fluorocitrate had no effect in the absence of gp120 (Fluorocitrate + Vehicle; herringbone bar), fluorocitrate attenuated the gp120-induced increase of IL-1 at this time (Fluorocitrate + gp120; striped bar).
Fluorocitrate attenuates gp120-induced increases in IL-1 in lumbosacral CSF
Lumbosacral intrathecal gp120 again greatly increased IL-1 protein in lumbosacral CSF (Fig. 9B, Vehicle + gp120 group). Intrathecal fluorocitrate primarily reduced this gp120-induced increase in IL-1 (Fig. 9B, Fluorocitrate + gp120 group). These differences resulted in reliable main effects of fluorocitrate [F(1,1)= 15.211; p < 0.001] and gp120 [F(1,1) = 40.819; p< 0.0001] and a reliable interaction of fluorocitrate and gp120 [F(1,21) = 5.535; p< 0.05]. Post hoc means comparisons revealed that gp120 (Fig. 9B, Vehicle + gp120 group) produced a reliable increase in IL-1 levels compared with all other treatment groups (Fig. 9B, Vehicle + Vehicle,Fluorocitrate + Vehicle, and Fluorocitrate + gp120 groups; p < 0.001 for each comparison). Fluorocitrate did not completely block gp120-induced IL-1 increases because fluorocitrate + gp120 increased IL-1 levels in CSF compared with fluorocitrate alone (Fig. 9B, Fluorocitrate + Vehicle group; p < 0.05).
Experiment 6: effect of intrathecal gp120 on microglia and astrocyte activation marker expression in lumbar dorsal spinal cord
We have reported previously that disrupting the action of spinal cord glia blocks gp120-induced exaggerated pain states (Milligan et al., 2000). The results of Experiments 1–5 provide further support for the involvement of glial cell activation in gp120-induced pain states by the release of IL-1 and TNF, factors known to be released by activated glia (Benveniste, 1997). To examine further the potential role of glia in intrathecal gp120-induced pain states, this experiment examined whether intrathecal gp120 increased the expression of activation markers by either microglia or astrocytes.
Astrocyte activation
Intrathecal gp120 caused a progressive increase in astrocyte activation markers at 4, 8, and 18 hr (Fig.10A,B). These increases resulted in a reliable main effect of gp120 [F(3,14) = 4.118; p< 0.05]. Post hoc means comparisons revealed that gp120 increased astrocyte activation at 18 hr compared with either the vehicle group or the group analyzed at 4 hr after gp120 (p < 0.05 for each comparison; Fig.10B). Photomicrographs of typical dorsal spinal cord sections from 8 and 18 hr control and gp120-treated groups are illustrated in Figure 11.
Fig. 10.

Astrocyte and microglial activation after gp120.A, C, Each graph was generated by analyzing every captured image at 0–100% threshold. By doing so, each image varied through the range of 100–0% of the field that was black. This allows the midrange of the functions to be determined for statistical analysis. The downward arrows in A andC indicate the point on the function that was statistically analyzed and graphically presented in Band D, respectively. B, Compared with vehicle controls (white bar), an increase in astrocyte activation (GFAP immunoreactivity) progressively occurred between 4 hr (black bar), 8 hr (herringbone bar), and 18 hr (striped bar) after intrathecal gp120.D, Increased microglial activation (OX-42 labeling) occurred after gp120 (time points as described above) compared with vehicle controls (white bar).
Fig. 11.

Photomicrographs of activation of dorsal lumbar spinal cord astrocytes by intrathecal gp120. A, C, GFAP labeling of lumbar dorsal horn at 8 and 18 hr, respectively, after intrathecal vehicle. B, D, GFAP labeling of lumbar dorsal horn at 8 and 18 hr, respectively, after intrathecal gp120. Scale bar, 50 μm. veh, Vehicle.
Microglial activation
Intrathecal gp120 caused an increase in microglia activation markers at 4, 8, and 18 hr (Fig. 10C,D). These differences resulted in a reliable main effect of gp120 [F(3,14) = 7.128; p< 0.005]. Post hoc means comparisons revealed that gp120 produced an increase in microglial activation at all time points compared with vehicle controls (p < 0.01 for each comparison; Fig. 10D). Photomicrographs of typical dorsal spinal cord sections from 8 and 18 hr control and gp120-treated groups are illustrated in Figure12.
Fig. 12.
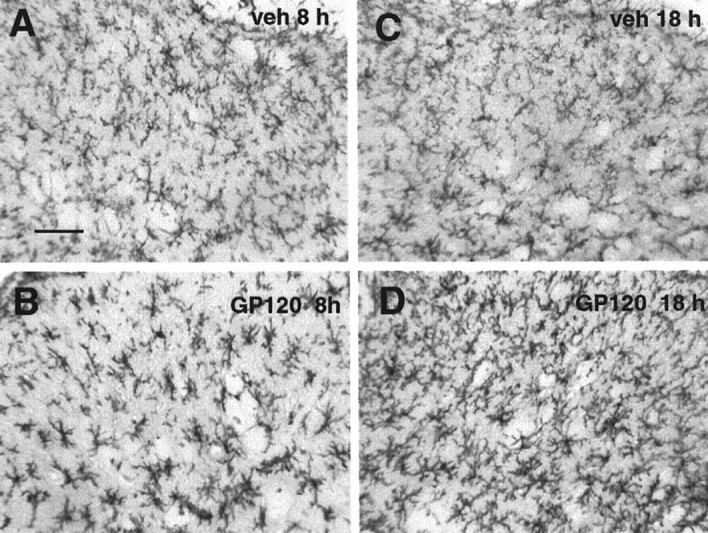
Photomicrographs of activation of dorsal lumbar spinal cord microglia by intrathecal gp120. A, C, OX-42 labeling of lumbar dorsal horn at 8 and 18 hr, respectively, after intrathecal vehicle. B, D, OX-42 labeling of lumbar dorsal horn at 8 and 18 hr, respectively, after intrathecal gp120. Scale bar, 50 μm.
DISCUSSION
We have shown recently that thermal hyperalgesia and mechanical allodynia are produced by intrathecal gp120 (Milligan et al., 2000). The present experiments implicate endogenous spinal proinflammatory cytokines (TNF and IL-1) as key mediators. Although previous studies linked spinal IL-1 to exaggerated pain (Watkins et al., 1997; Hammack et al., 1999; Sweitzer et al., 1999; Chacur et al., 2000), we demonstrate that endogenous spinal TNF also facilitates pain. Furthermore, this is the first study to document rapid in vivo spinal release of these proinflammatory cytokines by pain-enhancing stimuli. These studies demonstrate that (1) lumbosacral gp120 stimulates time-dependent, site-specific release of TNF and IL-1 from lumbar cord; (2) intrathecal IL-1ra blocks intrathecal gp120-induced mechanical allodynia and thermal hyperalgesia; (3) an intrathecal TNF functional antagonist (TNFbp) attenuates intrathecal gp120-induced mechanical allodynia (hyperalgesia not tested); (4) intrathecal gp120-induced production and release of spinal IL-1 is blocked by intrathecal pretreatment with IL-1ra, TNFbp, or fluorocitrate [glial inhibitor that blocks gp120-induced pain states (Milligan et al., 2000)]; and (5) intrathecal gp120 activates dorsal cord astrocytes and microglia as assessed by a new method for quantifying immunohistochemistry. Together, these experiments provide support that gp120-induced pain states are mediated by proinflammatory cytokines released from activated astrocytes and/or microglia in dorsal spinal cord.
Because gp120 actions in the CNS are predominantly via activation of astrocytes and microglia [for review, see Milligan et al. (2000)], how can gp120-induced exaggerated pain states result? First, IL-1 and TNF, released in response to gp120, may directly stimulate dorsal horn pain transmission neurons. Neurons in brain, at least, express receptors for IL-1 (Cunningham and De Souza, 1993) and TNF (Botchkina et al., 1997; Chambaut-Guerin et al., 1997). Supraspinally administered IL-1 rapidly creates pain behaviors (Oka et al., 1993, 1995; Watkins et al., 1994) and pain-specific excitation of trigeminal dorsal horn neurons (Oka and Hori, 1999). In cord, pain is facilitated by intrathecal IL-1 administered alone (Tadano et al., 1999) or with other cytokines (Meller et al., 1994), and blockade of endogenous spinal IL-1 (either alone or in combination with TNF blockade) attenuates various forms of pain facilitation (Watkins et al., 1997; Hammack et al., 1999;Sweitzer et al., 1999; Chacur et al., 2000). The present studies clearly document that endogenous spinal TNF also modulates pain. Intriguingly, it appears to do so at least in part by its influence on IL-1. That is, blocking the actions of endogenous TNF prevented the production and release of IL-1.
Indeed, because peripheral proinflammatory cytokines often act in cascades with TNF inducing the production and release of IL-1 (Watkins et al., 1999), the blockade of spinal IL-1 production by the TNF antagonist suggests that a similar cascade exists in spinal cord as well. Perhaps more surprising was that the IL-1 receptor antagonist also blocked the elevation of IL-1 normally observed 2 hr after intrathecal gp120. What is known from the peripheral proinflammatory cytokine literature is that “IL-1 begets IL-1”; that is, IL-1 stimulates its own further release (Watkins et al., 1999). Thus, although clearly speculative, it is plausible that IL-1ra blocks the ability of early released IL-1 to enhance further IL-1 production and release. Hence by 2 hr, gp120-induced IL-1 production is blocked.
It is also possible that IL-1 and TNF are not the proximate cause of pain. Of the neuroactive substances released directly or indirectly by gp120, NO is a prime candidate for creating pain facilitation. NO has been repeatedly implicated in exaggerated pain states (Meller et al., 1992b). Indeed, elevated NO is sufficient to induce pain facilitation (Shibuta et al., 1995). NO is principally generated by two major enzymes in the CNS, inducible nitric oxide synthase (iNOS) and constitutive NOS (cNOS). Gene activation and protein synthesis of iNOS are required before NO is produced. Hence, iNOS is not a likely mediator of gp120-induced pain phenomena because of the rapidity with which mechanical allodynia and thermal hyperalgesia appear after intrathecal gp120.
In contrast, cNOS is a likely mediator. Neurons (Murphy and Grzybicki, 1996), astrocytes (Murphy and Grzybicki, 1996;Togashi et al., 1997), and microglia all basally express cNOS. Indeed, cNOS is the principal isoform of NOS in both neurons (Murphy and Grzybicki, 1996) and astrocytes (Togashi et al., 1997). Only two prerequisites are required for NO production by cNOS: (1) intracellular l-arginine, the substrate for cNOS (Dreyer et al., 1999), and (2) elevation of intracellular calcium, which activates cNOS (Murphy and Grzybicki, 1996). gp120 (Corasaniti et al., 1995), IL-1 (Mollace et al., 1998), and TNF (Mollace et al., 1998) each activate the l-arginine transporter, thereby increasing intracellular l-arginine. Increased intracellular calcium can also occur rapidly after intrathecal gp120. First, gp120 increases extracellular excitatory amino acids (Lipton et al., 1994). These are agonists at dorsal horn NMDA receptors that, when activated, increase intracellular calcium in neurons (Dawson et al., 1993), microglia (Giulian et al., 1990), and astrocytes (Mollace et al., 1998). Second, the present studies show that gp120 rapidly releases IL-1. Neurons (Cunningham and De Souza, 1993) and glia (Ballestas and Benveniste, 1995; Pita et al., 1999) each express receptors for IL-1, and in tissues that have been tested to date, IL-1 binding increases intracellular calcium (Ballestas and Benveniste, 1995; Pita et al., 1999). Third, gp120 can activate chemokine receptor subtypes on astrocytes and microglia (Popik et al., 1998; Kaul and Lipton, 1999;Klein et al., 1999). This causes G-protein-linked increases in intracellular calcium (Madani et al., 1998; Bajetto et al., 1999). Together, these lines of evidence provide support that rapid NO production would be expected after intrathecal gp120. Indeed, we have found recently that a nonselective NOS inhibitor blocks intrathecal gp120 effects on pain (Holguin et al., 2000; Watkins et al., 2001); studies with cNOS- and iNOS-selective inhibitors are ongoing.
Although IL-1 and TNF protein are not classically thought to be constitutively expressed (Watkins et al., 1999), the rapidity of IL-1 and TNF release provides evidence of preexisting pools of these proteins in spinal cord. In support, IL-1 and TNF protein have been observed in normal rat spinal cord by the use of immunohistochemistry (DeLeo and Colburn, 1999), and basal levels of IL-1 have been reported in spinal cord tissue by ELISA (Wang et al., 1997; Nguyen et al., 2000). TNF has not yet been detected in spinal cord by ELISA because no appropriate ELISA yet exists for CNS tissues. However basal levels of TNF have been detected in spinal cord by bioassay (Covey et al., 2000). Furthermore, mRNA for TNF and IL-1 has been detected in cord under basal conditions (Wang et al., 1997; Hansen et al., 1999; Sweitzer et al., 1999; Le et al., 2000).
Although previous studies have documented rapid rises of IL-1 protein levels in the brain in response to various challenges (Nguyen et al., 1998, 2000), these studies were not able to prove that the IL-1 was released. Because IL-1 protein can be created but not released (Watkins et al., 1999), studies of CNS tissues have been compromised to date by the inability to prove that the IL-1 protein changes measured are physiologically meaningful. The present studies provide a clear demonstration that spinal IL-1 and TNF are released by gp120 challenge because both accumulate in CSF. Cytokines in lumbosacral CSF must reflect local release because (1) lumbosacral intrathecal gp120 failed to elevate IL-1 in cervical CSF or cervical dorsal spinal cord and (2) no gp120-induced increase in IL-1 was detected in systemic blood. Because spinal CSF can be readily assayed, spinal cord provides an excellent model system for examining dynamic changes in proinflammatory cytokines.
One shortcoming of ELISAs is that they cannot indicate which cell type(s) is creating the cytokine measured. Immunohistochemistry allows visualization of cytokine sources. We have found recently that IL-1 immunoreactivity is only expressed in astrocytes under either basal or gp120-stimulated conditions (Milligan et al., 1999a). Thus, to date, IL-1 release in response to gp120 appears to be from astrocytes.
The fact that IL-1 and TNF are both rapidly released by gp120 suggests that IL-1 and TNF may act in concert to create exaggerated pain states. Certainly, the fact that blockade of TNF actions prevented IL-1 production and release suggests that they work together to exaggerate pain. Beyond TNF simply influencing IL-1 release, peripheral IL-1 and TNF actions are also known to synergize frequently. Indeed, TNF–IL-1 synergies have been reported within the CNS as well (Benveniste, 1997;Bhat et al., 1999).
In summary, these studies suggest that exaggerated pain states can be created by immune challenge within spinal cord and that these exaggerated pain states are created by release of glial proinflammatory cytokines. These data suggest that spinal cord proinflammatory cytokines may be one source of clinical pain when pathogens (viruses, bacteria, etc.) invade the spinal cord. Using HIV-1 as the example, many HIV-1 variants are neurotropic (homing to the CNS) (Tyor et al., 1992) and concentrate in dorsal spinal cord (DiStefano et al., 1996). Because antiretroviral drugs used to treat HIV-1 do not cross the blood–brain barrier (VanLeeuwen et al., 1996), dorsal spinal cord infection would be expected to be unabated by current acquired immunodeficiency syndrome (AIDS) therapies. An examination of the clinical literature reveals that 80% of AIDS patients suffer from chronic pain and that approximately half of these pain conditions are of unknown origin. Whether a physical bodily reason for pain is identifiable or not, spinal cord glial activation would be predicted to exaggerate pain. If true, this would argue for developing drugs for clinical use that disrupt glial and/or proinflammatory cytokine actions. Such a strategy would represent a dramatic conceptual departure from all therapies currently used to treat these painful conditions.
Footnotes
This work was supported by National Institutes of Health Grants MH 01558, MH 00314, MH 45045, and NS 38020 and by the Undergraduate Research Opportunities Program at the University of Colorado at Boulder. We thank Marucia Chacur for her surgical assistance and Amgen for their gift of gp120, IL-1ra, TNFbp, and vehicles.
Correspondence should be addressed to Dr. Erin D. Milligan, Department of Psychology, Campus Box 345, University of Colorado at Boulder, Boulder, CO 80309-0345. E-mail: emilligan@psych.colorado.edu.
REFERENCES
- 1.Bajetto A, Bonavia R, Barbero S, Piccioli P, Costa A, Florio T, Schettini G. Glial and neuronal cells express functional chemokine receptor CXCR4 and its natural ligand stromal cell-derived factor 1. J Neurochem. 1999;73:2348–2357. doi: 10.1046/j.1471-4159.1999.0732348.x. [DOI] [PubMed] [Google Scholar]
- 2.Ballestas ME, Benveniste EN. Interleukin-1-beta and tumor necrosis factor-alpha mediated regulation of ICAM-1 gene expression in astrocytes requires protein kinase C activity. Glia. 1995;14:267–278. doi: 10.1002/glia.440140404. [DOI] [PubMed] [Google Scholar]
- 3.Benveniste EN. Cytokine expression in the nervous system. In: Keane RW, Hickey WF, editors. Immunology of the nervous system. Oxford UP; New York: 1997. pp. 419–459. [Google Scholar]
- 4.Bhat NR, Zhang P, Bhat AN. Cytokine induction of inducible nitric oxide synthase in an oligodendrocyte cell line: role of p38 mitogen-activated protein kinase activation. J Neurochem. 1999;72:472–478. doi: 10.1046/j.1471-4159.1999.0720472.x. [DOI] [PubMed] [Google Scholar]
- 5.Botchkina GI, Meistrell MEr, Botchkina IL, Tracey KJ. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol Med. 1997;3:765–781. [PMC free article] [PubMed] [Google Scholar]
- 6.Chacur M, Armstrong CB, Milligan ED, Myers RR, Gazda L, Martin D, Tracey KJ, Maier SF, Watkins LR. Exaggerated pain from sciatic inflammatory neuritis (SIN) is mediated by spinal cord glial activation. Soc Neurosci Abstr. 2000;26:1958. [Google Scholar]
- 7.Chambaut-Guerin AM, Rouher C, Gauthereau X. p55 tumour necrosis factor receptors distribution in neuroblastoma cells. NeuroReport. 1997;14:1451–1456. doi: 10.1097/00001756-199704140-00025. [DOI] [PubMed] [Google Scholar]
- 8.Chaplan S, Bach F, Pogrel J, Chung J, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 9.Corasaniti MT, Melino G, Navarra M, Garaci E, Finazzi-Agro A, Nistico G. Death of cultured human neuroblastoma cells induced by HIV-1 gp120 is prevented by NMDA receptor antagonists and inhibitors of nitric oxide and cyclooxygenase. Neurodegeneration. 1995;4:315–321. doi: 10.1016/1055-8330(95)90021-7. [DOI] [PubMed] [Google Scholar]
- 10.Covey WC, Ignatowski TA, Knight PR, Spengler RN. Brain-derived TNFα: involvement in neuroplastic changes implicated in the conscious perception of persistent pain. Brain Res. 2000;859:113–122. doi: 10.1016/s0006-8993(00)01965-x. [DOI] [PubMed] [Google Scholar]
- 11.Coyle DE. Partial peripheral nerve injury leads to activation of astroglia and microglia which parallels the development of allodynic behavior. Glia. 1998;23:75–83. [PubMed] [Google Scholar]
- 12.Cunningham ET, De Souza EB. Interleukin-1 receptors in the brain and endocrine tissues. Immunol Today. 1993;14:171–176. doi: 10.1016/0167-5699(93)90281-o. [DOI] [PubMed] [Google Scholar]
- 13.Dawson VL, Dawson TM, Uhl GR, Snyder SH. Human immunodeficiency virus type 1 coat protein neurotoxicity mediated by nitric oxide in primary cortical cultures. Proc Natl Acad Sci USA. 1993;90:3256–3259. doi: 10.1073/pnas.90.8.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeLeo JA, Colburn RW. Proinflammatory cytokines and glial cells: their role in neuropathic pain. In: Watkins LR, Maier SF, editors. Cytokines and pain. Birkhäuser; Basel: 1999. pp. 159–181. [Google Scholar]
- 15.DiStefano M, Gray F, Leitner T, Chiodi F. Analysis of ENV V3 sequences from HIV-1 infected brain indicates restrained virus expression throughout the disease. J Med Virol. 1996;49:41–48. doi: 10.1002/(SICI)1096-9071(199605)49:1<41::AID-JMV7>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 16.Dixon W. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- 17.Dreyer EB, Zurakowski D, Gorla M, Vorwerk CK, Lipton SA. The contribution of various NOS gene products to HIV-1 coat protein (gp120)-mediated retinal ganglion cell injury. Invest Ophthalmol Vis Sci. 1999;40:983–989. [PubMed] [Google Scholar]
- 18.Dutton G. Astrocyte amino acids: evidence for release and possible interactions with neurons. In: Murphy S, editor. Astrocytes: pharmacology and function. Academic; San Diego: 1993. pp. 173–192. [Google Scholar]
- 19.Fong Y, Tracey KJ, Moldawer LL, Hesse DG, Manogue KB, Kenny JS, Lee AT, Kuo GC, Allison AC, Lowry SF, Cerami A. Antibodies to cachectin/tumor necrosis factor reduce interleukin 1β and interleukin 6 appearance during lethal bacteremia. J Exp Med. 1989;170:1627–1633. doi: 10.1084/jem.170.5.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu K-Y, Light AR, Matsushima GK, Maixner W. Microglial reactions after subcutaneous formalin injection into the rat hind paw. Brain Res. 1999;825:59–67. doi: 10.1016/s0006-8993(99)01186-5. [DOI] [PubMed] [Google Scholar]
- 21.Garrison C, Dougherty P, Carlton S. GFAP expression in lumbar spinal cord of naive and neuropathic rats treated with MK-801. Exp Neurol. 1994;129:237–243. doi: 10.1006/exnr.1994.1165. [DOI] [PubMed] [Google Scholar]
- 22.Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. 1991;565:1–7. doi: 10.1016/0006-8993(91)91729-k. [DOI] [PubMed] [Google Scholar]
- 23.Giulian D, Vaca K, Noonan CA. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science. 1990;250:1593–1596. doi: 10.1126/science.2148832. [DOI] [PubMed] [Google Scholar]
- 24.Haley JE, Wilcox GL. Involvement of excitatory amino acids and peptides in the spinal mechanisms underlying hyperalgesia. In: Willis WD, editor. Hyperalgesia and allodynia. Raven; New York: 1992. pp. 281–293. [Google Scholar]
- 25.Hammack SE, Milligan ED, Gazda L, Martin D, Maier SF, Watkins LR. Spinal interleukin-1 receptor antagonist blocks allodynia from zymosan-induced neuritis. Soc Neurosci Abstr. 1999;25:1443. [Google Scholar]
- 26.Hansen MK, Nguyen KT, Fleshner M, Goehler LE, Gaykema RPA, Maier SF, Watkins LR. Interleukin-1beta protein and IL1beta, IL1 receptor type I and IL1 receptor accessory protein mRNA are present in normal brain and are modulated by LPS. Soc Neurosci Abstr. 1999;25:1446. [Google Scholar]
- 27.Hansen MK, Nguyen KT, Fleshner M, Goehler LE, Gaykema RPA, Maier SF, Watkins LR. Effects of vagotomy on serum endotoxin, cytokines, and corticosterone after intraperitoneal lipopolysaccharide. Am J Physiol Regul Integr Comp Physiol. 2000a;278:R331–R336. doi: 10.1152/ajpregu.2000.278.2.R331. [DOI] [PubMed] [Google Scholar]
- 28.Hansen MK, Nguyen KT, Goehler LE, Gaykema RPA, Fleshner M, Watkins LR, Maier SF. Effects of vagotomy on lipopolysaccharide-induced brain interleukin-1beta protein in rats. Auton Neurosci. 2000b;85:119–126. doi: 10.1016/s1566-0702(00)00230-7. [DOI] [PubMed] [Google Scholar]
- 29.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 30.Hartung HP, Heininger K, Schafer B, Toyka KV. Substance P stimulates release of arachidonic acid cyclooxygenation products from primary culture rat astrocytes. Ann NY Acad Sci. 1988;540:427–429. doi: 10.1111/j.1749-6632.1988.tb27123.x. [DOI] [PubMed] [Google Scholar]
- 31.Harvey LOJ. Efficient estimation of sensory thresholds. Behav Res Methods Instrum Comput. 1986;18:623–632. [Google Scholar]
- 32.Hassel B, Paulsen RE, Johnson A, Fonnum F. Selective inhibition of glial cell metabolism by fluorocitrate. Brain Res. 1992;249:120–124. doi: 10.1016/0006-8993(92)90616-h. [DOI] [PubMed] [Google Scholar]
- 33.Holguin A, Armstrong CB, Twinning C, Milligan ED, McGorry M, O'Connor K, Quan N, Martin D, Lappi DA, Maier SF, Watkins LR. Anatomical evidence for glial activation after intrathecal lumbosacral HIV-1 glycoprotein, gp120-induced allodynia. Soc Neurosci Abstr. 2000;26:1958. [Google Scholar]
- 34.Kaltschmidt C, Kaltschmidt B, Lannes-Vieira J, Kreutzberg G, Wekerle H, Baeuerle P, Gehrmann J. Transcription factor NF-kappa B is activated in microglia during experimental autoimmune encephalomyelitis. J Neuroimmunol. 1994;55:99–106. doi: 10.1016/0165-5728(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 35.Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci USA. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. [Google Scholar]
- 37.Klein RS, Williams KC, Alvarez-Hernandez X, Westmoreland S, Force T, Lackner AA, Luster AD. Chemokine receptor expression and signaling in macaque and human fetal neurons and astrocytes: implications for the neuropathogenesis of AIDS. J Immunol. 1999;163:1636–1646. [PubMed] [Google Scholar]
- 38.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 39.Le YL, Shih K, Bao P, Ghirnikar RS, Eng LF. Cytokine chemokine expression in contused rat spinal cord. Neurochem Int. 2000;36:417–425. doi: 10.1016/s0197-0186(99)00133-3. [DOI] [PubMed] [Google Scholar]
- 40.Lipton SA, Yeh M, Dreyer EB. Update on current models of HIV-related neuronal injury: platelet-activating factor, arachidonic acid and nitric oxide. Adv Neuroimmunol. 1994;4:181–188. doi: 10.1016/s0960-5428(06)80255-x. [DOI] [PubMed] [Google Scholar]
- 41.Madani N, Kozak SL, Kavanaugh MP, Kabat D. gp120 envelope glycoproteins of human immunodeficiency viruses competitively antagonize signaling by coreceptors CXCR4 and CCR5. Proc Natl Acad Sci USA. 1998;95:8005–8010. doi: 10.1073/pnas.95.14.8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marriott DR, Wilkin G, Wood JN. Substance P-induced release of prostaglandins from astrocytes: regional specialisation and correlation with phosphoinositol metabolism. J Neurochem. 1991;56:259–265. doi: 10.1111/j.1471-4159.1991.tb02590.x. [DOI] [PubMed] [Google Scholar]
- 43.Meller ST, Dykstra C, Gebhart GF. Production of endogenous nitric oxide and activation of soluble guanylate cyclase are required for N-methyl-d-aspartate-produced facilitation of the nociceptive tail-flick reflex. Eur J Pharmacol. 1992a;214:93–96. doi: 10.1016/0014-2999(92)90102-a. [DOI] [PubMed] [Google Scholar]
- 44.Meller ST, Pechman PS, Gebhart GF, Maves TJ. Nitric oxide mediates the thermal hyperalgesia produced in a model of neuropathic pain in the rat. Neuroscience. 1992b;50:7–10. doi: 10.1016/0306-4522(92)90377-e. [DOI] [PubMed] [Google Scholar]
- 45.Meller ST, Dykstra C, Grzbycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 46.Milligan ED, Nguyen K, Hansen MK, Martin D, Maier S, Watkins L. The human immunodeficiency virus-1 envelope glycoprotein, gp120, induces hyperalgesia and allodynia via glial activation and interleukin-1beta. Soc Neurosci Abstr. 1999a;25:1444. [Google Scholar]
- 47.Milligan ED, Hinde JL, Mehmert KK, Maier SF, Watkins LR. A method for increasing the viability of the external portion of the lumbar catheters placed in the spinal subarachnoid space of rats. J Neurosci Methods. 1999b;90:81–86. doi: 10.1016/s0165-0270(99)00075-8. [DOI] [PubMed] [Google Scholar]
- 48.Milligan ED, Mehmert KK, Hinde JL, Harvey LOJ, Martin D, Tracey KJ, Maier SF, Watkins LR. Thermal hyperalgesia and mechanical allodynia produced by intrathecal administration of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein, gp120. Brain Res. 2000;861:105–116. doi: 10.1016/s0006-8993(00)02050-3. [DOI] [PubMed] [Google Scholar]
- 49.Mollace V, Colasanti M, Muscoli C, Lauro GM, Iannone M, Rotiroti D, Nistico G. The effect of nitric oxide on cytokine-induced release of PGE2 by human cultured astroglial cells. Br J Pharmacol. 1998;124:742–746. doi: 10.1038/sj.bjp.0701852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murphy S, editor. Astrocytes: pharmacology and function. Academic; San Diego: 1993. [Google Scholar]
- 51.Murphy S, Grzybicki D. Glial NO: normal and pathological roles. The Neuroscientist. 1996;2:90–99. [Google Scholar]
- 52.Nguyen KT, Deak T, Owens SM, Kohno T, Fleshner M, Watkins LR, Maier SF. Exposure to acute stress induces brain interleukin-1beta protein in the rat. J Neurosci. 1998;18:2239–2246. doi: 10.1523/JNEUROSCI.18-06-02239.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nguyen KT, Deak T, Will MJ, Hansen M, Hunsaker BN, Fleshner M, Watkins LR, Maier SF. Timecourse and corticosterone sensitivity of the brain, pituitary, and serum interleukin-1b response to acute stress. Brain Res. 2000;859:193–201. doi: 10.1016/s0006-8993(99)02443-9. [DOI] [PubMed] [Google Scholar]
- 54.Oka T, Hori T. Brain cytokines and pain. In: Watkins LR, Maier SF, editors. Cytokines and pain. Birkhäuser; Basel: 1999. pp. 183–205. [Google Scholar]
- 55.Oka T, Aou S, Hori T. Intracerebroventricular injection of interleukin-1β induces hyperalgesia in rat. Brain Res. 1993;624:61–68. doi: 10.1016/0006-8993(93)90060-z. [DOI] [PubMed] [Google Scholar]
- 56.Oka T, Oka K, Hosoi M, Aou S, Hori T. The opposing effects of interleukin-1 beta microinjected into the preoptic hypothalamus and the ventromedial hypothalamus on nociceptive behavior in rats. Brain Res. 1995;700:271–278. doi: 10.1016/0006-8993(95)00980-5. [DOI] [PubMed] [Google Scholar]
- 57.Pita I, Jelaso AM, Ide CF. IL1beta increases intracellular calcium through an IL1 type 1 receptor mediated mechanism in C6 astrocytic cells. Int J Dev Neurosci. 1999;17:813–820. doi: 10.1016/s0736-5748(99)00063-5. [DOI] [PubMed] [Google Scholar]
- 58.Popik W, Hesselgesser JE, Pitha PM. Binding of human immunodeficiency virus type 1 to CD4 and CXCR4 receptors differentially regulates expression of inflammatory genes and activates the MEK/ERK signaling pathway. J Virol. 1998;72:6406–6413. doi: 10.1128/jvi.72.8.6406-6413.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shibuta S, Mashimo T, Ohara A, Zhang P, Yoshiya I. Intracerebroventricular administration of nitric oxide-releasing compound, NOC-18, produces thermal hyperalgesia in rats. Neurosci Lett. 1995;187:103–106. doi: 10.1016/0304-3940(95)11354-1. [DOI] [PubMed] [Google Scholar]
- 60.Sweitzer S, DeLeo JA, Martin D. Intrathecal interleukin-1 receptor antagonist and tumor necrosis factor binding protein exhibit an antiallodynic effect in a rat model of neuropathic pain. Soc Neurosci Abstr. 1999;25:1442. doi: 10.1016/s0306-4522(00)00574-1. [DOI] [PubMed] [Google Scholar]
- 61.Tabo E, Jinks SL, Eisele JHJ, Carstens E. Behavioral manifestations of neuropathic pain and mechanical allodynia, and changes in spinal dorsal horn neurons, following L4–L6 dorsal root constriction in rats. Pain. 1999;80:503–520. doi: 10.1016/S0304-3959(98)00243-7. [DOI] [PubMed] [Google Scholar]
- 62.Tadano T, Namiokka M, Nakagawasai O, Tan-No K, Matsushima K, Endo Y, Kisara K. Induction of nociceptive responses by intrathecal injection of interleukin-1 in mice. Life Sci. 1999;65:255–261. doi: 10.1016/s0024-3205(99)00244-1. [DOI] [PubMed] [Google Scholar]
- 63.Togashi H, Sasaki M, Frohman E, Taira E, Ratan RR, Dawson TM, Dawson VL. Neuronal (type I) nitric oxide synthase regulates nuclear factor kappaB activity and immunologic (type II) nitric oxide synthase expression. Proc Natl Acad Sci USA. 1997;94:2676–2680. doi: 10.1073/pnas.94.6.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Treutwein B, Strasburger H. Fitting the psychometric function. Percept Psychophys. 1999;61:87–106. doi: 10.3758/bf03211951. [DOI] [PubMed] [Google Scholar]
- 65.Tyor WR, Glas JD, Griffin J, Becker PS, McArtheru J, Bezman L, Griffin DE. Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann Neurol. 1992;31:349–360. doi: 10.1002/ana.410310402. [DOI] [PubMed] [Google Scholar]
- 66.VanLeeuwen R, Katlama C, Kitchen V, Boucher CA, Tubiana R. Evaluation of safety and efficacy of 3TC (lamivudine) in patients with asymptomatic or mildly symptomatic human immunodeficiency virus infection: a phase I/II study. J Infect Dis. 1996;171:1166–1171. doi: 10.1093/infdis/171.5.1166. [DOI] [PubMed] [Google Scholar]
- 67.Vitkovic L, Bockaert J, Jacque C. Inflammatory cytokines: neuromodulators in normal brain? J Neurochem. 2000;74:457–471. doi: 10.1046/j.1471-4159.2000.740457.x. [DOI] [PubMed] [Google Scholar]
- 68.Wang CX, Olschowka JA, Wrathall JR. Increase of interleukin-1beta mRNA and protein in the spinal cord following experimental traumatic injury of the rat. Brain Res. 1997;759:190–197. doi: 10.1016/s0006-8993(97)00254-0. [DOI] [PubMed] [Google Scholar]
- 69.Watkins LR, Wiertelak EP, Goehler LE, Smith KP, Martin D, Maier SF. Characterization of cytokine-induced hyperalgesia. Brain Res. 1994;654:15–26. doi: 10.1016/0006-8993(94)91566-0. [DOI] [PubMed] [Google Scholar]
- 70.Watkins LR, Deak T, Silbert L, Martinez J, Goehler L, Relton J, Martin D, Maier SF. Evidence for involvement of spinal cord glia in diverse models of hyperalgesia. Soc Neurosci Abstr. 1995a;21:897. [Google Scholar]
- 71.Watkins LR, Maier SF, Goehler LE. Immune activation: the role of proinflammatory cytokines in inflammation, illness responses, and pathological pain states. Pain. 1995b;63:289–302. doi: 10.1016/0304-3959(95)00186-7. [DOI] [PubMed] [Google Scholar]
- 72.Watkins LR, Martin D, Ulrich P, Tracey KJ, Maier SF. Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 1997;71:225–235. doi: 10.1016/s0304-3959(97)03369-1. [DOI] [PubMed] [Google Scholar]
- 73.Watkins LR, Hansen MK, Nguyen KT, Lee JE, Maier SF. Dynamic regulation of the proinflammatory cytokine, interleukin-1β: molecular biology for non-molecular biologists. Life Sci. 1999;65:449–481. doi: 10.1016/s0024-3205(99)00095-8. [DOI] [PubMed] [Google Scholar]
- 74. Watkins LR, Milligan ED, Maier SF. Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Immune mechanisms of analgesia Machelska H, Stein C. 2001, in press. New York: Landes. [PubMed]