

Abstract
The major goal of this study was to compare mechanisms of the neuroprotective potential of 17 β-estradiol in two models for oxidative stress-independent apoptotic neuronal cell death with that in necrotic neuronal cell death in primary neuronal cultures derived from rat hippocampus, septum, or cortex. Neuronal apoptosis was induced either by staurosporine or ethylcholine aziridinium (AF64A), as models for necrotic cell death glutamate exposure or oxygen–glucose deprivation (OGD) were applied. Long-term (20 hr) pretreatment (0.1 μm 17 β-estradiol) was neuroprotective in apoptotic neuronal cell death induced by AF64A (40 μm) only in hippocampal and septal neuronal cultures and not in cortical cultures. The neuroprotective effect was blocked by the estrogen antagonists ICI 182,780 and tamoxifen and the phosphatidylinositol 3-kinase (PI3-K) inhibitor LY294002. In glutamate and OGD-induced neuronal damage, long-term pretreatment was not effective. In contrast, short-term (1 hr) pretreatment with 17 β-estradiol in the dose range of 0.5–1.0 μm significantly reduced the release of lactate dehydrogenase and improved morphology of cortical cultures exposed to glutamate or OGD but was not effective in the AF64A model. Staurosporine-induced apoptosis was not prevented by either long- or short-term pretreatment. The strong expression of the estrogen receptor-α and the modulation of Bcl proteins by 17 β-estradiol in hippocampal and septal but not in cortical cultures indicates that the prevention of apoptotic, but not of necrotic, neuronal cell death by 17 β-estradiol possibly depends on the induction of Bcl proteins and the density of estrogen receptor-α.
Keywords: apoptosis, necrosis, estradiol, AF64A, staurosporine, oxygen–glucose deprivation, Bcl-2, primary neuronal cultures, estrogen receptor-α, hippocampus, cortex, septum, Alzheimer's disease
The neuroprotective potential of estrogens has gained increasing attention during the last years. Epidemiological evidence suggests that estrogen replacement therapy for postmenopausal women is associated with an improvement of some measures of cognitive performance, protection against cognitive deterioration, and decreased incidence of Alzheimer's disease (Paganini-Hill and Henderson, 1994; Robinson et al., 1994; Sherwin, 1994; Birge, 1996; Tang et al., 1996; Yaffe et al., 1998; Costa et al., 1999). Moreover, beneficial effects of estrogen on the mortality and morbidity associated with cerebral stroke have been demonstrated (Lafferty and Fiske, 1994; Grodstein et al., 1996; Hurn and MacRae, 2000). In a prospective 1 year study, however, estrogen failed to improve cognitive or functional outcomes of women with mild to moderate Alzheimer's disease (Mulnard et al., 2000).
The efficacy of estrogen has been shown in several models of neurodegeneration and ischemic injury in vivo and in vitro. In ovariectomized rats, physiological concentrations of estradiol attenuated the extent of brain damage caused by permanent cerebral ischemia (Dubal et al., 1998). Estradiol also alleviated stroke injury in reproductively senescent female rats (Alkayed et al., 2000). In primary neuronal cultures, organotypic hippocampal cultures and the hippocampal cell line HT22 estrogens attenuated neuronal injury caused by hypoxia, excitatory amino acids, superoxide anions, and hydrogen peroxide (Goodman et al., 1996; Singer et al., 1996; Behl et al., 1997; Regan and Guo, 1997; Weaver et al., 1997; Sawada et al., 1998). Moreover, estradiol reduced the neurotoxic effects of β-amyloid as well as the generation of β-amyloid in cell cultures (Gridley et al., 1997; Mook-Jung et al., 1997; Keller et al., 1997;Bonnefont et al., 1998; Xu et al., 1998; Pike, 1999). The overexpression of β-amyloid precursor protein mRNA after focal ischemia in female ovariectomized rats was attenuated by a single subcutaneous injection of 17 β-estradiol (100 μg/kg) 2 hr before middle cerebral artery occlusion (Shi et al., 1998).
Until now, the efficacy of estradiol has been investigated mainly in models of neurodegeneration associated with excitotoxicity and oxidative stress. The aim of the present study was to evaluate the neuroprotective effect of estradiol in apoptotic neuronal cell deathin vitro and to compare it with that in necrotic neuronal cell death. Apoptotic cell death in primary neuronal cell cultures derived from embryonic cortex, hippocampus, and septum was induced by ethylcholine aziridinium (AF64A) and staurosporine. AF64A, initially introduced as a model of cholinergic hypofunction (for review, seeHanin, 1996) has been shown to initiate neuronal cell death in vivo and in vitro by activating mechanisms of apoptosis (Rinner et al., 1997; Lautenschlager et al., 2000). The mycotoxin staurosporine activates programmed cell death in virtually all cells (Falcieri et al., 1993; Raff et al., 1993; Bertrand et al., 1994) and induces neuronal apoptosis in primary neuronal cultures (Koh et al., 1995; Wiesner and Dawson, 1996). Both AF64A and staurosporine have been demonstrated to initiate a caspase-dependent, free radical-independent apoptotic neuronal cell death in primary neuronal cultures (Harms et al., 2000). Either glutamate exposure or oxygen glucose deprivation (OGD) was used as a model for mainly necrotic types of neuro-degeneration.
MATERIALS AND METHODS
Materials. 17 β-Estradiol (cyclodextrin-encapsulated), staurosporine, tamoxifen, cycloheximide, DMSO, and enzyme standard for the kinetic lactate dehydrogenase (LDH)-test were obtained from Sigma (Deisenhofen, Germany); LY 294002 was from Calbiochem (Bad Soden, Germany); ICI 182,780 was from Tocris/Biotrend Chemikalien (Köln, Germany); neurobasal medium and supplement B27 were from Life Technologies/BRL (Eggenstein, Germany); modified Eagle‘s medium, PBS, HEPES buffer, trypsin/EDTA, penicillin–streptomycin,l-glutamine, collagen-G, and poly-l-lysine were from Biochrom (Berlin, Germany); multiwell plates were from Falcon (Franklin Lakes, NJ); Texas Red-labeled goat anti-mouse antibody were from Molecular Probes (Leiden, Holland); the mouse monoclonal antibody to estrogen receptor-α and the polyclonal (rabbit) antibody against estrogen receptor-β were from Alexis (Grünberg, Germany; no. 803-004-C050 and no. 210-135-CO50, respectively); ImmunoFluor mounting medium was from ICN (Eschwege, Germany); monoclonal antibodies to Bcl-2, Bcl-xL were from Transduction Laboratories (Lexington, KY); monoclonal antibodies to Bax were from Santa Cruz Biotechnology (Heidelberg, Germany) and to α-fodrin were from Biotrend Chemikalien; proteinase inhibitor cocktail and antibodies to β-tubulin were from Boehringer Mannheim (Mannheim, Germany); secondary anti-mouse horseradish peroxidase-linked antibody was from Jackson Immunoresearch (West Grove, PA); enhanced chemiluminescence kits were from Amersham and Pierce (Rockford, IL); x-ray films were from Kodak (Germany); high-range molecular weight standard was from Sigma (St. Louis, MO). AF64A was prepared from acetylethylcholine mustard (Research Biochemicals International, Natick, MA) according toFisher et al. (1982).
Primary neuronal cell cultures. Primary neuronal cultures of hippocampus, septum, and cerebral cortex were obtained from embryos (E) (E16–E18) of Wistar rats (Bundesinstitut für gesundheitlichen Verbraucherschutz und Veterinärmedizin, Berlin, Germany). Cultures were prepared according to Brewer (1995) with the following modifications. Septum, hippocampus, and cerebral cortex were dissected, incubated for 15 min in trypsin/EDTA (0.05/0.02% w/v in PBS) at 36.5°C, rinsed twice with PBS and once with dissociation medium (modified Eagle‘s medium with 10% fetal calf serum, 10 mm HEPES, 44 mm glucose, 100 U penicillin + streptomycin/ml, 2 mml-glutamine, 100 IE insulin/l), dissociated by Pasteur pipette in dissociation medium, pelleted by centrifugation (210 × g for 2 min at 21°C), redissociated in starter medium (Neurobasal medium with supplement B27, 100 U penicillin + streptomycin/ml, 0.5 mml-glutamine, 25 μmglutamate), and plated in 24-well plates or 6-well plates in a density of 200,000 cells per square centimeter. Wells were pretreated by incubation with poly-l-lysine (0.5% w/v in PBS) for 1 hr at room temperature, then rinsed with PBS, followed by incubation with coating medium (dissociation medium with 0.03‰ w/v collagen G) for 1 hr at 37°C, then rinsed twice with PBS before cells were seeded in starter medium. Cultures were kept at 36.5°C and 5% CO2 and fed beginning from 4 d in vitro (DIV) with cultivating medium (starter medium without glutamate) by replacing half of the medium twice a week. The cell culture media and supplement B27 were free of estrogens.
Injury paradigm. In all models the serum-free primary neuronal cultures were treated after 10–14 DIV. The condition of cells at various time points after treatment was determined morphologically by phase-contrast microscopy.
AF64A (40 μm) exposure was for 5 hr with subsequent rinsing and reapplication of conditioned medium. Controls received an equivalent amount of vehicle and were rinsed correspondingly.
Staurosporine was dissolved in DMSO (10 mm stock solution) and diluted with PBS to give the final concentrations of 100 and 300 nm in culture. The vehicle-treated cultures received the same amount of DMSO (in PBS) that was present in the highest dose of staurosporine.
For OGD, medium was removed from the cultures and preserved. Cultures were rinsed twice with PBS, then subjected to OGD for 120 min in a balanced salt solution at PO2 < 2 mmHg, followed by replacement of the preserved medium as described previously (Bruer et al., 1997). For standardization of the neuronal damage, the sister cultures were treated with an excess of glutamate (500 μm; full kill).
Glutamate exposure was performed with 100 μm glutamate for 30 min with subsequent rinsing and reapplication of conditioned medium.
Treatment with 17 β-estradiol, tamoxifen, ICI 182,780, LY 294002, and cycloheximide. Cyclodextrin-encapsulated 17 β-estradiol was dissolved in PBS and added to the cell cultures in a dose range of 30 nm to 10 μm (final concentration in the medium) either 20 hr (long-term pretreatment) or 1 hr (short-term pretreatment) before the initiation of the injury. In the case of long-term treatment with glutamate toxicity, cells were washed twice with PBS, and the conditioned medium without 17 β-estradiol was replaced. Tamoxifen was dissolved in ethanol (10 mm stock solution) and diluted in water (100 μm). The final concentrations in the medium were 1 μmtamoxifen and 0.01% ethanol. ICI 182,780 and LY 294002 were dissolved in DMSO (100 and 65 mm stock solution, respectively). Cycloheximide dissolved in medium was added to give a final concentration of 500 ng/ml medium. Tamoxifen, ICI 182,780, LY 294002, or cycloheximide was added at the same time points as estradiol.
Cell death assays. Neuronal injury was quantitatively assessed by the measurement of LDH in the medium (Koh and Choi, 1987) at 72 hr after AF64A application, at 48 hr after staurosporine application, and at 24 hr after OGD and glutamate exposure.
Western blots. Cells were rinsed with PBS and harvested in lysis buffer containing (in mm): 150 NaCl, 1 CaCl2, 1 MgCl2, 10 Tris-HCl, pH 7.8, 1% Triton X-100, and proteinase-inhibitor mixture, incubated for 15 min on ice, and centrifuged (12,000 rpm, 4°C). Protein concentration was determined using the BCA protein assay (Pierce), and samples were diluted in SDS sample buffer and boiled for 3 min. Ten micrograms of protein per lane were loaded on 10.0% SDS-polyacrylamide minigels followed by electrophoresis. Blocking was performed semidry onto polyvinyldifluoride membranes, blocked with 5% nonfat dry milk, and detected with primary monoclonal antibodies to Bcl-2 (1:250), Bcl-xL (1:250), Bax (1:250), α-fodrin (1:1000), or β-tubulin (1:4000). A secondary anti-mouse horseradish peroxidase-linked antibody, enhanced chemiluminescence kits, and x-ray films were used to visualize signals. β-Tubulin served as a positive control for protein loading, and high-range molecular weight standard was used to determine protein sizes. Cell lysates served as positive controls for Bcl-2 and Bcl-xL according to the manufacturer. To provide semiquantitative analysis of band intensity, band densitometry was determined from scanned images of nonsaturated immunoblot films, using Scion Image, version Beta 4.0.2 software (Scion Corporation). To compare at least three different experiments, for each protein and brain region the pixel intensity of the bands obtained in each experiment was added and set as 100%. The individual band was calculated as percentage of total signals.
Immunocytochemistry. For immunocytochemical analysis, cells were seeded onto glass coverslips and cultivated for 12 d. Cultures were then washed with PBS and fixed with freshly prepared 4% paraformaldehyde in PBS for 15 min at room temperature. Cells then were washed twice with PBS, permeabilized with 0.3% Triton X-100 in PBS, and exposed to blocking solution (PBS containing 10% goat serum and 1% BSA) for 30 min at room temperature. Cultures then were incubated with the mouse monoclonal antibody to estrogen receptor-α (5 μg/ml) or with the polyclonal (rabbit) antibody against estrogen receptor-β (5 μg/ml) for 48 hr at 4°C, washed three times in PBS, and developed with Texas Red-labeled goat anti-mouse antibody (dilution 1:500) for 30 min at room temperature. After being thoroughly rinsed with PBS, the cultures were placed on a glass slide in ImmunoFluor mounting medium (ICN, Eschwege, Germany) and observed by confocal microscopy. For negative controls, the primary antibody was omitted, resulting in no visible staining.
Confocal laser-scan microscopy and quantitative immunocytochemical evaluation. Quantitative immunocytochemical evaluation of cortical, septal, and hippocampal cultures was performed with an MRC600 confocal imaging system (Bio-Rad, Hemel Hempstead, UK) equipped with a Nikon Optiphot microscope as described previously (Gazzaley et al., 1996). Briefly, an argon–krypton laser was used to excite Texas Red at 568 nm. The image was visualized using a Leitz 63/1.30 oil immersion objective. All of the confocal parameters were kept constant throughout the evaluation to yield unbiased comparisons of different neuronal cultures. The investigator was blinded throughout the evaluation and did not know from which brain area the culture was derived. Fifty fields from each culture were randomly selected and scanned at slow speed. Each digitized image consisted of 768 × 512 pixels. The average pixel intensity of each cell in the field was selectively determined by a second blinded investigator using the CoMOS software program (Bio-Rad, Version 7.0a).
Terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling staining. Terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling (TUNEL) staining was performed with a commercially available ApopTag Kit (Qbiogene, Heidelberg, Germany) according to the manufacturer’s instructions. TUNEL-positive cells were analyzed for each injury paradigm in two independent preparations and were counted in a blinded manner in 20 randomly selected high-power fields. The investigator was not informed throughout the evaluation from which injury paradigm the culture was derived.
Data analyses. Data are shown as means ± SEM. To avoid possible variations of the cell cultures depending on the quality of dissection and seeding procedures, data were pooled from two to three representative experiments. For statistical analyses the one-way ANOVA was followed by Tukey's post hoc test.
RESULTS
17 β-Estradiol protects hippocampal but not cortical neuronal cultures against AF64A-induced neurodegeneration in primary neuronal cultures
Long-term pretreatment with a single addition of 17 β-estradiol 20 hr before AF64A was associated with a marked reduction in LDH release in hippocampal and septal neurons 72 hr after toxin application (Fig. 1). The neuroprotective effect of 17 β-estradiol in hippocampal and septal neurons was achieved only at the lower dose range of 0.1 μm and was not apparent at the higher dose of 1.0 μm. No protection, however, was observed in cortical neurons. The AF64A-induced increase in LDH release from cortical neurons (90.4 ± 9.3 U/ml) was not changed after pretreatment with 0.1 μm (80.8 ± 11.9) or 1.0 μm 17 β-estradiol (96.9 ± 8.8). In Figure2a–c, representative phase-contrast micrographs of primary neuronal cultures from hippocampus under control condition and after AF64A exposure without and with long-term pretreatment with 17 β-estradiol are summarized. AF64A triggered morphological signs of apoptosis including degeneration of neurites, shrinkage of cell bodies, and fragmentation into condensed particles, which were at least partly prevented by pretreatment with 17 β-estradiol. In contrast, short-term pretreatment of cultured neurons with various doses of 17 β-estradiol 1 hr before AF64A application did not result in a significant reduction in LDH release within 72 hr after toxin exposure. The missing protection was obvious in cultures obtained from cortex, hippocampus, and septum (data not shown).
Fig. 1.
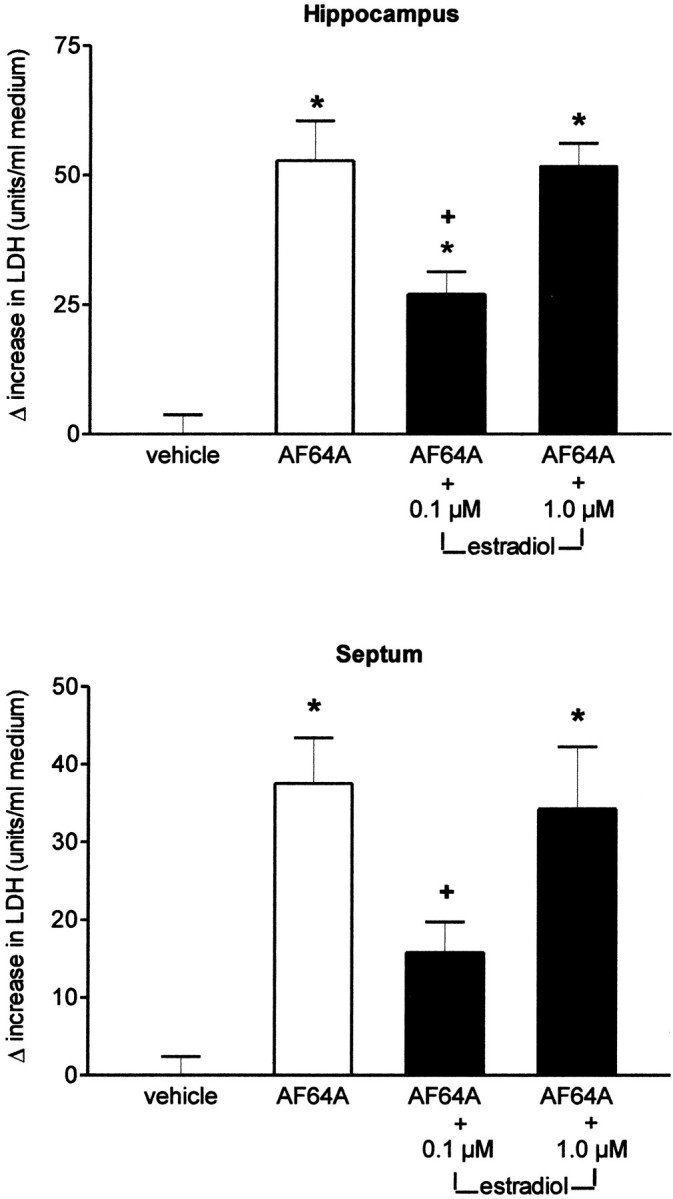
Effect of 17 β-estradiol on AF64A-induced LDH release in primary neuronal cultures of hippocampus and septum. At 10 DIV cells were pretreated with 0.1 or 1.0 μm 17 β-estradiol for 20 hr before the addition of 40 μmAF64A or the corresponding vehicle. LDH released into the medium was measured 72 hr after the addition of AF64A. Data are presented as Δ increase in LDH release as different from vehicle-treated sister cultures. The LDH activity in vehicle-treated sister cultures was 38.5 ± 3.7 and 34.1 ± 2.4 U/ml medium in hippocampal and septal neurons, respectively (n = 5–10 for each condition, pooled from 3 different sets of experiments; *p < 0.004 vs vehicle-treated cultures;+p < 0.05 vs AF64A).
Fig. 2.
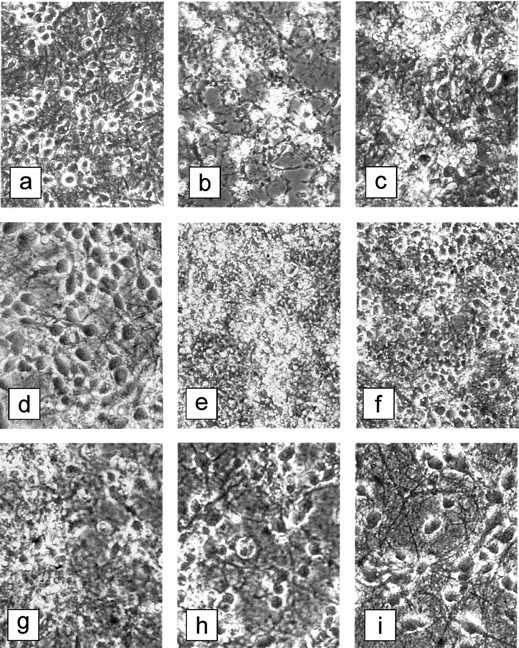
Phase-contrast microscopy of primary neuronal cultures after the various injury paradigms with and without 17 β-estradiol pretreatment (magnification 250×).a–c, The effect of long-term pretreatment with 17 β-estradiol on the AF64A-induced neurodegeneration in hippocampal cultures. The photographs were taken 72 hr after application of AF64A (40 μm).a, Vehicle-treated control cultures; b, AF64A-treated cultures; c, AF64A-treated cultures after pretreatment with 17β-estradiol (0.1 μm).d–i, The effect of short-term pretreatment with 17 β-estradiol on the glutamate- or OGD-induced neuronal cell death in cortical neurons 24 hr after the injury.d, Vehicle-treated control cultures; e, cultures exposed to glutamate (100 μm); f, cultures exposed to glutamate 1 hr after pretreatment with 17 β-estradiol (10.0 μm); g, cultures exposed to OGD (2 hr); h, cultures exposed to OGD 1 hr after pretreatment with 17 β-estradiol (0.1 μm); i, cultures exposed to OGD 1 hr after pretreatment with 17 β-estradiol (0.5 μm).
17 β-Estradiol does not prevent staurosporine-induced neurodegeneration
In staurosporine-induced apoptotic neuronal cell death, neither short-term (1 hr) nor long-term (20 hr) pretreatment with 0.1 or 1.0 μm 17 β-estradiol was effective in reducing the release of LDH. In the hippocampus the application of staurosporine (300 nm) induced an increase in LDH release from 77.2 ± 5 to 128.6 ± 8.2 U/ml 48 hr after addition (n = 6–12; p < 0.05). This increase was not significantly changed after short-term or long-term pretreatment with both doses of 17 β-estradiol (e.g., 138.7 ± 3.0 and 136.2 ± 11.8 after pretreatment with 0.1 μm 17 β-estradiol, 1 and 20 hr before, respectively; n = 6). Similarly in cortical cultures LDH release increased from 66 ± 6.8 to 119.2 ± 5.1 U/ml 48 hr after addition of staurosporine (300 nm; n = 6–12; p< 0.05) with no significant change of the increase after pretreatment with 17 β-estradiol (e.g., 121.9 ± 7.3 and 130.6 ± 9.6 U/ml after pretreatment with 0.1 μm 17 β-estradiol, 1 and 20 hr before, respectively; n = 6).
Only short-term pretreatment with 17 β-estradiol reduces LDH release from primary cortical cultures after glutamate exposure
Exposure of cortical neurons to 100 μm glutamate for 30 min induced marked neuronal cell death associated with a considerable increase in LDH release within 24 hr. Pretreatment of the cortical cultures with various doses of 17 β-estradiol 1 hr before glutamate attenuated the increase in LDH release. The neuroprotection was significant at 0.1 and 1.0 μm but started to disappear at higher doses (Fig.3A; Table1). In phase-contrast microscopy, the protective effect of 17 β-estradiol was clearly visible (Fig.2e,f). No neuroprotection was achieved after long-term pretreatment of the cultures with various doses of 17 β-estradiol (20 hr before 100 μm glutamate), whereas in glutamate-treated sister cultures short-term pretreatment with 17 β-estradiol significantly reduced LDH release (Table 1).
Fig. 3.
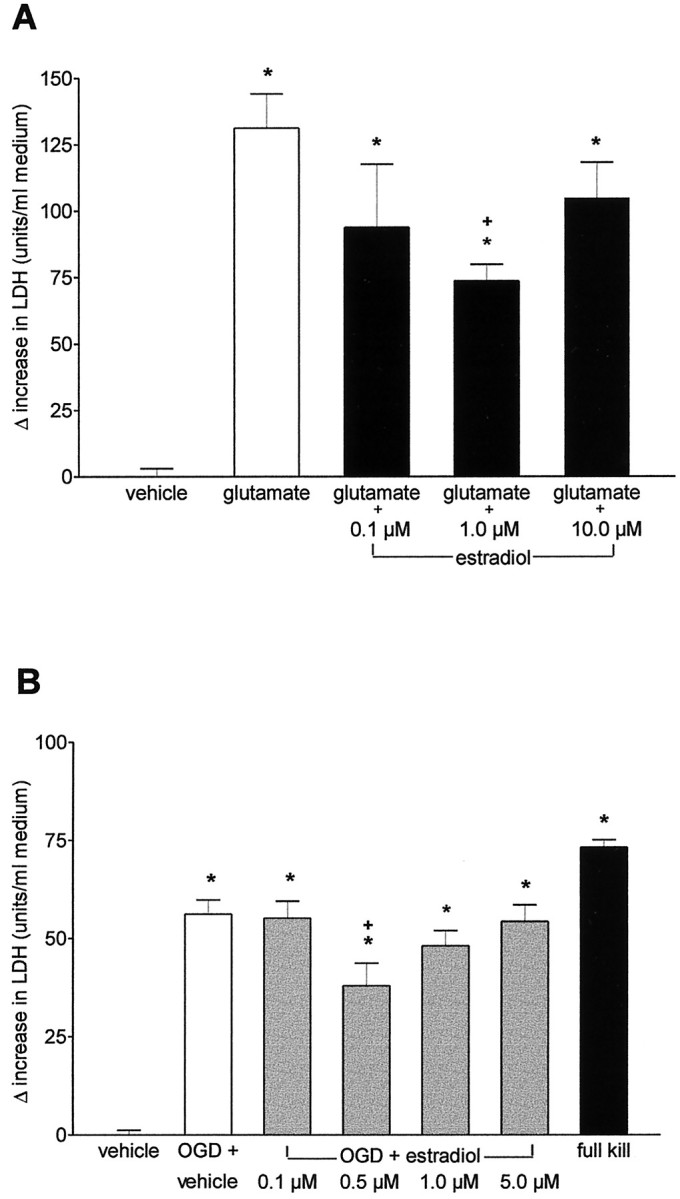
A, Effect of 17 β-estradiol on glutamate-induced LDH release in primary neuronal cultures of cortex. At 10 DIV, cells were pretreated with 0.1, 1.0, or 10.0 μm 17 β-estradiol for 1 hr before the addition of 100 μm glutamate or the corresponding vehicle. LDH released into the medium was measured after 24 hr. Data are presented as Δ increase in LDH release as different from vehicle-treated sister cultures (n = 6–8 for each condition, pooled from 2 different sets of experiments; *p < 0.001 vs vehicle-treated cultures; +p < 0.005 vs glutamate). B, Effect of 17 β-estradiol on LDH release after OGD in primary neuronal cultures of cortex. At 10 DIV, cells were pretreated with 0.1, 0.5, 1.0, or 5.0 μm 17 β-estradiol for 1 hr before OGD. LDH released into the medium was measured 24 hr after OGD. Data are presented as Δ increase in LDH release as different from vehicle-treated sister cultures. The LDH activity in vehicle-treated sister cultures was 63.8 ± 1.2 (n = 9–24 for each condition, pooled from 2 different sets of experiments; *p < 0.001 vs vehicle-treated cultures; +p < 0.05 vs OGD + vehicle).
Table 1.
Effect of short-term (1 hr) and long-term (20 hr) pretreatment with 17 β-estradiol on the increase in LDH release (Δ U/ml medium) in cortical and hippocampal primary neuronal cultures 24 hr after exposure to glutamate (100 μm)
| Estradiol pretreatment | Primary neuronal cultures | Glutamate + vehicle | Glutamate + 0.1 μmestradiol | Glutamate + 1.0 μm estradiol | Glutamate + 1.0 μm estradiol + 1.0 μmtamoxifen | Glutamate + 1.0 μm tamoxifen |
|---|---|---|---|---|---|---|
| 1 hr | Cortex | 76.1 ± 2.91-a | 45.4 ± 2.81-a,1-b | 43.7 ± 5.21-a,1-b | 48.6 ± 7.11-a,1-b | 66.0 ± 5.51-a |
| 20 hr | Cortex | 74.2 ± 4.31-a | 67.8 ± 5.41-a | 68.1 ± 3.71-a | n.d. | n.d. |
| Hippocampus | 50.7 ± 2.71-a | 42.0 ± 4.91-a | 45.8 ± 2.71-a | n.d. | n.d. |
n = 4–24; basal LDH release in vehicle-treated sister cultures was 61.5 ± 5.0 (1 hr) and 54.8 ± 4.9 U/ml medium (20 hr). n.d., Not done.
p < 0.001 versus vehicle-treated sister cultures.
p < 0.05 versus glutamate + vehicle.
Only short-term pretreatment with 17 β-estradiol protects against neurodegeneration in cortical cultures induced by oxygen–glucose deprivation
Comparable to its neuroprotective efficacy in glutamate-induced neuronal cell death, 17 β-estradiol was effective in OGD-related neuronal cell loss only after short-term pretreatment (1 hr). The effect was maximal in the dose range of 0.5–1.0 μm (Fig.3B). In the presence of 0.5 μm 17 β-estradiol, neurons appeared to be resistant against the influence of OGD (Fig. 2g–i). At the dose of 0.1 μm, which was effective as long-term pretreatment in the apoptotic model of neurodegeneration, 17 β-estradiol did not attenuate the release of LDH. No protection was achieved after long-term pretreatment with 17 β-estradiol (20 hr). In this experiment, the OGD-induced increase in LDH release from cortical neurons (59.7 ± 7.0 U/ml) was not changed after pretreatment with 0.1 μm (56.1 ± 3.7), 0.5 μm (64.5 ± 7.5), or 1.0 μm 17 β-estradiol (60.5 ± 4.8).
Modulation of Bcl-2 expression by 17 β-estradiol in primary neuronal cultures
In hippocampal cultures, 17 β-estradiol (30 and 100 nm) increased the levels of Bcl-2 24 and 48 hr after application (Fig. 4A). Increased Bcl-2 levels persisted up to 68 hr after the addition of 17 β-estradiol (Fig. 5A). At this late time point increased Bcl-xL levels were found. In contrast, we did not observe a change of the expression of Bcl-2 and Bcl-xL in cortical cultures after 17 β-estradiol treatment.
Fig. 4.

A, Effect of 17 β-estradiol (0.03 and 0.10 μm) on the expression of Bcl-2 in hippocampal and cortical neuronal cultures. Neuronal cultures derived from hippocampus or cortex were maintained for at least 10 DIV before a 24–48 hr exposure to 17 β-estradiol (30 or 100 nm) and then processed for Western blots. 1, Vehicle-treated cultures; 2, 24 hr exposure to 0.10 μm 17 β-estradiol; 3, 48 hr exposure to 0.10 μm 17 β-estradiol; 4, 48 hr exposure to 0.03 μm 17 β-estradiol. B, Antagonism of 17 β-estradiol-induced increase in Bcl-2 and Bcl-xL in hippocampal cultures by cycloheximide and tamoxifen. At 10 DIV, hippocampal cultures were treated for 48 hr and then processed for Western blots. a, Vehicle-treated cultures;b, cycloheximide (500 ng/ml medium)-treated cultures;c, tamoxifen (1 μm)-treated cultures;d, 17 β-estradiol (0.1 μm)/cycloheximide (500 ng/ml medium)-treated cultures; e, 17 β-estradiol (0.1 μm)/tamoxifen (1 μm)-treated cultures;f, 17 β-estradiol (0.1 μm)-treated cultures; g, 17 β-estradiol (1.0 μm)-treated cultures.
Fig. 5.
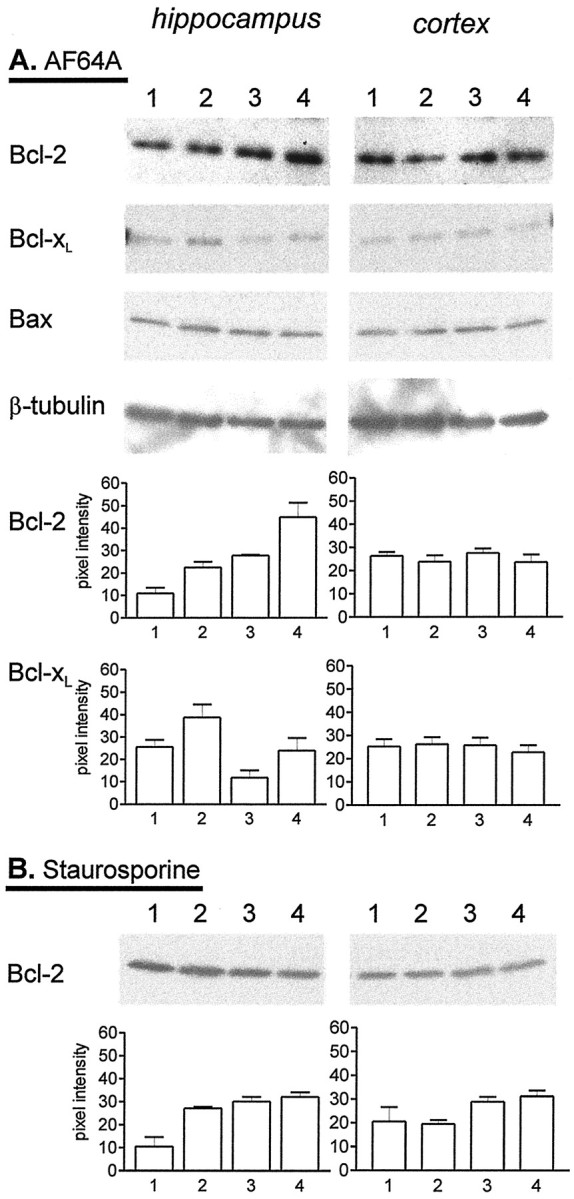
A, Effect of 17 β-estradiol (0.1 μm) and AF64A (40 μm) on the expression of Bcl-2, Bcl-xL, Bax, and β-tubulin in hippocampal and cortical neuronal cultures. Neuronal cultures derived from hippocampus or cortex were maintained for at least 10 DIV before the experiment. Cells were treated with vehicle or 17β-estradiol and 20 hr later with vehicle or AF64A. Cells were processed for Western blot 48 hr after application of AF64A. At least three separate experiments were performed. 1, Vehicle/vehicle-treated cultures;2, 17β-estradiol/vehicle-treated cultures;3, vehicle/AF64A-treated cultures; 4, 17β-estradiol/AF64A-treated cultures. The semiquantitative analysis of the Western blots of Bcl-2 and Bcl-xL by band densitometry from three separate experiments is shown.B, Effect of 17 β-estradiol (0.1 μm) and staurosporine (300 nm) on the expression of Bcl-2 in hippocampal and cortical neuronal cultures. Neuronal cultures derived from hippocampus or cortex were maintained for at least 10 DIV before the experiment. Cells were treated with vehicle or 17β-estradiol and 20 hr later with vehicle or staurosporine. Cells were processed for Western blot 24 hr after application of staurosporine.1, Vehicle/vehicle-treated cultures; 2, 17β-estradiol/vehicle-treated cultures; 3, vehicle/staurosporine-treated cultures; 4, 17β-estradiol/staurosporine-treated cultures. The semiquantitative analysis of the Western blots by band densitometry is shown from four separate experiments.
An increase in Bcl-2 expression also occurred 48 hr after the application of AF64A (40 μm) in the hippocampal cultures. The pretreatment with 17 β-estradiol resulted in a further synergistic increase in Bcl-2 levels (Fig. 5A). Levels of Bcl-xL and Bax were not altered 48 hr after AF64A treatment. Staurosporine rather suppressed the 17 β-estradiol-induced increase in Bcl-2 in the hippocampal neurons (Fig. 5B).
Antagonism of long-term neuroprotective effect of estradiol by tamoxifen, ICI 182,780, and cycloheximide
To show that the long-term effect of estradiol is based on a receptor-mediated, transcription-regulating mechanism, we additionally treated hippocampal cultures with tamoxifen, ICI 182,780, or cycloheximide. The neuroprotective effect of 17 β-estradiol on AF64A-induced apoptotic cell death was abolished by receptor blockade as well as by inhibition of protein synthesis. The results on LDH release are summarized in Table 2. In addition, both tamoxifen and cycloheximide reduced the increase in the expression of Bcl-2 and Bcl-xL observed 48 hr after treatment with 17 β-estradiol (Fig. 4B). Tamoxifen, however, which has a comparable relative affinity for estrogen receptor-α versus estrogen receptor-β [7 and 6, respectively; see Kuiper et al. (1997)], did not antagonize the neuroprotection by short-term treatment with 17 β-estradiol against glutamate exposure (Table 1).
Table 2.
Loss of the neuroprotective effect of 17-β estradiol on AF64A (40 μm)-induced neuronal cell death in hippocampal and septal cell cultures after pretreatment with tamoxifen, ICI 182,780, LY 294002, or cycloheximide
| Cell culture | AF64A + vehicle | AF64A + antagonist or inhibitor | AF64A + estradiol 0.1 μm | AF64A + estradiol + antagonist or inhibitor | |
|---|---|---|---|---|---|
| Tamoxifen 1 μm | Hippocampal | 41.9 ± 4.7 | 36.5 ± 4.1 | 21.0 ± 3.32-a | 44.8 ± 6.22-b |
| ICI 182,780 1 μm | Hippocampal | 65.5 ± 4.8 | 66.6 ± 3.8 | 31.8 ± 3.02-c | 68.7 ± 3.52-d |
| Septal | 49.4 ± 5.5 | 43.9 ± 1.9 | 24.9 ± 3.42-a | 50.7 ± 2.62-d | |
| LY 294002 5 μm | Hippocampal | 65.5 ± 4.8 | 61.3 ± 2.5 | 31.8 ± 3.02-c | 57.1 ± 5.12-d |
| Septal | 49.4 ± 5.5 | 49.2 ± 4.3 | 24.9 ± 3.42-a | 46.5 ± 6.12-b | |
| Cycloheximide 500 ng/ml | Hippocampal | 53.6 ± 4.2 | 54.5 ± 5.2 | 31.4 ± 3.42-a | 52.5 ± 4.22-b |
The values represent the AF64A-induced increase in LDH-release (Δ U/ml medium). The basal LDH-release in the corresponding control cultures was 43.3 ± 4.9 (hippocampal) and 41.7 ± 3.2 U/ml medium (septal). n = 4–9.
p < 0.05 versus AF64A + vehicle.
p < 0.05 versus AF64A + estradiol.
p < 0.001 versus AF64A + vehicle.
p < 0.001 versus AF64A + estradiol.
Lower expression of estrogen receptor-α in cortical neurons
To find out whether the neuroprotectivity of 17 β-estradiol against AF64A-induced apoptosis and the increase in Bcl expression depends on the presence of estrogen receptor-α, we compared the expression of both estrogen receptor-α and estrogen receptor-β in the septal, hippocampal, and cortical neuronal cultures by immunocytochemical methods. The immunostaining for estrogen receptor-α was considerably weaker in the cortical neurons than in hippocampal and septal neurons (Fig.6A). The intensity of immunoreactivity was quantified by means of confocal microscopy in 139 neurons of each culture system in a randomized, blinded manner. The mean pixel intensity was significantly higher in septal neurons (13.06 ± 0.57) and hippocampal neurons (11.37 ± 0.57) as compared with cortical neurons (7.62 ± 0.47; p < 0.05, one-way ANOVA on ranks followed by Tukey's post hoctest). In addition, the immunoreactive neurons were differentiated according to fluorescence intensity. The median pixel intensity of all counted cells (8.9) was taken as a border between cells with low or high intensity. In cortical cultures, neurons with low fluorescence intensity prevailed (100 of 139 neurons). The opposite distribution was found in septal and hippocampal cultures (Fig. 6C). In contrast, the immunostaining for estrogen receptor-β was more intense in the cortical neurons than in hippocampal and septal neurons (Fig.6B). The intensity of immunoreactivity was quantified by means of confocal microscopy in 140 neurons of each culture system in a randomized, blinded manner. The mean pixel intensity was significantly lower in septal neurons (7.85 ± 0.31) and hippocampal neurons (8.61 ± 0.44) as compared with cortical neurons (10.86 ± 0.39; p < 0.05, one-way ANOVA on ranks followed by Tukey's post hoc test). Again, the immunoreactive neurons were differentiated according to fluorescence intensity. The median pixel intensity of all counted cells (8.3) was taken as a border between cells with low or high intensity. In cortical cultures, neurons with high fluorescence intensity prevailed (91 of 140 neurons). The opposite distribution was found in septal and hippocampal cultures (Fig. 6C).
Fig. 6.
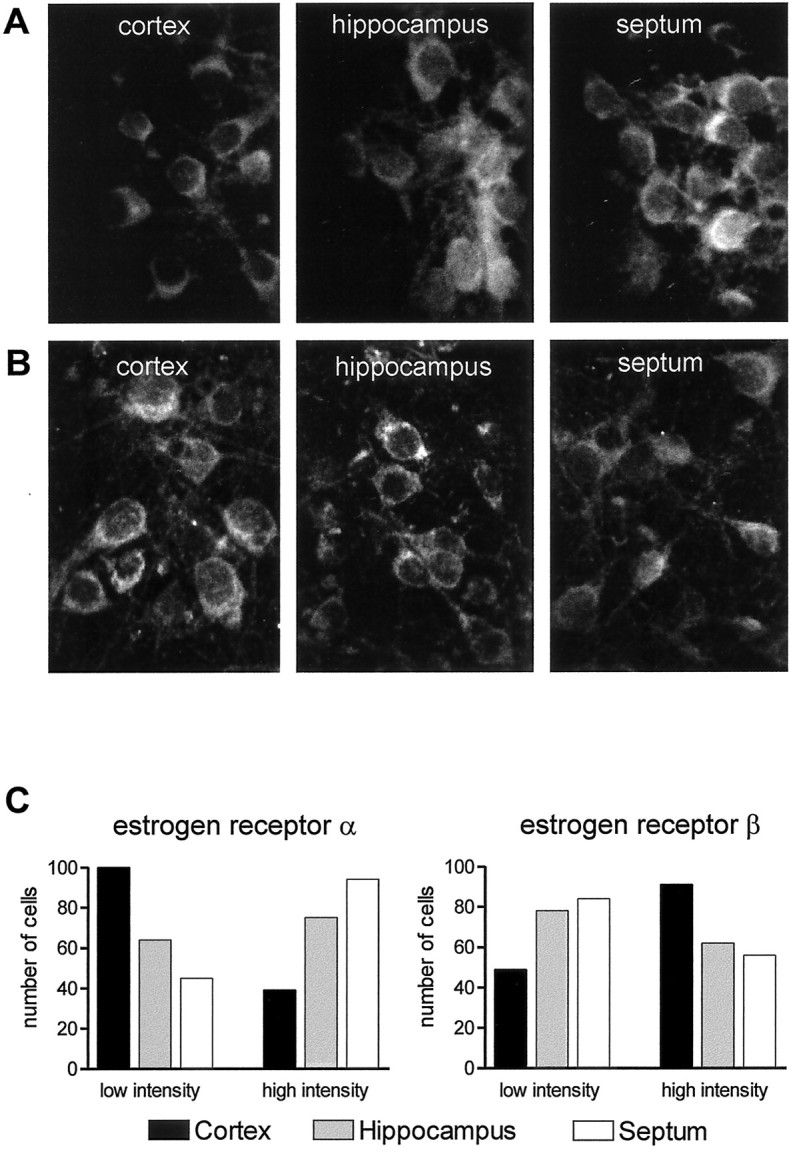
A, Immunocytochemical analysis on the localization of estrogen receptor-α (A) and estrogen receptor-β (B) in cortical, hippocampal, and septal neurons. Cells derived from cortex, hippocampus, or septum were cultivated for 12 d before they were fixed in 4% formaldehyde for 15 min at room temperature. After permeabilization with 0.3% Triton X-100 and exposure to blocking solution, cultures were incubated with the mouse monoclonal antibody to estrogen receptor-α or the polyclonal (rabbit) antibody against estrogen receptor-β for 48 hr at + 4°C and developed with Texas Red-labeled goat anti-mouse antibody (1:500) for 30 min at room temperature. Cultures were placed on a glass slide in ImmunoFluor mounting medium and observed by confocal microscopy (magnification 630×). C, Distribution of estrogen receptor-α- and estrogen receptor-β-immunoreactive neurons according to immunofluorescence intensity in cortical, hippocampal, and septal neuronal cultures. The quantitative immunocytochemical analysis was performed by means of confocal microscopy. The average pixel intensity was determined in 139 and 140 neurons, respectively, of each of the culture systems selected in a randomized, blinded manner. The median pixel intensity of all counted cells (8.9 and 8.3, respectively) was taken as a border between cells with low or high intensity.
Involvement of PI3-K cascade in the neuroprotectivity of 17 β-estradiol
As a further proof for the involvement of estrogen receptor-α, we tested the influence of the inhibitor of PI3-K, LY294002, on the neuroprotectivity of 17 β-estradiol in the AF64A model. Activation of PI3-K is an important step in the insulin-like growth factor 1 (IGF-1) receptor pathway. It has been demonstrated recently that 17 β-estradiol induces an activation of this pathway only by stimulation of the estrogen receptor-α and not of the estrogen receptor-β (Kahlert et al., 2000), When LY294002 was added (5 μm) concomitantly with 17 β-estradiol, the protective effect of 17 β-estradiol was abolished completely (Table 2).
Assessment of apoptosis versus necrosis
To clearly differentiate between the mode of neuronal cell death in the various types of injury, we used two different approaches in addition to previously published distinctions. First, we compared the number of TUNEL-positive cells in cortical cultures per high power field (n = 20) in the various conditions. The following numbers of TUNEL-positive cells were found: in control cultures, 6 ± 2; 24 hr after glutamate exposure, 7.5 ± 3; 24 hr after OGD, 3 ± 2; 24 hr after staurosporine, 34 ± 6; and 48 hr after addition of AF64A, 26 ± 4. As a second approach the breakdown pattern of α-fodrin was analyzed to distinguish between necrosis and apoptosis. According to Nath et al. (1996), α-fodrin is degraded to a 120 kDa fragment in apoptotic neurons but not in necrotic neurons (for review, see Wang, 2000). A considerable increase in the 120 kDa fragment of α-fodrin was only found in staurosporine-treated cells and time dependently after AF64A treatment but not after exposure to glutamate or OGD (Fig. 7). The results of both techniques clearly indicate that in the case of AF64A and staurosporine the apoptotic cell death predominates, whereas in the case of glutamate and OGD cell death is necrotic in nature.
Fig. 7.

α-Fodrin breakdown pattern as marker for caspase and calpain activation in the various models of neuronal death. Neuronal cultures derived from cortex were maintained for at least 10 DIV before the experiment. Cells were harvested for Western blot analysis as follows: 1, control-treated cultures;2, glutamate (100 μm)-treated cultures (24 hr after exposure); 3, cultures 24 hr after OGD;4, staurosporine (300 nm)-treated cultures (24 hr after application); 5, staurosporine (300 nm)-treated cultures (48 hr after application);6, AF64A (40 μm)-treated cultures (24 hr after application); 7, AF64A (40 μm)-treated cultures (48 hr after application);8, AF64A (40 μm)-treated cultures (72 hr after application). The semiquantitative analysis of the Western blots of the 120 kDa fragment of α-fodrin (p120) by band densitometry from three separate experiments is shown.
DISCUSSION
The present data provide in vitro evidence that different mechanisms are involved in the neuroprotective efficacy of estrogens depending on whether the injury model used is related to the apoptotic or necrotic type of neuronal cell death. Moreover, brain region-specific differences in neuroprotection by 17 β-estradiol were observed. In neuronal apoptotic cell death as induced by AF64A, neuroprotection was only achieved by long-term pretreatment with 17 β-estradiol in the 0.1 μm dose range in hippocampal and septal but not cortical primary cultures. The neuroprotection was reversed both by the estrogen receptor antagonists ICI 182,780 and tamoxifen and by inhibition of protein synthesis. These findings indicate that prevention of apoptotic neuronal cell death is probably associated with transcriptional regulation, mediated by activation of estrogen receptors. Similar evidence has been obtained for the estrogen neuroprotection against β-amyloid in cultured hippocampal neurons (Pike, 1999). Short-term mechanisms unrelated to estrogen receptors were not protective in the AF64A model for apoptotic neurodegeneration.
When apoptotic cell death was induced by the broad spectrum protein kinase inhibitor staurosporine, hippocampal, septal, and cortical neurons were not protected after either long-term or short-term pretreatment with 17 β-estradiol. The failure of 17 β-estradiol to protect against staurosporine-induced apoptosis is explained at least partly by the following findings. First, it has been demonstrated in the present study that staurosporine prevents the 17 β-estradiol-induced expression of Bcl-2. Second, the inhibition of protein kinases by staurosporine could further contribute to the inefficacy of 17 β-estradiol. Estrogen neuroprotection in primary cortical neurons after glutamate excitotoxicity is mediated by activation of the mitogen-activated protein kinase signaling pathway (Singer et al., 1999).
In neuronal injury caused by excitotoxicity or hypoxia, only short-term pretreatment and the use of higher doses of 17 β-estradiol (0.5–1.0 μm) provided neuroprotection that was not prevented by estrogen receptor blockade. This is in agreement with previous results demonstrated in cortical, hippocampal, and mesencephalic dopaminergic cultures (Goodman et al., 1996; Behl et al., 1997; Regan and Guo, 1997; Sawada et al., 1998). The independence of the neuroprotective effect of estrogen in excitotoxicity and hypoxia from estrogen receptor-mediated pathways or protein synthesis has been demonstrated by several groups (Sawada et al., 1998; Moosmann and Behl, 1999; Zaulyanov et al., 1999). These findings and the immediate efficacy of higher doses of 17 β-estradiol indicate that mechanisms unrelated to transcriptional regulation are involved, such as antioxidant properties of estrogens, direct inhibition of NMDA receptors (Weaver et al., 1997), rapid release of calcium from intracellular stores (Beyer and Raab, 1998), stimulation of guanylate cyclase activity (Chen et al., 1998), blockade of calcium entry via L-type calcium channels (Mermelstein et al., 1996), and stabilization of mitochondrial function (Mattson et al., 1997). In addition, the involvement of a possible membrane-binding site on neurons for various steroids including estrogens is discussed (for review, see Behl and Holsboer, 1999). Our finding of the inefficiency of 17 β-estradiol (0.1–5.0 μm) after long-term pretreatment in glutamate and OGD-induced cell death is in contrast to the significant degree of protection against excitotoxicity observed in primary cortical neurons or hippocampal slice cultures, when low doses of 17 β-estradiol (1–50 nm) were added 24 hr before the excitotoxin (Singer et al., 1996; Bi et al., 2000). In the hippocampal slice cultures, however, a comparable effect was observed whether the steroid was added 24 hr or 10 min before the excitotoxins (Bi et al., 2000). The discrepancies between the latter and our studies might be related to the shorter period of exposure to the excitotoxin (Singer et al., 1996) or to the lower dose of excitotoxin in a completely different culture system (Bi et al., 2000).
The inefficacy of short-term pretreatment with 17 β-estradiol in the AF64A and staurosporine model for apoptosis agrees very well with the previous findings that in the same models various antioxidants, including melatonin, N-tert-butyl-α-phenylnitrone, and dimethylthiourea, did not prevent neuronal cell death. In both models these antioxidants prevented the initial increase of malondialdehyde, indicating the accumulation of free radicals, but did not block the induction of apoptosis (Harms et al., 2000).
The brain region-specific differences in the neuroprotective efficacy of 17 β-estradiol in the case of apoptotic neuronal cell death are of considerable interest. We have observed that long-term pretreatment with 17 β-estradiol was effective only in hippocampal and septal cultures and did not attenuate the neuronal cell death induced by AF64A in cortical neuronal cultures. In contrast, cortical neurons pretreated with 17 β-estradiol for 1 hr became less vulnerable to excitotoxicity or OGD, an effect that was not antagonized by tamoxifen. Thus, receptor-independent effects of estradiol occur in cortical neurons, whereas modulations of gene activation do not exist or do not provide neuroprotection against AF64A in cortical cultures. One possible explanation is the differential distribution and regulation of estrogen receptor-α and estrogen receptor-β in the brain (Shughrue et al., 1997; Laflamme et al., 1998). However, it is still a matter of debate which estrogen receptor subtype mediates the neuroprotective efficacy of estrogens. According to Sawada et al. (2000), estradiol provides neuroprotection on nigral dopaminergic neurons by suppression of proapoptotic gene transcription through the AP-1 site via activation of estrogen receptor-β. Moreover, in experimental stroke models, estrogen receptor-α deficiency does not enhance tissue damage in female animals, suggesting that estrogen inhibits brain injury by mechanisms that do not depend on activation of estrogen receptor-α (Sampei et al., 2000). On the other hand, several lines of evidence support the involvement of estrogen receptor-α. Evidence for a link between estrogen receptor-α, Bcl-xL expression, and neuroprotection has been provided in cultured hippocampal neurons (Pike, 1999). Moreover, in estrogen receptor-α knock-out mice, estradiol did not protect the brain against ischemic injury, indicating that estrogen receptor-α is a critical link in estradiol-mediated neuroprotection (Dubal et al., 2000). Two findings of the present study also argue for a role of estrogen receptor-α. First, the lowest immunofluorescence intensity for estrogen receptor-α, but the highest immunofluorescence intensity for estrogen receptor-β, appeared in the cortical cultures as compared with hippocampal and septal cultures. This is in agreement with in vivo findings in the adult rat (Shughrue et al., 1997; Laflamme et al., 1998) and confirms the recent finding by Zhang et al. (2000) that the expression of estrogen receptor-α was barely detectable in cultured neurons from embryonic rat cortex (E14). Second, the neuroprotectivity of 17 β-estradiol was abolished by the PI3-K inhibitor LY29402, which also has been reported previously (Honda et al., 2000). This finding indicates a role of PI3-K in the estradiol-mediated neuroprotection and a possible involvement of the IGF-1 receptor pathway. This pathway is known to be activated by estrogen receptor-α but not by estrogen receptor-β (Kahlert et al., 2000). It is well established that although both estrogen receptors exhibit the same affinity for 17 β-estradiol (Kuiper et al., 1997), the two forms of receptors display different patterns of affinities for naturally occurring hormone response elements, suggesting a different pattern of gene activation by the two receptors (Hyder et al., 1999). This refers not only to the activation of IGF-1 receptor but also to the activation of the transcription at the AP-1 sites (Paech et al., 1997; Kahlert et al., 2000).
The ability of 17 β-estradiol to modulate the expression of Bcl proteins was demonstrated first in peripheral, non-neuronal tissue (human breast cancer cells, in uterine endometrium during the proliferative phase of menstrual cycle) (Bhargava et al., 1994; Otsuki et al., 1994; Teixeira et al., 1995). In the brain, an upregulation of Bcl-2 by estradiol was found in hypothalamic neurons of female ratsin vivo (Garcia-Segura et al., 1998). Estradiol prevents the downregulation of Bcl-2 expression in cerebral cortex of ovariectomized female rats induced by ischemic brain injury (Dubal et al., 1999). Moreover, 17 β-estradiol significantly increases Bcl-xL levels in cultured hippocampal neurons (Pike, 1999). In the present study we found an increase in Bcl-2 and Bcl-xL levels in cultured hippocampal and septal neurons up to 68 hr after treatment with 17 β-estradiol (30–100 nm), whereas the same treatment did not change Bcl-2 and Bcl-xL levels in cortical neurons. After treatment with AF64A an increase in Bcl-2 expression was observed in hippocampal cultures, and the effect of 17 β-estradiol on Bcl-2 expression was intensified.
Although the dose range of 17 β-estradiol used in the present experiments is above the physiological and possible therapeutic range, our data may to some extent help to explain clinical findings. In Alzheimer's disease, where the cerebral cortex is affected as well, the first controlled prospective clinical trial of adequate duration to determine the benefit of estrogen did not support a role of estrogen for the treatment of this disease (Mulnard et al., 2000). The lack of an effect of 17 β-estradiol in cortical neurons both on Bcl-2 and Bcl-xL expression and on protection against AF64A-induced apoptosis may provide an experimental basis for understanding the recently reported failure to slow disease progression in women with mild to moderate Alzheimer's disease.
Footnotes
This study was supported by Deutsche Forschungsgemeinschaft Grants INK 21/A1-1/B8 and SFB 507, by the Hermann and Lilly Schilling Stiftung, and by the Medical Faculty Charité, Humboldt University at Berlin. We are grateful to Hannelore Glatte for excellent technical assistance.
C.H. and M.L. contributed equally to this paper.
Correspondence should be addressed to Dr. Heide Hörtnagl, Institute of Pharmacology and Toxicology, Medical Faculty Charité, Dorotheenstrasse 94, D-10098 Berlin, Germany. E-mail:heide.hoertnagl@charite.de.
REFERENCES
- 1.Alkayed NJ, Murphy SJ, Traystman RJ, Hurn PD. Neuroprotective effects of female gonadal steroids in reproductively senescent female rats. Stroke. 2000;31:161–168. doi: 10.1161/01.str.31.1.161. [DOI] [PubMed] [Google Scholar]
- 2.Behl C, Holsboer F. The female sex hormone oestrogen as a neuroprotectant. Trends Pharmacol Sci. 1999;20:441–444. doi: 10.1016/s0165-6147(99)01392-9. [DOI] [PubMed] [Google Scholar]
- 3.Behl C, Skutella T, Lezoualc'h F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol. 1997;51:535–541. [PubMed] [Google Scholar]
- 4.Bertrand R, Solary E, O'Connor P, Kohn KW, Pommier Y. Induction of a common pathway of apoptosis by staurosporine. Exp Cell Res. 1994;211:314–321. doi: 10.1006/excr.1994.1093. [DOI] [PubMed] [Google Scholar]
- 5.Beyer C, Raab H. Nongenomic effects of oestrogen: embryonic mouse midbrain neurones respond with a rapid release of calcium from intracellular stores. Eur J Neurosci. 1998;10:255–262. doi: 10.1046/j.1460-9568.1998.00045.x. [DOI] [PubMed] [Google Scholar]
- 6.Bhargava V, Kell DL, van de Rijn M, Warnke RA. Bcl-2 immunoreactivity in breast carcinoma correlates with hormone receptor positively. Am J Pathol. 1994;145:535–540. [PMC free article] [PubMed] [Google Scholar]
- 7.Bi R, Broutman G, Foy MR, Thompson RF, Baudry M. The tyrosine kinase and mitogen-activated protein kinase pathways mediate multiple effects of estrogen in hippocampus. Proc Natl Acad Sci USA. 2000;97:3602–3607. doi: 10.1073/pnas.060034497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birge SJ. Is there a role for estrogen replacement therapy in the prevention and treatment of dementia? J Am Geriatr Soc. 1996;44:865–870. doi: 10.1111/j.1532-5415.1996.tb03749.x. [DOI] [PubMed] [Google Scholar]
- 9.Bonnefont AB, Munoz FJ, Inestrosa NC. Estrogen protects neuronal cells from the cytotoxicity induced by acetylcholinesterase-amyloid complexes. FEBS Lett. 1998;441:220–224. doi: 10.1016/s0014-5793(98)01552-x. [DOI] [PubMed] [Google Scholar]
- 10.Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum and dentate gyrus. J Neurosci Res. 1995;42:674–683. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- 11.Bruer U, Weih MK, Isaev NK, Meisel A, Ruscher K, Bergk A, Trendelenburg G, Wiegand F, Victorov IV, Dirnagl U. Induction of tolerance in rat cortical neurons: hypoxic preconditioning. FEBS Lett. 1997;414:117–121. doi: 10.1016/s0014-5793(97)00954-x. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z-J, Yu L, Chang C-H. Stimulation of membrane-bound guanylate cyclase activity by 17 β-estradiol. Biochem Biophys Res Commun. 1998;252:639–642. doi: 10.1006/bbrc.1998.9716. [DOI] [PubMed] [Google Scholar]
- 13.Costa MM, Reus VI, Wolkowitz OM, Manfredi F, Lieberman M. Estrogen replacement therapy and cognitive decline in memory-impaired post-menopausal women. Biol Psychiatry. 1999;46:182–188. doi: 10.1016/s0006-3223(98)00355-2. [DOI] [PubMed] [Google Scholar]
- 14.Dubal DB, Kashon ML, Pettigrew LC, Ren JM, Finklestein SP, Rau SW, Wise PM. Estradiol protects against ischemic injury. J Cereb Blood Flow Metab. 1998;18:1253–1258. doi: 10.1097/00004647-199811000-00012. [DOI] [PubMed] [Google Scholar]
- 15.Dubal DB, Shughrue PJ, Wilson ME, Merchenthaler I, Wise PM. Estradiol modulates bcl-2 in cerebral ischemia: a potential role for estrogen receptors. J Neurosci. 1999;19:6385–6393. doi: 10.1523/JNEUROSCI.19-15-06385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dubal DB, Zhu H, Yu J, Rau SW, Shugrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha (ERα) is critical in estradiol-mediated neuroprotection against stroke injury: insights from ERα knockout (ERKO) mice. Soc Neurosci Abstr. 2000;26:288.4. [Google Scholar]
- 17.Falcieri E, Martelli AM, Bareggi R, Cataldi A, Cocco L. The protein kinase inhibitor staurosporine induces morphological changes typical of apoptosis in MOLT-4 cells without concomitant DNA fragmentation. Biochem Biophys Res Commun. 1993;193:19–25. doi: 10.1006/bbrc.1993.1584. [DOI] [PubMed] [Google Scholar]
- 18.Fisher A, Mantione CR, Abraham DJ, Hanin I. Long-term central cholinergic hypofunction in mice by ethylcholine aziridinium ion (AF64A) in vivo. J Pharmacol Exp Ther. 1982;222:140–145. [PubMed] [Google Scholar]
- 19.Garcia-Segura LM, Cardona-Gomez P, Naftolin F, Chowen JA. Estradiol upregulates Bcl-2 expression in adult brain neurons. NeuroReport. 1998;9:593–597. doi: 10.1097/00001756-199803090-00006. [DOI] [PubMed] [Google Scholar]
- 20.Gazzaley AH, Weiland NG, McEwen BS, Morrison JH. Differential regulation of NMDAR1 mRNA and protein by estradiol in the rat hippocampus. J Neurosci. 1996;16:6830–6838. doi: 10.1523/JNEUROSCI.16-21-06830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosteron exacerbates excitotoxicity, oxidative injury, and amyloid β-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 22.Gridley KE, Green PS, Simpkins JW. Low concentrations of estradiol reduce β-amyloid (25–35)-induced neurotoxicity, lipid peroxidation and glucose utilization in human SK-N-SH neuroblastoma cells. Brain Res. 1997;778:158–165. doi: 10.1016/s0006-8993(97)01056-1. [DOI] [PubMed] [Google Scholar]
- 23.Grodstein F, Stampfer MJ, Manson JE, Colditz GA, Willet WC, Rosner B, Speizer FE, Hennekens CH. Postmenopausal estrogen and progestin use and the risk of cardiovascular disease. N Engl J Med. 1996;335:453–461. doi: 10.1056/NEJM199608153350701. [DOI] [PubMed] [Google Scholar]
- 24.Hanin I. The AF64A model of cholinergic hypofunction: an update. Life Sci. 1996;58:1955–1964. doi: 10.1016/0024-3205(96)00185-3. [DOI] [PubMed] [Google Scholar]
- 25.Harms C, Lautenschlager M, Bergk A, Freyer D, Weih M, Dirnagl U, Weber JR, Hörtnagl H. Melatonin is protective in necrotic but not in caspase-dependent, free radical-independent apoptotic neuronal cell death in primary neuronal cultures. FASEB J. 2000;14:1814–1824. doi: 10.1096/fj.99-0899com. [DOI] [PubMed] [Google Scholar]
- 26.Honda K, Sawada H, Kihara T, Urushitani M, Nakamizo T, Akaike A, Shimohama S. Phosphatidylinositol 3-kinase mediates neuroprotection by estrogen in cultured cortical neurons. J Neurosci Res. 2000;60:321–327. doi: 10.1002/(SICI)1097-4547(20000501)60:3<321::AID-JNR6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 27.Hurn PD, MacRae IM. Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab. 2000;20:631–652. doi: 10.1097/00004647-200004000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Hyder SM, Chiappetta C, Stancel GM. Interaction of human estrogen receptors α and β with the same naturally occurring estrogen response elements. Biochem Pharmacol. 1999;57:597–601. doi: 10.1016/s0006-2952(98)00355-4. [DOI] [PubMed] [Google Scholar]
- 29.Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohé C. Estrogen receptor α rapidly activates the IGF-1 receptor pathway. J Biol Chem. 2000;24:18447–18453. doi: 10.1074/jbc.M910345199. [DOI] [PubMed] [Google Scholar]
- 30.Keller JN, Germeyer A, Begley JG, Mattson MP. 17 β-Estradiol attenuates oxidative impairment of synaptic Na+/K+-ATPase activity, glucose transport, and glutamate transport induced by amyloid β-peptide and iron. J Neurosci Res. 1997;50:522–530. doi: 10.1002/(SICI)1097-4547(19971115)50:4<522::AID-JNR3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 31.Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 32.Koh JY, Wie MB, Gwag BJ, Seni SL, Canzoniero LM, Demaro J, Csernansky C, Choi DW. Staurosporine-induced neuronal apoptosis. Exp Neurol. 1995;135:153–159. doi: 10.1006/exnr.1995.1074. [DOI] [PubMed] [Google Scholar]
- 33.Kuiper GGJM, Carlsson B, Grandien K, Enmark E, Häggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 34.Lafferty FW, Fiske ME. Postmenopausal estrogen replacement: a long term cohort study. Am J Med. 1994;97:66–77. doi: 10.1016/0002-9343(94)90050-7. [DOI] [PubMed] [Google Scholar]
- 35.Laflamme N, Nappi RE, Drolet G, Labrie C, Rivest S. Expression and neuropeptidergic characterization of estrogen receptors (ERα and ERβ) throughout the rat brain: anatomical evidence of distinct roles of each subtype. J Neurobiol. 1998;36:357–378. doi: 10.1002/(sici)1097-4695(19980905)36:3<357::aid-neu5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 36.Lautenschlager M, Onufriev MV, Gulyaeva NV, Harms C, Freyer D, Sehmsdorf U-S, Ruscher K, Moiseeva YV, Arnswald A, Victorov I, Dirnagl U, Weber JR, Hörtnagl H. Role of nitric oxide in the ethylcholine aziridinium (AF64A) model of delayed apoptotic neurodegeneration in vivo and in vitro. Neuroscience. 2000;97:383–393. doi: 10.1016/s0306-4522(99)00599-0. [DOI] [PubMed] [Google Scholar]
- 37.Mattson MP, Robinson N, Guo Q. Estrogens stabilize mitochondrial function and protect neural cells against the pro-apoptotic action of mutant presenilin-1. NeuroReport. 1997;8:3817–3821. doi: 10.1097/00001756-199712010-00031. [DOI] [PubMed] [Google Scholar]
- 38.Mermelstein PG, Becker JB, Surmeier DJ. Estradiol reduces calcium currents in rat neostriatal neurons via a membrane receptor. J Neurosci. 1996;16:595–604. doi: 10.1523/JNEUROSCI.16-02-00595.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mook-Jung I, Joo I, Sohn S, Kwon HJ, Huh K, Jung MW. Estrogen blocks neurotoxic effects of β-amyloid (1–42) and induces neurite extension on B103 cells. Neurosci Lett. 1997;235:101–104. doi: 10.1016/s0304-3940(97)00632-0. [DOI] [PubMed] [Google Scholar]
- 40.Moosmann B, Behl C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc Natl Acad Sci USA. 1999;96:8867–8872. doi: 10.1073/pnas.96.16.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mulnard RA, Cotman CW, Kawas C, van Dyck CH, Sano M, Doody R, Koss E, Pfeiffer E, Jin S, Gamst A, Grundman M, Thomas R, Thal LJ. Estrogen replacement therapy for treatment of mild to moderate Alzheimer's disease. A randomized controlled trial. JAMA. 2000;283:1007–1015. doi: 10.1001/jama.283.8.1007. [DOI] [PubMed] [Google Scholar]
- 42.Nath R, Raser KJ, Stafford D, Hajimohammadreza I, Posener A, Allen H, Talanian RV, Yuen P, Gilbertsen RB, Wang KK. Non-erythroid α-spectrin breakdown by calpain and interleukin 1 β-converting-enzyme-like protease(s) in apoptotic cells: contributory roles of both protease families in neuronal apoptosis. Biochem J. 1996;319:683–690. doi: 10.1042/bj3190683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otsuki Y, Misaki O, Sugimoto O, Ito Y, Tsujimoto Y, Akao Y. Cyclic bcl-gene expression in humane uterine endometrium during menstrual cycle. Lancet. 1994;344:28–29. doi: 10.1016/s0140-6736(94)91051-0. [DOI] [PubMed] [Google Scholar]
- 44.Paech K, Webb P, Kuiper GGJM, Nilsson S, Gustafsson J-A, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERα and ERβ at AP1 site. Nature. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 45.Paganini-Hill A, Henderson VW. Estrogen deficiency and risk of Alzheimer's disease in women. Am J Epidemiol. 1994;140:256–261. doi: 10.1093/oxfordjournals.aje.a117244. [DOI] [PubMed] [Google Scholar]
- 46.Pike CJ. Estrogen modulates neuronal Bcl-xL expression and β-amyloid-induced apoptosis: relevance to Alzheimer's disease. J Neurochem. 1999;72:1552–1563. doi: 10.1046/j.1471-4159.1999.721552.x. [DOI] [PubMed] [Google Scholar]
- 47.Raff MC, Barres B, Burne J, Coles H, Ishizaki Y, Jacobson M. Programmed cell death and control of cell survival: lessons from the central nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- 48.Regan RF, Guo Y. Estrogens attenuate neuronal injury due to hemoglobin, chemical hypoxia, and excitatory amino acids in murine cortical cultures. Brain Res. 1997;764:133–140. doi: 10.1016/s0006-8993(97)00437-x. [DOI] [PubMed] [Google Scholar]
- 49.Rinner WA, Pifl C, Lassmann H, Hörtnagl H. The induction of apoptosis in vitro and in vivo by the cholinergic neurotoxin ethylcholine aziridinium. Neuroscience. 1997;79:535–542. doi: 10.1016/s0306-4522(96)00687-2. [DOI] [PubMed] [Google Scholar]
- 50.Robinson D, Friedman L, Marcus R, Tinklenberg J, Yesavage J. Estrogen replacement therapy and memory in older women. J Am Geriatr Soc. 1994;42:919–922. doi: 10.1111/j.1532-5415.1994.tb06580.x. [DOI] [PubMed] [Google Scholar]
- 51.Sampei K, Goto S, Alkayed NJ, Crain BJ, Korach KS, Traystman RJ, Demas GE, Nelson RJ, Hurn PD. Stroke in estrogen receptor-α-deficient mice. Stroke. 2000;31:738–744. doi: 10.1161/01.str.31.3.738. [DOI] [PubMed] [Google Scholar]
- 52.Sawada H, Ibi M, Kihara T, Urushitani M, Akaike A, Shimohama S. Estradiol protects mesencephalic dopaminergic neurons from oxidative stress-induced neuronal death. J Neurosci Res. 1998;54:707–719. doi: 10.1002/(SICI)1097-4547(19981201)54:5<707::AID-JNR16>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 53.Sawada H, Ibi M, Kihara T, Urushitani M, Honda K, Nakanishi M, Akaike A, Shimohama s. Mechanisms of antiapoptotic effects of estrogens in nigral dopaminergic neurons. FASEB J. 2000;14:1202–1214. doi: 10.1096/fasebj.14.9.1202. [DOI] [PubMed] [Google Scholar]
- 54.Sherwin BB. Estrogenic effects on memory in women. Ann NY Acad Sci. 1994;743:213–230. doi: 10.1111/j.1749-6632.1994.tb55794.x. [DOI] [PubMed] [Google Scholar]
- 55.Shi J, Panickar KS, Yang S-H, Rabbani O, Day AL, Simpkins JW. Estrogen attenuates over-expression of β-amyloid precursor protein messenger RNA in an animal model of focal ischemia. Brain Res. 1998;810:87–92. doi: 10.1016/s0006-8993(98)00888-9. [DOI] [PubMed] [Google Scholar]
- 56.Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-α and -β mRNA in the rat central nervous system. J Comp Neurol. 1997;388:507–525. doi: 10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 57.Singer CA, Rogers KL, Strickland TM, Dorsa DM. Estrogen protects primary cortical neurons from glutamate toxicity. Neurosci Lett. 1996;212:13–16. doi: 10.1016/0304-3940(96)12760-9. [DOI] [PubMed] [Google Scholar]
- 58.Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci. 1999;19:2455–2463. doi: 10.1523/JNEUROSCI.19-07-02455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang M-X, Jacobs D, Stern Y, Marder K, Schofield P, Gurland B, Andrews H, Mayeux R. Effect of oestrogen during menopause on risk and age at onset of Alzheimer's disease. Lancet. 1996;348:429–432. doi: 10.1016/S0140-6736(96)03356-9. [DOI] [PubMed] [Google Scholar]
- 60.Teixeira C, Reed JC, Pratt MA. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving Bcl-2 proto-oncogene expression in human breast cancer cells. Cancer Res. 1995;55:3902–3907. [PubMed] [Google Scholar]
- 61.Wang KKW. Calpain and caspase: can you tell the difference? Trends Neurosci. 2000;23:20–26. doi: 10.1016/s0166-2236(99)01479-4. [DOI] [PubMed] [Google Scholar]
- 62.Weaver CE, Jr, Park-Chung M, Gibbs TT, Farb DH. 17 β-Estradiol protects against NMDA-induced excitotoxicity by direct inhibition of NMDA receptors. Brain Res. 1997;761:338–341. doi: 10.1016/s0006-8993(97)00449-6. [DOI] [PubMed] [Google Scholar]
- 63.Wiesner DA, Dawson G. Staurosporine induces programmed cell death in embryonic neurons and activation of the ceramide pathway. J Neurochem. 1996;66:1418–1425. doi: 10.1046/j.1471-4159.1996.66041418.x. [DOI] [PubMed] [Google Scholar]
- 64.Xu H, Gouras GK, Greenfield JP, Vincent B, Naslund J, Mazzarelli L, Fried G, Jovanovic JN, Seeger M, Relkin NR, Liao F, Checler F, Buxbaum JD, Chait BT, Thinakaran G, Sisodia SS, Wang R, Greengard P, Gandy S. Estrogen reduces neuronal generation of Alzheimer β-amyloid peptides. Nat Med. 1998;4:447–451. doi: 10.1038/nm0498-447. [DOI] [PubMed] [Google Scholar]
- 65.Yaffe K, Sawaya G, Lieberburg I, Grady D. Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. JAMA. 1998;279:688–695. doi: 10.1001/jama.279.9.688. [DOI] [PubMed] [Google Scholar]
- 66.Zaulyanov LL, Green PS, Simpkins JW. Glutamate requirement for neuronal death from anoxia-reoxygenation: an in vitro model for assessment of the neuroprotective effects of estrogens. Cell Mol Neurobiol. 1999;19:705–718. doi: 10.1023/A:1006948921855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang L, Chang YH, Barker JL, Hu Q, Zhang L, Maric D, Li B-S, Rubinow DR. Testosterone and estrogen affect neuronal differentiation but not proliferation in early embryonic cortex of the rat: the possible roles of androgen and estrogen receptors. Neurosci Lett. 2000;281:57–60. doi: 10.1016/s0304-3940(99)00942-8. [DOI] [PubMed] [Google Scholar]