

Abstract
Muscle nicotinic acetylcholine receptors (AChRs) are pentamers that contain two α subunits a β, γ (or ε), and δ subunit. In this paper, we have characterized subunit processing and folding events leading to formation of the two AChR ligand binding sites. α subunit residues, 187–199, which are part of overlapping ACh and α-bungarotoxin (Bgt) binding sites on AChRs, were assayed using a monoclonal antibody (mAb) specific for these residues. We found that this region was inaccessible to the mAb early during AChR assembly but became accessible as the first of two Bgt binding sites formed later during assembly, indicating that the region changes conformation as the Bgt binding site appears. Without previous reduction, 20% of the α subunits could be alkylated by bromoacetylcholine bromide as the first ACh binding site formed, which further indicated that the disulfide bond between cysteines 192 and 193 does not form until the first ACh binding site appears soon after Bgt binding site formation. When α subunits were mutated to add a glycosylation site at residue 187, the number of Bgt binding sites increased threefold, AChRs assembled more efficiently, and 2.5-fold more AChRs reached the cell surface. Our results indicate that binding site formation involves a rate-limiting rearrangement of the α subunit that exposes the 187–199 region to the endoplasmic reticulum lumen and determines when cysteines 192 and 193 disulfide bond.
Keywords: nicotinic receptor, Torpedo, muscle, α-bungarotoxin, protein folding and assembly, acetylcholine binding site
Signal transduction by members of a family of neurotransmitter-gated ion channels, which include the acetylcholine (ACh), GABAA, glycine, and 5-HT3 receptors, is initiated by binding of neurotransmitters to specific sites on the receptors. Muscle-type nicotinic ACh receptors (AChRs), which include the receptor in fish electric organs, have long served as a model for the family (for review, see Changeux, 1995; Karlin and Akabas, 1995). AChRs are composed of four homologous subunits, α, β, γ (or ε), and δ, that assemble into pentamers with a stoichiometry of α2βγδ. There are two ligand binding sites per receptor. Affinity labeling and mutagenesis of the α subunit have identified six residues, Tyr-93, Trp-149, Tyr-190, Cys-192, Cys-193, and Tyr-198, in the vicinity of the binding site and have shown that the adjacent cysteines, Cys-192 and Cys-193, are disulfide-bonded. Residues on the γ and δ subunits have also been identified as contributing to the binding site, from which it was concluded that the two binding sites are found near the interfaces between an α subunit and either the γ or δ subunits (for review, see Karlin and Akabas, 1995; Tsigelny et al., 1997; Changeux and Edelstein, 1998).
Venom-derived α-peptide neurotoxins are potent AChR competitive antagonists and bind with high affinity to regions overlapping ACh binding sites. The best characterized of these neurotoxins is α-bungarotoxin (Bgt), for which the major region contributing to the binding site has been localized to α subunit residues 173–204 (Wilson et al., 1985; Tzartos and Remoundos, 1990). Neither Bgt nor ACh sites are present on nascent α subunits and only appear during AChR assembly (Merlie and Lindstrom, 1983; Green and Wanamaker, 1998). During AChR assembly, subunits first rapidly associate into αβγ trimers. Trimers slowly assemble with δ subunits into αβγδ tetramers and then with a second α subunit into α2βγδ pentamers (Green and Claudio, 1993). The first Bgt binding sites form on trimers just before δ subunits associate, after which the first ACh sites appear at the α–γ subunit interface on αβγδ tetramers. The second ACh and Bgt sites appear later at the α–δ interface on α2βγδ pentamers. This sequence of events suggests that the subunits undergo posttranslational changes after their assembly so that the Bgt and ACh binding sites form.
In the experiments presented, we have probed α subunit residues 187–199, which contribute to the Bgt and ACh binding sites, looking for posttranslational changes during AChR assembly. We demonstrate that this region is initially inaccessible to a monoclonal antibody (mAb) specific for the region and becomes accessible only as the Bgt binding site forms, implying that an intervening conformational change involving this region occurs during AChR assembly. Evidence is also presented that disulfide bonding of cysteine 192 and 193 requires the same conformational change. Finally, addition of an N-linked glycan to this region increases binding site formation and the efficiency of assembly. Our data suggest that the added glycan expedites a conformational change of residues 187–199 that is rate limiting, thereby increasing binding site formation and the assembly process.
MATERIALS AND METHODS
AChR subunit and mutant cDNAs. Torpedoα, β, γ, and δ subunit cDNAs and mouse α, β, ε, and δ subunit cDNAs subcloned into the pRBG4 expression vector (Lee et al., 1991) were a gift from Dr. S. Sine (Mayo Clinic, Rochester, MN). The mouse α subunit N-glycosylation site mutation cDNA, α187–189 or W187N/F189T (Kreienkamp et al., 1994) also cloned into pRBG4, was a gift from Dr. P. Taylor (University of California, San Diego, San Diego, CA).
Cell lines. Mouse L fibroblasts, stably transfected with theTorpedo subunit cDNAs under the control of SV 40 promoters (Claudio et al., 1987), were maintained in DMEM plus 10% calf serum and HAT (15 mg/ml hypoxanthine, 1 mg/ml aminopterin, and 5 mg/ml thymidine) at 37°C in 5% CO2. To enhance subunit expression, the DMEM was supplemented with 20 mm sodium butyrate (NB medium) 36 hr before the experiment.
Transient transfections. For transient transfections, the human embryonic kidney-derived tsA201 cells (Margolskee et al., 1993) were cultured in DMEM supplemented with 10% calf serum. A calcium phosphate protocol described previously (Eertmoed et al., 1998) was used for transfection. Because Torpedo subunits exhibit a temperature-dependent assembly, the temperature was dropped to 20°C for 2 d before the experiment. Cells transfected with mouse subunits were maintained at 37°C for 1 d before the experiment.
Metabolic labeling. Cultures (10 cm) of stably transfected cells (see Fig. 1) or 6 cm cultures of transiently transfected tsA201 cells (see Fig. 3) were labeled as described previously (Green and Claudio, 1993; Green and Wanamaker, 1997). Briefly, cultures were pulse labeled in 2 (see Fig. 1) or 1 (see Fig.3) ml of methionine-cysteine-free medium, supplemented with 333 (see Fig. 1) or 111 (see Fig. 3) μCi of a35S-methionine35S-cysteine mixture (NEN EXPE35S35S). The labeling was stopped with the addition of DMEM plus 5 mm methionine. To follow the subsequent changes in the labeled subunits, the cells were “chased” by incubation for the indicated times in regular medium at 20°C (see Fig. 1,Torpedo subunits) or 37°C (see Fig. 3, mouse subunits). All subsequent steps were performed at 4°C to prevent further subunit folding and assembly. The cells were solubilized in 1% LPC (1.83 mg/ml phosphatidylcholine and 1% Lubrol) (see Fig. 1) or 1% Triton (see Fig. 3) in lysis buffer (150 mm NaCl, 5 mm EDTA, 50 mm Tris, pH 7.4, and 0.02% NaN3) containing protease inhibitors (2 mm phenylmethylsulfonyl fluoride, 2 mm n-ethylmaleimide, and 10 μg/ml each of chymostatin, pepstatin, tosyl-lysine chloromethyl ketone, and leupeptin).
Fig. 1.

Accessibility of mAb 383c to α subunit residues 187–199. A, Binding of mAb 383c, which specifically recognizes α subunit residues 187–199, prevents 125I-Bgt binding to AChRs. Intact cells stably expressing TorpedoAChRs were incubated with the indicated amounts of mAb 383c (squares) or mAb 247g (circles) for 2 hr before performing 125I-Bgt binding to cell surface receptors. After 125I-Bgt labeling, cells were washed and counted. Points on the graph represent the mean of duplicate samples. B–D, AChR subunits were metabolically labeled for 30 min at 37°C and followed in culture for the indicated times at 20°C. Equal amounts of solubilized, labeled subunits were incubated with mAb 383c (B), Bgt-Sepharose (C), or a polyclonal α Ab (D) and then precipitated subunits were visualized by SDS-PAGE (7.5% gel) and autoradiography. Theband just above the δ subunit band (α′ in D) in the α subunit-specific AChR precipitations has been shown previously to be an unprocessed form of the α subunit (Green and Claudio, 1993; Green and Wanamaker, 1997). Both β and γ subunits migrate as doublet bands as a result of differential processing, possibly differences in disulfide bonding (Gelman and Prives, 1996) or oligosaccharide trimming (Chang et al., 1997). E, A comparison of mAb 383c epitope and Bgt binding site formation. Subunits precipitated with mAb 383c or Bgt-Sepharose were analyzed on SDS-PAGE as in B andC and quantified using phosphorimaging. The values for α and β subunits from both 383c and Bgt-Sepharose experiments are shown. The values for γ and δ subunits are displayed in Table 1. For the β and γ subunits, both doublet bands were included in the analysis.
Fig. 3.

Glycosylation of α subunit residue 187 alters AChR assembly. A, Effect of the glycosylation of α subunit residue 187 on formation of Bgt binding sites. Mouse mutated or wild-type α subunits were transiently transfected along with the mouse β, ε, and δ subunits into tsA201 cells, and125I-Bgt binding to intracellular AChR subunits was determined at the indicated times. The 125I-Bgt binding was plotted as the fraction of maximum binding for wild-type α-containing AChRs at 24 hr. All points on the graph represent the mean ± SD of three samples. Maximum binding was 69 ± 1.3 fmol. B, Effect of the glycosylation of α subunit residue 187 on AChR surface expression. In parallel to the experiments in Figure 3A, 125I-Bgt binding to surface AChRs was determined at the indicated times. The 125I-Bgt binding was plotted as in A. Again, the points on the graph represent the mean ± SD of three samples. Maximum binding was 132 ± 5.3 fmol. C, D, Subunit synthesis and formation of Bgt binding sites. Mouse mutated or wild-type α subunits transiently transfected with the β, ε, and δ subunits were metabolically labeled for 5 min and followed in culture for the indicated times at 37°C. Equal amounts of solubilized, labeled subunits were incubated with mouse α-specific mAb P22 (C) or Bgt-Sepharose (D) and analyzed by SDS-PAGE (7.5% gel) and autoradiography. E, Quantitation of the metabolically labeled mutated and wild-type α subunits. Metabolically labeled mutated and wild-type subunits were precipitated with the α-specific mAb, mAb P22, analyzed on SDS-PAGE as in C, and quantitated using a PhosphorImager. Points on the graph represent the mean ± SEM band intensities from five experiments.
Immunoprecipitation and Bgt-Sepharose precipitation.Solubilized AChR subunits were immunoprecipitated with mAb 383c (a gift from Dr. R. Fairclough, University of California, Davis, CA), which is specific for Torpedo α subunits, mAb P22 (a gift from Dr. V. Lennon, Mayo Clinic), which is specific for mouse α subunits, or a polyclonal Ab specific for both Torpedo and mouse α subunits (Ross et al., 1991). Immunoprecipitations were performed by overnight incubation at 4°C, and Ab-subunit complexes were precipitated by incubating with Protein G-Sepharose for 3 hr at 4°C. Alternatively, solubilized subunits were precipitated with a slurry of Bgt-Sepharose, which was prepared by coupling Bgt to cyanogen bromide-activated Sepharose according to the manufacturer's directions (Amersham Pharmacia Biotech, Arlington Heights, IL). mAb-Protein G- or Bgt-Sepharose-precipitated subunits and complexes were electrophoresed on 7.5% SDS polyacrylamide gels, fixed, enhanced for 30 min, dried on a gel dryer, and exposed to film at −70°C with an intensifying screen. To determine subunit band intensities, gels were exposed to a phosphor screen, developed using a PhosphorImager and quantified using ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
125I-Bgt binding. For cell surface125I-Bgt-binding, the cells were washed with PBS and incubated at room temperature in PBS containing 4 nm125I-Bgt for 2 hr. This incubation time is sufficient to saturate the binding. The cells were then washed three times in PBS and counted in a gamma counter. For intracellular125I-Bgt-binding, cell surface receptors were preblocked by incubation with 10 nmunlabeled Bgt in medium at 20°C for at least 1 hr before solubilization (see Fig. 2). Because Bgt dissociates more rapidly from the α187–189 mutated receptors, cell surface receptors were immunodepleted with mAb 35 [American Type Culture Collection (ATCC)] before intracellular receptor binding instead of an unlabeled Bgt preblock (see Fig. 3). For this, intact cells were incubated with saturating amounts of mAb 35 in PBS overnight at 4°C. Excess mAb was removed with three washes with PBS. The cells were then solubilized in 1% Triton X-100 in lysis buffer with protease inhibitors, and the mAb 35-bound surface receptors in the lysate were precipitated out with Protein G-Sepharose. The intracellular receptors remaining in the lysate were then incubated with 5 nm125I-Bgt and mAb 35 overnight at 4°C and precipitated with Protein G-Sepharose, followed by three washes with lysis buffer to remove excess Bgt. The 125I-Bgt bound was then counted in a gamma counter. For transiently transfected cells,125I-Bgt binding in parallel samples from sham-transfected cultures for which no cDNAs were included in the calcium phosphate solution estimated nonspecific binding. For stably transfected cells, nonspecific 125I-Bgt binding was measured by binding in the presence of 10 mm carbamylcholine.
Fig. 2.
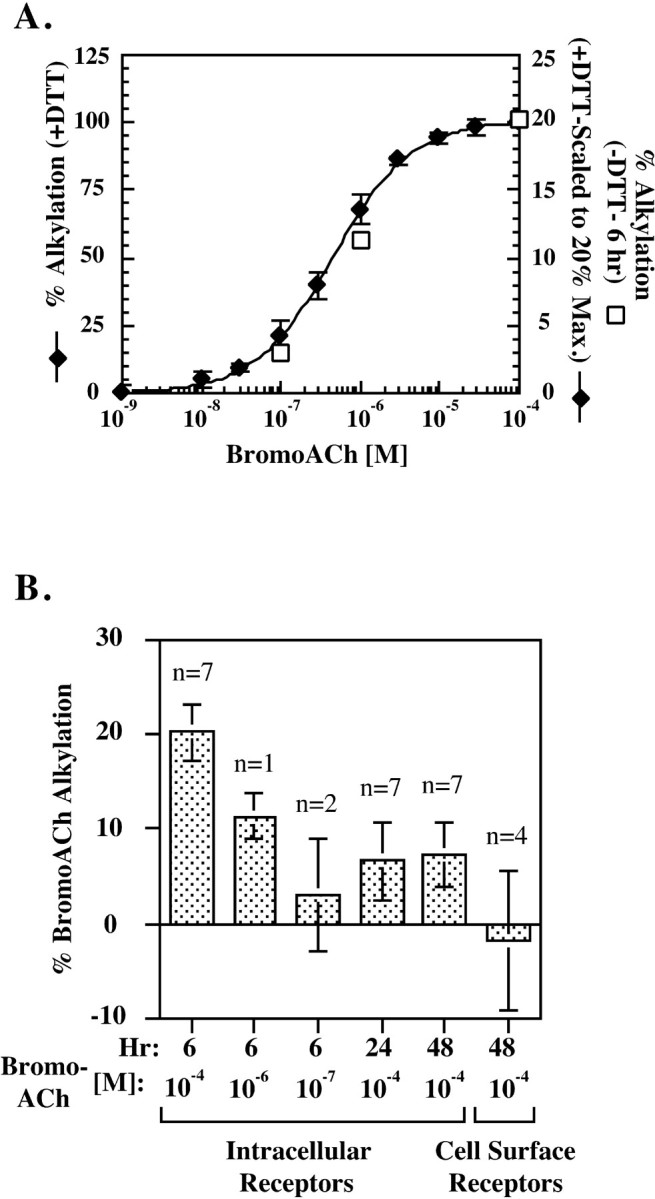
BromoACh alkylates assembling AChR subunits without previous reduction. A, Dose dependence of bromoACh alkylation of intracellular AChR complexes that were reduced with (filled diamonds) or without (open squares) DTT. Intracellular AChR complexes were immunoprecipitated with the γ and δ subunit-specific mAb 88b. Samples were then treated with or without 0.5 mm DTT and incubated with the indicated concentrations of bromoACh. The bromoACh alkylation was determined by measuring the loss of 125I-Bgt binding caused by alkylation. For the DTT-treated samples (y-axis on the left), the percentage of bromoACh alkylation was determined as the percentage of125I-Bgt binding blocked by bromoACh alkylation in the alkylated samples compared with samples not treated with bromoACh but treated with DTT. Shown are the data obtained 6 and 48 hr after initiation of assembly. Points on the graph represent the mean ± SD of four measurements, two at 6 hr and two at 48 hr. For the samples not treated with DTT (y-axis on the right), the bromoACh alkylation was performed 6 hr after initiation of assembly. The percentage of bromoACh alkylation was determined as the percentage of 125I-Bgt binding blocked by bromoACh alkylation compared with samples treated with 10−4m ACh instead of bromoACh and was maximally 20% of the total 125I-Bgt binding (for details, see Fig. 2B). B, BromoACh alkylation of intracellular and surface receptors without previous reduction. At the indicated times after the temperature shift from 37 to 20°C, intracellular AChR complexes were alkylated using the indicated concentration of bromoACh without previous reduction with DTT. In parallel, cell surface AChRs were incubated with 10−4m bromoACh before125I-Bgt binding. The percentage of bromoACh alkylation was determined as the percentage of 125I-Bgt binding blocked by bromoACh alkylation compared with samples treated with 10−4m ACh instead of bromoACh. For each experiment, the bromoACh and ACh treatment was performed in triplicate. The values shown represent the mean ± SEM ofn experiments. The values determined using 10−4, 10−6, and 10−7m bromoACh at 6 hr are replotted (open squares) in A. Statistical significance of the differences between the means for 10−4m bromoACh was estimated using thet test and the Mann–Whitney rank sum test. The 6 hr intracellular mean was significantly different from all those at 24 and 48 hr (t values ranging from 2.62 to 2.89; α of 0.05). The 24 and 48 hr values did not differ significantly from each other.
Alkylation with bromoacetylcholine bromide. For measuring bromoacetylcholine bromide (bromoACh) alkylation of intracellular receptors, surface receptors in 10 cm cultures of cells stably transfected with Torpedo receptor subunits were blocked by incubating in medium containing 10 nmBgt for 2 hr at 37°C, and then the temperature was lowered to 20°C to initiate assembly. After 6, 24, or 48 hr at 20°C, the unbound Bgt was washed away. The cells were harvested, solubilized in 1% LPC in lysis buffer with protease inhibitors, and immunoprecipitated with saturating amounts of mAb 88b (ATCC), which recognizes the γ and δ subunits. If receptors needed to be reduced before bromoACh alkylation (see Fig. 2A), the precipitated receptor complexes were incubated with 0.5 mm dithiothreitol (DTT) in lysis buffer for 30 min at room temperature and rapidly washed three times. The precipitated receptor complexes were incubated with the indicated concentration of bromoACh (see Fig. 2A) or with the saturating concentration of 10−4m (see Fig. 2B) for 30 min at room temperature. For the experiment in Figure 2B, 10−4m bromoACh was also included in the solubilization buffer. Unalkylated bromoACh was washed off the precipitated complexes with lysis buffer, and the bromoACh-treated receptors were bound with 10 nm125I-Bgt as described above, in lysis buffer containing 0.5% BSA. To control for block of125I-Bgt binding by unalkylated agonist, an equal number of samples were treated in parallel with 10−4m ACh instead of 10−4mbromoACh.
To measure bromoACh alkylation of surface receptors without DTT reduction, mAb 35 was bound to surface receptors by incubating intact cells in PBS with mAb 35 overnight at 4°C. The cells were solubilized and incubated with Protein G-Sepharose to precipitate the surface receptor–Ab complexes, which were then incubated with 10−4m bromoACh or 10−4m ACh, washed with lysis buffer, and bound with 125I-Bgt. The percentage of alkylated receptors was determined as above.
RESULTS
The α subunit 187–199 region changes conformation during binding site formation
A monoclonal antibody, mAb 383c (Gomez and Richman, 1983) generated against the Torpedo nicotinic AChR, was used to probe for conformational changes during formation of the AChR binding sites. The interaction between mAb 383c and Torpedo AChRs has been well characterized. Its epitope has been mapped to α subunit residues 187–199 (Fairclough et al., 1998a), a region that contributes to both the Bgt and ACh binding sites. In support of this region contributing to the Bgt binding site, mAb 383c binding specifically inhibits Bgt binding to native Torpedo AChRs (Mihovilovic and Richman, 1987). To test whether mAb 383c recognizesTorpedo AChRs heterologously expressed in mouse fibroblast cells, its effect on cell-surface 125I-Bgt binding was assayed. As shown in Figure1A, saturating concentrations of mAb 383c blocks virtually all surface125I-Bgt binding to intact fibroblasts. Shown for comparison in Figure 1A is the effect of mAb 247g on cell surface 125I-Bgt binding. mAb 247g binds to α subunits and blocks surface Bgt binding at only one of the two binding sites so that saturating concentrations inhibit only 50% of binding (Mihovilovic and Richman, 1987; Green and Wanamaker, 1998). Thus, mAb 383c appears to bind to α subunits in assembled receptors and block Bgt binding to both binding sites as observed previously for the native Torpedo AChRs.
The slow, temperature-dependent assembly of Torpedo AChRs was used to determine when during assembly mAb 383c recognizes α subunits. At 37°C, subunit assembly is halted at the earliest stages. Assembly can be initiated by lowering the temperature to 20°C and proceeds at this temperature at a rate more than an order of magnitude slower than for the mammalian subunits (Green and Claudio, 1993). Subunits were labeled at 37°C, the temperature was lowered to 20°C, and the labeled subunits were followed for the indicated times (Fig.1B-D). mAb 383c immunoprecipitated α subunits only after a delay of 3–6 hr (Fig. 1B). At the 3 and 6 hr time points, the ratio of α/β/γ/δ subunits precipitated by mAb 383c was ∼1:1:1:1 (Fig. 1E, Table1). This result indicates that the epitope is accessible to mAb 383 only after α subunits have assembled into complexes with the other three subunits. After the 3–6 hr delay, mAb 383c precipitated progressively more of the assembled complexes until ∼24 hr after the temperature shift, at which time the ratio of the α to the other subunits almost doubled (Fig.1E, Table 1). The lack of α subunit precipitation by mAb 383c at early time points is not caused by an absence of labeled subunits. Other Abs that recognize α subunits, such as the α subunit-specific polyclonal Ab used in Figure 1D, precipitate an excess of α subunits relative to the other subunits at the early time points. Because mAb 383c binds to denatured α subunits or linear stretches of the α subunit virtually as well as it binds to fully assembled receptors (Fairclough et al., 1998b), the delayed recognition of α subunits by mAb 383c indicates that the mAb 383c epitope is inaccessible at the early stages of AChR assembly. Thus, during AChR assembly, α subunits appear to change conformation from a state in which residues 187–199 are inaccessible to mAb 383c to a state in which this region is accessible to the mAb.
Table 1.
Quantitation and comparison of the time course of formation of the mAb 383c epitope and Bgt binding site during assembly of theTorpedo AChR
| Hours | mAb 383c | Bgt-Sepharose | ||||||
|---|---|---|---|---|---|---|---|---|
| α | β | γ | δ | α | β | γ | δ | |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | 0.35 ± 0.1 | 0.39 ± 0.09 | 0.43 ± 0.13 | 0.33 ± 0.12 | 0.26 ± 0.06 | 0.27 ± 0.08 | 0.26 ± 0.1 | 0.2 ± 0.11 |
| 6 | 0.64 ± 0.2 | 0.59 ± 0.18 | 0.63 ± 0.2 | 0.5 ± 0.16 | 0.6 ± 0.09 | 0.52 ± 0.11 | 0.52 ± 0.11 | 0.46 ± 0.08 |
| 24 | 1.67 ± 0.24 | 0.99 ± 0.02 | 1.09 ± 0.06 | 0.87 ± 0.14 | 1.6 ± 0.16 | 0.99 ± 0.03 | 1.06 ± 0.08 | 0.97 ± 0.09 |
| 48 | 1.71 ± 0.32 | 0.84 ± 0.11 | 0.96 ± 0.14 | 0.73 ± 0.14 | 1.7 ± 0.28 | 0.84 ± 0.09 | 0.96 ± 0.24 | 0.86 ± 0.16 |
Subunit band intensities from subunits precipitated with mAb 383c (as in Fig. 1B) or Bgt-Sepharose (as in Fig. 1C) were quantified using phosphorimaging. Shown are the means and SDs of scanned values from six experiments in which the mAb 383c and Bgt-Sepharose precipitations were performed in parallel from equal aliquots. The values are displayed as the fraction of the largest β subunit value that occurred at the 24 hr time point.
Because Bgt binding sites appear on α subunits during AChR assembly (Green and Claudio, 1993) and the mAb 383c epitope contributes to the Bgt binding site, we tested whether Bgt binding site formation parallels the appearance of the mAb 383c epitope. The formation of Bgt binding sites was assayed using Bgt-Sepharose, which can be used to recognize and precipitate assembling receptor complexes (Fig.1C) (Green and Wanamaker, 1998). Saturating amounts of either mAb 383c or Bgt-Sepharose were used to precipitate equal aliquots of the pulse-labeled subunits. We observed no significant difference in the amount of labeled subunits precipitated by mAb 383c or Bgt-Sepharose at any of the chase times (Fig.1B,C). The results from six different pulse-chase experiments, performed as in Figure 1,B and C, were quantified, and the means and SD of each subunit gel band are displayed in Figure1E and Table 1. The similar intensities of the subunit bands precipitated with mAb 383c or Bgt-Sepharose indicate that the mAb 383c epitope becomes accessible at approximately the same time as the first Bgt binding site appears during AChR assembly. Initially, the ∼1:1:1:1 ratio of α/β/γ/δ subunits precipitated by both mAb 383c and Bgt-Sepharose occurs because αβγδ tetramers are rapidly assembled immediately after the appearance of the first Bgt binding site on αβγ trimers (Green and Wanamaker, 1998). Later, the ratio of the α to the other subunits almost doubles as α2βγδ pentamers assemble (Green and Claudio, 1993). Because mAb 383c binds specifically to the 187–199 region (Fairclough et al., 1998a) and this region comprises a major part of the Bgt binding site, it appears that the 187–199 region becomes accessible to mAb 383c as part of the subunit folding that forms the first Bgt binding site. This interpretation of our data can explain why the Bgt binding site is not initially present after subunit synthesis, although Bgt can bind to the 173–204 peptide alone and to denatured α subunits (Wilson et al., 1988; Chaturvedi et al., 1992).
Affinity alkylation of α subunit cysteines 192 and/or 193 without previous reduction
Significant evidence indicates that most, if not all, of AChR assembly occurs before the subunits are released from the endoplasmic reticulum (ER) (Smith et al., 1987; Gu et al., 1989; Ross et al., 1991; Gelman et al., 1995). If the α187–199 region is in fact initially buried, as suggested by the inaccessibility of the mAb 383c epitope, then the disulfide bond between cysteines 192 and 193 would not be expected to form until this region changes conformation and becomes accessible to the oxidizing ER lumen (Hawkins et al., 1991;Hwang et al., 1992). Affinity alkylating agents, such as bromoACh, bind to the ACh binding site and specifically alkylate theTorpedo α subunit 192 and 193 cysteines after their reduction (Silman and Karlin, 1969; Damle et al., 1978; Kao and Karlin, 1986). BromoACh was used to determine when the 192–193 disulfide bond forms by testing at different times whether bromoACh could alkylate the cysteines without previous reduction of the disulfide bond. When solubilized intracellular AChR complexes were reduced with 0.5 mm DTT, bromoACh alkylated the binding sites and blocked all 125I-Bgt binding to the complexes in a dose-dependent manner (Fig.2A). Intracellular AChRs were separated from cell surface receptors by first blocking surface receptors with cold Bgt before solubilization. Without previous DTT reduction, bromoACh was unable to alkylate and block Bgt binding to cell surface receptors (Fig. 2B). However, a significant number of intracellular AChR complexes were alkylated by bromoACh in the absence of reduction (Fig. 2B). The ability to alkylate with bromoACh without reduction depended on when the cells expressing AChRs were harvested after a temperature shift from 37 to 20°C. Six hours after initiating assembly, a saturating concentration of bromoACh (10−4m) alkylated 20% of the intracellular complexes with no previous reduction of disulfide bonds (Fig.2B). At lower bromoACh concentrations, fewer complexes were alkylated without previous reduction, consistent with the dose dependence observed with reduction (Fig. 2A,open symbols, B). Six hours after the temperature shift, assembling subunit intermediates are either αβγ trimers that lack ACh binding sites or αβγδ tetramers on which the first of two ACh sites forms (Green and Wanamaker, 1998). At this time, if alkylation by bromoACh occurs with the specific binding of bromoACh to ACh binding sites, then we can conclude that bromoACh is alkylating α subunits assembled into αβγδ tetramers. Almost all newly assembled tetramers continue on to assemble into pentamers and are transported to the surface (Green and Wanamaker, 1998), indicating that the 20% of the Bgt binding sites alkylated by bromoACh in the absence of DTT are not on misfolded α subunits.
Twenty-four and 48 hr after the temperature shift to initiate AChR assembly, a much smaller percentage of the complexes were alkylated without previous reduction by a saturating concentration of bromoACh (Fig. 2B). Our ability to affinity alkylate cysteines 192 and 193 without reduction was transitory and was largest early in assembly when the first ACh binding site appears. The second ACh binding sites appear 24–48 hr after the temperature shift (Green and Wanamaker, 1998), and the much smaller percentage alkylated by bromoACh at these times could indicate that the second site forms differently in that the 192 and 193 cysteines are oxidized more rapidly than at the first site. However, an alternative explanation for the data are that, for assembling AChR subunits, events occur more synchronously early in assembly than later. The time when the 192 and 193 cysteines can be affinity alkylated without reduction occurs after the cysteines become accessible to the ER lumen and before the disulfide bond forms. Therefore, the ability to observe a significant amount of alkylation requires that the change in cysteine accessibility occur on a significant number of AChR complexes at approximately the same time. Because the formation of the second Bgt and ACh binding sites occurs 24–48 hr after the temperature shift, these events should be less synchronous and fewer cysteines would be expected to be available for alkylation at the same time.
Addition of an N-linked glycan to α subunit residue 187 increases assembly and binding site formation
To alter the initial conformation of the α subunit region 187–199, an N-linked glycosylation consensus site was added so that residue 187 was glycosylated. N-Linked glycosylation of this residue is of interest because it, in part, causes the resistance to Bgt binding that is observed in certain snakes and mongoose (Neumann et al., 1989;Barchan et al., 1992). We wanted to test whether the oligosaccharide would prevent the amino acids 187–199 from being buried within the α subunit and thereby expedite formation of the Bgt site. For theTorpedo α subunit N-linked glycosylated at residue 187 and coexpressed with the other subunits, the dissociation rate for Bgt from the Torpedo mutant was too rapid to quantitatively characterize the Torpedo mutant using our standard Bgt binding techniques (data not shown). As an alternative, a mutant mouse muscle α subunit with the glycosylation site at residue 187 was obtained (α187–189; a kind gift from Dr. P. Taylor). As with Torpedo receptors, Bgt dissociates more rapidly from receptors containing the mouse α187–189 mutant compared with wild-type receptors (Kreienkamp et al., 1994) but with a dissociation rate slower than for the Torpedo receptor. The slower dissociation of Bgt from the mouse α187–189 subunit makes it feasible to determine whether the rate at which Bgt binding sites form is affected by glycosylation of residue 187.
α187–189 mutant or wild-type mouse α subunits along with mouse β, ε, and δ subunits were transiently expressed in the human embryonic kidney cell line tsA201 to assay125I-Bgt binding. In parallel,125I-Bgt binding was performed on cell lysates to measure intracellular sites (Fig.3A) and on intact cells to measure surface sites (Fig. 3B). Because Bgt dissociates more rapidly from the α187–189 mutant subunit, cold Bgt could not be used to quantitatively block125I-Bgt binding to surface receptors as in Figure 2. To separate intracellular Bgt binding sites from the surface sites, saturating amounts of mAb 35 were bound to intact cells and the mAb 35-bound receptors were precipitated after solubilization. mAb 35, which is a conformation-specific mAb (Merlie and Lindstrom, 1983), was used because it appears to recognize and precipitate AChRs containing α187–189 subunits just as well as wild-type subunits. When metabolically labeled, α187–189 subunits were precipitated by mAb 35 just as well as wild-type subunits, and the same percentage of125I-Bgt-bound surface AChRs was precipitated by mAb 35 whether they contained α187–189 subunits or wild-type subunits (data not shown). Intracellular 125I-Bgt binding sites, precipitated with mAb 35 appeared 2 hr after the transfection, and their formation saturated after 24 hr. Over this time period, N-linked glycosylation of the mouse α187–189subunit resulted in a threefold increase in the formation of intracellular Bgt binding sites compared with wild-type α subunits. As observed previously (Kreienkamp et al., 1994), a 2.5-fold increase in 125I-Bgt binding to the cell surface was observed for AChRs containing α187–189subunits compared with AChRs containing wild-type α subunits. The appearance of 125I-Bgt binding sites on the surface lagged behind the appearance of intracellular sites by ∼2 hr. Because only fully assembled AChRs are released from the ER and transported to the surface, the lag in surface expression is consistent with the time for assembly and transport to the surface (Merlie and Lindstrom, 1983).
To further characterize the effects of the α187–189 mutation, subunits were metabolically labeled and precipitated with either α subunit-specific Abs (Fig.3C) or Bgt-Sepharose (Fig. 3D), which precipitates Bgt-binding α subunits. α187–189 or wild-type mouse α subunits were expressed along with mouse β, ε, and δ subunits and metabolically labeled for 5 min, after which maturation of the labeled subunits was followed in culture for the indicated times (Fig.3C,D). The α187–189subunits had an apparent molecular weight larger than wild-type subunits because of the addition of the oligosaccharide at residue 187. The addition of the glycan occurred as soon as the subunits were labeled, consistent with the cotranslational addition of the oligosaccharide. α subunit-specific Abs precipitated comparable amounts of labeled wild-type and mutated α subunits at all of the time points as shown from the quantitation of wild-type and mutated α subunit bands from five experiments (Fig. 3E). The rate of synthesis and the stability of the α187–189subunits are thus indistinguishable from that of the wild-type subunits. Furthermore, there were no differences in the levels of labeled β, ε, and δ subunits when coexpressed with the α187–189 subunits compared with their expression with the wild-type subunits (data not shown).
Significant differences were observed when α187–189 mutant and wild-type α subunits were precipitated with Bgt-Sepharose (Fig. 3D). Under identical conditions, Bgt binding sites formed more readily on the α187–189 subunits compared with wild-type subunits. As shown in Figure 3D, wild-type Bgt binding site formation reached saturating levels by 1 hr of chase, at which point the α187–189 still had threefold greater Bgt sites. Because the α187–189 subunit affinity for Bgt is less than that of wild-type, this estimate of the increase in Bgt binding site formation is perhaps an underestimate. However, the increase observed with the α187–189 subunit for Bgt-Sepharose precipitation and125I-Bgt binding to the total cell lysate was similar. The intensities of the α subunit gel bands in Figure3D plus four other separate experiments were estimated. These values were used to determine the ratio of the α187–189 subunit to wild-type averaged over all the time points. This ratio was 3.1 ± 0.3-fold (mean ± SEM) for Bgt-Sepharose precipitation of the α187–189 to wild-type α subunits. The ratio of α187–189 to wild-type α subunits for125I-Bgt binding to AChRs in the total cell lysate was also determined. The ratios were measured over four time points between 0 and 24 hr after transfection (i.e., until saturation of binding) in three separate experiments, and a ratio of 3.1 ± 0.8 (mean ± SEM), which is approximately the same as for Bgt-Sepharose binding, was observed. Because there was no apparent difference in α subunit protein levels between α187–189 and wild type (Fig. 3E), the difference in Bgt binding site formation must have resulted from an increased number of mutated α subunits with Bgt binding sites. Because the half-life for turnover of the surface receptors is 21 hr (Gu et al., 1990), the kinetics of surface expression are too fast (Fig. 3B) for a decrease in the mutated AChR turnover rate to contribute to the difference in surface expression, and we conclude that the difference in surface expression is caused by an increase in the efficiency of subunit assembly.
DISCUSSION
Little is known about how AChR α subunits are folded and processed as ligand binding sites form. Here, we have examined how one of the major determinants of ligand binding, α subunit amino acids 187–199, changes conformation as Bgt and ACh binding sites emerge during AChR assembly and how this change affects the disulfide bonding of cysteines 192 and 193. We presented evidence that this region changes conformation by showing that the region initially could not be bound by mAb 383c and became accessible only as the first Bgt binding site formed. The first Bgt binding site forms just before the addition of δ subunits to αβγ trimers to make αβγδ tetramers (Fig. 4) (Green and Wanamaker, 1998). The timing of the 187–199 conformational change, thus, indicates that it occurs while the α subunit is part of a αβγ trimer and precedes association of δ subunits to αβγ trimers. Before the formation of the Bgt binding site, a conformational change occurs on αβγ trimers in which the α subunit amino acids 128 to 142, initially accessible to a mAb specific for this region, becomes inaccessible (Green and Wanamaker, 1997). The conformational change of amino acids 128–142 occurs with a time course similar to the appearance of the first Bgt binding site but precedes it because the mAb does not recognize Bgt binding α subunits. Moreover, cysteines 128 and 142 are disulfide bonded before this conformational change can occur. As depicted in Figure 4, the conformational change of α subunit amino acids 187–199 (Fig. 4D) appears to be preceded first by the assembly of αβγ trimers and disulfide bonding of α subunit cysteines 128 and 142 (Fig. 4B) and second by the rearrangement of the 128–142 region (Fig. 4C). The series of events in Figure 4 are the simplest interpretation of our results. Other proteins, such as the ER chaperone proteins immunoglobulin heavy chain binding protein (Blount and Merlie, 1991; Paulson et al., 1991; Forsayeth et al., 1992) and calnexin (Gelman et al., 1995; Keller et al., 1996, 1998), can associate with AChR subunits. It is possible that the accessibility of the α subunit 187–199 region to mAb 383c changes because a chaperone protein that is bound to the region unbinds. This possibility would alter the model in Figure 4 in that the appearance of the mAb 383c epitope and the Bgt binding site would not result directly from a conformational change but from the unbinding of a chaperone protein.
Fig. 4.
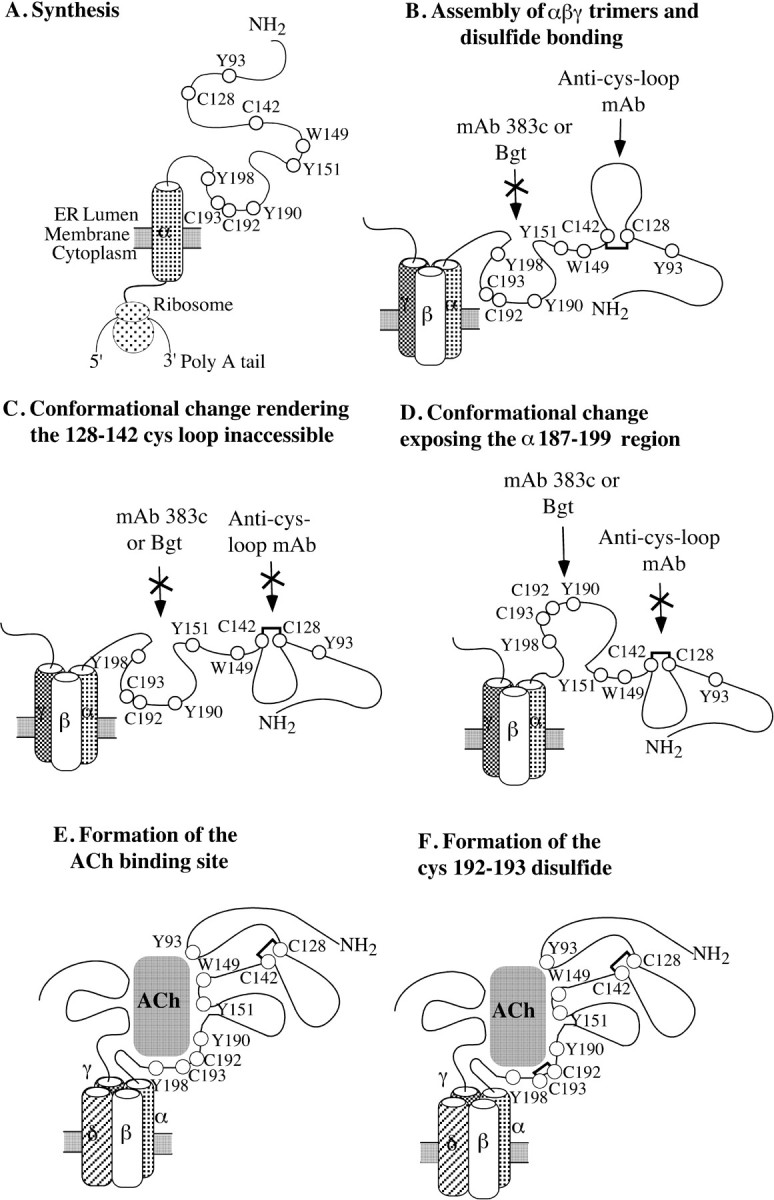
Sequence of α subunit conformational changes and processing events that occur during formation of the ligand binding sites. The model proposed in the figure is based on the interpretation of data presented in this paper together with previous data on AChR assembly and formation of the ligand binding sites. A, The α subunit rapidly folds during synthesis. B, Soon after, αβγ (or ε) trimers assemble and the 128–142 cystine loop forms. At this point, the 187–199 region is inaccessible to mAb 383c and Bgt, whereas the 128–142 region is accessible to its region-specific mAb. C, A conformational change causes the 128–142 cystine loop to become inaccessible to its region-specific mAb. D, A conformational change exposes the 187–199 region during formation of the Bgt binding site so that Bgt and mAb 383c can bind. E, The ACh binding site forms as the δ subunit assembles at the interface between the β and γ (or ε) subunits, and the α and γ (or ε) subunits fold bringing together all of the residues lining the ACh binding site. F, In the final step, cysteines 192 and 193 disulfide bond.
During AChR assembly, a significant delay occurs before δ subunits associate with αβγ trimers to assemble into αβγδ tetramers (Green and Claudio, 1993; Green and Wanamaker, 1998). The delay occurs despite an abundance of δ subunits and suggests that what is rate limiting are the folding events that precede the addition of the δ subunits. The effect of the α187–189 glycosylation provides additional evidence that a conformational change of the 187–199 region is part of a rate-limiting step at this stage of AChR assembly. We found that the α187–189 glycosylation increased Bgt binding site formation threefold and the efficiency of AChR assembly by at least twofold. Based on this and other results in this paper, it appears that the added oligosaccharide lowers the energy barrier for the 187–199 region conformational change, thereby expediting formation of the Bgt binding site and, as a consequence, the entire assembly process.
If the glycan added to residue 187 is, in fact, promoting the conformational change of the 187–199 region, we might expect Bgt binding sites to appear more rapidly on the mutated α subunits. We attempted to measure the kinetics of Bgt binding site formation during AChR assembly by briefly metabolically labeling the subunits and measuring Bgt binding site formation by subunit precipitation with Bgt-Sepharose (Fig. 3D). Based on quantitation of the bands from several experiments such as the ones shown in Figure3D, there was no significant difference in the rate in which the Bgt binding sites formed (data not shown), although Bgt binding sites did form on many more of the α187–189subunits compared with wild-type subunits. In another study, we found that transiently transfected AChR subunits assembled into receptors much less efficiently than when the subunits were stably transfected into cells (Eertmoed et al., 1998). The results of this study suggested that the low efficiency of assembly was caused by cell-to-cell variations in the amounts of each of the four subunits expressed, which resulted in much larger amounts of misfolded subunits and misassembled complexes. It is possible that differences in the kinetics of Bgt binding site formation are masked by the more rapid formation of misfolded α subunits on which Bgt binding sites can form. Consistent with this possibility is that Bgt binding sites formed more rapidly in the transiently transfected cells (Fig. 3D) than in other studies in which cells stably expressing the subunits were used (Merlie and Lindstrom, 1983; Blount and Merlie, 1988; Green and Claudio, 1993).
Subsequent to the association of δ subunits with αβγ trimers, the first ACh binding sites appear on αβγδ tetramers (Green and Wanamaker, 1998). Because other α subunit residues in addition to 187–199 lie within the ACh binding pocket, rearrangement of the α subunit to create ACh binding sites involves a large portion of the α subunit extracellular domain (Fig. 4E). The first ACh binding sites formed during assembly are located at or near the interface between α and γ subunits (Green and Wanamaker, 1998) and residues on the γ subunits also contribute to the sites (Karlin and Akabas, 1995; Tsigelny et al., 1997; Changeux and Edelstein, 1998;Chiara et al., 1999). δ subunits associate between the γ and β subunits in the αβγ trimer (Green and Wanamaker, 1998) and could alter the interface between α and γ subunits, as depicted in Figure4, D and E. The assembly of δ subunits with αβγ trimers may lower the energy barrier for and thus drive the formation of the ACh binding site. For bromoACh to bind and specifically alkylate cysteines 192 and 193, much of the first ACh binding site must be intact. Our ability to block125I-Bgt binding by bromoACh alkylation early in assembly indicates the 192–193 disulfide bond is not formed until after α and γ subunits have rearranged to bring together the residues that serve as the binding pocket for ACh, as depicted in Figure 4E. The last step in the formation of the first ACh binding sites appears to be the disulfide bonding of cysteines 192 and 193. Initially, this disulfide bonding may be prevented when the 187–199 region is inaccessible to the oxidizing potential of the ER lumen. Once this region is exposed to the ER lumen, disulfides should rapidly form. Our ability to alkylate these cysteines and block 20% of Bgt binding indicates that the cystine 192–193 does not form rapidly. What might slow the disulfide bonding of cysteines 192 and 193, and thus allow their alkylation by bromoACh, is the strain put on the peptide backbone by disulfide bonding adjacent cysteines (Kao and Karlin, 1986). Possibly, cysteines 192 and 193 are not placed into the proper positions to overcome the strain of disulfide bonding until after they are positioned in the ACh binding site, as shown in Figure 4, E and F.
Our experiments have only assayed changes involving the first ligand binding site, which forms at the α–γ (or ε) subunit interface (Green and Wanamaker, 1998). The second ligand binding site on the AChR appears to differ structurally from the first site. The second ligand binding site, which forms at the α–δ subunit interface, differs in its affinity for competitive antagonists (Neubig and Cohen, 1979; Sine and Taylor, 1981; Blount and Merlie, 1989; Pedersen and Cohen, 1990;Sine and Claudio, 1991) and is recognized by Abs different from those that recognize the first site (Mihovilovic and Richman, 1984; Dowding and Hall, 1987; Fairclough et al., 1998b). However, the same regions of the α subunit and regions on the δ subunit analogous to those on the γ (or ε) subunit appear to contribute to the second site. It is likely, therefore, that a series of events similar to those we have described for the first site occurs during formation of the second site.
The data presented in this paper suggest that formation of the 192–193 disulfide bond requires the occurrence of a conformational change so that cysteines 192 and 193 are exposed to the oxidizing ER lumen and that formation of this bond is one of the final steps in binding site formation. When cysteines 192 and 193 are mutated to serines, agonists bind with lower affinity, but the receptors are nonfunctional (Mishina et al., 1985). In other words, the ACh binding sites form in the absence of the 192–193 cystine, but ACh binding no longer activates the receptor. This finding further suggests that the ACh binding site is not functional until a series of steps is completed ending with the disulfide bonding of the 192 and 193 cysteines. A similar situation occurs during assembly of hen lysozyme (van den Berg et al., 1999). Of the four disulfide bonds present in the native state of hen lysozyme, one bond, between cysteines 76 and 94, is the last to form. After formation of the first three disulfide bonds of hen lysozyme, a native-like intermediate structure is formed without an active site. This intermediate then requires an additional structural rearrangement, which causes cysteines 76 and 94 to be repositioned and allows oxidizing agents to access Cys-76 and Cys-94. After this structural rearrangement and the consequent formation of the Cys-78–Cys-94 bond, a functional active site is formed. Thus, in hen lysozyme, as may be occurring in the ACh receptor, acquisition of functionality depends on the occurrence of a conformational change that allows formation of a disulfide bond. Formation of the disulfide bond constitutes one of the final events in the assembly process and plays the crucial role of conferring functionality to the protein.
Footnotes
This work was supported by grants from the National Institutes of Health (W.N.G) and the Brain Research Foundation (W.N.G.) and a training grant from the National Institute on Drug Abuse (M.M. and C.P.W.). We thank Dr. S. Sine for the AChR subunit cDNAs in pRBG4, Dr. P. Taylor for the α subunit mutation α187–198, Dr. R. Fairclough for mAb 383c, and Dr. V. Lennon for mAb P22. We also thank John C. Christianson, Dr. R. Fairclough, and members of the Green laboratory for discussions about this work, and Dr. D. Patneau for comments on this manuscript.
Correspondence should be addressed to William N. Green, Department of Neurobiology, Pharmacology, and Physiology, University of Chicago, 947 East 58th Street, Chicago, IL 60637. E-mail:wgreen@midway.uchicago.edu.
REFERENCES
- 1.Barchan D, Kachalsky S, Neumann D, Vogel Z, Ovadia M, Kochva E, Fuchs S. How the mongoose can fight the snake: the binding site of the mongoose acetylcholine receptor. Proc Natl Acad Sci USA. 1992;89:7717–7721. doi: 10.1073/pnas.89.16.7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blount P, Merlie JP. Native folding of an acetylcholine receptor alpha subunit expressed in the absence of other receptor subunits. J Biol Chem. 1988;263:1072–1080. [PubMed] [Google Scholar]
- 3.Blount P, Merlie JP. Molecular basis of the two nonequivalent ligand binding sites of the muscle nicotinic acetylcholine receptor. Neuron. 1989;3:349–357. doi: 10.1016/0896-6273(89)90259-6. [DOI] [PubMed] [Google Scholar]
- 4.Blount P, Merlie JP. BIP associates with newly synthesized subunits of the mouse muscle nicotinic receptor. J Cell Biol. 1991;113:1125–1132. doi: 10.1083/jcb.113.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang W, Gelman MS, Prives JM. Calnexin-dependent enhancement of nicotinic acetylcholine receptor assembly and surface expression. J Biol Chem. 1997;272:28925–28932. doi: 10.1074/jbc.272.46.28925. [DOI] [PubMed] [Google Scholar]
- 6.Changeux JP. Thudichum Medal Lecture. The acetylcholine receptor: a model for allosteric membrane proteins. Biochem Soc Trans. 1995;23:195–205. doi: 10.1042/bst0230195. [DOI] [PubMed] [Google Scholar]
- 7.Changeux JP, Edelstein SJ. Allosteric receptors after 30 years. Neuron. 1998;21:959–980. doi: 10.1016/s0896-6273(00)80616-9. [DOI] [PubMed] [Google Scholar]
- 8.Chaturvedi V, Donnelly-Roberts DL, Lentz TL. Substitution of Torpedo acetylcholine receptor alpha 1-subunit residues with snake alpha 1- and rat nerve alpha 3-subunit residues in recombinant fusion proteins: effect on alpha-bungarotoxin binding. Biochemistry. 1992;31:1370–1375. doi: 10.1021/bi00120a012. [DOI] [PubMed] [Google Scholar]
- 9.Chiara DC, Xie Y, Cohen JB. Structure of the agonist-binding sites of the Torpedo nicotinic acetylcholine receptor: affinity-labeling and mutational analyses identify gamma Tyr-111/delta Arg-113 as antagonist affinity determinants. Biochemistry. 1999;38:6689–6698. doi: 10.1021/bi9901735. [DOI] [PubMed] [Google Scholar]
- 10.Claudio T, Green WN, Hartman DS, Hayden D, Paulson HL, Sigworth FJ, Sine SM, Swedlund A. Genetic reconstitution of functional acetylcholine receptor channels in mouse fibroblasts. Science. 1987;238:1688–1694. doi: 10.1126/science.3686008. [DOI] [PubMed] [Google Scholar]
- 11.Damle VN, McLaughlin M, Karlin A. Bromoacetylcholine as an affinity label of the acetylcholine receptor from Torpedo californica. Biochem Biophys Res Commun. 1978;84:845–851. doi: 10.1016/0006-291x(78)91661-3. [DOI] [PubMed] [Google Scholar]
- 12.Dowding AJ, Hall ZW. Monoclonal antibodies specific for each of the two toxin-binding sites of Torpedo acetylcholine receptor. Biochemistry. 1987;26:6372–6381. doi: 10.1021/bi00394a010. [DOI] [PubMed] [Google Scholar]
- 13.Eertmoed AL, Vallejo YF, Green WN. Transient expression of heteromeric ion channels. Methods Enzymol. 1998;293:564–585. doi: 10.1016/s0076-6879(98)93034-8. [DOI] [PubMed] [Google Scholar]
- 14.Fairclough RH, Twaddle GM, Gudipati E, Stone RJ, Richman DP, Burkwall DA, Josephs R. Mapping the mAb 383C epitope to alpha 2(187–199) of the Torpedo acetylcholine receptor on the three-dimensional model. J Mol Biol. 1998a;282:301–315. doi: 10.1006/jmbi.1998.2000. [DOI] [PubMed] [Google Scholar]
- 15.Fairclough RH, Twaddle GM, Gudipati E, Lin MY, Richman DP. Differential surface accessibility of alpha(187–199) in the Torpedo acetylcholine receptor alpha subunits. J Mol Biol. 1998b;282:317–330. doi: 10.1006/jmbi.1998.2001. [DOI] [PubMed] [Google Scholar]
- 16.Forsayeth JR, Gu Y, Hall ZW. BiP forms stable complexes with unassembled subunits of the acetylcholine receptor in transfected COS cells and in C2 muscle cells. J Cell Biol. 1992;117:841–847. doi: 10.1083/jcb.117.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gelman MS, Prives JM. Arrest of subunit folding and assembly of nicotinic acetylcholine receptors in cultured muscle cells by dithiothreitol. J Biol Chem. 1996;271:10709–10714. doi: 10.1074/jbc.271.18.10709. [DOI] [PubMed] [Google Scholar]
- 18.Gelman MS, Chang W, Thomas DY, Bergeron JJ, Prives JM. Role of the endoplasmic reticulum chaperone calnexin in subunit folding and assembly of nicotinic acetylcholine receptors. J Biol Chem. 1995;270:15085–15092. doi: 10.1074/jbc.270.25.15085. [DOI] [PubMed] [Google Scholar]
- 19.Gomez CM, Richman DP. Anti-acetylcholine receptor antibodies directed against the alpha-bungarotoxin binding site induce a unique form of experimental myasthenia. Proc Natl Acad Sci USA. 1983;80:4089–4093. doi: 10.1073/pnas.80.13.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green WN, Claudio T. Acetylcholine receptor assembly: subunit folding and oligomerization occur sequentially. Cell. 1993;74:57–69. doi: 10.1016/0092-8674(93)90294-z. [DOI] [PubMed] [Google Scholar]
- 21.Green WN, Wanamaker CP. The role of the cystine loop in acetylcholine receptor assembly. J Biol Chem. 1997;272:20945–20953. doi: 10.1074/jbc.272.33.20945. [DOI] [PubMed] [Google Scholar]
- 22.Green WN, Wanamaker CP. Formation of the nicotinic acetylcholine receptor binding sites. J Neurosci. 1998;18:5555–5564. doi: 10.1523/JNEUROSCI.18-15-05555.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gu Y, Ralston E, Murphy-Erdosh C, Black RA, Hall ZW. Acetylcholine receptor in a C2 muscle cell variant is retained in the endoplasmic reticulum. J Cell Biol. 1989;109:729–738. doi: 10.1083/jcb.109.2.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gu Y, Franco A, Jr, Gardner PD, Lansman JB, Forsayeth JR, Hall ZW. Properties of embryonic and adult muscle acetylcholine receptors transiently expressed in COS cells. Neuron. 1990;5:147–157. doi: 10.1016/0896-6273(90)90305-y. [DOI] [PubMed] [Google Scholar]
- 25.Hawkins HC, de Nardi M, Freedman RB. Redox properties and cross-linking of the dithiol/disulphide active sites of mammalian protein disulphide-isomerase. Biochem J. 1991;275:341–348. doi: 10.1042/bj2750341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 27.Kao PN, Karlin A. Acetylcholine receptor binding site contains a disulfide cross-link between adjacent half-cystinyl residues. J Biol Chem. 1986;261:8085–8088. [PubMed] [Google Scholar]
- 28.Karlin A, Akabas MH. Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron. 1995;15:1231–1244. doi: 10.1016/0896-6273(95)90004-7. [DOI] [PubMed] [Google Scholar]
- 29.Keller SH, Lindstrom J, Taylor P. Involvement of the chaperone protein calnexin and the acetylcholine receptor beta-subunit in the assembly and cell surface expression of the receptor. J Biol Chem. 1996;271:22871–22877. doi: 10.1074/jbc.271.37.22871. [DOI] [PubMed] [Google Scholar]
- 30.Keller SH, Lindstrom J, Taylor P. Inhibition of glucose trimming with castanospermine reduces calnexin association and promotes proteasome degradation of the alpha-subunit of the nicotinic acetylcholine receptor. J Biol Chem. 1998;273:17064–17072. doi: 10.1074/jbc.273.27.17064. [DOI] [PubMed] [Google Scholar]
- 31.Kreienkamp HJ, Sine SM, Maeda RK, Taylor P. Glycosylation sites selectively interfere with alpha-toxin binding to the nicotinic acetylcholine receptor. J Biol Chem. 1994;269:8108–8114. [PubMed] [Google Scholar]
- 32.Lee BS, Gunn RB, Kopito RR. Functional differences among nonerythroid anion exchangers expressed in a transfected human cell line. J Biol Chem. 1991;266:11448–11454. [PubMed] [Google Scholar]
- 33.Margolskee RF, McHendry-Rinde B, Horn R. Panning transfected cells for electrophysiological studies. Biotechniques. 1993;15:906–911. [PubMed] [Google Scholar]
- 34.Merlie JP, Lindstrom J. Assembly in vivo of mouse muscle acetylcholine receptor: identification of an alpha subunit species that may be an assembly intermediate. Cell. 1983;34:747–757. doi: 10.1016/0092-8674(83)90531-7. [DOI] [PubMed] [Google Scholar]
- 35.Mihovilovic M, Richman DP. Modification of alpha-bungarotoxin and cholinergic ligand-binding properties of Torpedo acetylcholine receptor by a monoclonal anti-acetylcholine receptor antibody. J Biol Chem. 1984;259:15051–15059. [PubMed] [Google Scholar]
- 36.Mihovilovic M, Richman DP. Monoclonal antibodies as probes of the alpha-bungarotoxin and cholinergic binding regions of the acetylcholine receptor. J Biol Chem. 1987;262:4978–4986. [PubMed] [Google Scholar]
- 37.Mishina M, Tobimatsu T, Imoto K, Tanaka K, Fujita Y, Fukuda K, Kurasaki M, Takahashi H, Morimoto Y, Hirose T, Inayama S, Takahashi T, Kuno M, Numa S. Location of functional regions of acetylcholine receptor alpha-subunit by site-directed mutagenesis. Nature. 1985;313:364–369. doi: 10.1038/313364a0. [DOI] [PubMed] [Google Scholar]
- 38.Neubig RR, Cohen JB. Equilibrium binding of [3H]tubocurarine and [3H]acetylcholine by Torpedo postsynaptic membranes: stoichiometry and ligand interactions. Biochemistry. 1979;18:5464–5475. doi: 10.1021/bi00591a032. [DOI] [PubMed] [Google Scholar]
- 39.Neumann D, Barchan D, Horowitz M, Kochva E, Fuchs S. Snake acetylcholine receptor: cloning of the domain containing the four extracellular cysteines of the alpha subunit. Proc Natl Acad Sci USA. 1989;86:7255–7259. doi: 10.1073/pnas.86.18.7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paulson HL, Ross AF, Green WN, Claudio T. Analysis of early events in acetylcholine receptor assembly. J Cell Biol. 1991;113:1371–1384. doi: 10.1083/jcb.113.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pedersen SE, Cohen JB. d-Tubocurarine binding sites are located at alpha-gamma and alpha-delta subunit interfaces of the nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 1990;87:2785–2789. doi: 10.1073/pnas.87.7.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ross AF, Green WN, Hartman DS, Claudio T. Efficiency of acetylcholine receptor subunit assembly and its regulation by cAMP. J Cell Biol. 1991;113:623–636. doi: 10.1083/jcb.113.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silman I, Karlin A. Acetylcholine receptor: covalent attachment of depolarizing groups at the active site. Science. 1969;164:1420–1421. doi: 10.1126/science.164.3886.1420. [DOI] [PubMed] [Google Scholar]
- 44.Sine SM, Claudio T. Gamma- and delta-subunits regulate the affinity and the cooperativity of ligand binding to the acetylcholine receptor. J Biol Chem. 1991;266:19369–19377. [PubMed] [Google Scholar]
- 45.Sine SM, Taylor P. Relationship between reversible antagonist occupancy and the functional capacity of the acetylcholine receptor. J Biol Chem. 1981;256:6692–6699. [PubMed] [Google Scholar]
- 46. Smith MM, Lindstrom J, Merlie JP. Formation of the alpha-bungarotoxin binding site and assembly of the nicotinic acetylcholine receptor subunits occur in the endoplasmic reticulum. J Biol Chem 262 1987. 9428] 262:4367–4376 [Erratum (1987). [PubMed] [Google Scholar]
- 47.Tsigelny I, Sugiyama N, Sine SM, Taylor P. A model of the nicotinic receptor extracellular domain based on sequence identity and residue location. Biophys J. 1997;73:52–66. doi: 10.1016/S0006-3495(97)78047-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tzartos SJ, Remoundos MS. Fine localization of the major alpha-bungarotoxin binding site to residues alpha 189–195 of the Torpedo acetylcholine receptor. Residues 189, 190, and 195 are indispensable for binding. J Biol Chem. 1990;265:21462–21467. [PubMed] [Google Scholar]
- 49.van den Berg B, Chung EW, Robinson CV, Dobson CM. Characterisation of the dominant oxidative folding intermediate of hen lysozyme. J Mol Biol. 1999;290:781–796. doi: 10.1006/jmbi.1999.2915. [DOI] [PubMed] [Google Scholar]
- 50.Wilson PT, Lentz TL, Hawrot E. Determination of the primary amino acid sequence specifying the alpha-bungarotoxin binding site on the alpha subunit of the acetylcholine receptor from Torpedo californica. Proc Natl Acad Sci USA. 1985;82:8790–8794. doi: 10.1073/pnas.82.24.8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson PT, Hawrot E, Lentz TL. Distribution of alpha-bungarotoxin binding sites over residues 173–204 of the alpha subunit of the acetylcholine receptor. Mol Pharmacol. 1988;34:643–650. [PubMed] [Google Scholar]