

Abstract
Kainate receptor activation depresses synaptic release of neurotransmitter at a number of synapses in the CNS. The mechanism underlying this depression is controversial, and both ionotropic and metabotropic mechanisms have been suggested. We report here that the AMPA/kainate receptor agonists domoate (DA) and kainate (KA) cause a presynaptic depression of glutamatergic transmission at CA3→CA1 synapses in the hippocampus, which is not blocked by the AMPA receptor antagonist GYKI 53655 but is blocked by the AMPA/KA receptor antagonist CNQX. Neither a blockade of interneuronal discharge nor antagonists of several neuromodulators affect the depression, suggesting that it is not the result of indirect excitation and subsequent release of a neuromodulator. Presynaptic depolarization, achieved via increasing extracellular K+, caused a depression of the presynaptic fiber volley and an increase in the frequency of miniature EPSCs. Neither effect was observed with DA, suggesting that DA does not depress transmission via a presynaptic depolarization. However, the effects of DA were abolished by the G-protein inhibitors N-ethylmaleimide and pertussis toxin. These results suggest that KA receptor activation depresses synaptic transmission at this synapse via a direct, presynaptic, metabotropic action.
Keywords: domoate, kainate, metabotropic, presynaptic, hippocampus, CA1
Neurotransmitter receptors in the CNS can be separated into two broad classes: ionotropic receptors, which are coupled to ligand-gated conductances, and metabotropic receptors, which couple to GTP-binding proteins. Kainate receptors (KARs) are ionotropic glutamate receptors that, in addition to having conventional postsynaptic actions, are thought to act as presynaptic inhibitors of transmitter release at a number of synapses in the CNS (for review, see Frerking and Nicoll, 2000). The mechanisms underlying this depression are controversial. Both direct actions of presynaptic KARs (Chittajallu et al., 1996;Rodríguez-Moreno et al., 1997; Schmitz et al., 2000) and indirect actions of somatodendritic KARs (Frerking et al., 1999;Chergui et al., 2000; Schmitz et al., 2000) have been proposed to account for this presynaptic effect. The depression has also been suggested to be the downstream result of presynaptic depolarization via the conventional ionotropic actions of KARs (Chittajallu et al., 1996;Kamiya and Ozawa, 1998; Schmitz et al., 2000) or alternatively via a novel KAR-coupled metabotropic cascade (Rodríguez-Moreno and Lerma, 1998; Rodríguez-Moreno et al., 2000).
One difficulty in interpreting the effects of KAR activation is the frequent presence of somatodendritic KARs on the presynaptic neurons, which can lead to a number of different types of use-dependent depression. Glutamatergic CA3→CA1 synapses in the hippocampus provide a good system in which to study the KAR-induced depression, because the somatodendritic region of the presynaptic CA3 cells can be removed by microdissection. At these synapses, the KAR agonists kainate (KA) and domoate (DA) both induce a depression of glutamate release (Chittajallu et al., 1996; Kamiya and Ozawa, 1998; Vignes et al., 1998) that is not accompanied by changes in presynaptic excitability, as assessed by changes in the extracellular fiber volley (Kamiya and Ozawa, 1998; Vignes et al., 1998), but is accompanied by a decrease in presynaptic Ca2+ influx (Kamiya and Ozawa, 1998). However, these results do not exclude the possibility that the KAR-induced depression is the indirect result of neuromodulators released from CA1 interneurons, which fire intensely in response to KAR activation (Cossart et al., 1998; Frerking et al., 1998), nor do they address the relative contributions of ionotropic and metabotropic mechanisms to the depression.
In this study, we examine the mechanism underlying the KAR-induced presynaptic inhibition at glutamatergic CA3→CA1 synapses in the hippocampus. We confirm the previously described presynaptic actions of DA. We find that the depressant action of DA cannot be explained by the indirect release of a number of different neuromodulators and also cannot be explained by an increase in interneuronal discharge, suggesting that this presynaptic action is the direct result of KARs at or near the presynaptic terminal. We find that the selective action of DA on the field EPSP (fEPSP) is not mimicked by presynaptic depolarization with extracellular K+, which depresses the fEPSP and the fiber volley in parallel. However, the effects of DA are abolished by the G-protein inhibitorsN-ethylmaleimide (NEM) and pertussis toxin. These results suggest that KARs depress release at this synapse via a direct, metabotropic action.
MATERIALS AND METHODS
Slice preparation and recording techniques.Hippocampal slices 300–400 μm thick were prepared from 2- to 12-week-old Sprague Dawley rats as described (Frerking et al., 1998). After >1 hr of recovery time, a cut was made between area CA1 and area CA2/CA3 to remove somatic effects of KA on CA3 pyramidal cells, and slices were transferred to a submersion-type recording chamber perfused at room temperature with a solution consisting of (in mm): 119 NaCl, 26 NaHCO3, 10 glucose, 2.5 KCl, 4 CaCl2, 4 MgSO4, and 1.0 NaH2PO4, bubbled with 95% O2–5% CO2. For whole-cell recording, patch electrodes (2–5 MΩ) were filled with a solution adjusted to pH 7.2, 270–290 mOsm, containing (in mm): 117.5 Cs gluconate, 10 tetraethylammonium chloride, 10 HEPES, 8 NaCl, 5 QX-314Cl, 4 MgATP, 2.5 CsCl, 0.3 Na3GTP, and 0.2 EGTA. Patch electrodes were filled with the external solution to monitor extracellular field potentials. Field experiments were done both in the presence and absence of 100 μm picrotoxin, which had no effect on the depressant action of domoate (data not shown). All whole-cell experiments on glutamatergic transmission were done in the presence of 100 μm picrotoxin.
Stimulation and whole-cell recording techniques in slices were similar to those described in Frerking et al. (1998). EPSCs were evoked every 15–20 sec and were filtered at 2 kHz and digitized at 5 kHz. Data were analyzed on-line using Igor Pro (WaveMetrics, Inc., Lake Oswego, OR) and off-line using Igor Pro and Sigma Plot (Jandel Scientific, San Rafael, CA). K+-sensitive microelectrodes were manufactured and tested as described previously (Lux and Neher, 1973; Heinemann et al., 1977).
Data analysis. All data are presented as the mean ± SEM. Significance was assessed at p < 0.05, using the Student's t test and Pearson's product moment correlation. EPSC amplitudes were calculated by subtracting a baseline period preceding stimulation from a region of 2 msec during the peak of the EPSC. 1/CV2, where CV is the coefficient of variation, was calculated with noise subtraction. Paired-pulse facilitation was measured as the ratio of amplitudes of a second EPSC over the first, minus one, converted to a percentage. Miniature EPSCs were detected using the Mini Analysis Program (Synaptosoft, Inc., Leonia, NJ) and a detection threshold of 5–10 pA, depending on the noise level. Field EPSP (fEPSP) slope measurements were made using a time window of the initial 1–2 msec of the fEPSP, set at the start of the experiment; fiber volley amplitude measurements were made using the peak of the fiber volley. Experiments using pharmacological manipulations that did not affect the DA-induced depression were pooled together with control experiments and treated as a single population where appropriate. The depolarization caused by increasing extracellular K+ was estimated using the Goldman–Hodgkin–Katz voltage equation, assuming that the relative resting permeability of sodium in the bouton is ∼1/20th that of potassium and concentrations of K+ and Na+ in the bouton are 120 and 10 mm, respectively.
Pertussis toxin injections. Pertussis toxin was injected into the hippocampus of rats 2–3 d before slice preparation, as described (Pitler and Alger, 1994). Briefly, rats were anesthetized with sodium pentobarbital (50 mg/kg) and placed in a stereotaxic apparatus. A small hole was drilled through the skull over the dorsal right hippocampus, and three injections (1 μl each) of pertussis toxin (1 μg/ml) were made at the coordinates relative to bregma listed in Pitler and Alger (1994). The injections were made directly into the hippocampus, rather than into the ventricle, because this has been reported to lead to more effective penetration of pertussis toxin into the tissue without adverse side effects (Pitler and Alger, 1994). The rats were then allowed to recover, and slices from the injected hippocampus were made and used 2–3 d later. Only slices with track marks from the injection needle were used, to ensure that the pertussis toxin had been injected in the hippocampus near the site of recording.
RESULTS
Previous studies have reported that the AMPA/KA receptor agonist DA, when applied at a low dose of 200 nm, specifically activates kainate receptors (Bureau et al., 1999). We therefore examined this dose of DA on the AMPA receptor (AMPAR)-mediated EPSC recorded in CA1 pyramidal cells (Fig.1A; n = 6) and the AMPAR-mediated fEPSP recorded extracellularly (see Fig.2A; n = 8). The results from these two different measurements were indistinguishable (p > 0.2) and when pooled together gave an average depression of ∼40% (Fig. 1C; 39 ± 5%; n = 14) for AMPAR-mediated transmission, similar to that described by Kamiya and Ozawa (1998). To confirm that this low dose of DA was not causing the depression via activation of AMPARs, we examined the effects of DA on NMDA receptor (NMDAR)-mediated EPSCs in the presence of the AMPAR-selective antagonist GYKI 53655, at a concentration (10 μm) that blocks AMPAR function by > 90%, as assessed by blockade of the AMPAR-mediated fEPSP (n = 4; data not shown), but is widely agreed to have minimal effects on KARs (Paternain et al., 1995; Wilding and Huettner, 1995). DA under these conditions caused a depression of the NMDAR-mediated EPSC (Fig. 1B–D; n = 4) that was indistinguishable from the effects on AMPAR-mediated transmission in the absence of GYKI 53655 (p > 0.5), indicating that AMPARs are not involved in the depression induced by DA. However, in the presence of the AMPA/KA receptor antagonist CNQX (100 μm; n = 4), the effects of DA on the NMDAR-mediated EPSC were abolished (Fig. 1C;p < 0.02). The actions of DA were also not mediated via activation of other glutamate receptors, because neither the NMDAR antagonist APV (100 μm; n = 5) nor a combination of the metabotropic glutamate receptor antagonists methyl-4-carboxyphenylglycine (MCPG) and cyclopropyl-4-phosphonophenylglycine (CPPG) (1.3 and 0.2 mm, respectively; n = 4) had any effect on the depression (Fig. 1C).
Fig. 1.

KAR activation depresses glutamatergic transmission at CA3→CA1 synapses. A, Averaged, stimulus-evoked AMPAR-mediated EPSCs are depressed by 200 nm DA. B, Averaged, stimulus-evoked NMDAR-mediated EPSCs are depressed by 200 nm DA in the presence of 10 μm GYKI 53655 (GYKI). C, The depressant action of DA is not blocked by GYKI 53655, APV, or a combination of MCPG and CPPG but is blocked by CNQX. Except for the data in GYKI 53655, which examined NMDAR-mediated EPSCs, the results include data on AMPAR-mediated EPSCs and fEPSPs that were pooled together.D, The NMDAR-mediated EPSC amplitude (filled circles) is compared with the input resistance (open circles) as a function of time in the presence of 10 μm GYKI 53655. Domoate application is shown by the horizontal line. amp., Amplitude.
Fig. 2.

KAR activation reduces the probability of release.A, Top, DA has a larger effect on the first of two fEPSPs. Bottom traces show the paired-pulse responses after scaling to the first fEPSP. B, A summary of 23 experiments examining AMPAR-mediated transmission, pooled from both field and whole-cell recordings, shows the change in paired-pulse facilitation as a function of time. C, DA causes a decrease in 1/CV2 of the EPSC that is correlated with the decrease in mean EPSC amplitude. Individual experiments are shown. D, DA has no effect on mEPSC amplitude.
In previous studies, KAR activation has been reported to result in a substantial decrease in the input resistance of CA1 pyramidal cells (Frerking et al., 1999). However, the majority of this effect was removed by the GABAA receptor antagonist picrotoxin, which was present in all whole-cell recordings in the present experiments. To examine more closely a possible role of changes in the input resistance in the currently examined KAR-induced depression, we compared the effects of DA on the input resistance and the NMDAR-mediated EPSC recorded in the presence of GYKI 53655 (Fig.1D). Under these experimental conditions, the holding current changed by only 27 ± 16 pA, and there was no significant reduction in the input resistance (Fig. 1D,open circles) despite a robust decrease in the EPSC amplitude (Fig. 1D, filled circles). It therefore seems unlikely that the KAR-induced depression of synaptic transmission is mediated to any appreciable degree by changes in passive membrane properties. DA also did not cause any change in the frequency of spontaneous EPSCs (data not shown).
The KAR-induced depression has been reported to be presynaptic, primarily because of associated changes in short-term plasticity (Kamiya and Ozawa, 1998; Vignes et al., 1998). We confirmed that DA increases paired-pulse facilitation (Fig. 2A,B;p < 0.01; n = 23), consistent with a presynaptic locus of action. We also found that DA caused a substantial reduction in the quantal content contributing to the AMPAR-mediated EPSC, as assessed by changes in the 1/CV2ratio of the EPSC (Fig. 2C; p < 0.01;n = 6). To assess for possible postsynaptic effects, we examined the effects of DA on the amplitude of miniature EPSCs (mEPSCs); there was no change in the amplitude of mEPSCs after DA application (Fig. 2D; n = 5; see below). These results, collectively with the results in Figure 1showing that the NMDAR and AMPAR components of the EPSC are depressed to a similar extent and also showing that KAR activation does not cause a significant change in the input resistance under these conditions, provide strong evidence that the depressant effect of DA at this synapse is mainly, if not entirely, mediated via a presynaptic mechanism.
The results described thus far assess KAR activation via the use of the agonist DA and address the potential activation of AMPARs via the use of 10 μm GYKI 53655. To extend further the pharmacological correlation of the observed depression with the known properties of KARs, we examined the effects of 10 μm KA on NMDAR-mediated EPSCs. Because of the lower specificity of KA for KARs over AMPARs, we used a higher concentration of GYKI 53655 (100 μm) to remove completely the possibility of AMPAR activation. Under these conditions, we found that KA caused a substantial depression of the EPSC, similar in magnitude and time course to that induced by DA (Fig.3A,B, filled circles; n = 10). This depression, as expected, was blocked by CNQX (Fig. 3B, open circles;n = 4). Also, consistent with results using DA, KA caused a decrease in 1/CV2 (Fig.3C; n = 10). The depression of the NMDAR-mediated EPSC was also associated with changes in short-term plasticity (data not shown). The similarity of the actions of KA and DA, the sensitivity to CNQX, and the resistance to GYKI 53655 all provide strong support for the idea that the agonist-induced depression is mediated by KARs.
Fig. 3.
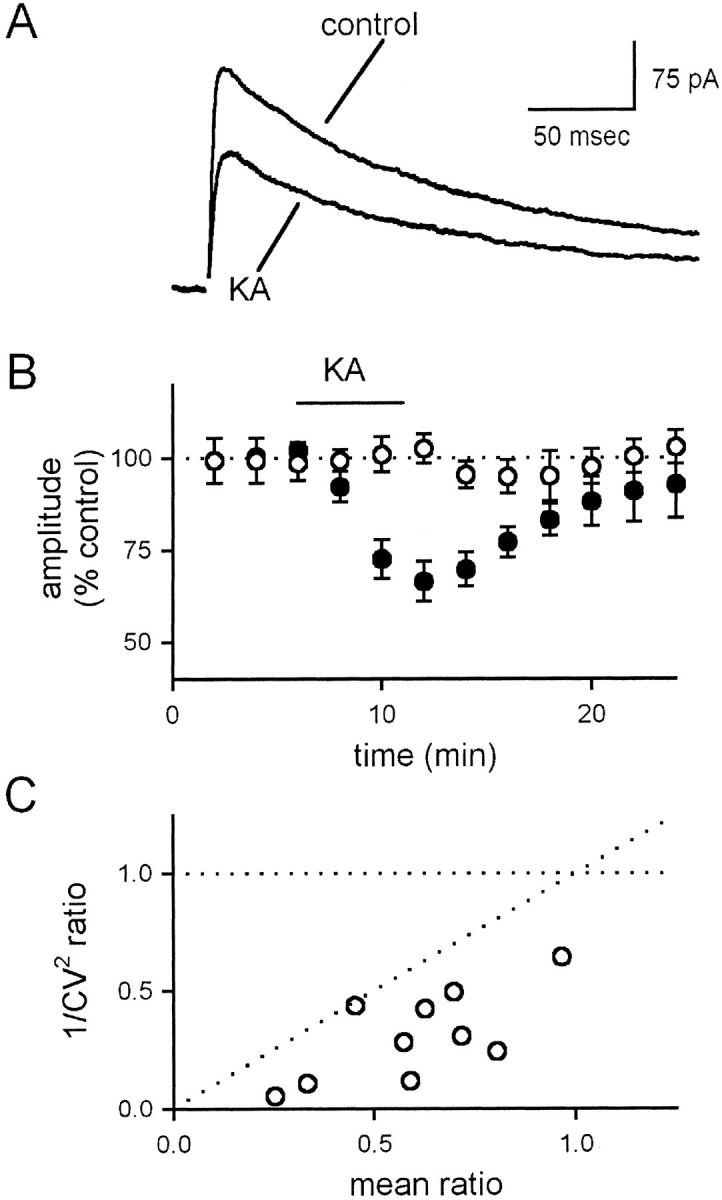
Kainate depresses the NMDA receptor-mediated EPSC.A, Averaged, stimulus-evoked NMDA receptor-mediated EPSCs are depressed by 10 μm KA in the presence of 100 μm GYKI 53655. B, The KA-induced depression is shown as a function of time under control conditions (filled circles) and in the presence of 100 μm CNQX (open circles). C, KA causes a decrease in 1/CV2 of the EPSC that is correlated with the decrease in mean EPSC amplitude. Individual experiments are shown.
Although the experiments performed so far indicate that the depression ultimately is caused by presynaptic expression mechanisms, it is not clear from these data whether or not the effects of KAR activation are caused by the direct activation of presynaptic KARs on the terminal or the indirect activation of somatodendritic KARs on some other cell population in the slice preparation that then releases a neuromodulator that acts heterosynaptically on the terminal. We therefore examined the sensitivity of the DA-induced depression to antagonists for a number of different neuromodulators known to affect synaptic transmission in area CA1. We found that neither a combination of 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) and SCH 50911 (to block adenosine 1A and GABAB receptors, respectively; n = 5) nor a combination of atropine, 1-(2-methoxyphenyl)-4-[4-(2-phthalimido)butyl]piperazine (NAN-190), sulpiride, and N-nitro-arginine [to block muscarinic acetylcholine receptors, 5-HT1A receptors, dopamine D2 receptors, and nitric oxide (NO) synthase, respectively; n = 5] had any effect on the depression of the fEPSP (Fig.4A).
Fig. 4.

KAR activation does not depress transmission via indirect interneuronal activation. A, A summary of experiments shows that the normal effect of DA (mean ± SEM shown as solid horizontal line ± dotted horizontal lines) on fEPSPs is unaffected by both a cocktail of antagonists for GABAB receptors (SCH 50911, 20 μm) and adenosine receptors (DPCPX, 10 μm) and a cocktail of antagonists for muscarinic receptors (atropine, 10 μm), NO synthase [N-nitro-l-arginine (l-NNA), 100 μm], dopamine receptors (sulpiride, 100 nm), and serotonin receptors (NAN-190, 50 μm). B, Spontaneous IPSCs are recorded at 0 mV. After addition of 200 nm DA, a large increase in sIPSC frequency is observed. In a solution containing high divalent concentrations (8 mm Ca and 17 mm Mg), the DA-induced increase in sIPSC frequency is absent in a different cell. C, The depressant action of DA on fEPSPs is unaffected by the solution with high divalents. D, The normal effect of DA on fEPSPs (filled circles) is unaffected by the solution with high divalents (open circles).
Although the absence of any effect of these antagonists increases confidence that the effect of DA on the fEPSP is direct, it remains possible that DA activates a population of neurons to release some neuromodulator that has not been blocked by these cocktails. Hippocampal interneurons in particular are known to release a wide variety of neuromodulatory substances upon activation and are also known to be activated by somatodendritic KARs. This activation has been reported to affect indirectly both inhibitory (Frerking et al., 1999) and excitatory (Schmitz et al., 2000) transmission at other synapses in the hippocampus. We therefore performed experiments to address more generally the possibility that the effects of DA are indirectly mediated by interneuronal activation. We found that, as expected if KARs cause interneuronal discharge, DA dramatically increased the spontaneous IPSC (sIPSC) frequency onto CA1 pyramidal cells (Fig.4B; 1066 ± 228% of control values;n = 4). It has been shown previously (Frerking et al., 1998) that KAR activation of interneuronal discharge can be prevented by a high divalent composition (8 mmCa2+ and 17 mmMg2+) of the extracellular solution without altering the release probability, presumably by increasing the charge screening. We confirmed that a solution containing 8 mm Ca2+ and 17 mm Mg2+ prevented the DA-induced increase in interneuronal discharge, as assessed by the increase in sIPSC frequency (Fig. 4B; 93 ± 13% of control values; n = 4). However, the high-divalent solution had no effect on the DA-induced depression of the fEPSP (Fig.4C,D; n = 7), demonstrating that this effect is not downstream of interneuronal discharge. We therefore conclude that the depression is not mediated indirectly via the release of any of the widely considered neuromodulators in the hippocampus and is independent of interneuronal activity, suggesting a direct action of DA on the presynaptic terminal.
Because KARs are ionotropic receptors and the effects of DA appear to be direct, we wondered whether or not the DA-induced depression could be explained by a presynaptic depolarization. One consequence of presynaptic depolarization that has been widely suggested is that a sufficiently large depolarization would inactivate sodium channels and diminish spike amplitude; however, KAR activation has been reported to depress transmission without affecting the presynaptic fiber volley, which is an extracellular measurement of the compound action potential (Kamiya and Ozawa, 1998; Vignes et al., 1998). One difficulty in interpreting these results, however, is that under normal conditions the fiber volley is much smaller than the fEPSP, making a comparison of these parameters difficult in the same experiment. We therefore examined the effects on the fiber volley and the fEPSP simultaneously in the continued presence of 1–2 μm GYKI 53655, to reduce the size of the fEPSP relative to that of the fiber volley. Under these conditions, DA caused a depression of the fEPSP, whereas the fiber volley was unaffected (Fig.5A1,B,D;n = 6). To test the relative sensitivities of the fiber volley and the fEPSP, we then added tetrodotoxin to the solution, which will depress Na channels in presynaptic axons and therefore also transmitter release; we found that TTX caused a parallel reduction of the fiber volley and the fEPSP (Fig.5A2,B,D; n = 6), confirming the sensitivity of the fiber volley under these conditions. We then performed a similar comparison of the fiber volley and fEPSP during stepwise increases in the extracellular K+ concentration, to depolarize the presynaptic terminals. We found that, in contrast to DA, extracellular K+ did not depress the fEPSP selectively; concentrations of extracellular K+ that depressed the fEPSP also depressed the fiber volley (Fig.5C,D; n = 6). The observation that extracellular K+ does not mimic DA suggests that the DA-induced depression is not mediated by a presynaptic depolarization.
Fig. 5.

KAR activation depresses synaptic transmission without affecting fiber excitability. A, The fEPSP was examined in the presence of submaximal concentrations of GYKI 53655 to enhance resolution of the fiber volley.A1, However, DA depressed the fEPSP without affecting the fiber volley.A2, In contrast, TTX depressed both the fEPSP and the fiber volley. B, A summary of the experiments in A is shown. The fEPSP (open circles) was depressed by both DA and TTX, but the fiber volley (filled circles) was affected only by TTX. Note that the fiber volley and fEPSP are decreased by TTX in parallel, suggesting that the depression of the fEPSP by TTX is the result of decreased fiber excitability rather than synaptic inhibition. fEPSP measurements shown after the break are renormalized to 100% at thearrow to correct for a slight (∼10%) overshoot in recovery after DA application (data not shown). C, A summary of experiments with elevated extracellular K+ is shown. The fEPSP (open circles) and fiber volley (filled circles) were similarly depressed by slowly increasing the extracellular KCl concentration in steps from 2.5 mm (control) to 5.5, 6.5, 7.5, 8.5, and 9.5 mm. The potentiation of the fEPSP after washout was seen consistently but not studied further. D, A summary of all experiments comparing the fEPSP and fiber volley is shown. Black circles are experiments with DA,white circles are experiments with TTX, and gray circles are experiments with KCl. The dotted lines represent selective effects on the synapse (horizontal line) or on the fiber (diagonal line). fv, Fiber volley.
To perform a more general test for a DA-induced presynaptic depolarization, we examined the effects of DA on mEPSC frequency. It is well established that presynaptic depolarization increases mEPSC frequency. The relationship between presynaptic depolarization and spontaneous release is steep starting at approximately −50 mV (Frerking et al., 1997); however, under normal conditions the presynaptic voltage is well below this threshold, and thus small depolarizations will have little effect on mEPSC frequency. We therefore examined mEPSCs in the presence of 15 mmextracellular K+ to bring the presynaptic voltage closer to this sensitive range of voltages. Under these conditions, DA did not have any resolvable effect on mEPSC frequency (Fig. 6A,C;n = 5). To ensure that this assay is in fact sensitive to small presynaptic depolarizations, an additional 5 mm K+ was applied, which would be expected to cause a presynaptic depolarization of ∼5 mV (see Materials and Methods). Unlike DA, this modest increase in K+ caused a rapid and easily resolvable increase in the frequency of mEPSCs (Fig. 6B,C;n = 4). We conclude that DA under these conditions does not resolvably depolarize the presynaptic terminal.
Fig. 6.
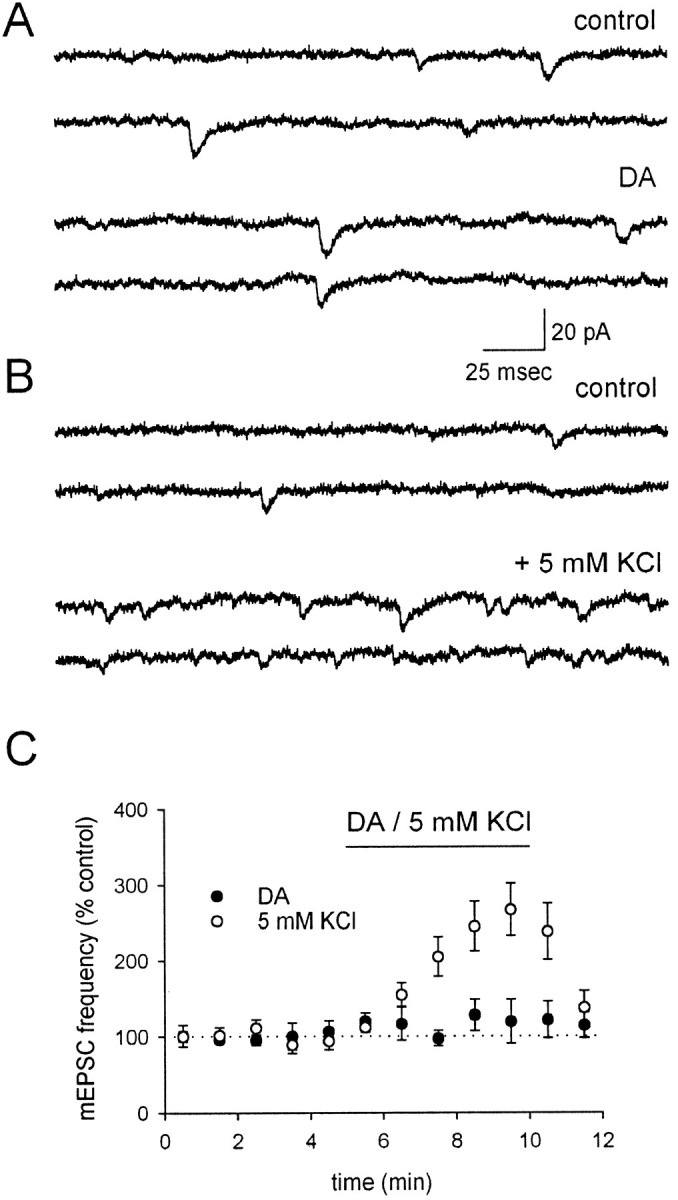
KAR activation by 200 nm DA does not resolvably depolarize the presynaptic terminal. A, mEPSCs are shown in control conditions, in which the concentration of K+ has been increased to 15 mm to bring the presynaptic membrane potential into a range in which small depolarizations will cause an increase in Ca-dependent mEPSCs (see text). DA has no effect on the mEPSC frequency. B, Addition of 5 mm KCl, corresponding to an ∼5 mV depolarization, causes a pronounced increase in the frequency of mEPSCs in a different cell. C, A summary of experiments comparing the effects of DA (filled circles) and 5 mm KCl (open circles) on mEPSC frequency is shown.
If KARs are in fact present on the terminal, they should affect the fiber volley with sufficient activation. We therefore examined the fiber volley during application of 20 μm DA, in the presence of 100 μm GYKI 53655 to remove completely the possibility of AMPA receptor activation at this high dose and also in the presence of 100 μm cadmium to block calcium influx and thereby prevent all depolarization-evoked transmitter or modulator release. Under these conditions, DA causes a strong reduction of the fiber volley (Fig. 7A,B;n = 5). Although this effect could be attributable to a direct depolarizing action of KARs on the presynaptic terminal, an alternative is that somatodendritic KAR activation causes an increase in K+ efflux that then causes the depolarization. We therefore measured the extracellular K+ levels in response to DA application using K+-sensitive microelectrodes. The K+ levels rose by ∼4 mm in response to the DA application (Fig.7C1; n = 4); however, bath application of 4 mm KCl did not mimic the effects of DA on the fiber volley (Fig. 7C2;n = 5), indicating that the DA-induced increase in extracellular K+ cannot explain the DA-induced reduction of the fiber volley. These results indicate that the presynaptic terminals do have kainate receptors, which with sufficient activation cause substantial depolarization of the terminal; however, lower doses of DA do not cause a resolvable depolarization but nevertheless depress transmitter release.
Fig. 7.

Strong KAR activation depresses the fiber volley.A, a, The fiber volley was recorded in the presence of 100 μm GYKI 53655 and 100 μmCdCl2. b, After application of 20 μm DA, the fiber volley was substantially depressed.c, The fiber volley recovered fully after washout.B, A summary of the experiments in A is shown. C1, The depression of the fiber volley cannot be explained by K+ efflux. DA (20 μm) caused an increase in K+levels of ∼4 mm, as measured by K+-sensitive microelectrodes.C2, Application of 4 mm KCl under these conditions has no effect on the fEPSP.
If DA depresses transmitter release without causing a detectable depolarization, what is the mechanism mediating this inhibitory action?Rodríguez-Moreno and Lerma (1998) have suggested that presynaptic kainate receptors can depress transmitter release from interneurons in the hippocampus via the depolarization-independent activation of a G-protein-coupled cascade. We therefore tested the effects of the G-protein inhibitor NEM (200 μm) on the DA-induced depression of the fEPSP. NEM has complex effects on baseline synaptic transmission (Fig.8A; n = 6), causing first an increase, probably because of inhibition of the tonic depressant action of adenosine, and then a decrease, probably because of a lower-affinity inhibition of the exocytotic proteinN-ethylmaleimide-sensitive factor, which is required for transmitter release. Because of these complex effects, the effect of NEM on the DA-induced depression of the fEPSP was assessed by comparing the fEPSP as a function of time during DA application with the fEPSP as a function of time during application of vehicle at the same time window, after 15 min of NEM incubation (Fig.8A, shaded box). During this time period, the fEPSP runs down by ∼20% in NEM (Fig. 8B,C,filled circles; n = 6). Application of DA did not have any further effect on the fEPSP (Fig.8B,C, open circles; n = 6), indicating that NEM prevents the effects of DA. To illustrate this point more clearly, we corrected the fEPSP measurements in NEM during the DA application by subtracting the effects of NEM alone during the same time period (Fig. 8D, filled circles). The corrected values show no effect of DA on the fEPSP in NEM, and this is clearly different from the depressant action of DA in the absence of NEM (Fig. 8D, open circles).
Fig. 8.

NEM blocks the depressant action of 200 nm DA. A, After a control period (a), NEM affects baseline synaptic transmission, causing first an increase (b), and then a decrease (c). During the time indicated by the shaded box, either DA or vehicle was applied. B, fEPSPs run down during the vehicle application as a function of time in NEM (top traces), and no further effect is seen when DA is applied instead of the vehicle solution (bottom traces).C, A summary of experiments, in which the fEPSP slope is normalized to that during the 5 min preceding application of DA or vehicle. Experiments in which DA was applied (open circles) are no different from those in which vehicle was applied (filled circles). The total time window in C corresponds to the region enclosed byvertical dotted lines in A.D, After removal of the baseline effects of NEM on synaptic transmission by subtracting the depression caused by DA from the depression caused by the vehicle, DA has no effect on the fEPSP in NEM-treated slices (filled circles). For comparison, the normal effect of DA in control conditions from Figure4D is shown (open circles).
To test further for the involvement of G-proteins, we examined the effects of the G-protein inhibitor pertussis toxin on the DA-induced depression of the fEPSP. Pertussis toxin, which selectively blocks Gi/Go-mediated processes, is reported to have very limited penetration in intact tissue, and previous reports have indicated that injection of pertussis toxin directly into the hippocampus 1–2 d before experiments is required to obtain full effectiveness of this toxin in brain slices. We therefore injected pertussis toxin into the hippocampus of 8- to 12-week-old rats at three injection sites 2–3 d before experiments. We found, consistent with the results using NEM, that DA had no effect on fEPSPs in slices from pertussis toxin-injected rats (Fig.9, filled circles;n = 7); in contrast, DA caused a robust depression in age-matched controls (Fig. 9, open circles;n = 6). We conclude that activation of an NEM- and pertussis toxin-sensitive G-protein is required for the DA-induced depression of glutamate release.
Fig. 9.
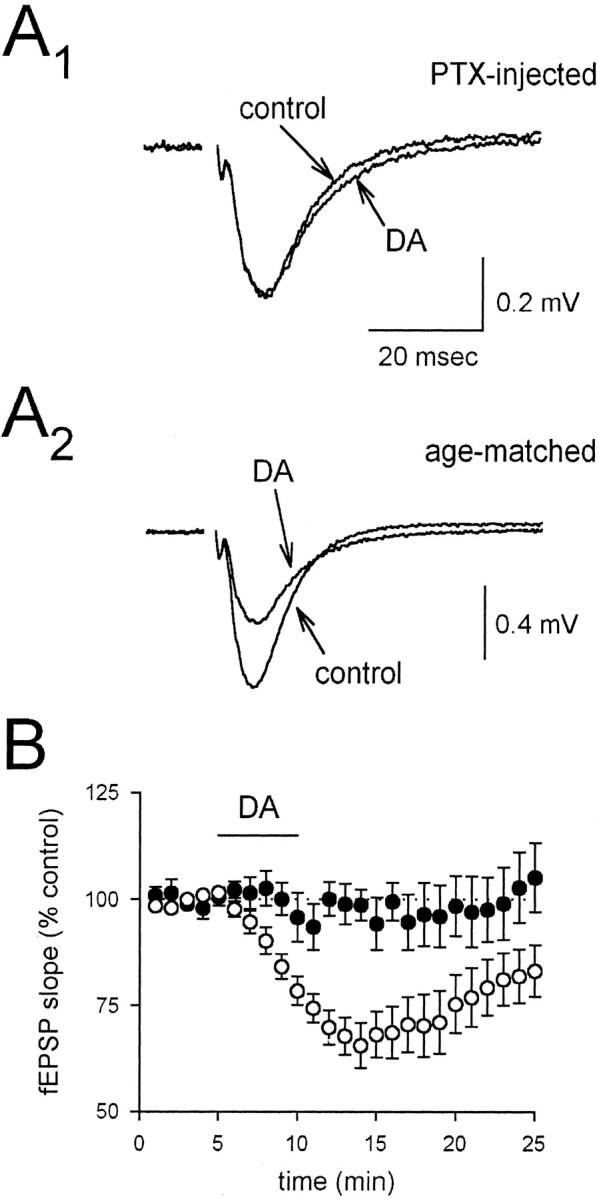
Pertussis toxin blocks the depressant action of 200 nm DA. A, Averaged fEPSPs in slices from pertussis toxin-injected rats (A1) and age-matched controls (A2) are shown in the presence and absence of DA. B, A summary of experiments using slices from pertussis toxin-injected rats (filled circles) and slices from age-matched controls (open circles) is shown. PTX, Pertussis toxin.
DA-induced G-protein activation could lead to a depression of synaptic transmission via the well described, direct interaction between the βγ G-protein subunits and calcium channels; alternatively,Rodríguez-Moreno and Lerma (1998) have suggested that a PKC activation downstream of G-protein activity is essential for the actions of KARs at inhibitory synapses. We therefore examined the effects of preincubating the slices with the broad-spectrum serine/threonine protein kinase inhibitor 1-(5-iso-quinolinesulfonyl)-2-methylpiperazine (H-7) and found that it had no effect on the depressant action of DA [Fig.10A,B; control slices (n = 8), filled circles; treated slices (n = 6), open circles]. To ensure that the H-7 was effectively blocking protein kinase activity, we examined its effects on the presynaptic potentiation induced by the adenylate cyclase activator forskolin, coapplied with the phosphodiesterase inhibitor IBMX (Chavez-Noriega and Stevens, 1994). As reported previously, forskolin and IBMX caused a substantial potentiation of the fEPSP under control conditions (Fig. 10C,D, filled circles; n = 5); however, after preincubation with H-7, this effect was strongly reduced (Fig. 10C,D,open circles; n = 5), confirming the effectiveness of H-7 in the present experiments. We conclude that protein kinases do not appear to be required for the DA-induced depression of glutamate release.
Fig. 10.

Protein kinases are not involved in the DA-induced depression of synaptic transmission. A, Averaged fEPSPs are shown before and after application of 200 nm DA in control conditions (top traces) and after incubation in the protein kinase inhibitor H-7 (bottom traces). B, A summary of the experiments inA is shown. The DA-induced depression in the presence of H-7 (open circles) is not different from the depression in control experiments (filled circles).C, Averaged fEPSPs are shown before and after application of forskolin and IBMX in control and H-7-treated slices. Forskolin and IBMX together (each at 150 μm) cause a potentiation of the fEPSP, which is strongly reduced in H-7-treated slices. D, A summary of experiments shows that the enhancement caused by forskolin and IBMX (filled circles) is greatly reduced by H-7 (open circles).
DISCUSSION
We have performed experiments to elucidate the mechanism by which KAR activation depresses excitatory synaptic transmission at CA3→CA1 synapses in the hippocampus. It has been reported previously that KARs cause a presynaptic depression of transmitter release, because KAR activation depresses release from synaptosomes (Chittajallu et al., 1996), acts on both the AMPAR- and NMDAR-mediated components of the EPSC, alters short-term plasticity (Kamiya and Ozawa, 1998; Vignes et al., 1998), and depresses presynaptic Ca2+ influx (Kamiya and Ozawa, 1998). Our results are entirely in agreement with this previous literature. We have confirmed that KAR agonists can depress both components of the EPSC and alter short-term plasticity, and we have also found that this depression is associated with a reduction in quantal content, as assessed by 1/CV2 measurements. The absence of any KAR-induced change in input resistance, coupled with the absence of any effect of KAR activation on mEPSC amplitude, suggests that little, if any, of the depressant action of KARs on excitatory transmission is postsynaptic.
The observation that KARs cause a presynaptic depression of synaptic transmission does not necessarily indicate that KARs are located on the presynaptic terminal; activation of KARs on other cells could cause the release of a presynaptic neuromodulator that acts heterosynaptically. This has in fact been a proposed mechanism of the KAR-induced depressant action in a few systems (Frerking et al., 1999; Chergui et al., 2000; Schmitz et al., 2000) (but see Rodríguez-Moreno et al., 2000). Three broad lines of experimental evidence argue against such a scenario in the present experiments. First, the depression induced by DA is unaffected by antagonism of a number of neuromodulators in the hippocampus. Second, the depression is unaffected by increasing the divalent composition, a manipulation that reduces cell excitability and prevents KAR-dependent interneuronal spiking. Finally, strong KAR activation via a high dose of DA can affect presynaptic fiber excitability even in the presence of cadmium to block calcium influx-dependent neuromodulator release, providing direct evidence of the existence of presynaptic KARs in this system. These experiments strongly suggest that the KAR-induced depression of glutamate release at CA3→CA1 synapses is caused by the direct activation of presynaptic KARs.
One possible indirect action of KARs that our experiments have not addressed is the release of neuromodulators because of calcium influx via calcium-permeable KARs, located either at the soma or on the terminals of some other population of synapses. However, the somatodendritic KARs on interneurons do not have the characteristically nonlinear I–V relations of calcium-permeable KARs (Frerking et al., 1998), making their involvement in such a cascade unlikely. Moreover, if KARs on presynaptic terminals were calcium permeable, their activation would lead to a large increase in the frequency of miniature PSCs; such an increase has not been reported for mEPSCs (this study) or miniature IPSCs (Rodríguez-Moreno et al., 1997;Cossart et al., 1998; Frerking et al., 1998; Bureau et al., 1999) onto CA1 pyramidal cells. We therefore think this scenario unlikely, although we cannot exclude the possibility that some unidentified population of cells that do not synapse onto CA1 pyramidal cells could have these receptors and release a modulator that causes the effect.
Because of the evidence supporting a direct presynaptic action of KARs, it is surprising that the effects of KAR activation, at least at low doses, are distinct from those of presynaptic depolarization via increasing extracellular K+. Although DA depresses synaptic transmission without affecting the presynaptic fiber volley, presynaptic depolarization affects both in parallel (see Fig.5C). Moreover, even modest presynaptic depolarizations (∼5 mV) cause a detectable change in mEPSC frequency in the presence of 15 mm extracellular K+, but DA does not. However, a higher dose of DA robustly depresses the fiber volley, suggesting that strong activation of KARs can depolarize the terminal sufficiently to have presynaptic ionotropic effects. These results indicate that low doses of DA that are sufficient to depress synaptic transmission cause no detectable depolarization of the presynaptic terminal and that the depression of release does not occur via an ionotropic mechanism. Moreover, the observation that elevated K+ depressed fiber excitability to the same extent that it depressed transmission suggests that presynaptic depolarization at this synapse has little direct effect on the synapse but instead acts mainly by preventing spike initiation and/or propagation. This would occur if axonal Na channels were inactivated at more hyperpolarized potentials than were presynaptic Ca channels. Strikingly, different results have been observed at mossy fiber synapses in the hippocampus, where synaptic transmission is depressed by levels of depolarization that enhance the fiber volley (Schmitz et al., 2000).
The evidence that KARs do not depress release via ionotropic mechanisms suggests that KARs activate a metabotropic signaling pathway, and the observation that G-protein inhibitors block the KAR-dependent synaptic depression is consistent with this suggestion. The sensitivity of the KAR-induced depression to pertussis toxin implicates the Gi/Go subtype of G-protein in the effect. It is noteworthy that binding studies have suggested a functional coupling between this subclass of G-protein and KARs in hippocampal membranes (Cunha et al., 1999). AMPARs have also been shown to couple to G-proteins (Wang et al., 1997; Kawai and Sterling, 1999). A major unresolved issue is how these receptors might activate G-proteins, because they have little structural similarity to the classical metabotropic receptors. A frequently proposed solution is that an accessory protein acts as a link between G-proteins and KARs, but so far a protein with this role has not been identified. Another caveat concerning the involvement of G-proteins is that the pharmacological blockade of the effect by G-protein inhibitors could indicate that KARs activate a G-protein but could alternatively result from a nonspecific action of the inhibitors or a dependence of KAR activity on G-proteins. Direct biochemical evidence will be required to establish unequivocally KARs as activators of G-proteins and to identify the protein–protein interactions that are involved.
We propose that presynaptic KARs couple to Gi/Go G-proteins, which causes the G-protein to dissociate into the α and β/γ subunits. Because our results indicate that protein kinases are not required in the signaling cascade, we suggest that the β/γ subunits directly bind to and inhibit presynaptic Ca2+channels and thereby depress release. This would be consistent with the observation that KAR activation depresses stimulus-evoked presynaptic Ca2+ transients at this synapse (Kamiya and Ozawa, 1998). We further suggest that presynaptic KARs may be sparsely distributed presynaptically, so that strong activation of KARs is required to resolve the ionotropic activity of the KAR, but weak activation appears to be sufficient to generate resolvable metabotropic activity. This higher sensitivity of the metabotropic pathway is presumably caused by the substantial biochemical amplification that takes place at the level of receptor–G-protein coupling, although we cannot at present exclude the alternative that separate, high-affinity metabotropic and low-affinity ionotropic KARs are present.
The degree to which this model can be extrapolated to other systems is unclear. KARs have been reported to activate a metabotropic cascade to depress release at inhibitory synapses onto CA1 pyramidal cells (Rodríguez-Moreno and Lerma, 1998; Cunha et al., 2000;Rodríguez-Moreno et al., 2000). However, because GABAergic interneurons are strongly excited by KAR activation (Cossart et al., 1998; Frerking et al., 1998; Bureau et al., 1999) and at least some of the inhibition induced by KARs has been proposed to occur via the indirect activation of metabotropic GABABreceptors caused by that excitation (Frerking et al., 1998, 1999), to what extent a direct presynaptic metabotropic action accounts for the KA-induced depression at these synapses is controversial. KAR activation is known to activate indirectly metabotropic cascades at other synapses (Chergui et al., 2000; Schmitz et al., 2000). Presynaptic KAR activation has also been reported to modulate transmitter release via ionotropic mechanisms (Kamiya and Ozawa, 2000;Schmitz et al., 2000). It therefore seems likely that KAR-dependent modulation of transmitter release at different synapses operates via different mechanisms.
In conclusion, we have provided evidence that presynaptic KARs depress glutamate release at CA3→CA1 synapses via a depolarization-independent metabotropic cascade. The conditions under which these receptors are activated and the role that they play remain unclear. However, an autoreceptive function seems likely because of the presynaptic location of these receptors at terminals releasing their endogenous agonist. KARs are widely thought to play a role in epileptiform activity, particularly in area CA3 of the hippocampus. The depressant action of the presynaptic receptors described here will tend to reduce transmission of epileptiform activity from CA3 to CA1, in conjunction with the previously described “overinhibitory” action of KARs on interneurons (Cossart et al., 1998). These results suggest that KARs, depending on their location and mechanism of action, both promote and reduce excitability in the hippocampus.
Footnotes
R.A.N. is a member of the Keck Center for Integrative Neuroscience and the Silvio Conte Center for Neuroscience Research and is supported by grants from the National Institutes of Health. D.S. is supported by a grant from the Deutsche Forschungsgemeinschaft (Emmy-Noether-Programm). We thank the members of the Nicoll laboratory for useful discussions.
Correspondence should be addressed to Dr. Roger A. Nicoll, Department of Cellular and Molecular Pharmacology, University of California, San Francisco, CA 94143-0450. E-mail: nicoll@phy.ucsf.edu.
REFERENCES
- 1.Bureau I, Bischoff S, Heinemann SF, Mulle C. Kainate receptor-mediated responses in the CA1 field of wild-type and GluR6-deficient mice. J Neurosci. 1999;19:653–663. doi: 10.1523/JNEUROSCI.19-02-00653.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chergui K, Bouron A, Normand E, Mulle C. Functional GluR6 kainate receptors in the striatum: indirect downregulation of synaptic transmission. J Neurosci. 2000;20:2175–2182. doi: 10.1523/JNEUROSCI.20-06-02175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chittajallu R, Vignes M, Dev KK, Barnes JM, Collingridge GL, Henley JM. Regulation of glutamate release by presynaptic kainate receptors in the hippocampus. Nature. 1996;379:78–81. doi: 10.1038/379078a0. [DOI] [PubMed] [Google Scholar]
- 5.Cossart R, Esclapez M, Hirsch J, Bernard C, Ben-Ari Y. GluR5 kainate receptor activation in interneurons increases tonic inhibition of pyramidal cells. Nat Neurosci. 1998;1:470–478. doi: 10.1038/2185. [DOI] [PubMed] [Google Scholar]
- 6.Cunha RA, Malva JO, Ribeiro JA. Kainate receptors coupled to G(i)/G(o) proteins in the rat hippocampus. Mol Pharmacol. 1999;56:429–433. doi: 10.1124/mol.56.2.429. [DOI] [PubMed] [Google Scholar]
- 7.Cunha RA, Malva JO, Ribeiro JA. Pertussis toxin prevents presynaptic inhibition by kainate receptors of rat hippocampal [(3)H]GABA release. FEBS Lett. 2000;469:159–162. doi: 10.1016/s0014-5793(00)01272-2. [DOI] [PubMed] [Google Scholar]
- 8.Frerking M, Nicoll RA. Synaptic kainate receptors. Curr Opin Neurobiol. 2000;10:342–351. doi: 10.1016/s0959-4388(00)00094-5. [DOI] [PubMed] [Google Scholar]
- 9.Frerking M, Borges S, Wilson M. Are some minis multiquantal? J Neurophysiol. 1997;78:1293–1304. doi: 10.1152/jn.1997.78.3.1293. [DOI] [PubMed] [Google Scholar]
- 10.Frerking M, Malenka RC, Nicoll RA. Synaptic activation of kainate receptors on hippocampal interneurons. Nat Neurosci. 1998;1:479–486. doi: 10.1038/2194. [DOI] [PubMed] [Google Scholar]
- 11.Frerking M, Petersen CCH, Nicoll RA. Mechanisms underlying kainate receptor-mediated disinhibition in the hippocampus. Proc Natl Acad Sci USA. 1999;96:12917–12922. doi: 10.1073/pnas.96.22.12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinemann U, Lux HD, Gutnick MJ. Extracellular free calcium and potassium during paroxysmal activity in the cerebral cortex of the cat. Exp Brain Res. 1977;27:237–243. doi: 10.1007/BF00235500. [DOI] [PubMed] [Google Scholar]
- 13.Kamiya H, Ozawa S. Kainate receptor-mediated inhibition of presynaptic Ca2+ influx and EPSP in area CA1 of the rat hippocampus. J Physiol (Lond) 1998;509:833–845. doi: 10.1111/j.1469-7793.1998.833bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamiya H, Ozawa S. Kainate receptor-mediated presynaptic inhibition at the mouse hippocampal mossy fibre synapse. J Physiol (Lond) 2000;523:653–665. doi: 10.1111/j.1469-7793.2000.t01-1-00653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawai F, Sterling P. AMPA receptor activates a G-protein that suppresses a cGMP-gated current. J Neurosci. 1999;19:2954–2959. doi: 10.1523/JNEUROSCI.19-08-02954.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lux HD, Neher E. The equilibration time course of (K+)o in cat cortex. Exp Brain Res. 1973;17:190–205. doi: 10.1007/BF00235028. [DOI] [PubMed] [Google Scholar]
- 17.Paternain AV, Morales M, Lerma J. Selective antagonism of AMPA receptors unmasks kainate receptor-mediated responses in hippocampal neurons. Neuron. 1995;14:185–189. doi: 10.1016/0896-6273(95)90253-8. [DOI] [PubMed] [Google Scholar]
- 18.Pitler TA, Alger BE. Differences between presynaptic and postsynaptic GABAB mechanisms in rat hippocampal pyramidal cells. J Neurophysiol. 1994;72:2317–2327. doi: 10.1152/jn.1994.72.5.2317. [DOI] [PubMed] [Google Scholar]
- 19.Rodríguez-Moreno A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 1998;20:1211–1218. doi: 10.1016/s0896-6273(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 20.Rodríguez-Moreno A, Herreras O, Lerma J. Kainate receptors presynaptically downregulate GABAergic inhibition in the rat hippocampus. Neuron. 1997;19:893–901. doi: 10.1016/s0896-6273(00)80970-8. [DOI] [PubMed] [Google Scholar]
- 21.Rodríguez-Moreno A, López-García JC, Lerma J. Two populations of kainate receptors with separate signaling mechanisms in hippocampal interneurons. Proc Natl Acad Sci USA. 2000;97:1293–1298. doi: 10.1073/pnas.97.3.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmitz D, Frerking M, Nicoll RA. Synaptic activation of presynaptic kainate receptors on hippocampal mossy fiber synapses. Neuron. 2000;27:327–338. doi: 10.1016/s0896-6273(00)00040-4. [DOI] [PubMed] [Google Scholar]
- 23.Vignes M, Clarke VR, Parry MJ, Bleakman D, Lodge D, Ornstein PL, Collingridge GL. The GluR5 subtype of kainate receptor regulates excitatory synaptic transmission in areas CA1 and CA3 of the rat hippocampus. Neuropharmacology. 1998;37:1269–1277. doi: 10.1016/s0028-3908(98)00148-8. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Small DL, Stanimirovic DB, Morley P, Durkin JP. AMPA receptor-mediated regulation of a Gi-protein in cortical neurons. Nature. 1997;389:502–504. doi: 10.1038/39062. [DOI] [PubMed] [Google Scholar]
- 25.Wilding TJ, Huettner JE. Differential antagonism of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-preferring and kainate-preferring receptors by 2,3-benzodiazepines. Mol Pharmacol. 1995;47:582–587. [PubMed] [Google Scholar]