

Abstract
The glial cell line-derived neurotrophic factor (GDNF) is first characterized for its trophic activity on dopaminergic neurons. Recent data suggested that GDNF could modulate the neuronal death induced by ischemia. The purpose of this study was to characterize the influence of GDNF on cultured cortical neurons subjected to two paradigms of injury (necrosis and apoptosis) that have been identified during cerebral ischemia and to determine the molecular mechanisms involved. First, we demonstrated that both neurons and astrocytes express the mRNA and the protein for GDNF and its receptor complex (GFRα-1 and c-Ret). Next, we showed that the application of recombinant human GDNF to cortical neurons and astrocytes induces the activation of the MAP kinase (MAPK) pathway, as visualized by an increase in the phosphorylated forms of extracellular signal-regulated kinases (ERKs). Thereafter, we demonstrated that GDNF fails to prevent apoptotic neuronal death but selectively attenuates slowly triggered NMDA-induced excitotoxic neuronal death via a direct effect on cortical neurons. To further characterize the neuroprotective mechanisms of GDNF against NMDA-mediated neuronal death, we showed that a pretreatment with GDNF reduces NMDA-induced calcium influx. This effect likely results from a reduction of NMDA receptor activity rather than an enhanced buffering or extrusion capacity for calcium. Finally, we also demonstrated that an ERKs activation pathway is necessary for GDNF-mediated reduction of the NMDA-induced calcium response. Together, these results describe a novel mechanism by which the activation of MAPK induced by GDNF modulates NMDA receptor activity, a mechanism that could be responsible for the neuroprotective effect of GDNF in acute brain injury.
Keywords: GDNF, excitotoxicity, apoptosis, NMDA receptor, Fura-2, MAPK
Ischemia-induced neuronal injury is thought to result from an excessive release of excitatory amino acids and the subsequent activation of their postsynaptic receptors. This excitotoxic pathway may also contribute to brain and spinal cord cell loss after various acute insults. The first step in the excitotoxic cascade is mediated by the activation of ion channel-linked glutamate receptors, especially NMDA receptors, probably because of their high calcium (Ca2+) permeability (Choi, 1995). The subsequent activation of cytoplasmic Ca2+-dependent enzymes has been proposed to mediate the final excitotoxicity. More recently, various clues have emerged to suggest that both global and focal ischemia may have an apoptotic component (Linnik et al., 1993; MacManus et al., 1993).
Because of their capacity to promote neuronal survival during embryogenesis, the use of growth factors has been proposed to limit the consequences of cerebral ischemia. Among these cytokines, a structurally related family has gained prominence because of its high level of expression in biopsies of patients with acute brain injury or neurodegenerative diseases, i.e. the transforming growth factor-βs (TGF-βs) (Pratt and McPherson, 1997). Although the beneficial effects of TGF-β have been well studied (Buisson et al., 1998), questions have arisen about the role of a distant member of the TGF-β family, the GDNF, during brain injury.
The GDNF transcript has been detected in all major brain regions (Schaar et al., 1993). Based on an expression strategy to identify high affinity binding proteins, the GDNF receptor α-1 (GFRα-1) has been cloned (Jing et al., 1996; Treanor et al., 1996). It is a protein anchored to the cell surface by a glycosyl-phosphatidyl inositol (GPI) devoid of an associated kinase activity. The signaling component of the GDNF receptor has been identified as the tyrosine kinase receptor encoded by the proto-oncogene c-Ret (Durbec et al., 1996; Trupp et al., 1996). After binding to its specific receptor complex, GDNF activates several downstream intracellular pathways, including mitogen-activated protein kinase (MAPK) (Worby et al., 1996) and PI-3 kinase (Soler et al., 1999), resulting in long-term changes in gene expression (Messer et al., 1999).
GDNF was first characterized as a potent neurotrophic factor for dopaminergic and motor neurons (Lin et al., 1993; Henderson et al., 1994). To date, several studies have revealed a neuroprotective influence of GDNF against various toxic challenges (Krieglstein et al., 1995). Recently, it has been suggested that GDNF could also modulate neuronal death induced by acute brain injury (Wang et al., 1997). In this study, these authors demonstrated that, after middle cerebral artery occlusion (MCAo) in rodents, the application of GDNF reduces the infarcted volume. Although the neuroprotective activity of GDNF against ischemia-induced neuronal death is now well established (Abe et al., 1997; Kitagawa et al., 1998), little is known about the mechanisms that underpin this neuroprotection. The purpose of the present study was to determine the influence of GDNF on apoptosis and necrosis, two different paradigms of neuronal death that have been identified during cerebral ischemia, and to explore the molecular mechanisms by which GDNF exerts its effect.
MATERIALS AND METHODS
PCR-reverse transcriptase system kits, Wizard Plus Minipreps DNA Purification System, and U0126 were purchased from Promega (Charbonnières, France). DMEM, poly-d-lysine, laminin, cytosine β-d-arabinofuranoside (Ara-C), staurosporine (Stp), ExtrAvidin, and cycloheximide were obtained from Sigma (L'Isle D'Abeau, France). Horse serum and fetal bovine serum were from Life Technologies (Cergy Pontoise, France). Recombinant human GDNF (rhGDNF) was obtained from R&D Systems (Oxon, UK), and antibodies raised against GDNF, GFRα-1, c-Ret, extracellular signal-regulated kinase 1 (ERK1), phospho-ERKs (p-ERKs), p-c-Jun N-terminal protein kinase (p-JNK), and p-38 were purchased from Santa Cruz (Heidelberg, Germany). NMDA, AMPA, kainate, and (+)5-methyl-10,11-dihydro-5H-dibenzo(a,d)cyclohepten-5,10-imine maleate (MK-801) were from Tocris (Bristol, UK).
Mixed cortical cultures
Mixed cortical cultures containing both neurons and astrocytes were prepared from fetal OF1 mice at 14–16 d gestation, as described by Rose et al. (1993). Briefly, dissociated cortical cells were plated in 24 wells on a layer of confluent astrocytes, using a medium stock (MS; MEM with 25 mm glucose) supplemented with 5% horse serum, 5% fetal bovine serum, and 2 mm glutamine. After 7 d in vitro (DIV), non-neuronal cell division was halted by a 1–3 d exposure to 10 μm Ara-C. Subsequent partial medium replacement was performed twice a week, and after 12 DIV, cultures were shifted to a maintenance medium identical to plating medium but lacking serum. Cultures were used after 14 DIV.
Glial cultures
Glial cell cultures were prepared from postnatal mice (1–3 d after birth), as described previously (Rose et al., 1993). Dissociated cortical cells were grown in multiwell vessels using a plating medium of MS supplemented with 10% horse serum, 10% fetal bovine serum, and 2 mm glutamine. Cultures were kept at 37°C in a humidified 5% CO2-containing atmosphere until they reached confluency (7–14 DIV). Confluent cultures were used then as a support for mixed cultures.
Near pure neuronal cell cultures
These cultures containing <5% astrocytes were prepared as detailed previously (Rose et al., 1993). Dissociated cortical cells in MS supplemented with 5% fetal bovine, 5% horse serum, and 2 mm glutamine were plated in multiwell vessels that had been coated previously with poly-d-lysine and laminin. After 3 DIV, non-neuronal cell division was halted by an exposure to 10 μm Ara-C. There was no further exchange of the media. After 12 DIV, cultures did not need the presence of serum to survive. They were shifted to a maintenance medium identical to plating medium but lacking serum. Cultures were used after 7 DIV for serum deprivation (SD) and after 13–14 DIV for excitotoxic injury.
Reverse transcription and PCR
Total RNAs were isolated from mouse cerebral cortices and murine cell cultures through the use of either the RNAxel kit (Eurobio, Les Ulis, France) or the Rneasy Kit (Qiagen, Courtaboeuf, France). One microgram of total RNAs was reverse transcribed into cDNA using poly-dT oligonucleotides. Then, an aliquot of the cDNA libraries was amplified using sense and antisense primers for GDNF (640 bp, 45 cycles), GFRα-1 (351 bp, 38 cycles), and c-Ret (615 bp, 40 cycles). The number of PCR cycles was chosen, corresponding to 50% of the saturating curve of each PCR product. The following constructs were used: GDNF sense oligonucleotide, ATG AAG TTA TGG GAT GTC GT, and antisense oligonucleotide, CAG GGT CAG ATA CAT CCA CA; GFRα-1 sense oligonucleotide, CAT GTT CCT AGC CAC TCT GT, and antisense oligonucleotide, TCCAGT AGG TCA TTT CCC TG; c-Ret sense oligonucleotide, TGT ATG TAG ACC AGC CAG CT, and antisense oligonucleotide, ACT ATG CAC AAA GCC TCC AG. Amplified products were separated by 1.5% agarose gel electrophoresis and visualized by ethidium bromide staining.
SDS-PAGE and Western blot
The cerebral cortex of adult mouse and the murine-cultured cortical neurons (DIV 14) and astrocytes (confluent cultures) were lysed in Tris–NaCl–Triton buffer and centrifuged for 5 min (2500 rpm) to obtain whole-cell extracts. Then, an SDS-PAGE (polyacrylamide percentage, 20% for GDNF and 12% for GFRα-1 and c-Ret) was performed before immobilization onto a polyvinylidene difluoride membrane. Blots were exposed for 1 hr at room temperature to the primary antibody (anti-Ret, 1:100; anti-GFRα-1, 1:100; anti-GDNF, 1:100) in a blocking reagent [Tris-buffered saline (TBS): Tris 10 mm and NaCl 200 mm, pH 7.4] containing 0.1% Tween 20, 5% dry milk. The membranes were washed and incubated for 1 hr with the appropriate secondary biotin-conjugated antibody (1:1000) and then for 1 hr with ExtrAvidin (1:2000) before revelation using a chemiluminescence kit (NEN, Paris, France).
Immunocytochemistry
Murine cortical cultures (DIV 14) were fixed in 4% paraformaldehyde in PBS, pH 7.4, for 30 min. After two rinsings in PBS, the preparations were incubated overnight with the primary antibody raised against either GDNF or GDNF receptors (GDNF, GFRα-1, and c-Ret, 1:100 in PBS, plus 1% BSA and 0.1% Tween). Cells were then washed and incubated for 1 hr with the appropriate secondary biotin-conjugated antibody. Antibody–antigen complexes were amplified with avidin (Vectastain ABC Kit; Vector Laboratories, Burlingame, CA) and revealed by H2O2–peroxidase reaction.
Analysis of p-ERKs
Murine-cultured cortical neurons (DIV 14) and astrocytes (confluent cultures) were lysed in a buffer containing 1 mmsodium orthovanadate and phosphatase inhibitors mixture (Sigma, L'Isle D'Abeau, France). After homogenization, the protein concentration was measured by Bradford's method, using BSA as the standard. Equivalent amounts of protein for each sample were resolved in 15% SDS-PAGE that was blotted electrophoretically to polyvinylidene difluoride membranes. Blots were incubated with the p-ERK antibody (1:200), followed by incubation with the appropriate secondary biotin-conjugated antibody (1:1000) and then with ExtrAvidin (1:2000) before revelation using a chemiluminescence kit (NEN, Paris, France). The blots were then incubated in stripping buffer (62 mmTris HCl, pH 6.8, 2% SDS, and 100 mm β-mercaptoethanol) for 30 min at 50°C, followed by incubation with TBS containing 0.1% Tween 20, 5% dry milk. The blots were then incubated with an ERK1 polyclonal antibody (1:200) that binds to ERK1 and ERK2.
Excitotoxicity
Slowly triggered excitotoxicity was induced at 37°C by 24 hr exposure to 12.5 μm NMDA, 10 μm AMPA, or 50 μm kainate in MS supplemented with 10 μmglycine (Choi, 1992). MK-801 (10 μm) was always added concurrently with AMPA or kainate to block secondary NMDA receptor activation. rhGDNF was coapplied with the excitotoxin and left in the bathing media for 24 hr. Neuronal death was estimated by examination of the cultures under phase-contrast microscopy and quantified by the measurement of lactate dehydrogenase (LDH) release from damaged cells into the bathing medium 1 d after the onset of excitotoxin exposure (Koh and Choi, 1987). The LDH level corresponding to complete neuronal death (without glial death) was determined in sister cultures exposed to 200 μm NMDA. Background LDH levels were determined in sister cultures subjected to sham wash and subtracted from experimental values to yield the signal specific for experimentally induced injury.
Apoptosis
Serum deprivation. SD was initiated by transferring pure neuronal cultures (DIV 7), which require serum to survive for 24 hr, into growth medium (MS) lacking serum (Martin et al., 1988) in the presence or absence of rhGDNF. The control consisted of sham wash with serum-containing medium. Secondary NMDA receptor activation was blocked by addition of MK-801 to the bathing medium. Neuronal cell death was assessed by phase-contrast cell counting after staining with 0.4% trypan blue dye.
Staurosporine-induced apoptosis. Stp exposure was performed at 37°C for 24 hr by transferring mixed cortical cell cultures (DIV 13–14) in MS supplemented with glycine (10 μm), containing 200 nmStp (Koh et al., 1995) with or without rhGDNF. To confirm the neuronal death as apoptotic, the protein synthesis inhibitor cycloheximide (1 μg/ml) was administered with Stp in an additional set of cultured cells. MK-801 (10 μm) was always added concurrently with Stp to block secondary NMDA receptor activation. Neuronal cell death was assessed by the measurement of LDH release.
Analysis of DNA fragmentation in agarose gel. Cortical cells were lysed at 4°C for 30 min in a buffer containing 0.2% SDS, 200 μg/ml proteinase K, 50 μg/ml RNase A. After treatment, DNA was extracted with Wizard Plus Minipreps DNA Purification System. DNA samples were loaded onto a 1.4% agarose gel and run at 90 V for 1 hr.
Intracellular free Ca2+measurement. Cell cultures were loaded with fura-2 (30 min, room temperature) in 5 μm fura-2 AM plus 0.1% pluronic F-127 (Molecular Probes, Leiden, the Netherlands) and incubated for an additional 30 min in an HEPES-buffered saline solution. Experiments were performed at room temperature, on the stage of a Nikon Eclipse inverted microscope equipped with a 75 W xenon lamp and a Nikon 40×, 1.3 numerical aperture (NA) epifluorescence oil immersion objective. Fura-2 (excitation of 340 and 380 nm; emission of 510 nm) ratio images were acquired with a CCD camera (Princeton Instruments, Trenton, NJ) and digitized (256 × 512 pixels) using Metafluor software (Universal Imaging Corporation, Chester, PA). Fluorescence ratios (340/380 nm) were converted to intracellular Ca2+ concentrations using the following formula: [Ca2+]i =Kd[(R −Rmin)/(Rmax− R)]F0/Fs, where R is the ratio for observed 340/380 fluorescence ratio, Rmin is the ratio for a Ca2+-free solution,Rmax is the ratio for a saturated Ca2+ solution,Kd = 135 nm (the dissociation constant for fura-2), F0 is the intensity of a Ca2+-free solution at 380 nm, and Fs is the intensity of a saturated Ca2+ solution at 380 nm. We analyzed 20 neurons per coverslip from three different dissections containing both neurons and astrocytes.
Statistical analysis. Results are expressed as mean ± SEM. When n = 12 is indicated, this value corresponds to 12 different well pools derived from three different dissections. Statistical analysis consisted of one-way ANOVA, followed by Bonferroni–Dunn's test. For the fura-2 experiments, statistical analysis consisted of multiple ways ANOVA followed by Bonferroni–Dunn's test.
RESULTS
Cultured cortical astrocytes and neurons both display GDNF and GDNF receptors
The expression of GDNF and the GDNF receptor complex in murine cerebral cortex and murine cortical cultures was determined at the mRNA level. Each set of oligonucleotides used for reverse transcription-PCR studies gave the PCR products of the expected size (640 bp for GDNF, 351 bp for GFRα-1, 615 bp for c-Ret), as illustrated in Figure1, A,D, and G. To demonstrate the specificity of the PCR products, digest controls through the use of the internal restriction sites of the PCR products were performed (data not shown). These studies revealed that mRNAs for GDNF (Fig. 1A), GFRα-1 (Fig. 1D), and c-Ret (Fig. 1G) are expressed in murine cerebral cortex, cortical neurons, and astrocytes.
Fig. 1.
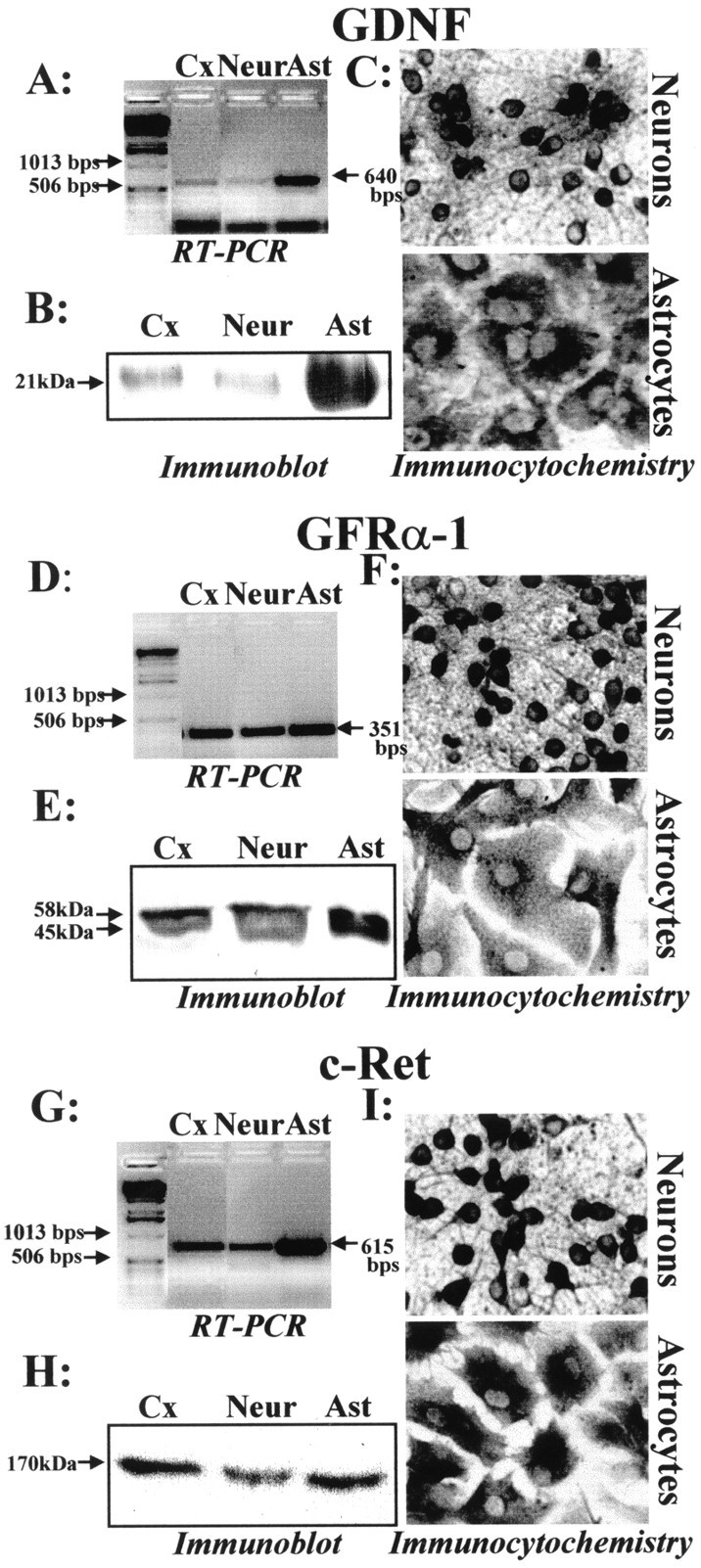
Comparison of protein expression of GDNF and GDNF receptor complex between murine cerebral cortex and cultured cortical neurons and astrocytes. Total RNAs of murine astrocytic (Ast) and neuronal (Neur) cortical cultures and murine cerebral cortex (Cx) were extracted and reverse transcribed using a poly-dT primer. A,D, G, PCR amplification was performed then using a specific set of primers for GDNF (A), GFRα-1 (D), and c-Ret (G). B, E,H, Western blot was performed on a lysate derived from murine cerebral cortex, neurons, or astrocyte cultures showing GDNF (B) and GDNF receptor expression (E, H). C,F, I, Immunocytochemistry analysis of GDNF and GDNF receptor components in cultured astrocytes and neurons. Bright-field photomicrographs of either pure cortical astrocytes or pure cortical neurons in culture, after fixation and peroxidase staining for antibodies raised against GDNF (C), GFRα-1 (F), and c-Ret (I).
The expression of GDNF and GDNF receptor complex was characterized further at the protein level by immunoblotting and immunocytochemistry. Immunoblot that is performed from cerebral cortex lysate, cultured neurons, or astrocyte lysates with an anti-GDNF antibody reveals a single band (Fig. 1B) at the expected molecular weight (20 kDa) (Krieglstein et al., 1998). Similarly, immunoblot that is performed with an anti-GFRα-1 antibody reveals the presence of two bands at the expected molecular weights (58 and 45 kDa) (Fig.1E), corresponding to the glycosylated and nonglycosylated forms of GFRα-1 (Jing et al., 1996). Immunoblot that is performed with an anti-c-Ret antibody reveals the presence of a band (Fig. 1H), as previously described by Van Weering et al. (1998), corresponding to a 170 kDa mature form at the plasma membrane. Together, these results reveal the expression of GDNF and GDNF receptor proteins in mice cerebral cortex, cortical neurons, and cortical astrocyte cultures. In the next experiment, we performed immunocytochemistry studies on pure cultured cortical neurons and astrocytes. We showed that both neurons and astrocytes in culture express GDNF and the GDNF receptor complex (GFRα-1 and c-Ret) (Fig.1C,F,I). When the same experiments were performed without adding the primary antibody, we did not observe any nonspecific labeling (data not shown).
Neurons and astrocytes express a functional receptor complex
It has been demonstrated previously in dopaminergic neurons that the GDNF transduction pathway involves MAPK (Worby et al., 1996). However, there is no information available about the transduction pathway in cortical neurons. Here, we performed an immunoblotting study with antibodies raised against the phosphorylated forms of ERKs (p-ERKs) on cortical neurons and astrocytes exposed to rhGDNF. Then, aliquots of whole-cell lysates were separated by SDS-PAGE and incubated with the anti-p-ERKs antibody. A representative temporal pattern of ERKs phosphorylation under GDNF exposure is shown in neurons (Fig. 2A) and in astrocytes (Fig. 2B). In both cortical neurons and astrocytes, addition of rhGDNF (10 ng/ml) resulted in a rapid increase (15 min) in the phosphorylation of p44 (ERK1) and p42 (ERK2) that was maintained for the next 60 min of rhGDNF exposure to cortical neurons. The same blots were stripped and reprobed for total ERKs, showing that the treatment with rhGDNF modifies the phosphorylation level of ERKs but not the total level of ERK protein (Fig.2A,B).
Fig. 2.

GDNF induces a phosphorylation of ERKs. Mouse primary cortical neurons (A) or astrocytes (B) were treated with rhGDNF (10 ng/ml) for the indicated times. Aliquots of whole-cell lysates were separated by SDS-PAGE, transferred to membranes, and incubated with the anti-p-ERKs antibody or anti-total ERK antibody. The immunoblots are representative of three independent experiments.
GDNF protects neurons against NMDA-induced cell death
To test whether GDNF modulates neuronal outcome after an ischemic insult, we studied the effects of rhGDNF on murine cortical cell cultures exposed to different paradigms of cell death (apoptosis and necrosis) identified during cerebral ischemia.
To induce apoptosis, near pure cortical neuronal cultures after 7 DIV were transferred to a serum-deficient medium (Martin et al., 1988), which provoked the death of ∼80% of neurons over 24 hr. This paradigm evidenced three features typical of apoptosis: (1) neurons exhibited a gradual shrinkage of the cell body; (2) death was almost completely abolished by the addition of cycloheximide (1 μg/ml), a protein synthesis inhibitor (Fig.3C,D); and (3) death was accompanied by a nuclear condensation (Fig. 3A) and appearance of DNA fragmentation (Fig. 3B). Addition of rhGDNF to the bathing medium (1–10 ng/ml) failed to modify the progression and the extent of neuronal degeneration (Fig.3C). Neuronal apoptosis was also induced by exposing mixed neuron–glia cultures (14 DIV) to staurosporine (200 nm), a nonspecific protein kinase inhibitor, which caused a neuronal degeneration evolving over 24 hr (Koh et al., 1995). Although cycloheximide treatment blocked the staurosporine-induced neuronal death, the addition of rhGDNF (1–10 ng/ml) was without effect (Fig. 3D).
Fig. 3.
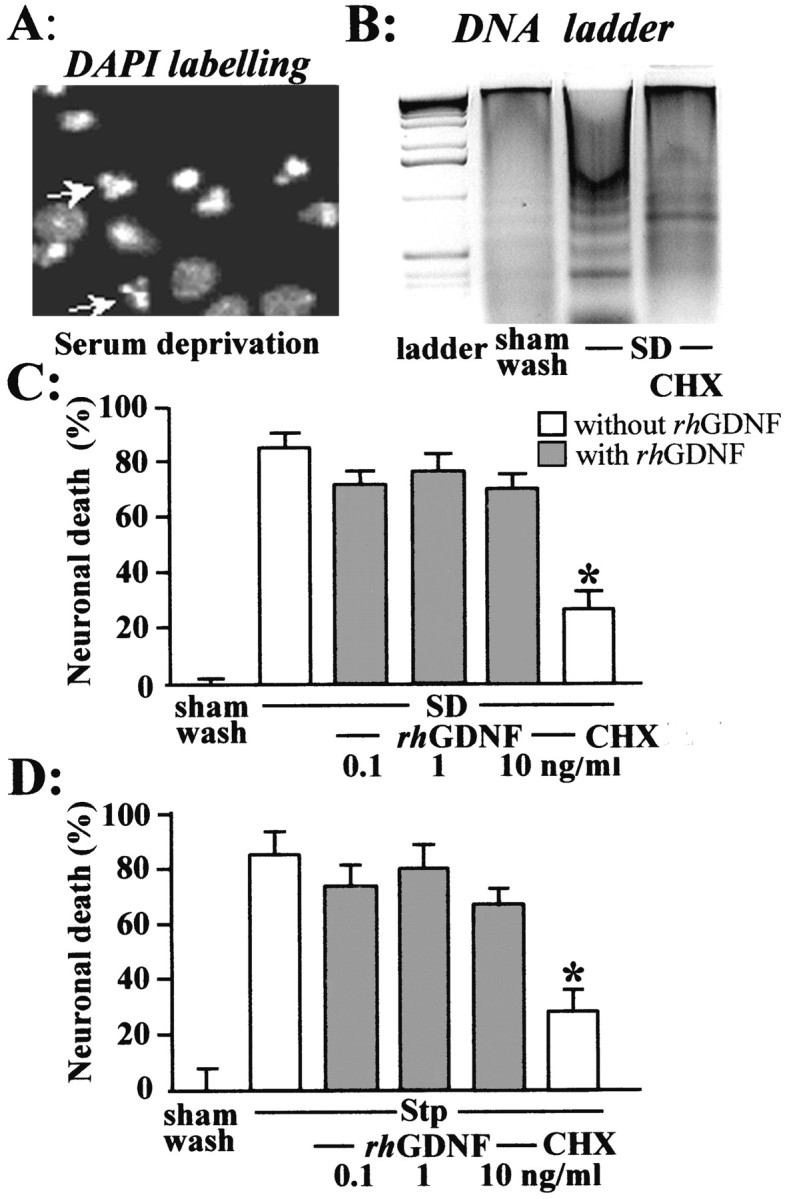
rhGDNF does not protect neurons against apoptotic cell death. To induce apoptosis, near pure cortical neuronal cultures (DIV 7) were transferred to a serum-deficient medium (SD). A, B, 24 hr after serum deprivation, the neurons were fixed and stained with DAPI to observe DNA condensation (A) or the DNA of cortical neurons was extracted and loaded onto an agarose gel to evidence DNA fragmentation (B). C,D, Neuronal death percentage was assessed after 24 hr by trypan blue dye staining (mean ± SEM; n = 12) after SD (C) or by LDH release (mean ± SEM;n = 12) after exposure of mixed cortical cultures (14 DIV) to staurosporine 200 nm (Stp) (D). SD and Stp exposure were performed in the presence (gray bars) or absence (white bars) of rhGDNF and with or without 1 μg/ml cycloheximide (CHX). In both apoptotic paradigms, MK-801 was systematically added to block the secondary activation of NMDA receptors. * indicates significantly different from SD or STP by ANOVA, followed by Bonferroni–Dunn's test (p < 0.05).
Thereafter, we investigated the influence of GDNF on excitotoxic necrosis, a process morphologically distinct from apoptosis and characterized by a prominent and early cell swelling. A 24 hr incubation with the ionotropic glutamatergic agonists NMDA, AMPA, and kainate induced necrosis (Rose et al., 1993). Exposure of mixed cortical neuron–glia cultures (14 DIV) to these agonists produced acute swelling of neuronal cell bodies that was followed 24 hr later by a widespread neuronal degeneration, whereas the glia remained intact. Although the rhGDNF (1–10 ng/ml) failed to produce any neuroprotective effect against neuronal death induced by the exposure to non-NMDA agonists (AMPA and kainate) (Fig.4A) (n= 10), this growth factor significantly reduced NMDA-induced necrotic neuronal death in mixed neuron–glia cultures. This neuroprotective effect was dose dependent (Fig. 4B) (n = 12; p < 0.05). We further investigated the influence of rhGDNF on excitotoxic neuronal death by applying NMDA (12.5 μm) for 24 hr in near pure neuronal cultures (14 DIV) containing <5% of astrocytes, as determined by GFAP immunostaining (data not shown). In these cultures, rhGDNF (10 ng/ml) showed a neuroprotective activity against NMDA-induced necrotic death similar to that observed in mixed neuron–glia cultures (Fig. 4C) (n = 12;p < 0.05).
Fig. 4.
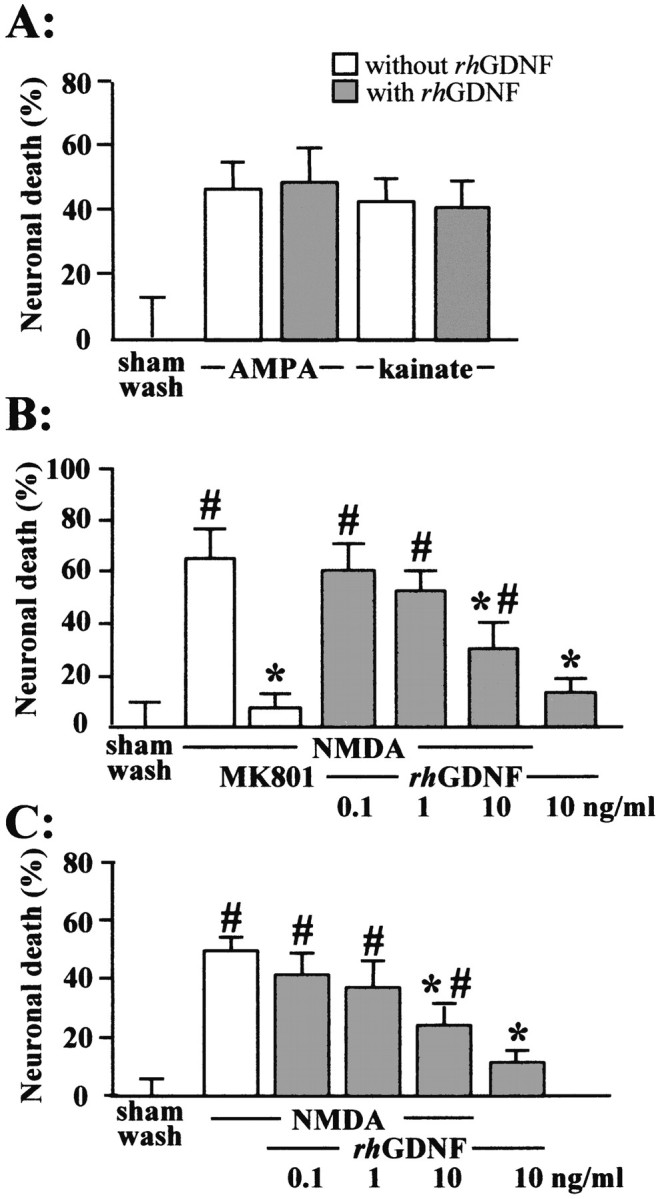
rhGDNF is neuroprotective against NMDA-induced neuronal death. A, Cultures were exposed for 24 hr to AMPA (10 μm) or kainate (50 μm) (with 10 μm MK-801 added to block secondary NMDA receptor-mediated toxicity), without (white bars) or with (gray bars) 10 ng/ml rhGDNF. Medium LDH was assessed at the end of the exposure to excitotoxins (mean ± SEM; n = 10 cultures per condition). B, C, Mixed neuron–glia cortical cultures (B) or near pure cortical neuron cultures (C) were exposed for 24 hr to NMDA (12.5 μm) without (white bars) or with (gray bars) increasing concentration of rhGDNF or MK-801 (10 μm). Medium LDH was assessed at the end of the exposure to excitotoxin (mean ± SEM;n = 12 cultures per condition. * indicates significantly different from NMDA; # indicates significantly different from control (sham wash) by ANOVA, followed by Bonferronni–Dunn's test (p < 0.05).
A GPI-linked protein mediates the neuroprotective activity of GDNF
To further characterize the neuroprotective activity of GDNF, we attempted to demonstrate whether the GFRα-1 receptor, a GPI-anchored protein, mediates the neuroprotective effect of GDNF. Mixed glial cultures were treated with phosphoinositide-specific phospholipase C (PIPLC; 0.3 IU/ml), an enzyme that cleaves the GPI moiety to the cell surface. As illustrated in Figure5A, cultures exposed to NMDA (12.5 μm for 24 hr) exhibited 50% neuronal death. The coincubation with rhGDNF (10 ng/ml) reduced by ∼50% the NMDA-induced neuronal death (Fig. 5A). The PIPLC pretreatment (0.3 IU/ml for 2 hr at 37°C) abolished the neuroprotective effect of rhGDNF (Fig.5A,B) (n = 12;p < 0.05). PIPLC pretreatment alone did not affect neuronal survival and was without effect on the NMDA-induced neuronal death (Fig. 5B). Overall, these results suggest that a GPI-linked protein mediates the neuroprotective activity of GDNF.
Fig. 5.

A GPI-linked protein mediates the neuroprotective activity of rhGDNF. A, Phase-contrast photomicrographs of mixed neuron–glia cortical cultures pretreated or not with PIPLC (0.3 IU/ml for 2 hr at 37°C) after NMDA application (12.5 μm) in the presence or absence of rhGDNF. Top left, Sham-washed cultures; top right, NMDA-treated cells; bottom left, NMDA-treated cells coincubated with rhGDNF (10 ng/ml); bottom right, cells pretreated with PIPLC and incubated with NMDA and rhGDNF.B, Neuronal death percentage was estimated by LDH release (mean ± SEM; n = 12) after a 24 hr exposure to NMDA (12.5 μm) in mixed neuron–glia cortical cultures in the presence (gray bars) or absence (white bars) of rhGDNF and pretreated or not pretreated with 0.3 IU/ml PIPLC for 2 hr at 37°C. * indicates significantly different from NMDA; # indicates significantly different from NMDA plus GDNF by ANOVA, followed by Bonferroni–Dunn's test (p < 0.05).
The neuroprotective effect of GDNF is independent of an antioxidant effect of GDNF treatment
The characteristic features of glutamate receptor activation are an intracellular accumulation of Ca2+ that induces the production of cytotoxic molecules, including nitric oxide (Dawson et al., 1991), and the production of free radicals (Coyle and Puttfarcken, 1993; Lafon-Cazal et al., 1993). To determine the mechanisms by which GDNF reduces the NMDA-induced neurotoxicity, we tested the influence of rhGDNF incubation against free radical-mediated injury.
To induce free radical injury, mixed cultures of cortical neurons and glia (14 DIV) were exposed continuously for 24 hr to 35 μm FeCl2. The Fe2+ treatment induces the production of hydroxyl free radicals through the Fenton reaction (Halliwell, 1992). As previously reported (Gwag et al., 1995), mixed cortical cell cultures exhibit a marked neuronal cell body swelling that resulted in a widespread neuronal death 24 hr later, whereas the glia remained intact. The coincubation of Trolox (100 μm), an antioxidant agent, totally blocked this Fe2+-mediated neuronal death, as reported by Chow et al. (1994). In contrast, cotreatment with 10 ng/ml rhGDNF did not reduce neuronal death (Fig. 6) (n = 12). This result suggests that the GDNF neuroprotective activity on NMDA-mediated neuronal death is not caused by a free radical scavenging effect.
Fig. 6.

GDNF does not protect against neuronal death induced by reactive oxygen species. Mixed neuron–glia cortical cultures were exposed for 24 hr to FeCl2 (35 μm) without (white bars) or with (gray bars) 10 ng/ml rhGDNF and with or without Trolox (100 μm). Medium LDH was assessed at the end of FeCl2 exposure (mean ± SEM; n = 12 cultures per condition). * indicates significantly different from FeCl2 by ANOVA, followed by Bonferroni–Dunn's test (p < 0.05).
The neuroprotective effect of GDNF results from a decrease of the NMDA-induced Ca2+ response in cortical neurons
Because the massive Ca2+ influx through the glutamate receptors has been shown to be a critical event in excitotoxic neuronal death (MacDermott et al., 1986), we examined the question of whether rhGDNF reduces NMDA and non-NMDA-induced Ca2+ influx through the use of fura-2 fluorescence videomicroscopy.
To investigate the effect of rhGDNF on the NMDA response, we performed a 10 min exposure to NMDA (100 μm) which produced a rapid increase in neuronal [Ca2+]i that quickly achieved a plateau. The incubation with rhGDNF (10 ng/ml for 45 min at 37°C) before NMDA application reduced the plateau value (Fig.7A,B) (three experiments; 60 cells; p < 0.05). This GDNF-mediated reduction of the NMDA-induced intracellular Ca2+ rise was abolished by a PIPLC pretreatment (0.3 IU/ml, 2 hr at 37°C) (Fig. 7B). To test whether the GDNF-mediated reduction in the intracellular Ca2+ rise might be the consequence of an enhanced Ca2+ buffering capacity, we studied the effect of rhGDNF pretreatment on the KCl-induced Ca2+ influx in the presence of an NMDA antagonist (MK-801; 10 μm), a paradigm that induces an intracellular Ca2+ rise independent of the NMDA receptor. A 50 mm KCl exposure produced a rapid increase in [Ca2+]i influx into neurons with a peak value similar to that observed after an NMDA exposure (Fig. 7C). The rhGDNF treatment before the KCl exposure failed to modify the rise in intracellular Ca2+ influx. Together, these results suggest that the GDNF neuroprotective effect might be attributable to a specific modulation of NMDA-induced Ca2+influx.
Fig. 7.
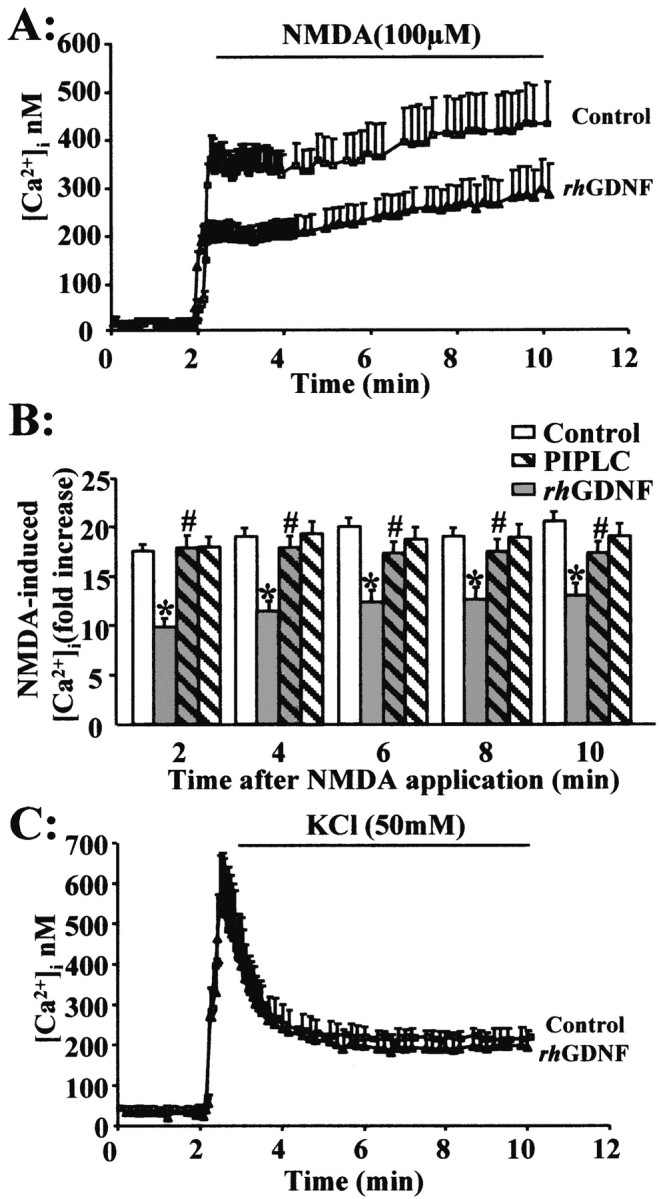
rhGDNF pretreatment reduces the NMDA-evoked calcium increase in cortical neurons. A, Intracellular free Ca2+([Ca2+]i) was measured using fura-2 fluorescence videomicroscopy. Neurons (13–14 DIV) were exposed to a serum-free medium for 45 min in the absence (control, vehicle treated) or presence of rhGDNF (10 ng/ml). Resting [Ca2+ ]i was recorded, and NMDA (100 μm) was applied. [Ca2+]i was measured and analyzed as described in Materials and Methods. B, Representative graph of NMDA-induced Ca2+ response in neurons pretreated with rhGDNF (gray bars) or vehicle (control; white bars) for 45 min. Time course of the effect of rhGDNF on the NMDA-induced [Ca2+] increase, with (hatched bars) or without (unhatched bars) pretreatment with 0.3 IU/ml PIPLC for 2 hr. Data are expressed as an NMDA-induced fold increase in [Ca2+]i. Results represent the mean ± SEM of three separate preparations (3 coverslips; 20 neurons being imaged per coverslip in a single microscopic field). * indicates significantly different from NMDA; # indicates significantly different from NMDA plus GDNF by ANOVA for repeated measures, followed by a Bonferroni–Dunn's test for multiple comparisons (p < 0.05). C, Neurons (13–14 DIV) were exposed to serum-free medium containing MK-801 (10 μm) for 45 min in the absence (control; vehicle treated) or presence of rhGDNF (10 ng/ml). Resting [Ca2+ ]i was recorded, and KCl (50 mm) was applied. Representative graph of KCl-induced Ca2+ response in neurons pretreated with rhGDNF or vehicle (control) for 45 min.
The ERKs signaling pathway is necessary for GDNF attenuation of NMDA-induced Ca2+ response
To determine whether the activation of the ERKs pathway by GDNF (Fig. 2B) mediates the neuroprotection against NMDA-induced neuronal death, we treated mixed cortical neuron–glia cultures with the MAPK kinase (MEK) inhibitor, U0126. This compound has been described as a specific inhibitor of MEK without any effect on other kinases (Favata et al., 1998). We showed that, although the addition of U0126 alone does not significantly affect cell viability in the presence or absence of NMDA, the neuroprotective activity of GDNF is blocked by the addition of U0126 (1 μm) (Fig. 8B) (n = 12; p < 0.05). Concurrently, an immunoblotting study revealed that a treatment with U0126 (1 μm) blocks the appearance of p-ERKs induced by a 15 min application of rhGDNF (10 ng/ml) (Fig. 8A) without modifying the level of p-P38 or p-JNK (data not shown). We have determined the p-ERK Western blot signal for GDNF in the presence of NMDA with or without U0126. In these experiments, we showed that NMDA plus GDNF induces a greater increase in p-ERKs than NMDA alone at 15 min (Fig. 8A) and 1 hr (data not shown). This effect is blocked by the addition of the inhibitor of ERK activation, U0126.
Fig. 8.
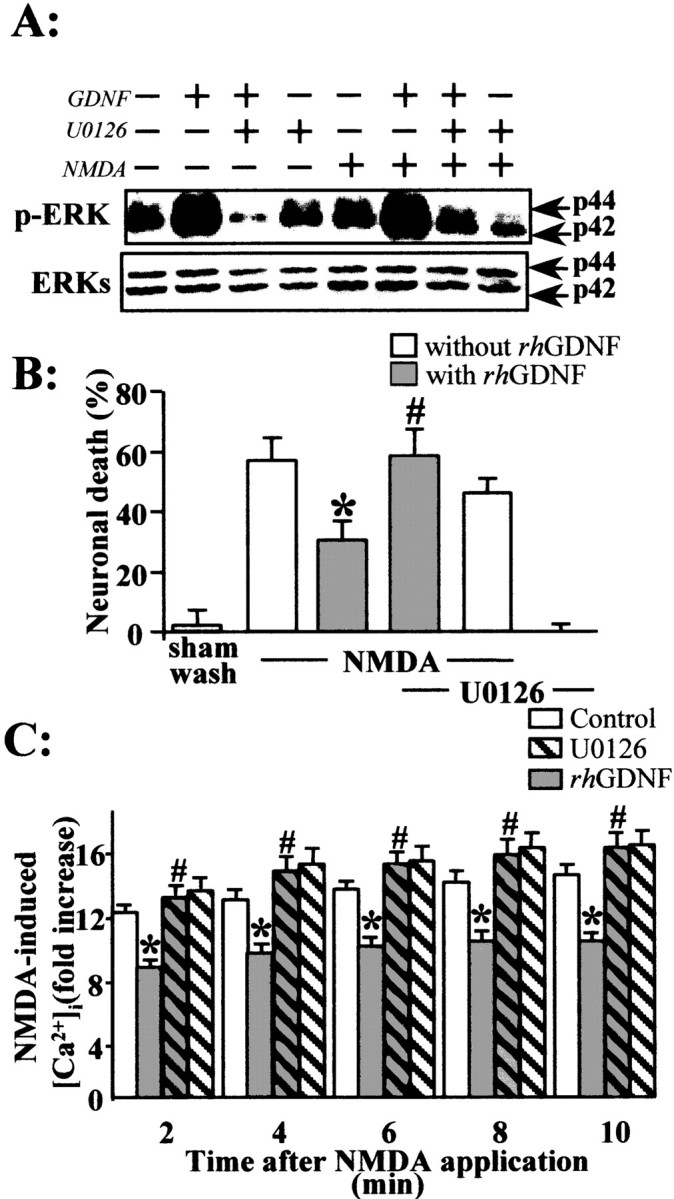
The MEK kinase inhibitor U0126 blocks the rhGDNF-induced reduction of NMDA-evoked calcium influx.A, Mouse primary cortical neurons were treated for 15 min with rhGDNF (10 ng/ml) in the presence of U0126 (1 μm) and with or without NMDA (12.5 μm). Aliquots of whole-cell lysates were separated by SDS-PAGE, transferred to membranes, and incubated with the anti-MAPK antibody (p-ERKs, ERKs). Representative immunoblots are shown, demonstrating the specific blockade of ERKs phosphorylation by U0126 treatment. B, Neuronal death percentage was estimated by LDH release (mean ± SEM;n = 12) after a 24 hr exposure to NMDA (12.5 μm) in mixed neuron–glia cortical cultures in the presence (gray bars) or absence (white bars) of rhGDNF and treated or not treated with 1 μm U0126. * indicates significantly different from NMDA alone; # indicates significantly different from NMDA plus GDNF by ANOVA, followed by Bonferroni–Dunn's test (p < 0.05). C, Calcium imaging was performed as described in the legend to Figure 7. Neurons (13–14 DIV) were exposed to serum-free medium for 45 min in the absence (control; vehicle treated; white bars) or presence (gray bars) of rhGDNF (10 ng/ml) and with (hatched bars) or without (unhatched bars) U0126 (1 μm). Time course of the effect of rhGDNF on the NMDA-induced [Ca2+] increase, with or without treatment with 1 μm of U0126. Data are expressed as an NMDA-induced fold [Ca2+]i. Results represent the mean ± SEM of three separate preparations (3 coverslips; 20 neurons being imaged per coverslip in a single microscopic field). * indicates significantly different from NMDA; # indicates significantly different from NMDA plus GDNF by ANOVA for repeated measures followed by a Bonferroni–Dunn's test for multiple comparisons (p < 0.05).
Finally, to further demonstrate that the ERKs phosphorylation induced by GDNF mediates the neuroprotective activity of GDNF, we studied the effect of the treatment with U0126 on the intracellular Ca2+ influx induced by NMDA application (100 μm). Although U0126 alone (1 μm for 45 min before NMDA application) did not modify the NMDA-induced Ca2+ response, this MEK inhibitor totally abolished the reduction of the NMDA-induced intracellular Ca2+ influx that followed incubation with rhGDNF (10 ng/ml for 45 min) (Fig. 8C).
DISCUSSION
Among the multiple roles played by TGF-β in the CNS, recent studies have suggested that it could be synthesized in response to different types of injury and act as an endogenous neuroprotective factor (Krieglstein and Krieglstein, 1998; Ruocco et al., 1999). According to this hypothesis, GDNF, a distant member of the TGF-β family, the expression of which is increased in the cortex after MCAo (Abe et al., 1997), has been identified as a potent neuroprotective agent. Indeed, various studies have revealed that the application of GDNF reduces the extent of neuronal injury induced by MCAo in rats (Abe et al., 1997; Wang et al., 1997; Kitagawa et al., 1998). However, little is known about the molecular mechanisms involved in this effect. Here, we characterize the influence of GDNF on cortical neurons subjected to two paradigms of injury (necrosis and apoptosis) (Choi, 1992) that have been identified during cerebral ischemia and determine the intracellular mechanism(s) involved. The major findings are as follows: (1) in cortical neurons, GDNF exerts a selective neuroprotective activity against NMDA-induced neuronal death; (2) this neuroprotective activity of GDNF is caused by a reduction of the NMDA-induced Ca2+ influx; and (3) the neuroprotective effect of GDNF involves the activation of ERKs.
Cortical neurons and astrocytes express functional GDNF receptor complex
To investigate the role of GDNF during ischemic brain injury, we used cortical murine cultures (Goldberg and Choi, 1993). First, we checked whether this culture system was suitable for the study of the effect of GDNF. We demonstrated that both cultured neurons and astrocytes express the mRNA and the protein for both GDNF and its receptor complex, GFRα-1 and c-Ret. The profile of expression identified in cortical cultures is similar to that observed in the adult murine cerebral cortex. These data are in accordance with previous studies (Kitagawa et al., 1999; Wei et al., 2000) and validate the use of cortical murine cultures to study the effect of GDNF. Our results also suggest that cortical neurons and astrocytes express the receptor complex necessary for the transduction of GDNF signaling. This possibility was confirmed by showing that a GDNF application induces a phosphorylation-mediated activation of ERKs in both cortical astrocytes and neurons.
GDNF reduces NMDA-induced neuronal death
To investigate the influence of GDNF during cerebral ischemia, cultured cortical neurons and astrocytes were submitted to apoptotis or excitotoxicity. To examine the effects of GDNF on apoptosis, cortical cell cultures were exposed to two apoptotic challenges (serum deprivation and staurosporine exposure). In both cases, the application of rhGDNF failed to prevent neuronal death, whereas cycloheximide succeeded. Because various reports have described survival-promoting properties of GDNF on dopaminergic and peripheral sensory neurons (Lin et al., 1993; Henderson et al., 1994; Yan et al., 1995), this lack of neuroprotective effect of rhGDNF against apoptosis in cortical cultures is surprising. However, Sawada et al. (2000)recently described a similar lack of neuroprotection exerted by GDNF on mesencephalic neurons exposed to serum deprivation. To the best of our knowledge, there is no direct proof of an antiapoptotic effect of GDNF on cortical neurons in the literature. In vivo, although the neuroprotective effect of GDNF evidenced in rats subjected to focal cerebral ischemia was accompanied by a reduction of apoptotic markers, there was also an important reduction in edema formation, one of the landmarks of the necrotic pathway. These authors suggested that GDNF, by attenuating necrosis, could reduce the severity of the insult and, consequently, minimize the appearance of apoptotic markers (Abe et al., 1997). For these reasons, we determined the influence of rhGDNF incubation on cortical neurons exposed to an excitotoxic insult. Accordingly, cortical neuronal cultures were exposed to different glutamatergic ionotropic receptor agonists. Although the application of rhGDNF failed to prevent AMPA/kainate receptor-mediated neuronal death, it did protect neurons against NMDA receptor-induced necrosis. Astrocytes do not seem to be necessary for the antiexcitotoxic activity of GDNF, because the beneficial effects against NMDA-induced neuronal death were still observed in pure neuronal cultures. We thus validated the previous observation of an antiexcitotoxic effect of GDNF demonstrated in a model of excitotoxicity induced by injection of glutamatergic agonists (Martin et al., 1995; Perez-Navarro et al., 1996; Ho et al., 2000). We demonstrated that rhGDNF directly targets neurons to protect them against NMDA-induced insult.
We further characterized the influence of GDNF on excitotoxicity by demonstrating that pretreatment with PIPLC, an enzyme that specifically cleaves GPI linkages, blocks the beneficial effect of rhGDNF on NMDA-induced neuronal death. These results underline the implication of a GPI-anchored protein in the neuroprotection exerted by GDNF against excitotoxicity. The GDNF receptor, GFRα-1, is a protein attached to the cell surface by a GPI link. Based on the above, it can be suggested that GFRα-1 is necessary for GDNF to exert its neuroprotective activity against NMDA-induced neuronal death.
Molecular mechanisms of GDNF neuroprotective activity
Among the characteristic features of NMDA receptor activation, two have been particularly implicated in excitotoxicity: first, the initial rise in intracellular calcium concentrations (MacDermott et al., 1986); and second, the consequent production of free radicals (Coyle and Puttfarcken, 1993; Lafon-Cazal et al., 1993). To investigate the mechanisms by which GDNF is neuroprotective, we tested the effect of rhGDNF application against free radical-induced neuronal death. rhGDNF failed to exert any effect against Fe2+-induced neuronal death, a paradigm of neuronal death totally blocked by the coincubation of the free radical scavenger: the cell permeant derivative of vitamin E, Trolox. The absence of effect of GDNF treatment on Fe2+-induced neurotoxicity demonstrates that GDNF is not neuroprotective by directly scavenging free radicals. However, we cannot exclude that GDNF is protective by reducing intracellular free radical production.
Our next step was to analyze whether rhGDNF incubation modulates calcium influx induced by the application of NMDA. When rhGDNF was applied to cortical neurons, the Ca2+influx induced by NMDA was reduced. Even after washing out GDNF, the NMDA receptor function is reduced, suggesting that the GDNF application triggered an intracellular signaling responsible for the modulation of the NMDA receptor function. This effect likely results from a reduction of NMDA receptor activity rather than from an enhanced Ca2+ buffering or extrusion, because the same rhGDNF pretreatment did not modify KCl-induced Ca2+ influx. These results are in accordance with an increasing set of data that show a tight regulation of glutamatergic receptor function by growth factors (Cheng et al., 1995; Boxer et al., 1999). Recent data have demonstrated that the activation of tyrosine kinase-coupled platelet-derived growth factor receptors produces a long-lasting inhibition of NMDA receptor function (Valenzuela et al., 1996). Because the extent of neuronal injury is directly correlated with the intracellular concentration of Ca2+ (Hartley et al., 1993), our results may reflect the mechanism through which GDNF protects neurons against the toxicity mediated by the NMDA receptor overstimulation.
Finally, we investigated the intracellular signaling pathways involved in the neuroprotective effect of GDNF. Many effects of GDNF are known to be mediated by the activation of the Ras-MAPK pathway (Messer et al., 1999; Ren et al., 1999). Accordingly, we hypothesized that the neuroprotective effect of GDNF might be mediated by the activation of the ERKs pathway. To test this, we blocked the ERKs pathway by using a selective inhibitor of MEK, U0126. We demonstrated that the coapplication of U0126 blocks the activation of the ERKs and the neuroprotection induced by GDNF on an NMDA-mediated insult, as well as the GDNF-related reduction of the NMDA-induced calcium influx. The present observation that activation of ERKs may lead to neuronal survival is consistent with the previous report of Singer et al. (1999)in which the MAPK pathway mediates estrogen-induced neuroprotection after glutamate toxicity in primary cortical neurons. Together, these results demonstrate that GDNF, through the activation of the ERKs pathway, modulates the level of activity of the NMDA receptors. This modulation leads to a reduction of Ca2+influx, which consequently helps to restore Ca2+ homeostasis and prevents neuronal death.
In conclusion, the present study reveals that the binding of GDNF to its neuronal receptors induces the activation of ERKs, leading to a reduction of the NMDA-mediated Ca2+influx. Further investigations are required to determine the link between ERKs activation and the down-modulation of NMDA receptor activity. Regarding the neuroprotective profile of GDNF on apoptosis and necrosis described in this study, it can be suggested that to be beneficial in the treatment of cerebral ischemia, GDNF would have to be administered in the early phase of stroke, when excitotoxicity is predominant. An increase in GDNF expression has been detected in both CNS and PNS, not only in several paradigms of acute injury such as cerebral ischemia (Abe and Hayashi, 1997) but also in models reproducing neurological diseases such as Huntington's disease and amyotrophic lateral sclerosis (Yamamoto et al., 1998; Grundstrom et al., 1999). Consequently, the inhibitory actions of GDNF on NMDA receptor function, as evidenced in the present study, could represent an efficient way to reduce the extent of excitotoxic neuronal death in these pathophysiological situations.
Footnotes
This work was supported by grants from the Centre National de la Recherche Scientifique and the University of Caen. Doctoral bursaries were provided by Comissariat à l'Energie Atomique (to O.N.), the Regional Council of Lower Normandy (to F.D.), and the Ministry of Education and Research (to C.A.). The investigation was performed within the framework of the Functional Neuroimaging Institute (IFR 47).
Correspondence should be addressed to Drs. Alain Buisson and Denis Vivien, Université de Caen-Centre National de la Recherche Scientifique, Unité Mixte de Recherche 6551, Cyceron, 14074 Caen Cedex, France. E-mail: a.buisson@neuro.unicaen.fr ord.vivien@neuro.unicaen.fr.
REFERENCES
- 1.Abe K, Hayashi T. Expression of the glial cell line-derived neurotrophic factor gene in rat brain after transient MCA occlusion. Brain Res. 1997;776:230–234. doi: 10.1016/s0006-8993(97)01041-x. [DOI] [PubMed] [Google Scholar]
- 2.Abe K, Hayashi T, Itoyama Y. Amelioration of brain edema by topical application of glial cell line-derived neurotrophic factor in reperfused rat brain. Neurosci Lett. 1997;231:37–40. doi: 10.1016/s0304-3940(97)00517-x. [DOI] [PubMed] [Google Scholar]
- 3.Boxer AL, Moreno H, Rudy B, Ziff EB. FGF-2 potentiates Ca(2+)-dependent inactivation of NMDA receptor currents in hippocampal neurons. J Neurophysiol. 1999;82:3367–3377. doi: 10.1152/jn.1999.82.6.3367. [DOI] [PubMed] [Google Scholar]
- 4.Buisson A, Nicole O, Docagne F, Sartelet H, MacKenzie ET, Vivien D. Up-regulation of a serine protease inhibitor in astrocytes mediates the neuroprotective activity of transforming growth factor beta1. FASEB J. 1998;12:1683–1691. [PubMed] [Google Scholar]
- 5.Cheng B, Furukawa K, O'Keefe JA, Goodman Y, Kihiko M, Fabian T, Mattson MP. Basic fibroblast growth factor selectively increases AMPA-receptor subunit GluR1 protein level and differentially modulates Ca2+ responses to AMPA and NMDA in hippocampal neurons. J Neurochem. 1995;65:2525–2536. doi: 10.1046/j.1471-4159.1995.65062525.x. [DOI] [PubMed] [Google Scholar]
- 6.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 7.Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 8.Chow HS, Lynch JJ, 3rd, Rose K, Choi DW. Trolox attenuates cortical neuronal injury induced by iron, ultraviolet light, glucose deprivation, or AMPA. Brain Res. 1994;639:102–108. doi: 10.1016/0006-8993(94)91769-8. [DOI] [PubMed] [Google Scholar]
- 9.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 10.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durbec P, Marcos-Gutierrez CV, Kilkenny C, Grigoriou M, Wartiowaara K, Suvanto P, Smith D, Ponder B, Costantini F, Saarma M. GDNF signalling through the Ret receptor tyrosine kinase. Nature. 1996;381:789–793. doi: 10.1038/381789a0. [DOI] [PubMed] [Google Scholar]
- 12.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 13.Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grundstrom E, Askmark H, Lindeberg J, Nygren I, Ebendal T, Aquilonius SM. Increased expression of glial cell line-derived neurotrophic factor mRNA in muscle biopsies from patients with amyotrophic lateral sclerosis. J Neurol Sci. 1999;162:169–173. doi: 10.1016/s0022-510x(98)00333-5. [DOI] [PubMed] [Google Scholar]
- 15.Gwag BJ, Koh JY, Chen MM, Dugan LL, Behrens MM, Lobner D, Choi DW. BDNF or IGF-I potentiates free radical-mediated injury in cortical cell cultures. NeuroReport. 1995;7:93–96. [PubMed] [Google Scholar]
- 16.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 17.Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Simmons L, Moffet B, Vandlen RA. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266:1062–1064. doi: 10.1126/science.7973664. [DOI] [PubMed] [Google Scholar]
- 19.Ho TW, Bristol LA, Coccia C, Li Y, Milbrandt J, Johnson E, Jin L, Bar-Peled O, Griffin JW, Rothstein JD. TGF beta trophic factors differentially modulate motor axon outgrowth and protection from excitotoxicity. Exp Neurol. 2000;161:664–675. doi: 10.1006/exnr.1999.7290. [DOI] [PubMed] [Google Scholar]
- 20.Jing S, Wen D, Yu Y, Holst PL, Luo Y, Fang M, Tamir R, Antonio L, Hu Z, Cupples R, Louis JC, Hu S, Altrock BW, Fox GM. GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell. 1996;85:1113–1124. doi: 10.1016/s0092-8674(00)81311-2. [DOI] [PubMed] [Google Scholar]
- 21.Kitagawa H, Hayashi T, Mitsumoto Y, Koga N, Itoyama Y, Abe K. Reduction of ischemic brain injury by topical application of glial cell line-derived neurotrophic factor after permanent middle cerebral artery occlusion in rats. Stroke. 1998;29:1417–1422. doi: 10.1161/01.str.29.7.1417. [DOI] [PubMed] [Google Scholar]
- 22.Kitagawa H, Sasaki C, Sakai K, Mori A, Mitsumoto Y, Mori T, Fukuchi Y, Setoguchi Y, Abe K. Adenovirus-mediated gene transfer of glial cell line-derived neurotrophic factor prevents ischemic brain injury after transient middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 1999;19:1336–1344. doi: 10.1097/00004647-199912000-00007. [DOI] [PubMed] [Google Scholar]
- 23.Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 24.Koh JY, Wie MB, Gwag BJ, Sensi SL, Canzoniero LM, Demaro J, Csernansky C, Choi DW. Staurosporine-induced neuronal apoptosis. Exp Neurol. 1995;135:153–159. doi: 10.1006/exnr.1995.1074. [DOI] [PubMed] [Google Scholar]
- 25.Krieglstein K, Krieglstein J. Neuroprotection signal transduction (Matson MP, ed), pp 119–144. Humana; Totowa, NJ: 1998. [Google Scholar]
- 26.Krieglstein K, Suter-Crazzolara C, Fischer WH, Unsicker K. TGF-beta superfamily members promote survival of midbrain dopaminergic neurons and protect them against MPP+ toxicity. EMBO J. 1995;14:736–742. doi: 10.1002/j.1460-2075.1995.tb07052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krieglstein K, Henheik P, Farkas L, Jaszai J, Galter D, Krohn K, Unsicker K. Glial cell line-derived neurotrophic factor requires transforming growth factor-beta for exerting its full neurotrophic potential on peripheral and CNS neurons. J Neurosci. 1998;18:9822–9834. doi: 10.1523/JNEUROSCI.18-23-09822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 29.Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- 30.Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002–2008. doi: 10.1161/01.str.24.12.2002. [DOI] [PubMed] [Google Scholar]
- 31.MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 32.MacManus JP, Buchan AM, Hill IE, Rasquinha I, Preston E. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neurosci Lett. 1993;164:89–92. doi: 10.1016/0304-3940(93)90864-h. [DOI] [PubMed] [Google Scholar]
- 33.Martin D, Miller G, Rosendahl M, Russell DA. Potent inhibitory effects of glial derived neurotrophic factor against kainic acid mediated seizures in the rat. Brain Res. 1995;683:172–178. doi: 10.1016/0006-8993(95)00369-2. [DOI] [PubMed] [Google Scholar]
- 34.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM., Jr Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Messer CJ, Son JH, Joh TH, Beck KD, Nestler EJ. Regulation of tyrosine hydroxylase gene transcription in ventral midbrain by glial cell line-derived neurotrophic factor. Synapse. 1999;34:241–243. doi: 10.1002/(SICI)1098-2396(19991201)34:3<241::AID-SYN8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 36.Perez-Navarro E, Arenas E, Reiriz J, Calvo N, Alberch J. Glial cell line-derived neurotrophic factor protects striatal calbindin-immunoreactive neurons from excitotoxic damage. Neuroscience. 1996;75:345–352. doi: 10.1016/0306-4522(96)00336-3. [DOI] [PubMed] [Google Scholar]
- 37.Pratt BM, McPherson JM. TGF-beta in the central nervous system: potential roles in ischemic injury and neurodegenerative diseases. Cytokine Growth Factor Rev. 1997;8:267–292. doi: 10.1016/s1359-6101(97)00018-x. [DOI] [PubMed] [Google Scholar]
- 38.Ren RF, Lah JJ, Diehlmann A, Kim ES, Hawver DB, Levey AI, Beyreuther K, Flanders KC. Differential effects of transforming growth factor-beta(s) and glial cell line-derived neurotrophic factor on gene expression of presenilin-1 in human post-mitotic neurons and astrocytes. Neuroscience. 1999;93:1041–1049. doi: 10.1016/s0306-4522(99)00215-8. [DOI] [PubMed] [Google Scholar]
- 39.Rose K, Goldberg MP, Choi DW. In vitro biological methods (Tyson CA, Frazier JM, eds), pp 46–60. Academic; San Diego: 1993. [Google Scholar]
- 40.Ruocco A, Nicole O, Docagne F, Ali C, Chazalviel L, Komesli S, Yablonsky F, Roussel S, MacKenzie ET, Vivien D, Buisson A. A transforming growth factor-beta antagonist unmasks the neuroprotective role of this endogenous cytokine in excitotoxic and ischemic brain injury. J Cereb Blood Flow Metab. 1999;19:1345–1353. doi: 10.1097/00004647-199912000-00008. [DOI] [PubMed] [Google Scholar]
- 41.Sawada H, Ibi M, Kihara T, Urushitani M, Nakanishi M, Akaike A, Shimohama S. Neuroprotective mechanism of glial cell line-derived neurotrophic factor in mesencephalic neurons. J Neurochem. 2000;74:1175–1184. doi: 10.1046/j.1471-4159.2000.741175.x. [DOI] [PubMed] [Google Scholar]
- 42.Schaar DG, Sieber BA, Dreyfus CF, Black IB. Regional and cell-specific expression of GDNF in rat brain. Exp Neurol. 1993;124:368–371. doi: 10.1006/exnr.1993.1207. [DOI] [PubMed] [Google Scholar]
- 43.Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci. 1999;19:2455–2463. doi: 10.1523/JNEUROSCI.19-07-02455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soler RM, Dolcet X, Encinas M, Egea J, Bayascas JR, Comella JX. Receptors of the glial cell line-derived neurotrophic factor family of neurotrophic factors signal cell survival through the phosphatidylinositol 3-kinase pathway in spinal cord motoneurons. J Neurosci. 1999;19:9160–9169. doi: 10.1523/JNEUROSCI.19-21-09160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Treanor JJ, Goodman L, de Sauvage F, Stone DM, Poulsen KT, Beck CD, Gray C, Armanini MP, Pollock RA, Hefti F, Phillips HS, Goddard A, Moore MW, Buj-Bello A, Davies AM, Asai N, Takahashi M, Vandlen R, Henderson CE, Rosenthal A. Characterization of a multicomponent receptor for GDNF. Nature. 1996;382:80–83. doi: 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- 46.Trupp M, Arenas E, Fainzilber M, Nilsson AS, Sieber BA, Grigoriou M, Kilkenny C, Salazar-Grueso E, Pachnis V, Arumae U. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature. 1996;381:785–789. doi: 10.1038/381785a0. [DOI] [PubMed] [Google Scholar]
- 47.Valenzuela CF, Xiong Z, MacDonald JF, Weiner JL, Frazier CJ, Dunwiddie TV, Kazlauskas A, Whiting PJ, Harris RA. Platelet-derived growth factor induces a long-term inhibition of N-methyl-d-aspartate receptor function. J Biol Chem. 1996;271:16151–16159. doi: 10.1074/jbc.271.27.16151. [DOI] [PubMed] [Google Scholar]
- 48.Van Weering DJ, Moen TC, Braakman I, Baas PD, Bos JL. Expression of the receptor tyrosine kinase ret on the plasma membrane is dependent on calcium. J Biol Chem. 1998;20:12077–12081. doi: 10.1074/jbc.273.20.12077. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Lin SZ, Chiou AL, Williams LR, Hoffer BJ. Glial cell line-derived neurotrophic factor protects against ischemia-induced injury in the cerebral cortex. J Neurosci. 1997;17:4341–4348. doi: 10.1523/JNEUROSCI.17-11-04341.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei G, Wu G, Cao X. Dynamic expression of glial cell line-derived neurotrophic factor after cerebral ischemia. NeuroReport. 2000;11:1177–11783. doi: 10.1097/00001756-200004270-00007. [DOI] [PubMed] [Google Scholar]
- 51.Worby CA, Vega QC, Zhao Y, Chao HHJ, Seasholtz AF, Dixon JE. Glial cell line-derived neurotrophic factor signals through the RET receptor and activates mitogen-activated protein kinase. J Biol Chem. 1996;271:23619–23622. doi: 10.1074/jbc.271.39.23619. [DOI] [PubMed] [Google Scholar]
- 52.Yamamoto M, Mitsuma N, Ito Y, Hattori N, Nagamatsu M, Li M, Mitsuma T, Sobue G. Expression of glial cell line-derived neurotrophic factor and GDNFR-alpha mRNAs in human peripheral neuropathies. Brain Res. 1998;809:175–181. doi: 10.1016/s0006-8993(98)00858-0. [DOI] [PubMed] [Google Scholar]
- 53.Yan Q, Matheson C, Lopez OT. In vivo neurotrophic effects of GDNF on neonatal and adult facial motor neurons. Nature. 1995;373:341–344. doi: 10.1038/373341a0. [DOI] [PubMed] [Google Scholar]