

Abstract
Unitary IPSCs elicited by fast-spiking (FS) interneurons in layer V pyramidal cells of the neocortex were studied by means of dual whole-cell recordings in acute slices. FS to pyramidal cell unitary IPSCs were depressed by (RS)-S-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) (ATPA), a kainate (KA) receptor agonist, and by the endogenous agonist l-glutamate in the presence of AMPA, NMDA, mGluR, and GABAB receptor antagonists. This effect was accompanied by an increase in failure rate of synaptic transmission, in the coefficient of variation, and in the paired pulse ratio, indicating a presynaptic origin of the IPSC depression. Pairing the activation of the presynaptic neuron with a depolarization of the postsynaptic cell mimicked the decrease of unitary IPSCs, and this effect persisted when postsynaptic sodium action potentials were blocked with the local anesthetic QX314. The effects of ATPA, glutamate, and of the pairing protocol were almost totally blocked by CNQX. These data suggest that KA receptors located on presynaptic FS cell terminals decrease the release of GABA and can be activated by glutamate released from the somatodendritic compartment of the postsynaptic pyramidal cells.
Keywords: GYKI, kainate receptors, ATPA, neocortical pyramidal cells, neocortical fast spiking interneurons, IPSC, EPSC
Inhibitory interneurons play an important role in regulating excitation in cortical circuits. This regulation is disrupted during epileptic seizures, which can be induced by activating kainate (KA) receptors with exogenously applied kainate (Sloviter and Damiano, 1981; Westbrook and Lothman, 1983; Fisher and Alger, 1984). Therefore, KA receptors may play a major role in the balance between excitation and inhibition.
Almost all of the fast glutamatergic excitatory synaptic transmission is mediated by AMPA and NMDA receptors, and only recently it has been shown that a small contribution to excitatory input made to specific targets is mediated by KA receptors (Castillo et al., 1997; Vignes and Collinridge, 1997; Cossart et al., 1998, Frerking et al., 1998;DeVries and Schwartz, 1999; Kidd and Isaac, 1999; Li et al., 1999). KA receptors have also been proposed to modulate synaptic excitation (Kamiya and Ozawa, 1998, 2000; Chittajallu et al., 1999; Kerchner et al., 2001) and inhibition (Clarke et al., 1997; Rodriguez-Moreno et al., 1997; Rodriguez-Moreno and Lerma, 1998). In the hippocampus, the inference that presynaptic KA receptors regulate inhibition was made on the basis that electrically elicited IPSCs could be depressed with either exogenous application of kainate (Clarke et al., 1997;Rodriguez-Moreno et al., 1997, 2000; Rodriguez-Moreno and Lerma, 1998;Cossart et al., 1998; Frerking et al., 1998, 1999; Bureau et al., 1999) or by a direct activation of glutamatergic pathways (Min et al., 1999). However, some interneurons also express functional somatodendritic KA receptors, which after activation will enhance the spontaneous firing rates of these cells, therefore increasing the frequency of spontaneous IPSCs (Frerking et al., 1998, 1999; Bureau et al., 1999). This has lead to the proposal that the depression of stimulus-elicited IPSCs during application of KA receptor agonists could be attributable to secondary effects of the excess GABA acting on both presynaptic and postsynaptic cells rather than to the activation of presynaptic KA receptors (Frerking et al., 1998, 1999). One way to demonstrate the involvement of KA receptors on presynaptic terminals was to look at action potential-independent miniature IPSCs. However, although some studies have reported a decrease in the frequency of miniature IPSC (Rodriguez-Moreno et al., 1997), others have disagreed (Frerking et al., 1998, 1999; Bureau et al., 1999). Thus, the debate of whether KA receptors are located presynaptically to modulated inhibition remains in doubt.
To determine whether KA receptors located presynaptically modulate GABA release and the intrinsic mechanism governing any modulation, paired whole-cell recordings between fast-spiking GABAergic interneurons and pyramidal cells were performed in the motor cortex of acute slices of 17- to 22-d-old rats. The data obtained suggest that a somatodendritic release of a retrograde messenger, the most likely candidate being glutamate, activates presynaptic KA receptors located on presynaptic FS cells to decrease GABA release, thus inhibiting unitary IPSCs.
MATERIALS AND METHODS
Slice preparation. Wistar rats (17–22 postnatal days) were anesthetized intraperitoneally with ketamine (65 mg/kg) and xylazine (14 mg/kg). After decapitation, the brain was rapidly removed under ice-cold conditions, and 300-μm-thick coronal sections of cerebral motor cortex were obtained. The slices were incubated for 1 hr in a physiological extracellular saline solution containing (in mm): 121 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 20 glucose, and 5 pyruvate, and bubbled perfused with a mixture of 95% O2 and 5% CO2. For recordings, they were transferred to a chamber and perfused at 1–2 ml/min with the same physiological extracellular solution at room temperature (20–24°C).
Electrophysiological recordings. Simultaneous dual whole recordings were made in voltage clamp in layer V of rat motor cortex from FS cells somata synaptically connected with pyramidal cells. The cell types were selected using videomicroscopy with Nomarski optics under infrared illumination. Presynaptic cells were selected with round somata and further characterized from their firing properties recorded in current-clamp fast mode of the Axopatch 200A amplifier (see Cauli et al., 1997 for criteria used for classification). Experiments were conducted at room temperature with patch pipettes (resistance 3–5 MΩ) pulled from borosilicate glass tubing and filled with an internal solution containing (in mm): 144 K-gluconate, 3 MgCl2, 0.2 EGTA, 10 HEPES, 2 Na2-ATP, 0.2 Na2-GTP, and 0.02% w/v of biocytin, pH 7.2–7.4, 300 mOsm. Both cells were clamped at −70 mV initially, and all membrane potentials were corrected for a junction potential of −10 mV. Series resistance, which was monitored continuously, ranged between 10 and 30 MΩ and was not compensated.
Data were collected in the presence of bath-applied GYKI 53655 (a gift from EGIS, Hungary) and d-AP-5 (50 μm), MCPG (1 mm), CPPG (100 μm), and CPG55845 (100 μm) to block AMPA, NMDA, mGluR, and GABAB receptors. In experiments aimed at measuring miniature IPSCs, tetrodotoxin (TTX; 1 μm) was also added to the extracellular saline solution, and K-gluconate was replaced by K-chloride in the internal solution. In experiments in which l-glutamate was used as an agonist, the following drugs, naloxone (100 μm), atropine sulfate (50 μm), and DPCPX (1 μm) were added to the drug cocktail mentioned above to block opioid, muscarinic, and adenosine receptors. During paired recordings, unitary IPSCs were elicited by double presynaptic action potentials (APs) with an interspike interval of 50 msec or a train of APs by eliciting depolarizing pulses of 50 mV lasting for 0.5 msec. Single sweep data were collected at rate of 0.2 Hz using a patch-clamp amplifier (Axopatch 200A; Axon Instruments, Foster City, CA) and filtered at 5 and 2 kHz.
Conditioning protocol. In control conditions the postsynaptic membrane potential was stepped for 1.5 sec to −40 mV to increase chloride driving force, and two presynaptic APs were elicited with an interval of 50 msec before the end of the postsynaptic step. During conditioning, the postsynaptic membrane potential was depolarized to −10 mV for 200 msec and maintained at −40 mV for 1.5 sec. The two postsynaptic APs were elicited only 900 msec after the end of the step of −10 mV to allow the complete relaxation of the postsynaptic membrane currents.
Data analysis. Digitized data were acquired and analyzed using Acquis1 software (Gérard Sadoc, Centre National de la Recherche Scientifique, Gif-sur-Yvette, France). Averages are given as means ± SD from 50–150 sweeps. The rise times (10–90%) and the time constant (fitted with a single exponential) were calculated from average IPSCs elicited by single AP. Apparent failures of synaptic transmission were measured manually. IPSC amplitude in the range of the synaptic noise were taken as failures. The wraparound procedure for measuring synaptic failures was also used, and the results obtained did not deviate >2% from the manual analysis. Statistical significance was analyzed by Student's paired t test. For coefficient of variation (CV) analysis, mean IPSC amplitudes and CVs were calculated from single sweep data of 50–150 sweeps, which had reached steady state in the condition in which they were studied.
Morphology. Slices containing biocytin-filled cells were fixed overnight in 4% paraformaldehyde and 0.2% glutaraldehyde in 0.1m phosphate buffer (PB) at 4°C. Then, they were rinsed extensively with PB, including an intermediate step with 1% H2O2 (in PB) to block endogenous peroxidase activity. Next, they were incubated in a cryoprotectant (25% saccharose and 10% glycerol in 0.01m PB) for 1 hr. Then, the slices were freeze-thawed three times over liquid nitrogen. After three rinses in PB, the slices were incubated overnight with ABC (1:200; Vector Laboratories) at 4°C. Thereafter, 1 mg/ml 3,3′ diaminobenzidine (DAB; Sigma) was preincubated for 10 min, and then the peroxidase was revealed by starting the reaction with 0.01% H2O2. The reaction was stopped by rinsing with PB. The slices were intensified with 1% OsO4 (in PB) for 1 hr and dehydrated in an ascending series of ethanol (including contrasting with 1% uranyl acetate in 70% ethanol for 45 min). After immersion in propylene oxide, the sections were flat-embedded in Durcupan ACM (Fluka, Buchs, Switzerland) and cured for 16 hr at 60°C.
The slices were examined with a light microscope (Eclipse 800; Nikon, Ratingen, Germany) connected to the computerized reconstruction system Neurolucida (Microbrightfield, Colchester, VT). The soma and the dendrites of cells, which appeared to be sufficiently well labeled, were three-dimensionally and quantitatively reconstructed. Some neurons were digitally photographed by using the capability of Neurolucida to acquire Z-scans.
RESULTS
Characterization of unitary connections between fast-spiking cells and pyramidal cells in layer V of the motor cortex
In layer V of the rat motor cortex, presynaptic FS interneurons were targeted under infrared DIC, with small, usually round somata (11.0–16.4 μm, largest diameter) close to a pyramidal cell. The firing characteristics of these cells were immediately recorded in current clamp at a membrane potential of −70 mV and resembled classical FS behavior, an example of which is shown in Figure1C (see also Connors and Gutnick, 1990; Cauli et al., 1997; Angulo et al., 1999b). The APs of FS cells displayed an average amplitude of 69 ± 9.5 mV (from threshold to peak), were narrow (average, 0.5 ± 0.16 msec at half amplitude), and terminated with a deep, fast spike afterhyperpolarization (−17 ± 03.4 mV from spike threshold to peak and 6 ± 2 msec; width at half amplitude), and trains of spikes showed little accommodation or adaptation. The FS cells had low input resistance (239 ± 32 MΩ) and brief time constant (τ = 12.7 ± 4.7 msec) and a linear current–voltage relationship, at a potential more negative than resting membrane potential.
Fig. 1.
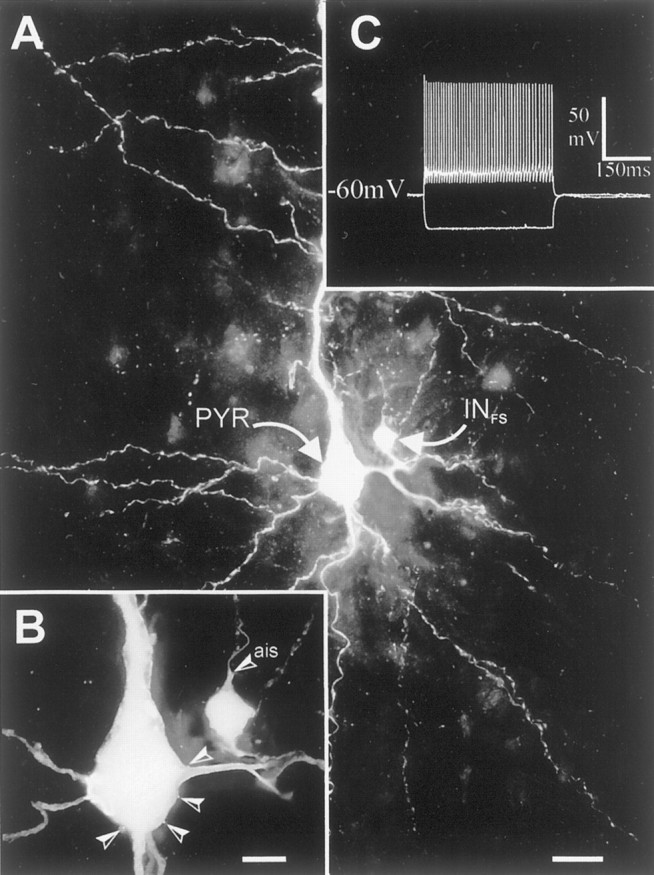
Extended-focus view reconstructions of a fast-spiking interneuron (INFS) and pyramidal cell (PYR) located in layer V of rat neocortex. A, Synaptically coupled FS interneuron to pyramidal cell. Note the small multipolar interneuron that gives rise to a dense axonal plexus (punctuate labeling) around the extremely large pyramidal cell. B, Higher magnification of the two cells. The axon initial segment (ais) of the interneuron is pointing toward the pial surface and emits a collateral, which is contacting the pyramidal cell four times (arrowheads), forming putative somatic synapses. This is an extended-focus view covering 50 μm in depth. C, Voltage responses of the presynaptic FS cell to −200 and +100 pA. The synaptic physiology of this pair is shown in Figure 2. Scale bars: A, 25 μm;B, 10 μm.
From 22 FS cells recorded, eight were recovered histologically. Their somata were round or oval and extended 1–6 primary smooth and varicose dendrites. The axons of these cells ramified with a preferential horizontal elongation in layer V with the postsynaptic pyramidal cell soma always located in the core terminal field. The postsynaptic pyramidal cells consistently possessed an apical dendrite with a terminal tuft in layer I. Figure 1 illustrates an example of an “extended-focus view” reconstruction of an FS and pyramidal cell, which were synaptically connected. The insert (C) shows the intrinsic membrane properties of this FS cell.
Unitary IPSCs were elicited in postsynaptic pyramidal cells (n = 22) by single, double, or trains of presynaptic FS cell action potentials at 0.2 Hz (Fig.2B). The average amplitude, including failures of the unitary IPSCs ranged from 13 to 172 pA at a postsynaptic membrane potential of −30 mV. All FS to pyramidal cell connection studied showed paired pulse and brief train depression. The example shown in Figure 2A shows the fluctuation in the peak amplitude of the first and second IPSCs elicited at an interspike interval of 50 msec. The paired pulse ratio (PPR) defined as the ratio of mean IPSC2amplitude/mean IPSC1 amplitude had a value of 0.65 at this connection and an average value of 0.6 ± 0.024 in 22 pairs. Apparent failures of transmission were observed only at connections displaying small-amplitude average IPSCs of 13–50 pA. At these connections the mean failure rate for first and second IPSCs was 4.07 ± 8.3 and 9.8 ± 10%, respectively (n= 14). The coefficients of variation were typically smaller for the first IPSCs (CV range, 0.14–0.4) than for the second IPSCs (CV range, 0.2–0.75), indicating a greater fluctuation around the mean for the second IPSCs.
Fig. 2.
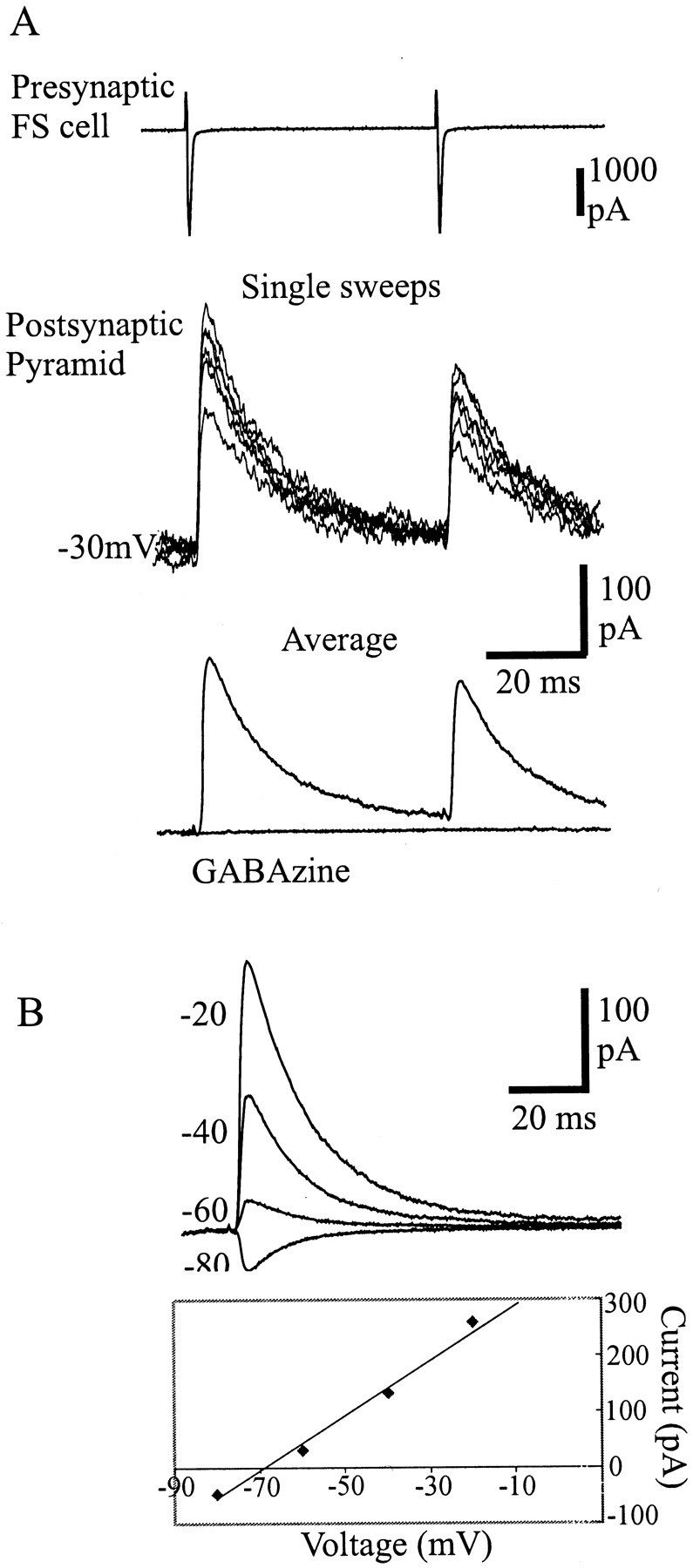
Characterization of a unitary connection between a fast-spiking cell and a pyramidal cell in layer V of the motor cortex.A, Top traces, Single sweep variation of IPSCs elicited in the pyramidal cell held at −30 mV by pairs of APs evoked in the FS cell. Bottom traces, Average IPSCs in control and in GABAzine (10 μm), a GABAA receptor antagonist. Note that the FS to pyramidal cell connections studied in layer V of the motor cortex typically displayed synaptic depression, which was frequency-dependent. B, Current–voltage relation of the unitary IPSCs characterized by a reversal potential of −70 mV.
The 10–90% rise times of these IPSCs were fast (1.6 ± 0.8 msec;n = 22) with fast decay time constant that could be fitted with a single exponential (τ = 13.6 ± 4 msec;n = 22). As shown in the example Figure2B, the current–voltage relation reversed close to −70 mV, and all the connections studied were totally blocked by GABAA receptor antagonist GABAzine (10 μm), indicating that these connections involved GABAA receptors.
Activation of KA receptors depresses unitary IPSCs
After obtaining a connected pair, all experiments were continued in the presence of GYKI 53655 (50 μm), d-AP-5 (50 μm), MCPG (1 mm), CPPG (100 μm), and CPG55845 (100 μm) to block AMPA, NMDA, mGluR, and GABAB receptors, respectively. Previous studies have shown that the GluR5 subunit is expressed specifically in interneurons (Bahn et al., 1994; Cauli et al., 2000), therefore to activate KA receptors on FS cells, bath application of (RS)-S-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) (ATPA), which has been proposed to be a GluR5 subunit-selective agonist (Clarke et al., 1997), was used. Figure3A illustrates an example of the action of ATPA (1 μm) that reversibly decreased the unitary IPSCs. On average, ATPA decreased the amplitude of first and second IPSCs to 59 ± 14.5 and 69.7 ± 19%, respectively, of their control values (paired t test;p = 0.01; n = 7). ATPA also caused a small inward current (20–50 pA) and an increase in the frequency of spontaneous IPSCs in the FS and pyramidal cells (data not shown). However, when experiments were performed to examine action potential-independent miniature IPSCs (mIPSCs) in layer V pyramidal cells in the presence of TTX (1 μm; see Materials and Methods), ATPA (1 μm) did not significantly change their frequency (2.07 ± 1.19 and 1.92 ± 0.66 Hz in control and ATPA, respectively; pairedt test p = 0.58; n = 7).
Fig. 3.

Depression of unitary IPSCs by ATPA andl-glutamate. A, The selective kainate receptor agonist ATPA (1 μm) decreased the amplitude of the unitary IPSCs elicited in pyramidal cells (gray, bold trace). B, For another connection, the depression of unitary IPSCs was observed after application of the endogenous agonist l-glutamate (10 μm). All experiments were performed in the presence of GYKI 53655(50 μm) and d-AP-5 (50 μm), MCPG (1 mm), CPPG (100 μm), and CPG 55845 (100 μm) to block AMPA, NMDA, mGluR, and GABAB receptors, respectively. In B, naloxone (100 μm), atropine sulfate (50 μm), and DPCPX (1 μm) were also added to the drug cocktail to block opioid, muscarinic, and adenosine receptors, respectively. C,Plot of the peak amplitude of the first IPSCs and the input resistance of the postsynaptic cell for the connection shown in Bduring the time course of the experiment. Note that the input resistance did not significantly change during agonist application. Subsequent addition of CNQX (30 μm) almost completely abolished the suppression of the IPSCs induced byl-glutamate. Both connections in A andB were totally blocked by GABAzine.
The effect of the endogenous agonist l-glutamate (10 μm) was also tested on unitary IPSCs.l-glutamate mimicked the action of ATPA in reducing the IPSCs but without changing the frequency of spontaneous IPSCs or the holding current in both presynaptic and postsynaptic cells. These experiments were performed in the presence of naloxone (100 μm), atropine sulfate (50 μm), and DPCPX (1 μm) in addition to the drug cocktail mentioned above to block opioid, muscarinic, and adenosine receptors, respectively.l-glutamate decreased the unitary IPSCs to 61 ± 12.4 and 66.8 ± 16.5% (p = 0.01;n = 6) of the control values of the first and second IPSCs, respectively. The effect of l-glutamate on inhibitory synaptic transmission was not accompanied by any significant change in the input resistance of the postsynaptic pyramidal cells, which was 518 ± 46.28 and 517.5 ± 44.56 MΩ during control and l-glutamate application, respectively (n = 6). Figure 3C is a plot of peak amplitude of the first IPSCs and the input resistance of the postsynaptic pyramidal cell during the time course of the experiment for the connection shown in Figure 3B. The depression of the IPSCs by l-glutamate and ATPA was reversible and could be repeatedly induced. Subsequent addition of the broad-spectrum non-NMDA receptor antagonist CNQX (30 μm) almost completely prevented the depressant action on unitary IPSCs. In CNQX, ATPA and l-glutamate reduced the IPSCs to 94 ± 5.6 (n = 4) and 94 ± 5% (n = 4) of average control first IPSCs, respectively.
Endogenous mechanism inducing depression of unitary IPSCs
To determine whether presynaptic KA receptors could be activated by the release of endogenous glutamate instead of bath application of exogenous agonists, the effect of large depolarizing steps applied to the postsynaptic pyramidal cells was tested. Similar conditioning protocols have been used previously in the cerebellum, hippocampus, and neocortex to elicit the release of transmitter from somatodendritic component of postsynaptic neurons (Llano et al., 1991; Pitler and Alger, 1995; Zilberter et al., 1999). Unitary IPSCs were elicited in pyramidal cells by double or train of presynaptic FS cell APs in control or 900 msec after a conditioning depolarizing voltage step induced in the postsynaptic pyramidal cells. The same antagonist cocktail used when l-glutamate was exogenously applied was also used here. The results obtained were comparable with the depression induced by ATPA and l-glutamate and could be repeatedly activated in the same pair. During conditioning the mean first and second unitary IPSCs were reduced to 63 ± 11.3 and 69.4 ± 15% (p = 0.01; n = 5) of control values, respectively (Fig.4A). These data suggest that depolarization of postsynaptic pyramidal cells caused the release of an endogenous messenger, most likely glutamate, which activated presynaptic KA receptors on FS cells and induced the depression of unitary IPSCs. Glutamate could be released from the soma and dendrites or the axon collaterals of the pyramidal cells. To determine whether the propagation of sodium-dependent action potentials were needed to release glutamate the local anesthetic, QX-314 (2.5 mm) was included in the postsynaptic recording electrode. In the presence of QX-314, the conditioning protocol still induced a decrease of unitary IPSCs. In four pairs the mean amplitude of the first and second IPSCs decreased to 64 ± 14 and 75 ± 15% (p = 0.01) of control value, respectively. Figure 4 shows two examples of conditioning experiments in normal intracellular solution and with loading QX-314 in the postsynaptic electrode. The plots represent the peak amplitudes of the first IPSCs from single sweeps during the time course of the experiment, and the average IPSCs are highlighted for each condition. The dotted lines indicate the onset and offset of the conditioning. Repeating the conditioning protocol after subsequent addition of CNQX, the average first IPSC decreased to 98 ± 2.5% (n = 4) of control first IPSC after conditioning.
Fig. 4.

Endogenous release of l-glutamate from the postsynaptic cell induced a depression of unitary IPSCs.A, B, Plot of the peak amplitude of the first IPSCs throughout experiments during which unitary synaptic responses were elicited in pyramidal cells alternatively in control conditions (control) and 900 msec after a conditioning depolarization of the postsynaptic pyramidal cells (conditioning). Insets illustrate the average unitary IPSCs elicited by pairs (A) or trains (B) of APs at different epochs of the experiments. Conditioning protocols with standard intracellular solution (A) or in the presence of QX-314 in the patch pipette (B) induced a decrease of the unitary IPSCs, which was prevented after subsequent addition of CNQX.
Origin of synaptic depression of unitary IPSCs
Figure 5 illustrates the summary of the data obtained with bath application of ATPA andl-glutamate and with conditioning protocols. The IPSC amplitude distribution and the cumulative amplitude histogram before and during l-glutamate for a connection are illustrated in Figure 5A. Because only a relatively small number of events for the first and second IPSCs are included in the plot, no attempt was made here to distinguish peaks, and data are binned coarsely. The amplitude distribution is relatively evenly distributed around the mean in each condition. There is clear shift of the amplitude distribution toward smaller values after agonist application in the example shown in Figure 5A, which suggests that multiple release sites may have been involved.
Fig. 5.

Origin of synaptic depression of unitary IPSCs. A, Example of the amplitude distribution and cumulative plot of first (top) and second (bottom) IPSCs in control (black) and during l-glutamate application (gray); open bars indicate noise. Single-sweep IPSC amplitudes are binned coarsely, and no peaks are distinguished. There is a clear shift in the mean amplitude after the application of l-glutamate. Billustrates the average change in failure rate and CV of first and second IPSCs and the PPR (IPSC2/IPSC1) before and during agonist application or during conditioning protocol. There was a greater increase in failure rate and CV for the first IPSCs compared with the second IPSCs. This resulted in an increase in PPR after agonist application and conditioning. C, To investigate the origin of the depression of IPSCs, a plot of normalized CV−2(CV−2 during KA receptor activation/control CV−2) against the normalized mean, M (KA receptor activation IPSC/control IPSC), for the first (black symbols) and second (gray symbols) IPSC amplitude for 18 of the FS to pyramidal cell connections, assuming a simple binomial model. Each data point represents a pair in ATPA (diamonds) and l-glutamate (circles). A change of the amplitude without a change in the in CV−2 would be caused by a change in q (quantal amplitude), represented by the zero slope dashed line. An equivalent change in the CV−2 and M, represented by the line with the slope of 1, could be a change caused by n (number of release sites) and both the probability (p) and q. A greater change in CV-2 than in M would be attributable to a change in p with a slope >1. Most of the data points fall between these two lines (slope 0 and 1), indicating a change caused by a presynaptic origin, p.
This shift toward smaller values after agonist application may be presynaptic or postsynaptic in origin. To determine whether the change was caused by presynaptic mechanisms, the rate of apparent failures of synaptic transmission was measured. Figure 5B illustrates the average change in the failure rate and CV for the first and second IPSCs after agonist application and conditioning. The increase in failure rate was also proportionally larger for first IPSCs, which increased to 232 ± 50, 700 ± 43, and 440 ± 100% of control after ATPA, l-glutamate application, and conditioning, respectively (p = 0.01;n = 4, for both agonist and n = 7 for conditioning). In comparison, the second IPSCs, increased to 150 ± 26, 278 ± 100, and 150 ± 89% of control after ATPA,l-glutamate application and conditioning, respectively (p = 0.01; n = 4, for both, agonist and n = 7 for conditioning). In all pairs studied the significant reduction in the mean IPSCs was accompanied by an increase in CV during agonist application, which was proportionally larger for the first IPSCs compared with the second IPSCs. The average increase in CV for first and second IPSCs was 167 ± 31 and 124.9 ± 30% after ATPA application (p = 0.01 for both; n = 6) and 136 ± 38 and 110 ± 19% afterl-glutamate application (p= 0.01 for both; n = 6). The PPR increased in the range of 106–113% for both, after agonist application (p = 0.01 for both; n = 13) and conditioning (p = 0.01; n = 9), as illustrated in Figure 5B, which shows the percentage change in PPR in control and after agonist application and conditioning.
Assuming that the data do not significantly depart from the binomial model of synaptic release, comparisons of changes in CV−2 and mean amplitude (M) may also indicate whether the depression of unitary IPSCs observed was of presynaptic or postsynaptic origin (Bekkers et al., 1990; Clements, 1990; Faber and Korn, 1991; Larkman et al., 1991). In a simple binomial model of transmission, an increase in quantal amplitude (q) results in an equivalent proportional increase in M [npq], but no change in CV−2[np/(1 − p), where n is equal to the number of release sites, and p is the probability of release]. A change in n alone results in an equivalent proportional change in both M and CV−2, whereas a change in p results in a greater proportional change in CV−2 than in M. Figure 5C is a plot of normalized CV−2 (test CV−2/control CV−2) against normalized M (test IPSC/control IPSC). As illustrated in Figure5C, majority of the data points (each point represents a pair), indicate that the change in first and second IPSCs after the application of ATPA and l-glutamate was associated with larger proportional increase in CV−2 than in M (slope > 1). Therefore, the depression of unitary IPSCs observed is predominantly of presynaptic origin caused by a change inn and/or p.
DISCUSSION
The present study describes a novel mechanism by which presynaptic KA receptors are activated at connections between FS cells and pyramidal cells in the layer V of the rat motor cortex. This mechanism involved the release of a retrograde messenger, which is likely to be glutamate, from pyramidal cell soma and/or dendrites. Glutamate then activates presynaptic KA receptors and induces the depression of unitary IPSCs by decreasing GABA release.
Presynaptic KA receptors decrease unitary IPSCs
Our results show a reduction of unitary IPSCs induced by KA receptor agonists at FS to pyramidal cell synapses, and this observation is consistent with the depression of stimulus-elicited IPSCs observed in hippocampal slices (Min et al., 1999;Rodriguez-Moreno et al., 2000). There are several lines of evidence supporting the occurrence of a presynaptic mechanism and the involvement of KA receptors from the present study. First, ATPA andl-glutamate were probably acting on KA receptors only because all experiments were performed in the presence of a potent selective AMPA receptor antagonist GYKI 53655, and NMDA, group I, II, and III mGlu receptors were also pharmacologically blocked. In addition, subsequent addition of the broad-spectrum non-NMDA receptor antagonist CNQX almost completely blocked the reduction of unitary IPSCs induced by ATPA, l-glutamate, or conditioning. Second, although we did not observe a significant modification of miniature IPSCs with KA receptor agonists, the reduction of unitary IPSCs observed in paired recordings during KA receptor agonist application seem to involve a presynaptic mechanism. Indeed, all FS cell to pyramidal cell connections displayed paired pulse and brief train depression, and during agonist application the paired pulse ratio increased. There was also a greater proportional increase in CVs for first IPSCs compared with second IPSCs. Finally, CV−2analysis indicated that the change in the amplitude of unitary IPSCs is predominantly caused by a change of presynaptic origin.
These observations could be explained by the activation of KA receptors located on presynaptic inhibitory terminals, but also if these receptors were on the soma of FS interneurons or expressed by a third partner that could release another transmitter that reduces GABA release. We used ATPA in the present study because it has been proposed to be a selective agonist of GluR5 subunit-containing receptors (Clarke et al., 1997; but see also Patermain et al., 2000), which are predominantly expressed by interneurons in the cortex (Bahn et al., 1994; Cauli et al., 2000). Indeed, in previous studies performed in the hippocampus (Cossart et al., 1998), ATPA was shown to induce a postsynaptic membrane depolarization of inhibitory interneurons, leading to an increase in the frequency of spontaneous IPSCs in pyramidal cells. It has been proposed that this increase of action potential-dependent release of GABA may explain the suppression of electrically evoked IPSCs (Frerking et al., 1999). By activating presynaptic GABAB receptors, excess GABA will ultimately decrease the probability of releasing GABA. In addition, activation of postsynaptic GABAAreceptors during the large increase in spontaneous IPSC frequency recorded in pyramidal cells will shunt the postsynaptic membrane, thereby contributing to decreasing the amplitude of measured elicited IPSCs. These mechanisms, however, do not seem to apply at inhibitory synapses between FS interneurons and pyramidal cells in the neocortex because the reduction of unitary IPSCs by KA receptor agonists was observed in the presence of a potent GABABreceptor antagonist CPG55845 and without significant effects on the input resistance of the postsynaptic membrane. In addition, the low concentration of l-glutamate that reduced unitary IPSCs had no effect on the holding current of both presynaptic and postsynaptic neurons (see also Min et al., 1999; Rodriguez-Moreno et al., 2000). Furthermore, the depression of the unitary IPSCs, the changes in failure rate, and the changes in the paired pulse ratio induced by KA receptor agonists were mimicked by the conditioning protocol, which is not expected to induce excessive action potential firing in presynaptic inhibitory cells, especially when the local anesthetic QX314 was included in the postsynaptic pipette. Finally, duringl-glutamate applications and conditioning experiments, opioid, muscarinic, and adenosine receptor antagonists were also added to the cocktail of glutamate and GABA receptor antagonists, which makes the occurrence of indirect heterosynaptic effects observed in other systems unlikely (Frerking et al., 1999; Schmitz et al., 2000). Therefore, we favor the hypothesis that the reduction of unitary IPSCs observed in the present study is mediated by the activation of KA receptors located on the terminal of inhibitory FS interneurons. We cannot, however, totally exclude the involvement an unidentified heterosynaptic mechanism that would be responsible for the reduction of the GABA release.
How does then the activation of presynaptic KA receptors decrease the release of GABA from FS cell terminals? Previous studies have proposed an inactivation of voltage-gated sodium or calcium channels at the terminals induced by the depolarization of ionotropic receptors (MacDermott et al., 1999; Kamiya and Ozawa, 2000; Schmitz et al., 2000). However, results obtained from hippocampal slices have also provided evidence for a presynaptic inhibitory action of KA primarily caused by inhibition of calcium influx into presynaptic terminals (Kamiya and Ozawa, 1998). Furthermore, KA caused the depression of calcium-dependent GABA release from isolated presynaptic terminals (Cunha et al., 1997; Perkinton and Sihra, 1999). This regulatory mechanism may involve G-proteins because the decrease in GABA release induced by KA is affected by PTx-sensitive G-protein and PKC activation (Rodriguez-Moreno and Lerma, 1998; Rodriguez-Moreno et al., 2000), and it has been suggested that there is a physical link between KA receptors and G-protein involved in the process (Rodriguez-Moreno and Lerma, 1998).
Activation of KA receptors by somatodendritic release of glutamate
Depolarization of the postsynaptic pyramidal cell induced a transient reduction of unitary IPSC amplitude mediated by the activation of presynaptic KA receptors. It is therefore most likely that the messenger linking the postsynaptic depolarization to the modulation of presynaptic release is glutamate. Because the depression of IPSCs was still apparent when QX-314 was included in the postsynaptic pipette, glutamate release probably occurs from the soma and/or the dendrites of pyramidal cells. Modulation of inhibitory transmission by the release of glutamate from postsynaptic principal neurons has been already described. In Purkinje cells of the cerebellum and in pyramidal cells of the hippocampus, membrane depolarization induces a calcium-dependent release of glutamate, which depresses spontaneous IPSCs recorded in these neurons (Llano et al., 1991; Pitler and Alger, 1992) (for review, see Alger and Pitler, 1995; Marty and Llano, 1995). At these synapses, however, the effect of glutamate is mediated by the activation of presynaptic metabotropic receptors (Glitsch et al., 1996; Morishita et al., 1997), and in the hippocampus it does not involve KA receptors (Morishita and Alger, 1999). In layer II/III of the neocortex, it has been recently reported that a calcium-dependent release of glutamate from the dendrites of pyramidal cells also depresses unitary IPSCs through the activation of presynaptic metabotropic receptors (Zilberter, 2000). Our own results indicate that glutamate is also released from the dendrites of layer V pyramidal cells and modulates the release of GABA through the activation of presynaptic KA receptors. However, they do not exclude a possible modulation of the GABA release by presynaptic metabotropic receptors also in layer V because all our experiments were performed in the presence of metabotropic receptor blockers.
It has been shown in layer II/III of the neocortex that backpropagating dendritic APs in bitufted interneurons cause the release of GABA, which acts as a retrograde messenger to depress EPSPs from layer II/II pyramidal cells (Zilberter et al., 1999). Therefore in the upper layers of the neocortex, both glutamate and GABA can be released from dendrites to modulate synaptic transmission, and in both cases this dendritic release leads, directly or indirectly, to decrease the impact of inhibition. In layer V, FS interneurons receive direct axonal inputs from pyramidal cells (Angulo et al., 1999a,b), and these two cell types are often reciprocally connected (A. Ali and E. Audinat, unpublished results), forming an inhibitory feedback loop. Our present study provides evidence for a dendritic release of glutamate from layer V pyramidal cells that also leads to a disinhibition through the activation of presynaptic KA. It would be interesting to know whether the dendrites of FS cells can also release GABA to decrease the excitatory inputs from pyramidal cells. In that case, the same pair of neurons interacting reciprocally through axosomatic or axodendritic synapses would also interact reciprocally through dendroaxonal relations, and the two types of interactions would have opposite effect on the activity of layer V pyramidal cells; i.e., on the output of the neocortex. The balance between these mechanisms will be probably differentially regulated in different physiological and pathological conditions.
Footnotes
This work was supported by the Wellcome Trust (London) and European Community Grant QLRT 1999 00649. A.B.A. is a Wellcome Trust international postdoctoral fellow.
Correspondence should be sent to Dr. Afia B. Ali at her present address: University Laboratory of Physiology, University of Oxford, Parks Road, Oxford OX1 3PT, UK. E-mail:afia.ali@physiol.ox.ac.uk.
Dr. Audinat's present address: Laboratoire de Neurophysiologie, ESPCI, Institut National de la Santé et de la Recherche Médicale, EPI 00-02, 10 rue Vauquelin, 75005 Paris, France.
REFERENCES
- 1.Alger BE, Pitler TA. Retrograde signalling at GABAA-receptor synapses in the mammalian CNS. Trends Neurosci. 1995;18:333–340. doi: 10.1016/0166-2236(95)93923-l. [DOI] [PubMed] [Google Scholar]
- 2.Angulo MC, Rossier J, Audinat E. Postsynaptic glutamate receptors and integrative properties of fast-spiking interneurons in the rat neocortex. J Neurophysiol. 1999a;82:1295–1302. doi: 10.1152/jn.1999.82.3.1295. [DOI] [PubMed] [Google Scholar]
- 3.Angulo MC, Staiger JF, Rossier J, Audinat E. Developmental synaptic changes increase the range of integrative capabilities of an identified excitatory neocortical connection. J Neurosci. 1999b;19:1566–1576. doi: 10.1523/JNEUROSCI.19-05-01566.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahn S, Volk B, Wisden W. Kainate receptor gene expression in the developing rat brain. J Neurosci. 1994;14:5525–5547. doi: 10.1523/JNEUROSCI.14-09-05525.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bekkers JM, Richerson GB, Stevens CF. Origin of variability in quantal size in cultured hippocampal neurons and hippocampal slices. Proc Natl Acad Sci USA. 1990;87:5359–5362. doi: 10.1073/pnas.87.14.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bureau I, Bischoff S, Heinemann SF, Mulle C. Kainate receptor-mediated responses in the CA1 field of wild-type and GluR6-deficient mice. J Neurosci. 1999;19:653–663. doi: 10.1523/JNEUROSCI.19-02-00653.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurones. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- 8.Cauli B, Audinat E, Lambolez B, Angulo M-C, Ropert N, Tsuzuki K, Hestrin S, Rossier J. Molecular and physiological diversity of cortical nonpyramidal cells. J Neurosci. 1997;17:3894–3906. doi: 10.1523/JNEUROSCI.17-10-03894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cauli B, Porter JT, Tsuzuki K, Lambolez B, Rossier J, Quenet B, Audinat E. Classification of fusiform neocortical interneurons based on unsupervised clustering. Proc Natl Acad Sci USA. 2000;97:6144–6149. doi: 10.1073/pnas.97.11.6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chittajallu R, Braithwaite SP, Clarke VR, Henley JM. Kainate receptors: subunits, synaptic localization and function. Trends Pharmacol Sci. 1999;20:26–35. doi: 10.1016/s0165-6147(98)01286-3. [DOI] [PubMed] [Google Scholar]
- 11.Clarke VRJ, Ballyk BA, Hoo KH, Mandelzys A, Pellizzari A, Bath CP, Thomas J, Sharpe EF, Davies CH, Ornstein PL, Schoepp DD, Kamboj RK, Collingridge GL, Lodge D, Bleakman D. A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature. 1997;389:599–603. doi: 10.1038/39315. [DOI] [PubMed] [Google Scholar]
- 12.Clements JD. A statistical test for demonstrating a presynaptic site of action for a modulator of synaptic amplitude. J Neurosci Methods. 1990;31:75–88. doi: 10.1016/0165-0270(90)90012-5. [DOI] [PubMed] [Google Scholar]
- 13.Connors BW, Gutnick MJ. Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci. 1990;13:99–104. doi: 10.1016/0166-2236(90)90185-d. [DOI] [PubMed] [Google Scholar]
- 14.Cossart R, Esclapez M, Hirsch JC, Bernard C, Ben-Ari Y. GluR5 kainate receptor activation in interneurones increase tonic inhibition of pyramidal cells. Nat Neurosci. 1998;6:470–478. doi: 10.1038/2185. [DOI] [PubMed] [Google Scholar]
- 15.Cunha RA, Constantino MD, Ribeiro JA. Inhibition of [3H] gamma-aminobutyric acid release by kainate receptor activation in rat hippocampal synaptosomes. Eur J Pharmacol. 1997;323:167–172. doi: 10.1016/s0014-2999(97)00043-5. [DOI] [PubMed] [Google Scholar]
- 16.DeVries SH, Schwartz EA. Kainate receptors mediate synaptic transmission between cones and “off” bipolar cells in a mammalian retina. Nature. 1999;397:157–160. doi: 10.1038/16462. [DOI] [PubMed] [Google Scholar]
- 17.Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J. 1991;60:1288–1294. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fisher RS, Alger BE. Electrophysiological mechanisms of kainic acid-induced epileptiform activity in the rat hippocampal slices. J Neurosci. 1984;4:1312–1323. doi: 10.1523/JNEUROSCI.04-05-01312.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frerking M, Malenka RC, Nicoll RA. Synaptic activation of kainate receptors on hippocampal interneurones. Nat Neurosci. 1998;6:479–486. doi: 10.1038/2194. [DOI] [PubMed] [Google Scholar]
- 20.Frerking M, Petersen CC, Nicoll RA. Mechanisms underlying kainate receptor-mediated disinhibition in the hippocampus. Proc Natl Acad Sci USA. 1999;96:12917–12922. doi: 10.1073/pnas.96.22.12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glitsch M, Llano I, Marty A. Glutamate as a candidate retrograde messenger at interneurone-Purkinje cell synapses of rat cerebellum. J Physiol (Lond) 1996;497:531–537. doi: 10.1113/jphysiol.1996.sp021786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamiya H, Ozawa S. Kainate receptor-mediated inhibition of presynaptic Ca2+ influx and EPSP in area CA1 of the rat hippocampus. J Physiol (Lond) 1998;509:833–845. doi: 10.1111/j.1469-7793.1998.833bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamiya H, Ozawa S. Kainate receptor-mediated presynaptic inhibition at the mouse hippocampal mossy fibre synapse. J Physiol (Lond) 2000;523:653–665. doi: 10.1111/j.1469-7793.2000.t01-1-00653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerchner GA, Wilding TJ, Li P, Zhuo M, Huettner JE. Presynaptic kainate receptors regulate spinal sensory transmission. J Neurosci. 2001;21:59–66. doi: 10.1523/JNEUROSCI.21-01-00059.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kidd FL, Isaac JTR. Developmental and activity dependent regulation of kainate receptors at thalamocortical synapse. Nature. 1999;400:569–573. doi: 10.1038/23040. [DOI] [PubMed] [Google Scholar]
- 26.Larkman A, Stratford K, Jack J. Quantal analysis of excitatory synaptic action and depression in hippocampal slices. Nature. 1991;350:344–347. doi: 10.1038/350344a0. [DOI] [PubMed] [Google Scholar]
- 27.Li P, Wilding TJ, Kim SJ, Calejesan AA, Huettner JE, Zhuo M. Kainate-receptor-mediated sensory synaptic transmission in mammalian spinal cord. Nature. 1999;397:161–164. doi: 10.1038/16469. [DOI] [PubMed] [Google Scholar]
- 28.Llano I, Leresche N, Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- 29.MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- 30.Marty A, Llano I. Modulation of inhibitory synapses in the mammalian brain. Curr Opin Neurobiol. 1995;5:335–341. doi: 10.1016/0959-4388(95)80046-8. [DOI] [PubMed] [Google Scholar]
- 31.Min MY, Melyan Z, Kullmann DM. Synaptically released glutamate reduces gamma-aminobutyric acid (GABA)ergic inhibition in the hippocampus via kainate receptors. Proc Natl Acad Sci USA. 1999;96:9932–9937. doi: 10.1073/pnas.96.17.9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morishita W, Alger BE. Evidence for endogenous excitatory amino acids as mediators in DSI of GABA(A)ergic transmission in hippocampal CA1. J Neurophysiol. 1999;82:2556–2564. doi: 10.1152/jn.1999.82.5.2556. [DOI] [PubMed] [Google Scholar]
- 33.Morishita W, Kirov SA, Pitler TA, Martin LA, Lenz RA, Alger BE. N-ethylmaleimide blocks depolarization-induced suppression of inhibition and enhances GABA release in the rat hippocampal slice in vitro. J Neurosci. 1997;17:941–950. doi: 10.1523/JNEUROSCI.17-03-00941.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patermain AV, Herrera MT, Nieto MA, Lerma J. GluR5 and GluR6 kainate receptor subunits coexist in hippocampal neurons and coassemble to form functional receptors. J Neurosci. 2000;20:196–205. doi: 10.1523/JNEUROSCI.20-01-00196.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perkinton MS, Sihra TS. A high-affinity presynaptic kainate-type glutamate receptor facilitates glutamate exocytosis from cerebral cortex nerve terminals (synaptosomes). Neuroscience. 1999;90:1281–1292. doi: 10.1016/s0306-4522(98)00573-9. [DOI] [PubMed] [Google Scholar]
- 36.Pitler TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci. 1992;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez-Moreno A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 1998;20:1211–1218. doi: 10.1016/s0896-6273(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez-Moreno A, Herreras O, Lerma J. Kainate receptors presynaptically downregulate GABAergic inhibition in the rat hippocampus. Neuron. 1997;19:893–901. doi: 10.1016/s0896-6273(00)80970-8. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez-Moreno A, Lopez-Garcia JC, Lerma J. Two populations of kainate receptors with separate signaling mechanisms in hippocampal interneurons. Proc Natl Acad Sci USA. 2000;97:1293–1298. doi: 10.1073/pnas.97.3.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmitz D, Frerking M, Nicoll RA. Synaptic activation of presynaptic kainate receptors on hippocampal mossy fiber synapses. Neuron. 2000;27:327–338. doi: 10.1016/s0896-6273(00)00040-4. [DOI] [PubMed] [Google Scholar]
- 41.Sloviter RS, Damiano BP. On the relationship between kainic acid-induced epileptiform activity and hippocampal neuronal damage. Neuropharmacology. 1981;20:1003–1011. doi: 10.1016/0028-3908(81)90088-5. [DOI] [PubMed] [Google Scholar]
- 42.Vignes M, Collingridge GL. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- 43.Westbrook GL, Lothman EW. Cellular and synaptic basis of kainic acid-induced hippocampal epileptiform activity. Brain Res. 1983;273:97–109. doi: 10.1016/0006-8993(83)91098-3. [DOI] [PubMed] [Google Scholar]
- 44.Zilberter Y. Dendritic release of glutamate suppresses synaptic inhibition of pyramidal neurons in rat neocortex. J Physiol (Lond) 2000;528.3:489–496. doi: 10.1111/j.1469-7793.2000.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zilberter Y, Kaiser KM, Sakmann B. Dendritic GABA release depresses excitatory transmission between layer 2/3 pyramidal and bitufted neurons in rat neocortex. Neuron. 1999;24:979–988. doi: 10.1016/s0896-6273(00)81044-2. [DOI] [PubMed] [Google Scholar]