

Abstract
We have reported recently that syntaxin 1A mediates two effects on N-type channels transiently expressed in tsA-201 cells: a hyperpolarizing shift in the steady-state inactivation curve as well as a tonic inhibition of the channel by G-protein βγ subunits (Jarvis et al., 2000). Here we have examined some of the molecular determinants and factors that modulate the action of syntaxin 1A on N-type calcium channels. With the additional coexpression of SNAP25, the syntaxin 1A-induced G-protein modulation of the channel became reduced in magnitude by ∼50% but nonetheless remained significantly higher than the low levels of background inhibition seen with N-type channels alone. In contrast, coexpression of nSec-1 did not reduce the syntaxin 1A-mediated G-protein inhibition; however, interestingly, nSec-1 was able to induce tonic G-protein inhibition even in the absence of syntaxin 1A. Both SNAP25 and nSec-1 blocked the negative shift in half-inactivation potential that was induced by syntaxin 1A. Activation of protein kinase C via phorbol esters or site-directed mutagenesis of three putative PKC consensus sites in the syntaxin 1A binding region of the channel (S802, S896, S898) to glutamic acid (to mimic a permanently phosphorylated state) did not affect the syntaxin 1A-mediated G-protein modulation of the channel. However, in the S896E and S898E mutants, or after PKC-dependent phosphorylation of the wild-type channels, the susceptibility of the channel to undergo shifts in half-inactivation potential was removed. Thus, separate molecular determinants govern the ability of syntaxin 1A to affect N-type channel gating and its modulation by G-proteins.
Keywords: SNARE proteins, protein kinase C, Gβγ, phosphorylation, calcium channels, site-directed mutagenesis
The interaction of presynaptic calcium channels with the vesicle release core complex is of fundamental importance for neurotransmitter release (Pevsner et al., 1994a; Mochida et al., 1996; Stanley, 1997) (for review, see Linial and Bledi, 2000). In the presynaptic nerve terminal, calcium currents are generated predominantly by P/Q-type and N-type calcium channels (Westenbroek et al., 1992, 1995). They are colocalized with synaptic vesicles by calcium-dependent binding of the t-SNARE proteins syntaxin 1A and SNAP25 to the domain II/III linker region of the N-type and P/Q-type calcium channel α1 subunits [termed synaptic protein interaction (synprint) site; residues 718–963 of the α1B subunit] and by direct interaction of these two proteins with the v-SNARE proteins synaptotagmin and synaptobrevin (Sheng et al., 1994, 1996; Rettig et al., 1996, 1997;Walker and De Waard, 1998) (for review, see Lin and Scheller, 2000). The amount of calcium entry via the channel and, thus, neurotransmitter release is modulated with the activation of certain second messenger systems, including protein kinase C (PKC) and G-protein βγ subunits (Dunlap and Fischbach, 1981; Swartz, 1993; Swartz et al., 1993; Zamponi et al., 1997; Hamid et al., 1999). Moreover, the interactions between the channels and the SNARE protein complex can be modulated by phosphorylation of the syntaxin 1A binding site on the channel (Yokoyama et al., 1997) and by other types of presynaptic proteins such as cysteine string protein (Umbach and Gunderson, 1997; Wu et al., 1999).
The interaction between syntaxin 1A and the N-type calcium channel α1 subunit has two major functional consequences. First, with the coexpression of syntaxin 1A and α1B N-type calcium channels, the channels undergo a 15–20 mV negative shift in half-inactivation potential (Bezprozvanny et al., 1995, 2001; Jarvis et al., 2000) as well as enhanced slow inactivation (Degtiar et al., 2000). Furthermore, in the presence of syntaxin 1A the channels become subject to a large tonic inhibition by G-protein βγ subunits, which can be relieved with the application of strong depolarizing prepulses (Jarvis et al., 2000). Although the basis of the syntaxin 1A effect on channel inactivation is not yet understood, G-protein inhibition may occur via a syntaxin 1A-mediated colocalization of the calcium channel α1 subunit and endogenous Gβγ (Jarvis et al., 2000).
Here we identify novel factors that affect the ability of syntaxin 1A to promote G-protein modulation and voltage-dependent inactivation of N-type calcium channels. We show that coexpression of either SNAP25 or neuronal Sec-1 (nSec-1) antagonizes the ability of syntaxin 1A to inactivate N-type calcium channels, whereas the ability of syntaxin 1A to enhance tonic G-protein inhibition is retained in the presence of SNAP25 and nSec-1. PKC-dependent phosphorylation of the channel induced by the application of phorbol esters or point mutations mimicking the effects of protein kinase C-dependent phosphorylation of the synprint region also preclude the inactivation effect of syntaxin 1A while leaving the G-protein effects intact. Thus, the molecular determinants that govern the two syntaxin 1A-mediated phenomena are distinct in nature. Moreover, our data may suggest that syntaxin 1A might have only a transient effect on N-type channel inactivationin vivo, depending on the presence of other SNARE proteins and the activity of PKC.
MATERIALS AND METHODS
Molecular biology
Scoring of PKC consensus sites
The likelihood of PKC-dependent phosphorylation of PKC consensus sites was obtained by using the probability values reported by Kennelly and Krebs (1991). Scores were assigned depending on the proximity of basic (i.e., arginine or lysine) residues in positions −1 to −3 and +1 to +3 around threonine and serine residues as follows: R/K−3, 25; R/K−2, 31; R/K−1, 9; R/K+1, 5; R/K+2, 34; R/K+3, 24. For S/T residues bracketed on either side by basic residues on either side, the scores were added. For S/T sites flanked by more than one basic residue on the same side, the higher score of the two was assigned. CaMKII sites were assigned on the basis of the sequence R-X-X-S/T (seeKennelly and Krebs, 1991).
Mutagenesis of α1B synprint phosphorylation sites
The pSL1180 vector was digested withStuI and SmaI to remove a 151 bp section of the polycloning site (PCS), creating pSL1180(ΔStu–Sma). The full-length α1B calcium channel clone (Dubel et al., 1992) in a cytomegalovirus (CMV) mammalian expression vector, kindly provided by Dr. T. P. Snutch (University of British Columbia, Vancouver, Canada), was digested with SpeI and BsiWI, liberating a fragment coding for the first 969 amino acids of α1B plus 700 bp of CMV vector sequence, which was ligated into pSL1180(ΔStu–Sma) to give pSL1180(α1B SpeI, BsiWI). This construct was digested by AgeI to liberate a fragment coding for amino acids 438–951, which was inserted into the PCS of pSL1180 to give pSL1180(α1B AgeI). This construct consequently was used as the template in mutagenesis reactions.
Mutagenesis was performed with QuikChange Site-Directed Mutagenesis (Stratagene, La Jolla, CA). Reaction conditions in a volume of 50 μl consisted of 5 μl of 10× reaction buffer, 1 μl dNTP mix, 2.5 U ofPfuTurbo DNA polymerase, 5 ng of pSL1180–AgeI (or subsequent mutated constructs), and 125 ng of each forward and reverse primer (University of Calgary Core DNA services). Using a PTC-100HB thermal cycler (MJ Research, Watertown, MA), we hot-started the reaction and held it at 95°C for 30 sec. We conducted 16 cycles, which consisted of denaturation for 30 sec at 95°C, annealing for 1 min at 55°C, and extension for 16 min at 68°C. The reaction product was cooled to 37°C and digested withDpnI for 2 hr. Then the DpnI-digested reaction was transformed into DH5α supercompetent cells (Life Technologies, Burlington, Ontario, Canada). DNA was isolated from colonies with a QIAprep Spin Miniprep (Qiagen, Chatsworth, CA) and sequenced. The mutated pSL1180(α1B AgeI) construct was digested with AgeI, and the 1513 bp fragment was isolated and ligated into the original AgeI-digested pSL1180(α1B SpeI, BsiWI) construct. Correct orientation was confirmed by checking theSacI digestion pattern. Then the construct was digested withSpeI and BsiWI, and the fragment containing the mutations was ligated into the original CMV α1Bconstruct.
All restriction enzymes were purchased from New England Biolabs (Mississauga, Ontario, Canada), unless otherwise stated. All digested DNA was run on 0.8% agarose gels, extracted, and purified with QIAquick Gel Extraction columns (Qiagen). T4 DNA ligase (Roche–Boehringer Mannheim, Laval, Quebec, Canada) was used for all ligations. Bacterial alkaline phosphatase (BAP) and all PCR reagents and primers were purchased from Life Technologies, unless otherwise stated. Rat brain cDNA (oligo-dT-primed) was kindly provided by Bob Winkfein (University of Clagary, Calgary, Canada). PCR and mutagenesis reactions were performed in a PTC-100HB thermal cycler (MJ Research).
Cloning of SNARE proteins
The syntaxin 1A construct used here was the same as that described in Jarvis et al. (2000). The cloning of syntaxin 1B was performed via RT-PCR, as described (Lu et al., 2001).
Cloning of SNAP25. A forward primer containing aNotI restriction site and an ideal Kozak sequence and a reverse primer containing a ClaI restriction site downstream of the stop codon were used to clone SNAP25 via PCR from rat brain cDNA. The primer sequences were as follows: forward 5′-GAGCGGCCGCCATGGCCGAGGACGCAGACATGC-3′ and reverse 5′-GGAGAGCACAGCAGAAGGATCGATTTAACCACTTCCCAGC-3′.
The PCR reaction solution, in a 50 μl volume, consisted of (in mm) 20 Tris-HCl, pH 8.4, 50 KCl, dNTPs (0.2 each), and 1.5 MgCl2 plus 2.5 U of platinum Taq DNA polymerase, 20 pmol of each primer, and 50 ng of cDNA. The reaction was hot-started and held at 94°C for 2 min. We conducted 34 cycles, which consisted of denaturation for 30 sec at 94°C, annealing for 45 sec at 60°C, and extension for 1.15 min at 72°C. The resultant SNAP25b DNA product was ligated into a pGEM T-Easy vector (Promega, Madison, WI) and sequenced. The SNAP25–T-Easy construct was digested withNotI and ClaI; the liberated SNAP25 fragment was ligated with T4 ligase (Roche–Boehringer Mannheim) into pMT2sx for subsequent expression studies.
Cloning of nSec-1. A forward primer containing aSpeI restriction site and an ideal Kozak sequence and a reverse primer containing a ClaI restriction site downstream of the stop codon were used to clone nSec-1 from rat cDNA with the following primer sequences: forward 5′-CGGAGACTAGTAGGCCGCCATGGCCC-3′ and reverse 5′-GCCAGGGTTTTGGAGGATCGATATTTTAACTGCTTATTTCTTCG-3′.
The PCR reaction solution, in a 50 μl volume, consisted of (in mm) 20 Tris-HCl, pH 8.8, 10 KCl, 1.5 MgCl2, 2 MgSO4, and 10 (NH4)2SO4plus 0.1% Triton X-100, 5 μg BSA, dNTPs (0.2 mm each), 2.5 U of PfuTurbo (Stratagene), 20 pmol of each primer, and 50 ng of cDNA. The reaction was hot-started and held at 94°C for 2 min. We conducted 30 cycles, which consisted of denaturation for 30 sec at 94°C, annealing for 45 sec at 60°C, and extension for 5 min at 74°C. The resultant blunt-ended DNA product was ligated into pCR-Blunt II-TOPO (Invitrogen, San Diego, CA) and sequenced. The nSec-1–pCR-Blunt II construct was digested with SpeI andClaI; the liberated nSec-1 fragment was ligated into pMT2sx for expression studies.
Biochemistry
Generation and preparation of fusion proteins
6xHis synprint fusion proteins were generated and prepared as described previously (Jarvis et al., 2000). Briefly, residues 718–963 from the II/III linker of the α1B calcium channel (wild type and mutants) were amplified and subcloned into pTrcHisC (Invitrogen), followed by transformation intoEscherichia coli TOP10. Induction and preparation of purified fusion proteins were performed by using conditions adapted from the manufacturer and described in detail in Jarvis et al. (2000).
Immunoblots for SNARE proteins in human embryonic kidney (HEK) cells
Primary antibodies were purchased from StressGen (Victoria, British Columbia). Secondary antibodies were purchased from Amersham Pharmacia Biotech (Buckinghamshire, UK). tsA-201 cells were transfected with SHAM, SNAP-25, nSec1, or syntaxin 1A as described below and prepared for Western blotting as described previously in Jarvis et al. (2000). Anti-syntaxin 1A and anti-SNAP25 monoclonal antibodies were used at 1:1000 and 1:2000, respectively; anti-nSec1 polyclonal was used at 1:1000 in a 5% solution of skim milk powder in PBS supplemented with 0.1% Triton X-100 (PBST), all for 2 hr at room temperature. Secondary antibodies were used at 1:2000 in a 2% solution of skim milk powder in PBST for 1 hr at room temperature.
In vitro phosphorylation of the II/III synprint peptide and syntaxin binding assay
6xHis-tagged synprint peptides (wild type and EEE) were prepared as described previously (Jarvis et al., 2000). Purified synprint peptides were phosphorylated by 0.25 μg of PKC (Sigma, Oakville, Ontario, Canada) for 3 hr at 30°C in a solution containing (in mm) 20 HEPES, pH 7.4, 10 MgCl2, 10 DTT, 0.15 CaCl2, and 0.4 ATP plus 20% mixed micelles (PS and 1,2-DG). After phosphorylation the peptides were washed three times with TBS and quantified by a Bradford assay. Syntaxin 1A–glutathione S-transferase (GST) was purified as described previously (Jarvis et al., 2000). The GST tag was removed by cleavage with thrombin (Sigma), and the purified syntaxin 1A was dialyzed overnight against Tris-buffered saline (TBS; 10 mmTris-HCl, pH 7.4, and 150 mm NaCl). Syntaxin 1A and the synprint peptides were quantified by using a Bradford assay against a standard BSA curve.
Synprint peptides were immobilized on Ni2+-NTA beads, and syntaxin 1A was added in ratios ranging from 1:8 to 1:1. The proteins were incubated in TBS supplemented with 0.1% Triton X-100 (TBST) at 4°C with end-over-end rotation for 3 hr. After incubation the beads were washed with 30 bed volumes of TBST. Laemmli sample buffer (2×) was added to the washed beads, which then were boiled for 2 min at 90°C and shaken at 4°C for 2 hr to ensure the dissociation of the complexes. The protein samples subsequently were subjected to SDS-PAGE; Western blots were performed as described previously (Jarvis et al., 2000).
Tissue culture and transient transfection of tsA-201 cells
HEK cells were grown to 80% confluence in DMEM medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (v/v). Cells were split and plated on glass coverslips at 10% confluence 12 hr before transfection. Immediately before transfection the medium was renewed, and a calcium phosphate transfection procedure was used to transfect cDNAs encoding for wild-type or mutant α1B, α2-δ, and β1bsubunits and the reporter gene EGFP (Clontech, Cambridge, UK). Cells were washed after 12 hr and maintained at 37°C for an additional 12 hr before being moved into a CO2 incubator set at 28°C. The cells were maintained under those conditions for 24–72 hr before recording.
Patch-clamp recordings
Glass coverslips carrying transfected cells were transferred to a 3 cm culture dish and bathed in recording solution consisting of (in mm) 20 BaCl2, 1 MgCl2, 10 HEPES, 40 tetraethylammonium chloride (TEA-Cl), 10 glucose, and 65 CsCl (pH 7.2 with TEA-OH). Whole-cell patch-clamp recordings were performed with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA) linked to a personal computer equipped with pClamp version 6.0. Patch pipettes (Sutter borosilicate glass, BF150-86-15) were pulled with a Sutter P-87 microelectrode puller and fire-polished; they showed typical resistances of 3–4 MΩ. The internal pipette solution contained (in mm) 108 cesium methane sulfonate, 4 MgCl2, 9 EGTA, and 9 HEPES, pH 7.2. Data were filtered at 1 kHz and recorded directly onto the hard drive of the computer. Unless stated otherwise, currents were evoked by stepping from −100 mV to a test potential of +20 mV. G-protein inhibition was assessed by the application of a strong depolarizing (+150 mV) prepulse (PP) of 50 msec duration preceding the test depolarization by 10 msec. Steady-state inactivation curves were obtained by maintaining the cells at various holding potentials for 5 sec before stepping to a test depolarization of +10 mV.
The raw data were analyzed with Clampfit software. Steady-state inactivation curves were fit with standard Boltzmann relations, using Sigmaplot software (Jandel Scientific, San Rafael, CA). All figures were generated in Sigmaplot. The numbers in parentheses reflect individual experiments that typically were obtained in multiple transfections. The error bars that are given reflect SE; pvalues reflect Student's t tests, except in Figure2C in which we used a one-way ANOVA test (Student–Newman–Keuls method).
Fig. 2.
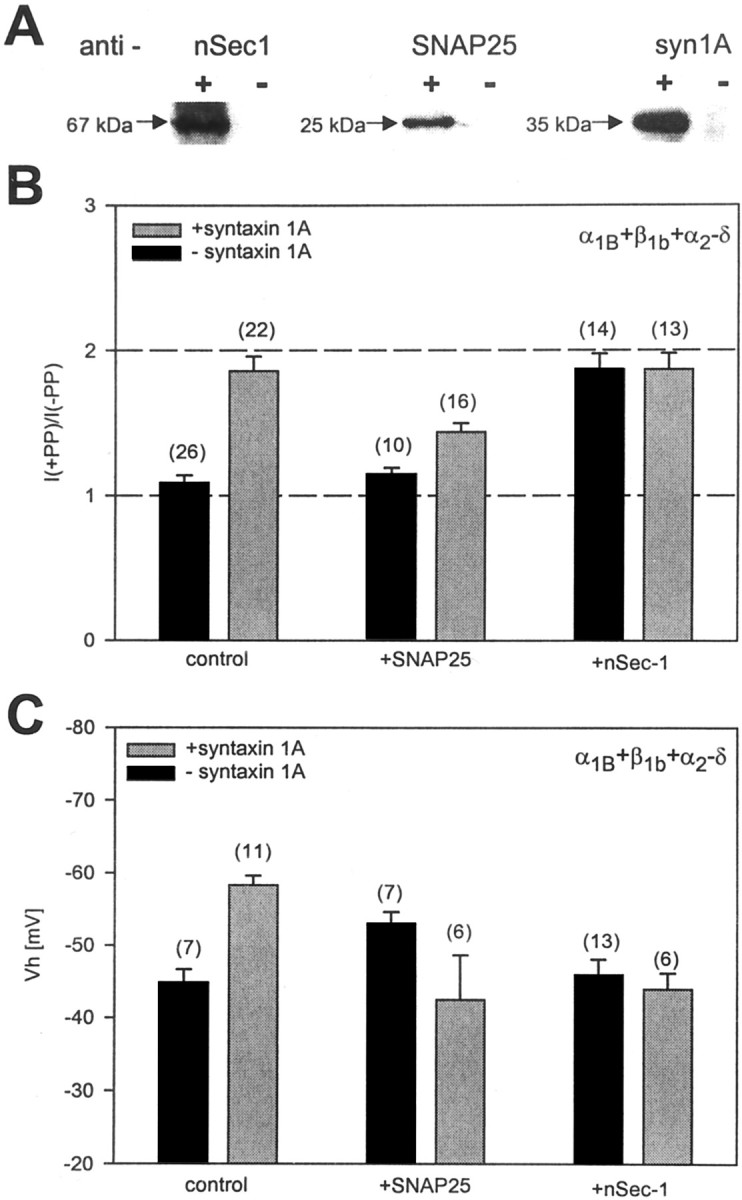
Effect of SNAP25 and nSec-1 on syntaxin 1A-mediated effects on N-type calcium channels. A, Immunoblots from tsA-201 cell lysates obtained from sham-transfected cells (−) or cells transfected with syntaxin 1A, SNAP25, or nSec-1 (+). Note that the three proteins are detected only when exogenously expressed. B, Degree of PP relief obtained in various combinations of syntaxin 1A, SNAP25, and nSec-1. Note that SNAP25 reduces the degree of syntaxin 1A-mediated G-protein inhibition and that coexpression of the channel with nSec-1 per se results in pronounced PP relief. C, Effect of SNAP25 and nSec-1 on the syntaxin 1A-mediated shift in half-inactivation potential. Similar to what is observed with syntaxin 1A, SNAP25 mediates a hyperpolarizing shift in half-inactivation potential; however, in the presence of both syntaxin 1A and SNAP25, or syntaxin 1A and nSec-1, the half-inactivation potential returns to control levels. The data obtained with SNAP25 plus syntaxin 1A differed from those obtained with either syntaxin 1A or SNAP25 alone, whereas no statistical difference was found in conditions in which the two proteins were present individually (one-way ANOVA). Error bars denote SE; thenumbers in parentheses indicate the numbers of experiments. The control and syntaxin 1A data inB and C are the same as in Figure1.
RESULTS
Syntaxin 1A exerts dual effects on N-type channel function
We have shown previously that syntaxin 1A promotes G-protein inhibition of N-type channels, which may occur by a syntaxin 1A-mediated colocalization of Gβγ and the N-type channel α1 subunit (Jarvis et al., 2000). This effect of syntaxin 1A is illustrated in Figure1A in the form of current records. With the coexpression of syntaxin 1A, N-type (α1B+β1b+α2-δ) currents can be enhanced dramatically after the application of a strong depolarizing prepulse (PP), consistent with tonic G-protein inhibition. To provide additional support for the involvement of G-protein βγ subunits, we coexpressed syntaxin 1A and the channel with the C-terminal fragment of the β-adrenergic receptor kinase (β-ARKct, residues 495–689; see Magga et al., 2000) to create a Gβγ sink (Koch et al., 1994). As shown in Figure 1A, in the presence of the fragment the effect of syntaxin 1A became greatly diminished from 86 ± 10 to 29 ± 3% PP relief (<0.05) (Fig.1B), further supporting a role of Gβγ in the PP effect. To demonstrate that the effect of syntaxin 1A-mediated G-protein inhibition was dependent on a physical interaction between the channel and syntaxin 1A, we overexpressed a peptide of the synprint region on the N-type channel (residues 718–963; see Magga et al., 2000) to inhibit syntaxin 1A binding competitively to the channel. As seen in Figure 1B, coexpression with the synprint peptide resulted in a significant reduction in the degree of PP relief (28 ± 4%), indicating that the effect of syntaxin 1A on G-protein inhibition of the channel is indeed dependent on syntaxin 1A binding to the channel rather than on a diffuse activation of G-protein signaling in the tsA-201 cells. If the syntaxin 1A-mediated enhancement of G-protein inhibition instead had been attributable to a syntaxin 1A-mediated transport of Gβγ to the plasma membrane, the synprint peptide should not have been able to interfere with such a process because syntaxin 1A can interact concomitantly with Gβγ and the synprint region (Jarvis et al., 2000).
Fig. 1.

Effect of syntaxin 1A on N-type channel function.A, Current records obtained from N-type (α1B + β1b + α2-δ) calcium channels coexpressed with either syntaxin 1A (top) or syntaxin 1A plus the C-terminal fragment of the β-adrenergic receptor kinase (β-ARKct;bottom). In the presence of syntaxin 1A the channels undergo a pronounced tonic inhibition, which is reversed with the application of a strong depolarizing prepulse (PP). In the presence of β-ARKct the PP effect is attenuated, consistent with the involvement of Gβγ. B, Comparison of the degree of PP relief obtained in the absence of syntaxin 1A, in the presence of syntaxin 1A alone, or in combination with β-ARKct or a peptide fragment of the synprint region of the channel. C, Steady-state inactivation curves obtained from transiently expressed N-type channels in the absence and presence of syntaxin 1A. The data were fit with the Boltzmann relation; the half-inactivation potentials obtained from the fits were −44.2 and −59.7 mV in the absence and presence of syntaxin 1A, respectively, with respective slope factors of 3.9 and 3.0. Inset, Half-inactivation potentials obtained in the absence of syntaxin 1A, in the presence of syntaxin 1A alone, or with syntaxin 1A plus either β-ARKct or the synprint peptide. The numbers inparentheses indicate the numbers of experiments; error bars denote SE.
A second action of syntaxin 1A is illustrated in Figure 1C. With the coexpression of syntaxin 1A the half-inactivation potential observed with wild-type α1B(β1b+α2-δ) calcium channels was shifted toward more hyperpolarized potentials by ∼15 mV, which is consistent with previous results obtained inXenopus oocytes (Bezprozvanny et al., 1995, 2001). Unlike in the case of the G-protein effect, the coexpression of the β-ARKct fragment did not affect the syntaxin 1A-mediated shift in half-inactivation potential significantly (p > 0.05), indicating that the effect of syntaxin 1A on channel inactivation occurs independently of Gβγ. As expected, the coexpression of the synprint peptide completely blocked the shift in half-inactivation potential. Overall, the data presented in Figure 1illustrate two separate actions of syntaxin 1A on N-type channel gating and modulation by G-proteins and show that the two effects are not coupled to each other.
Coexpression of nSec-1 and SNAP25 affects syntaxin 1A action
Besides syntaxin 1A, the N-type calcium channel domain II/III linker also interacts with SNAP25 (Yokoyama et al., 1997). In addition, syntaxin 1A tightly binds to nSec-1 (Hata et al., 1993; Pevsner et al., 1994b). This raises the possibility that the effects of syntaxin 1A on calcium channel function may be affected by these two proteins. To test this possibility, we cloned nSec-1 and SNAP25 from rat brain, coexpressed them individually with the N-type calcium channel and syntaxin 1A in tsA-201 cells, and assessed the level of tonic G-protein inhibition and inactivation properties of the channel.
To ensure that our SNAP25 and nSec-1 constructs were expressed in tsA-201 cells, we performed Western blot analysis of the transfected cells. As seen in Figure2A, whereas tsA-201 cells did not express the syntaxin 1A, nSec-1, or SNAP25 endogenously, transient transfection of the appropriate cDNA constructs resulted in bands that were detected by specific antibodies to the three proteins. Hence, we conclude that our cDNA constructs are expressed effectively in our experimental system and only when exogenously transfected.
Figure 2B examines the degree of prepulse relief that is obtained with transiently expressed N-type calcium channels under several experimental conditions. As already shown in Figure 1, in the absence of any exogenously expressed SNARE proteins the N-type calcium channels showed only a low degree of tonic G-protein inhibition (9 ± 5% relief) that was enhanced dramatically after the coexpression of syntaxin 1A. Coexpression of the channel with SNAP25 alone did not induce G-protein inhibition (15 ± 4% PP relief;p > 0.05 vs control) but was able to reduce the syntaxin 1A-mediated effect significantly, although substantial modulation (44 ± 6% relief) remained. nSec-1 did not appear to affect the syntaxin 1A-mediated G-protein inhibition; however, surprisingly, nSec-1 alone produced a large tonic inhibition of the channel, thus precluding us from ruling out the possibility that nSec-1 could interfere functionally with the syntaxin 1A-mediated inhibition. Commonly, it is thought that nSec-1 does not interact directly with the channel but rather with syntaxin 1A (Misura et al., 2000); hence, unlike syntaxin 1A, nSec-1 by itself is unlikely to be able to mediate a colocalization of the channel and Gβγ. Furthermore, Western blots indicate that nSec-1 expression does not induce the expression of syntaxin 1A (data not shown). One possible explanation of this effect could be a direct interaction between nSec-1 and Gα, thereby creating a Gα sink and making endogenous Gβγ available for modulating the channel, but additional biochemical work will be needed to substantiate this possibility. Nonetheless, our data show that syntaxin 1A-mediated tonic G-protein inhibition of the channel is maintained in the presence of other SNARE proteins.
To test whether the presence of SNAP25 or nSec-1 could affect the ability of syntaxin 1A to promote voltage-dependent inactivation of the channel, we recorded steady-state inactivation curves from transiently expressed N-type channels either in the absence of or in the presence of various combinations of SNARE proteins. As shown in Figure2C, qualitatively similar to syntaxin 1A, SNAP25 significantly shifted the half-inactivation potential of the channel toward more depolarized potentials (from −44.9 ± 1.8 to −53 ± 1.6 mV). In contrast, the coexpression of nSec-1 did not affect channel gating per se but ablated the syntaxin 1A-mediated shift in half-inactivation potential. Interestingly, despite the finding that syntaxin 1A and SNAP25 both affected inactivation when coexpressed with the channels individually, no shift was observed when both proteins were present concomitantly (Vh = −42.5 ± 6 mV). This may be consistent with the idea that SNAP25 and syntaxin 1A form a highly stable complex (Söllner et al., 1993; Hayashi et al., 1994) in which these proteins may adopt a different conformation that is not conducive to inducing shifts in half-inactivation potentials. Thus, the full effects of syntaxin 1A on N-type channel inactivation are dependent on the absence of other types of SNARE proteins and thus may occur in intact neurons only under certain specific conditions. Furthermore, the data presented in Figure2 indicate that separate determinants govern the effects of syntaxin 1A on channel inactivation and on G-protein modulation.
PKC-dependent phosphorylation affects syntaxin 1A action
Work by Yokoyama et al. (1997) indicates that phosphorylation of the synprint site (residues 718–963 of the α1Bsubunit) of the N-type calcium channel domain II/III linker region by either PKC or CaMKII completely abolishes syntaxin 1A binding. In contrast, neither PKA nor PKG was effective (Yokoyama et al., 1997). In view of this evidence, we hypothesized that PKC-dependent phosphorylation of the appropriate sites in the synprint region should result in the removal of the syntaxin 1A-mediated effect on inactivation and G-protein inhibition of the channel.
To test this hypothesis, we coexpressed N-type calcium channels with syntaxin 1A and then applied a 30 nm concentration of the phorbol ester phorbol-12-myristate 13-acetate (PMA) to activate protein kinase C during whole-cell patch-clamp recordings (see Hamid et al., 1999) and to assess any putative effects on steady-state inactivation of the channel. As shown in Figure3A, the syntaxin 1A-mediated negative shift in half-activation potential was abolished after PMA treatment (Vh = −40.4 ± 2 mV;n = 6), indicating that protein kinase C-dependent phosphorylation of the channel interferes with the functional interaction between syntaxin 1A and the N-type channel. Application of PMA per se did not affect the half-inactivation potential of N-type channels that were expressed in the absence of syntaxin 1A (Vh = −45.9 ± 1.6 mV;n = 5), suggesting that PKC-dependent phosphorylation in itself does not affect steady-state inactivation of N-type channels.
Fig. 3.
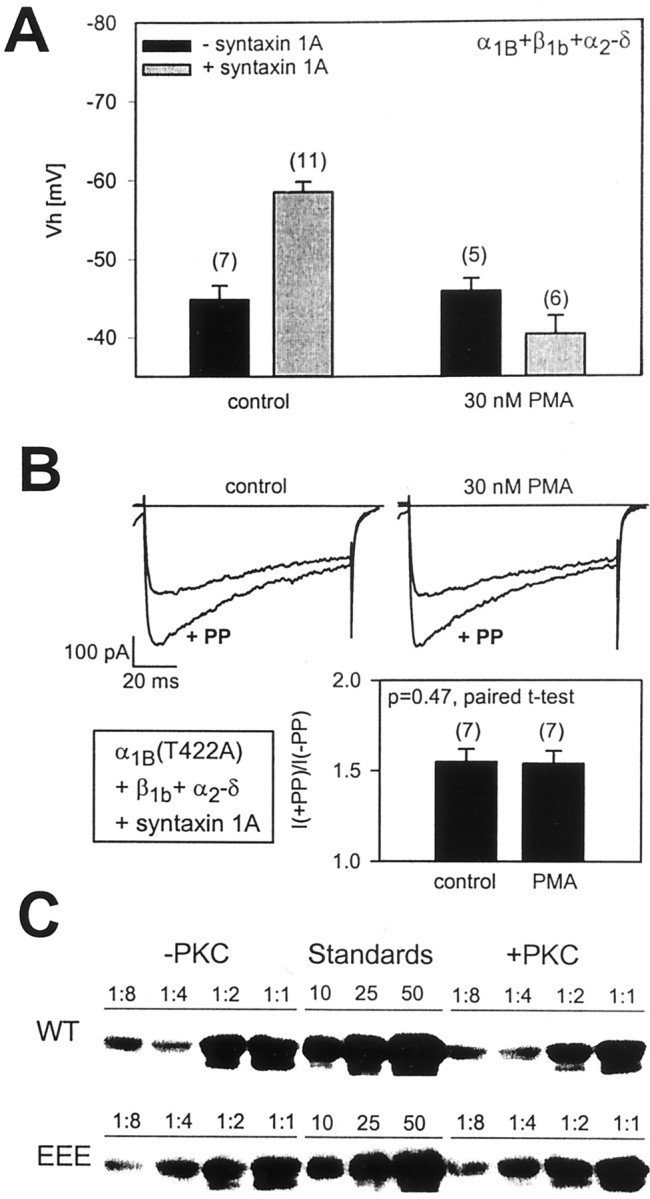
Effect of protein kinase C-dependent phosphorylation on the ability of syntaxin 1A to modulate steady-state inactivation and G-protein inhibition of N-type calcium channels.A, Effect of the application of 30 nm PMA on half-inactivation potentials obtained with N-type calcium channels in the presence and absence of syntaxin 1A. Note that PMA treatment precludes the syntaxin 1A-mediated shift in half-inactivation potential but does not affect the inactivation of N-type calcium channels per se. The half-inactivation potentials obtained with PMA plus syntaxin 1A did not differ significantly from those obtained in the presence of PMA alone (p = 0.08). B, Protein kinase C-dependent phosphorylation does not affect G-protein inhibition of mutant N-type calcium channels in which the I/II linker residue T422 has been replaced with alanine (to prevent PKC–G-protein crosstalk) (Hamid et al., 1999). In this case, data in the absence and presence of PMA (30 nm for 90 sec) (Cooper et al., 2000) were obtained from the same cell. C, The effect of phosphorylation on the in vitro binding of syntaxin 1A to the α1B synprint peptide. Protein kinase C-dependent phosphorylation of the synprint peptide does not inhibit the binding of syntaxin 1A under our experimental conditions. In all, 1 nmol SHAM or PKC-treated synprint peptide (wild type and mutated S802, S896, S898E) immobilized on Ni2+-NTA agarose was incubated with increasing amounts of purified syntaxin 1A. Syntaxin was added in increasing ratios from 1:8 to 1:1. The two bands in the doublet represent nonphosphorylated and phosphorylated syntaxin 1A, which is a result of bacterial post-translational modification. Note that syntaxin 1A phosphorylation does not interfere with binding to the synprint motif. The control lanes show 10, 25, and 50 pmol of purified syntaxin 1A, which allows for comparisons between the WT and EEE blots.
It is not possible to use wild-type N-type channels to determine whether PKC-dependent phosphorylation is able to antagonize the syntaxin 1A-mediated G-protein inhibition of the channel because of the well documented antagonistic effects of PKC-dependent phosphorylation on G-protein inhibition of the channel (Swartz, 1993; Zamponi et al., 1997), which occurs via phosphorylation of residue T422 in the Gβγ binding region of the N-type calcium channel domain I/II linker (Hamid et al., 1999;Cooper et al., 2000). We have, however, shown previously that replacement of residue 422 with alanine renders the channels insensitive to this PKC–G-protein crosstalk (Hamid et al., 1999). Hence, to isolate any putative effects of PKC-dependent phosphorylation on the ability of syntaxin 1A to induce tonic G-protein inhibition of the channel, we coexpressed the α1B(T422A) mutant with β1b, α2-δ, and syntaxin 1A and assessed the ability of syntaxin 1A to induce G-protein inhibition before and after the stimulation of PKC with 30 nm PMA. In the absence of PMA the T422A mutant displayed robust G-protein inhibition after coexpression with syntaxin 1A (55 ± 7% PP relief), albeit to a somewhat smaller degree as compared with the wild-type channels, which is consistent with our previous observations that used a somatostatin receptor-mediated inhibition of this mutant (Hamid et al., 1999). More importantly, as shown in Figure 3B, PMA application did not affect the syntaxin 1A-mediated G-protein inhibition of the channel significantly (54 ± 7% PP relief; p = 0.48, paired t test). Hence, PKC-dependent phosphorylation selectively interferes with the ability of syntaxin 1A to affect N-type channel inactivation while leaving the syntaxin 1A-induced G-protein inhibition intact.
PKC-dependent phosphorylation does not abolish syntaxin 1A binding to the synprint motif
We have suggested previously that the effect of syntaxin 1A on G-protein inhibition might be attributable to a syntaxin 1A-mediated colocalization of Gβγand the channel (Jarvis et al., 2000). Within the framework of this model a retention of the syntaxin 1A effect on G-protein inhibition of the channel after PKC-dependent phosphorylation would imply that syntaxin 1A should maintain some ability to bind to the channel. To test this hypothesis, we performed an in vitro binding assay involving recombinant syntaxin 1A and fusion proteins of the N-type calcium channel synprint motif. As shown in Figure 3C, syntaxin 1A binds to the synprint motif in a dose-dependent manner, consistent with previous work (Sheng et al., 1994; Jarvis et al., 2000). Interestingly, however, under our conditions PKC-dependent phosphorylation of the synprint fusion proteins did not affect their ability to interact with syntaxin 1A. This was observed in seven experiments, whereas no binding of syntaxin 1A to histidine beads could be detected (data not shown). We note that, in the experiments shown in Figure 3C, immobilized synprint fusion proteins were phosphorylated with commercially available purified PKC and recombinant syntaxin 1A subsequently was allowed to bind, whereas Yokoyama et al. (1997) phosphorylated the synprint peptides in solution (i.e., after elution) and then bound them to immobilized GST–syntaxin 1A. In two additional experiments in which we followed a reverse paradigm (i.e., binding of phosphorylated synprint peptide to immobilized syntaxin 1A), synprint binding was reduced, albeit not eliminated, after PKC-dependent phosphorylation (data not shown). Hence, it appears that the previously reported PKC-mediated ablation of the syntaxin 1A–synprint interaction in vitro perhaps may be highly dependent on the experimental paradigm that is used. Nonetheless, under physiological conditions syntaxin 1A binding to the domain II/III linker of N-type calcium channels likely occurs even after PKC-dependent phosphorylation of this region because PMA treatment did not appear to affect syntaxin 1A-induced G-protein inhibition of the channel. Yet, the data shown in Figure 3A clearly support a role of protein kinase C in the modulation of at least some of the effects of syntaxin 1A on N-type calcium channels, consistent with the work of Yokoyama et al. (1997).
The effect of PKC on syntaxin 1A action is mimicked by point mutations in the synprint motif
To identify the molecular basis of the effects of PKC on syntaxin 1A-mediated inactivation of N-type channels, we considered a mutagenesis strategy of possible PKC sites in the synprint motif of N-type channels. This region contains a total of 18 serines and 13 threonine residues (Fig. 4). Of those, nine residues were not flanked by basic residues in positions −1 to −3 or +1 to +3, leaving 22 possible phosphorylation sites. To help identify the best candidate sites, we estimated the likelihood of PKC- and CaMKII-dependent phosphorylation of each of the remaining residues according to Kennelly and Krebs (1991). As shown in Figure 4, we identified three sites that scored well above the other sites (S774, S802, S896), of which S802 was also a substrate for CaMKII and S896 was located immediately adjacent to another CaMKII site, S898. For the purposes of mutagenesis, we therefore first considered serines in positions 774, 802, 896, and 898 while being aware that this may not encompass all of the possible residues in the synprint region that might contribute to the antagonistic effect of phosphorylation on the actions of syntaxin 1A.
Fig. 4.

Likelihood of PKC-dependent phosphorylation of serine and threonine residues in the synprint region of the N-type calcium channel α1 subunit according to Kennelly and Krebs (1991). Asterisks indicate substrates for CaMKII. Of the three best PKC consensus sites, only S802 forms a CaMKII site, but S896 is immediately adjacent to a CaMKII consensus motif. S921 is the only PKA consensus site within the synprint region.
To mimic the effects of phosphorylation permanently, we substituted these serine residues for glutamic acid either individually or in combination and assessed their effects on the ability of syntaxin 1A to shift the steady-state inactivation curve and to promote G-protein inhibition. The S774E construct yielded current levels that were too low to permit meaningful recordings. Circular dichroism spectral analysis on wild-type and the S774E mutant synprint fusion proteins suggests that this mutation may result in the disruption of an α-helix (data not shown); hence, this residue was not considered further. However, mutant channels in which residues 802, 896, and 898 were replaced individually or simultaneously with glutamic acid exhibited robust expression. As shown in Figure5A, after individual or even concomitant substitution of S802, S896, and S898 the channels underwent a large tonic G-protein inhibition in the presence of syntaxin 1A, which did not differ significantly from that observed with the wild-type channel, consistent with the data shown in Figure 3,B and C.
Fig. 5.

Effect of point mutations in PKC consensus sites of the synprint motif on syntaxin 1A-mediated changes in channel function. A, Effect of point mutations on the degree of syntaxin 1A-induced PP facilitation of the channel. Note that, even in the triple S802,896,898E mutant, the G-protein effect remains evident.B, Effect of the mutations on the ability of syntaxin 1A to mediate shifts in half-inactivation potential. Note that the syntaxin 1A-mediated shifts in the half-inactivation potential were abolished with the mutagenesis of residues 896 and 898, but not with the substitution of residue 802. The numbers inparentheses denote the numbers of experiments; error bars are SE. The control and syntaxin 1A data are the same as in Figure1.
Figure 5B examines the effect of the triple mutation on the position of the steady-state inactivation curve along the voltage axis in the absence and presence of syntaxin 1A. As seen from Figure5B, the triple mutant exhibited a slightly more negative half-inactivation potential (−49.7 ± 2 mV) as compared with the wild-type channel but was no longer susceptible to further negative shifts in Vh with the coexpression of syntaxin 1A. Site-directed mutagenesis of the individual serine residues reveals that the syntaxin 1A-induced shift inVh was blocked by “permanent phosphorylation” of either residue 896 or 898, but not by residue 802. We also created a channel in which residues 896 and 898 were replaced concomitantly by alanine to test whether the removal of these two PKC consensus sites could block the effects of PMA on N-type channel inactivation, but this construct could not be expressed functionally in tsA-201 cells. Thus, we cannot rule out the possibility that phosphorylation of one of the remaining serine/threonine residues within the synprint region perhaps also might be able to interfere with the syntaxin 1A-mediated inactivation of the channel.
Nonetheless, the data shown in Figure 5 are consistent with a mechanism by which phosphorylation of residues 896 and/or 898 by protein kinase C is sufficient to prevent the effects of syntaxin 1A on voltage-dependent inactivation of N-type calcium channels. Additionally, these observations further support the notion that distinct structural determinants govern the ability of syntaxin 1A to modulate G-protein inhibition and inactivation of the N-type calcium channel.
Syntaxin 1B selectively affects steady-state inactivation of N-type channels
We have shown recently that, in contrast with syntaxin 1A, the expression of syntaxin 1B does not result in a tonic G-protein inhibition of transiently expressed N-type calcium channels (Lu et al., 2001) To assess whether syntaxin 1B was able to shift the half-inactivation potential of N-type calcium channels, we coexpressed syntaxin 1B with α1B(+β1b+α2-δ) calcium channels and recorded a series of steady-state inactivation curves. As shown in Figure 6, syntaxin 1B was able to induce a negative shift in half-inactivation potential (Vh = −55.7 ± 2.4 mV), which was similar in magnitude to that observed with syntaxin 1A. Hence, syntaxin 1B affects steady-state inactivation of the channels without promoting their inhibition by G-proteins, indicating that separate syntaxin structural requirements underlie these two phenomena.
Fig. 6.

Syntaxin 1B induces a negative shift in half-inactivation potential. Shown is an ensemble of seven steady-state inactivation curves recorded from N-type calcium channels in the presence of syntaxin 1B. The data were fit with the Boltzmann equation. The half-inactivation potential obtained from the fit was −55.6 mV.Inset, Comparison of the half-inactivation potentials obtained in the absence and in the presence of syntaxin 1B. Individual inactivation curves were fit with the Boltzmann equation, and the means plus SEM values are plotted in the form of bar graphs. Thenumbers in parentheses denote the numbers of experiments.
DISCUSSION
Modulation of syntaxin 1A action by other SNARE proteins
Both syntaxin 1A and SNAP25 have been shown to interact with the domain II/III linker regions of P/Q-type and N-type calcium channels, and both proteins can bind to this region simultaneously (Sheng et al., 1994; Rettig et al., 1996; Yokoyama et al., 1997). The bound proteins appear to participate in the interaction with v-SNARES, and it is known that syntaxin 1A forms tight interactions with nSec-1 (Hata et al., 1993; Pevsner et al., 1994b). One therefore might expect that the changes in calcium channel function observed with the coexpression of the channel with only syntaxin 1A in transient expression systems (Bezprozvanny et al., 1995; Jarvis et al., 2000) may not be truly indicative of the events occurring in neurons. Indeed, coexpression of either nSec-1 or SNAP25 removed the ability of syntaxin 1A to mediate a negative shift in Vh while leaving a significant portion (∼50%) of its effect on G-protein modulation intact, which is consistent with the notion that the interactions of syntaxin 1A with SNAP25/nSec-1 and the synprint region are not mutually exclusive. Thus, depending on which proteins are associated with the channel at a given point in time, calcium channels would have altered inactivation properties. Although our experiments were designed primarily to elucidate molecular determinants that govern the two distinct effects of syntaxin 1A on N-type calcium channel function, it is tempting to speculate on possible physiological implications of our results. For example, in the presence of only syntaxin 1A or SNAP25, vesicles cannot dock to the channel; thus, such a channel would be unlikely to participate in fast neurotransmitter release. The negative shift in half-inactivation potential and the concomitant tonic G-protein inhibition perhaps could reduce calcium entry through this channel and thus prevent unnecessary calcium overload (Bezprozvanny et al., 1995). With the binding of both SNAP25 and syntaxin 1A, the docking of the presynaptic vesicle would become possible, and concomitantly the level of tonic G-protein inhibition would become reduced and the availability for opening increased because of the removal of the syntaxin 1A-induced hyperpolarizing shift inVh. In principle, such a mechanism perhaps could provide for selective calcium entry through channels bound to docked vesicles. However, unlike tsA-201 cells an intact synapse likely expresses a number of other binding partners for SNARE proteins, which could influence the effects of syntaxin 1A on both G-protein modulation and voltage-dependent inactivation of the channel plus the regulation of these effects by PKC. Thus, our findings should not be extrapolated firmly to what might occur in intact neurons. Indeed, our observations with nSec-1 and the recently reported effects of cysteine string proteins on N-type channel regulation by G-proteins (Magga et al., 2000) may be indicative of such added complexity.
Phosphorylation dependence of syntaxin 1A effects
The interaction between SNARE proteins and presynaptic calcium channels is considered a key step in the fast release of neurotransmitter, and factors that modulate either the entry of calcium or the association of the SNARE protein/calcium channel complex have the propensity to regulate neurotransmission (Wheeler et al., 1994;Dunlap et al., 1995; Mochida et al., 1996; Stanley, 1997). It is well established that Gβγsubunits and activation of PKC are important determinants of the regulation of calcium channel activity (Swartz et al., 1993; Stea et al., 1995; Herlitze et al., 1996; Ikeda, 1996; De Waard et al., 1997;Page et al., 1997; Zamponi et al., 1997; Hamid et al., 1999; Arnot et al., 2000). In addition, there appears to be a complex interplay between calcium channel modulation by second messengers and the association of the channel with SNARE proteins (Stanley and Mirotznik, 1997; Yokoyama et al., 1997; Jarvis et al., 2000). At least within the confines of a recombinant system, our present work adds further intricacy to these interactions.
Our functional electrophysiological data show that activation of protein kinase C with PMA continued to result in the syntaxin 1A-mediated tonic G-protein inhibition of the channel. We note that disruption of syntaxin 1A binding to the channel by other means (i.e., overexpression of synprint peptides) was able to depress the extent of G-protein inhibition dramatically. It therefore seems unlikely that syntaxin 1A would be dislodged fully from the channel after PKC-dependent phosphorylation under physiological conditions, although we must acknowledge the possibility that PKC-dependent phosphorylation of synprint peptides in vitro may be capable of abolishing their interactions with syntaxin 1A completely under certain circumstances (Yokoyama et al., 1997). Under physiological conditions, however, syntaxin 1A well may associate more tightly with the channel because of the presence of the plasma membrane and possibly weak interactions with other channel regions that may contribute to the maintained G-protein effect despite PKC-dependent phosphorylation.
In contrast, the syntaxin 1A-mediated shift in half-inactivation potential was removed after activation of PKC or by site-directed mutagenesis of putative PKC consensus sites. Together with the observation that coexpression of either SNAP25 or nSec-1 also prevented the ability of syntaxin 1A to shift the half-inactivation potential, this may indicate that the conformation of the domain II/III linker, which results in the negative shifts inVh, is disrupted easily. Although individual substitutions of residues 896 and 898 with glutamic acid were sufficient to mimic the effect of protein kinase C-dependent phosphorylation on voltage-dependent inactivation of the channel, we cannot rule out the possibility that phosphorylation of one or more of the other serine/threonine residues contained in the N-type channel domain II/III linker perhaps could result in a similar loss of the effects of syntaxin 1A on N-type channel inactivation.
We have shown previously that activation of PKC results in two effects on N-type calcium channels (Zamponi et al., 1997; Hamid et al., 1999). First, PKC-dependent phosphorylation of either residues T422 or S425 in the N-type calcium channel domain I/II linker region results in an upregulation of current activity. In addition, selective PKC-dependent phosphorylation of T422 antagonizes G-protein binding to the channel (see also Swartz et al., 1993; Barrett and Rittenhouse, 2000; Cooper et al., 2000). The observation that PKC-dependent phosphorylation of residues S896 or S898 is sufficient to block the negative shift in half-inactivation potential would suggest a further increase in the availability of the channels despite the presence of syntaxin 1A. This built-in redundancy, together with the notion that the activity of N-type calcium channel can be increased via multiple avenues after phosphorylation, may support a critical role of the phosphorylation process in the control of calcium entry.
Possible mechanism underlying the modulation of syntaxin 1A action
One common theme emerging from the present study is the notion that the molecular determinants that govern the effect of syntaxin 1A on channel inactivation differ from those underlying the syntaxin 1A-induced G-protein inhibition. Neither of the point mutations of PKC consensus sites in the synprint motif nor activation of PKC with PMA nor the coexpression of SNAP25 or nSec-1 eliminated the tonic G-protein inhibition mediated by syntaxin 1A, whereas either one of these factors could abolish the effects of syntaxin 1A on inactivation of the channel. In contrast, syntaxin 1B promoted N-type channel inactivation without mediating a G-protein effect (Lu et al., 2001), which suggests that syntaxin 1B is capable of interacting with the channel, but perhaps not with Gβγ.
Our observation that PKC-dependent phosphorylation did not appear to be capable of removing syntaxin 1A from the synprint site under physiological conditions would be consistent with the presence of multiple syntaxin micro-binding sites within the synprint region. PKC-dependent phosphorylation or site-directed mutagenesis of S896 and S898 might result in the loss of one of the contact points between syntaxin 1A and the channel, but syntaxin 1A nonetheless could remain attached to other subregions of the channel that are not affected by phosphorylation. The possibility of a weak interaction between syntaxin 1A and a portion of the synprint motif that is linked to calcium channel inactivation perhaps could explain the observation that the H3 domain of syntaxin 1A, which appears to be required for the inactivation effect, does not exhibit detectable binding to the domain II/III linker of the N-type channel in vitro (Bezprozvanny et al., 2001). Along these lines, the coexpression of nSec-1 or SNAP25 either could hinder the interaction of syntaxin 1A with the regions responsible for the inactivation effect sterically or perhaps could allosterically preclude the translation of syntaxin 1A binding into a change in channel gating. However, ultimately for these possibilities to be substantiated, more detailed structural information, such as a crystal structure of the synprint region in the presence and absence of SNARE proteins and G-proteins, will be required.
Overall, our data provide novel insights into the interactions among N-type calcium channels, SNARE proteins, and cytoplasmic messengers at the molecular level. To appreciate fully the significance of these interactions for neurotransmission is a daunting task, particularly in light of recent evidence that the cysteine string protein also may interact with the synprint region to produce functional consequences on G-protein modulation (Leveque et al., 1998; Magga et al., 2000) and that the expression of P/Q-type calcium channels can mediate calcium-dependent gene transcription of syntaxin 1A (Sutton et al., 1999). The sheer complexity of these interactions suggests that the mammalian brain has devised multiple avenues for the precise control of events resulting in calcium homeostasis and vesicle release in the presynaptic nerve terminal.
Footnotes
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR). G.W.Z. holds faculty scholarships from the CIHR, the Alberta Heritage Foundation for Medical Research (AHFMR), and The EJLB Foundation; he is the Novartis Investigator in Schizophrenia Research. S.E.J. is the recipient of studentship awards from the AHFMR and the Health Research Foundation of Canada. We thank Dr. Terry Snutch for providing cDNA encoding for wild-type calcium channel subunits and Dr. William Catterall for discussion. We also thank Dr. Janice Braun for helpful discussion and for donating a syntaxin 1A–GST construct and antibodies to SNARE proteins. Finally, we thank Dr. Jawed Hamid for providing the T422A mutant, Bob Winkfein for providing rat brain cDNA, and Clinton Doering for technical assistance.
Correspondence should be addressed to Dr. Gerald W. Zamponi, Department of Physiology and Biophysics, University of Calgary, 3330 Hospital Drive NW, Calgary, T2N 4N1, Canada. E-mail: Zamponi@ucalgary.ca.
REFERENCES
- 1.Arnot MI, Stotz SC, Jarvis SE, Zamponi GW. Differential modulation of N-type a1B and P/Q-type a1A calcium channels by different G-protein β-subunit isoforms. J Physiol (Lond) 2000;527:203–212. doi: 10.1111/j.1469-7793.2000.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett CF, Rittenhouse AR. Modulation of N-type calcium channel activity by G-proteins and protein kinase C. J Gen Physiol. 2000;115:277–286. doi: 10.1085/jgp.115.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- 4.Bezprozvanny I, Zhong P, Scheller RH, Tsien RW. Molecular determinants of the functional interaction between syntaxin and N-type Ca2+ channel gating. Proc Natl Acad Sci USA. 2001;97:13943–13948. doi: 10.1073/pnas.220389697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper CB, Arnot MI, Feng Z-P, Jarvis SE, Hamid J, Zamponi GW. Crosstalk between G-protein and PKC modulation of N-type calcium channels is dependent on the G-protein β-subunit isoform. J Biol Chem. 2000;275:40777–40781. doi: 10.1074/jbc.C000673200. [DOI] [PubMed] [Google Scholar]
- 6.Degtiar VE, Scheller RH, Tsien RW. Syntaxin modulation of slow inactivation of N-type calcium channels. J Neurosci. 2000;20:4355–4367. doi: 10.1523/JNEUROSCI.20-12-04355.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP. Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- 8.Dubel SJ, Starr TVB, Hell J, Ahlijanian MK, Enyeart JJ, Catterall WA, Snutch TP. Molecular cloning of the α1 subunit of an ω-conotoxin-sensitive calcium channel. Proc Natl Acad Sci USA. 1992;89:5058–5062. doi: 10.1073/pnas.89.11.5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunlap K, Fischbach GD. Neurotransmitters decrease the calcium conductance activated by depolarization of embryonic chick sensory neurones. J Physiol (Lond) 1981;317:519–535. doi: 10.1113/jphysiol.1981.sp013841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunlap K, Luebke JL, Turner TJ. Exocytotic Ca2+ channels in the mammalian central nervous system. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 11.Hamid J, Nelson D, Spaetgens R, Dubel SJ, Snutch TP, Zamponi GW. Identification of an integration center for crosstalk between PKC and G-protein modulation of N-type calcium channels. J Biol Chem. 1999;278:6195–6202. doi: 10.1074/jbc.274.10.6195. [DOI] [PubMed] [Google Scholar]
- 12.Hata Y, Slaughter CA, Sudhof TC. Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature. 1993;366:347–351. doi: 10.1038/366347a0. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Sudhof TC, Niemann H. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 15.Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 16.Jarvis SE, Magga J, Beedle A, Braun J, Zamponi GW. G-protein modulation of N-type calcium channels is facilitated by physical interactions between syntaxin 1A and Gβγ. J Biol Chem. 2000;275:6388–6394. doi: 10.1074/jbc.275.9.6388. [DOI] [PubMed] [Google Scholar]
- 17.Kennelly PJ, Krebs EG. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J Biol Chem. 1991;266:15555–15558. [PubMed] [Google Scholar]
- 18.Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. Cellular expression of the carboxyl terminus of a G-protein-coupled receptor kinase attenuates Gβγ-mediated signaling. J Biol Chem. 1994;269:6193–6197. [PubMed] [Google Scholar]
- 19.Leveque C, Pupier S, Marqueze B, Geslin L, Kataoka M, Takahashi M, De Waard M, Seagar M. Interaction of cysteine string proteins with the α1A subunit of the P/Q-type calcium channel. J Biol Chem. 1998;273:13488–13492. doi: 10.1074/jbc.273.22.13488. [DOI] [PubMed] [Google Scholar]
- 20.Lin RC, Scheller RH. Mechanisms of synaptic vesicle exocytosis. Annu Rev Cell Dev Biol. 2000;16:19–49. doi: 10.1146/annurev.cellbio.16.1.19. [DOI] [PubMed] [Google Scholar]
- 21.Linial M, Bledi Y. Syntaxin the pillar of secretion. Modulator. 2000;12:11–13. [Google Scholar]
- 22.Lu Q, Atkisson MS, Jarvis SE, Feng Z-P, Zamponi GW, Dunlap KD. Syntaxin 1A supports voltage-dependent inhibition of α1B Ca2+ channels by Gβγ in chick sensory neurons. J Neurosci. 2001;21:2949–2957. doi: 10.1523/JNEUROSCI.21-09-02949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magga JM, Jarvis SE, Arnot MI, Zamponi GW, Braun JEA. Cysteine string protein regulates G-protein modulation of N-type calcium channels. Neuron. 2000;28:195–204. doi: 10.1016/s0896-6273(00)00096-9. [DOI] [PubMed] [Google Scholar]
- 24.Misura KMS, Scheller RH, Weis WI. Three-dimensional structure of the neuronal Sec1–syntaxin 1A complex. Nature. 2000;404:355–362. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- 25.Mochida S, Sheng Z-H, Baker C, Kobayashi H, Catterall WA. Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2+ channels. Neuron. 1996;17:781–788. doi: 10.1016/s0896-6273(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 26.Page KM, Stephens GJ, Berrow NS, Dolphin AC. The intracellular loop between domains I and II of the B-type calcium channel confers aspects of G-protein sensitivity to the E-type calcium channel. J Neurosci. 1997;17:1330–1338. doi: 10.1523/JNEUROSCI.17-04-01330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pevsner J, Hsu SC, Braun JE, Calakos N, Ting AE, Bennett M, Scheller RH. Specificity and regulation of a synaptic vesicle docking complex. Neuron. 1994a;13:353–361. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- 28.Pevsner J, Hsu SC, Scheller RH. nSec-1: a neuronal-specific syntaxin binding protein. Proc Natl Acad Sci USA. 1994b;91:1445–1449. doi: 10.1073/pnas.91.4.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA. Isoform-specific interaction of the α1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP25. Proc Natl Acad Sci USA. 1996;93:7363–7368. doi: 10.1073/pnas.93.14.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rettig J, Heinemann C, Ashery U, Sheng ZH, Yokoyama CT, Catterall WA, Neher E. Alteration of Ca2+ dependence of neurotransmitter release by disruption of Ca2+channel/syntaxin interaction. J Neurosci. 1997;17:6647–6656. doi: 10.1523/JNEUROSCI.17-17-06647.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheng Z, Rettig T, Takahashi M, Catterall WA. Identification of a syntaxin-binding site on N-type calcium channels. Neuron. 1994;13:1303–1313. doi: 10.1016/0896-6273(94)90417-0. [DOI] [PubMed] [Google Scholar]
- 32.Sheng Z-H, Rettig J, Cook T, Catterall WA. Calcium-dependent interaction of N-type calcium channels with the synaptic core complex. Nature. 1996;379:451–454. doi: 10.1038/379451a0. [DOI] [PubMed] [Google Scholar]
- 33.Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 34.Stanley EF. The calcium channel and the organization of the presynaptic transmitter release face. Trends Neurosci. 1997;20:404–409. doi: 10.1016/s0166-2236(97)01091-6. [DOI] [PubMed] [Google Scholar]
- 35.Stanley EF, Mirotznik RR. Cleavage of syntaxin prevents G-protein regulation of presynaptic calcium channels. Nature. 1997;385:340–343. doi: 10.1038/385340a0. [DOI] [PubMed] [Google Scholar]
- 36.Stea A, Soong TW, Snutch TP. Determinants of PKC-dependent modulation of a family of neuronal calcium channels. Neuron. 1995;5:929–940. doi: 10.1016/0896-6273(95)90183-3. [DOI] [PubMed] [Google Scholar]
- 37.Sutton KG, McRory JE, Guthrie H, Murphy TH, Snutch TP. P/Q-type calcium channels mediate the activity-dependent feedback of syntaxin 1A. Nature. 1999;401:800–804. doi: 10.1038/44586. [DOI] [PubMed] [Google Scholar]
- 38.Swartz KJ. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: disruption of G-protein-mediated inhibition. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- 39.Swartz KJ, Merrit A, Bean BP, Lovinger DM. Protein kinase C modulates glutamate receptor inhibition of Ca2+ channels and synaptic transmission. Nature. 1993;361:165–168. doi: 10.1038/361165a0. [DOI] [PubMed] [Google Scholar]
- 40.Umbach JA, Gunderson CB. Evidence that cysteine string proteins regulate an early step in Ca2+-dependent secretion of neurotransmitter at Drosophila neuromuscular junctions. J Neurosci. 1997;17:7203–7209. doi: 10.1523/JNEUROSCI.17-19-07203.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker D, De Waard M. Subunit interaction sites in voltage-dependent Ca2+ channels: role in channel function. Trends Neurosci. 1998;21:148–154. doi: 10.1016/s0166-2236(97)01200-9. [DOI] [PubMed] [Google Scholar]
- 42.Westenbroek RE, Hell JW, Warberm C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel α1 subunit. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- 43.Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP, Catterall WA. Immunochemical identification and subcellular distribution of the α1A subunits of brain calcium channels. J Neurosci. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- 45.Wu MN, Fergestad T, Lloyd TE, He Y, Broadie K, Bellen HJ. Syntaxin 1A interacts with multiple exocytotic proteins to regulate neurotransmitter release in vivo. Neuron. 1999;23:593–605. doi: 10.1016/s0896-6273(00)80811-9. [DOI] [PubMed] [Google Scholar]
- 46.Yokoyama CT, Sheng Z, Catterall WA. Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J Neurosci. 1997;17:6929–6938. doi: 10.1523/JNEUROSCI.17-18-06929.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G-proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 1997;385:242–246. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]