

Abstract
Desensitization is a common property of glutamate and other ligand-gated ion channels, yet its molecular mechanism is unknown. For glutamate receptors, agonist binding involves interactions with identified amino acids from two lobes and may result in stabilizing the lobes in a closed “clamshell” conformation. The present studies demonstrate that two structures, β-strands 7 and 8 and α-helices J and K, functionally interact with each other and likely form hinges between the two lobes, influencing the coupling between agonist binding and desensitization. Two amino acids identified within these regions form a solvent-exposed interface with a third amino acid, a mutation of which was shown previously to block receptor desensitization (L507 in glutamate receptor 3). This interface may regulate a concerted conformational shift of the AMPA subtype of glutamate receptor subunits to the desensitized state.
Keywords: AMPA receptors, glutamate receptors, cyclothiazide, desensitization, electrophysiology, ion channels, mutagenesis
Glutamate receptors mediate rapid excitatory synaptic transmission in the CNS. This signaling occurs via activation, deactivation, and desensitization gating transitions, which control ion permeation on a millisecond time scale (Edmonds et al., 1995; Trussell and Otis, 1996). Although the molecular events underlying coupling between the agonist-binding site and the control of ion permeation are unclear, a 38 amino acid “flip/flop” domain that lies between transmembrane segments M3 and M4 of all AMPA receptors is involved (Sommer et al., 1990). A single residue within flip/flop [S750 in flip or N750 in flop; amino acids are numbered according to the mature protein (Bettler et al., 1992)] determines differential sensitivity to allosteric modulators (Partin et al., 1996).
The agonist-binding domain of the glutamate receptor 2 (GluR2) subunit of AMPA receptors is formed by two globular lobes (domains 1 and 2) consisting of highly ordered α-helices and β-sheets that are connected by two polypeptide strands (crossovers 1 and 2) (Armstrong et al., 1999). Domain 1 is made up of residues mostly from the “S1” region upstream of membrane segment 1 (M1), whereas domain 2 is made up mostly of residues from the “S2” region between M2 and M3 (Stern-Bach et al., 1994). Glutamate receptors share structural homology with the prokaryotic amino acid-binding proteins, lysine/arginine/ornithine-binding protein (LAOBP) (Oh et al., 1993), and glutamine-binding protein (QBP) (Nakanishi et al., 1990; O'Hara et al., 1993; Hsiao et al., 1996). For LAOBP, bound ligand stabilizes a protein conformation consisting of a rotation of one lobe with respect to the other, in what has been described as a closed “Venus fly-trap” or “clamshell” conformation. The closed conformation results in a large shift in the torsion angle of the N-Cα peptide bond (ψ) of one amino acid (A90) and smaller shifts of φ or ψ for four other amino acids within the two connecting strands (Oh et al., 1993). For glutamate receptors it is assumed that the same type of conformational shift occurs (Mano et al., 1996; Sutcliffe et al., 1996; Swanson et al., 1997; Paas, 1998). However, although amino acid-binding proteins contain sequences homologous to the N-terminal portion of flip/flop in glutamate receptors, the receptors contain additional C-terminal sequences that continue through and beyond this region of homology. The additional residues contain a cysteine that forms a disulfide bond with a cysteine in domain 2, allowing flip/flop (overlapping α-helices J and K; see Fig. 1) to serve as a third connecting strand between the two lobes.
Fig. 1.

Sequence and structure of GluR6 chimeras.a, The molecular structure of GluR2 S1/S2 (Armstrong et al., 1999), with BETA 7,8 shown in red and the flip/flop region shown in green, is in accordance with thecolor scheme shown for the primary sequence below.b, GluR1 amino acids swapped into GluR6 are indicated with red and green lines for the following chimeras (GluR1 residue numbers are indicated in parentheses in the following and refer to the mature GluR1 protein):GluR6-BETA 7,8 (489–502), -Flip(722–781), -Helix J (742–752), -Helix K(750–763), and -FlipC17 (764–780). Structural elements of the GluR2 agonist-binding domain (Armstrong et al., 1999) are indicated in gray; horizontal bars represent α-helices, and arrows represent β-strands. Other symbols indicate residues known to be important in desensitization: the filled diamondindicates the amino acid that, when mutated, completely blocks desensitization [L507 in GluR3 (Stern-Bach et al., 1998)]; the filled triangle marks a site of glycosylation that is important for modulation by Concanavalin A of GluR6 desensitization (Everts et al., 1999); the filled square represents a residue thought to form an important exposed patch within a hydrophobic domain (Chen et al., 1999); andfilled circles indicate residues that differ as a result of alternative splicing within the flip/flop domain of GluR1 (Sommer et al., 1990). Residues in bold represent amino acids that are divergent between GluR1 and GluR6. c, Spatial relationship between GluR2 amino acids homologous to GluR1 residues S493 (within BETA 7,8), N750 (S750within GluR1 flip), and L479 [L507 of GluR3 (Stern-Bach et al., 1998)] is shown. This view of the molecule is rotated, with respect to a, to show better the side chains of residues used in this study. d, An identical view of the molecule is shown in a space-filling representation. The BETA 7,8 domain is shown in red, the flip/flop domain is shown in green, and the three critical residues are shown in pink. BETA 7,8, β-Strands 7 and 8; C, C terminal; N, N terminal.
The goal of the present study was to determine whether residues within the S1/S2 structure interact with flip/flop to control desensitization and its modulation by drugs, by the use of chimeric receptors. A better understanding of the molecular details of allosteric modulation may provide a rational basis for the development of new drugs to downregulate receptor activity during episodes of hyperexcitability (Rogawski, 1993) or to upregulate glutamate receptor activity, enhancing learning and memory after loss of glutamatergic neurons after stroke or brain injury (Yamada, 1998).
MATERIALS AND METHODS
Recombinant DNA. Plasmids encoding the cDNA for the rat flip variant of wild-type GluRA (GluR1), as well as the cDNAs for green fluorescent protein (GFP) and GluR6, were a gift of Dr. Peter Seeburg (MPI Medical Research, Heidelberg, Germany). The wild-type glutamate receptor cDNAs were subcloned into pBlueScript II (Stratagene, La Jolla, CA). Point mutations in GluR1 were constructed by the use of QuikChange (Stratagene), whereas GluR6/GluR1 chimeras were made by using overlapping PCR. All mutations were confirmed by sequencing (Macromolecular Resources Facility, Colorado State University, Fort Collins, CO). Purified plasmid DNA was restricted with EcoRI and was then used as a template forin vitro transcription by T7 polymerase (Ambion, Austin, TX). Mutations were numbered according to the mature (truncated) GluR1 or GluR6 protein (Bettler et al., 1992).
Oocyte electrophysiology. Oocytes were harvested fromXenopus laevis as described previously (Cotton and Partin, 2000). Animal care and surgical procedures conformed to the institutional animal care and use committee standards and practices. Forty-six nanoliters of cRNA at 0.5–1.0 μg/μl were injected into each oocyte cytoplasm. Experiments on oocytes were performed under two-electrode voltage clamp with an Axoclamp 2A (Axon Instruments, Foster City, CA) at a holding potential of −60 mV, in a continuously perfused chamber of ∼5 μl volume. The extracellular solution contained modified Barth's solution [88 mmNaCl, 1 mm KCl, 2.4 mmNaHCO3, 0.3 mmBa(NO3)2, 0.41 mm BaCl2, 0.82 mm MgSO4, and 15 mm HEPES, pH 7.6], to which was added glutamate or kainate along with cyclothiazide (20 mm stock solution dissolved in DMSO). Salts and drugs, including kainate and cyclothiazide, were purchased from Sigma (St. Louis, MO). DMSO was added so that all solutions contained equivalent amounts of vehicle. Solution exchange was controlled via an electronic BPS-8 valve control system (ALA Scientific, Westbury, NY) and electronic valves (The Lee Company, Westbrook, CT). Electrodes of 0.1–3 MΩ resistance were filled with 1 m CsCl and 5 mm EGTA. Current responses were filtered at 100 Hz (Cygnus Technology, Delaware Water Gap, PA) and acquired by a Power Macintosh 7600/132 computer with an Instrutech ITC-16 interface (Great Neck, NY) that was controlled by the program Synapse (Synergistic Research Systems, Silver Springs, MD).
Whole-cell electrophysiology. Human embryonic kidney fibroblasts (HEK293 cells; CRL 1573) from American Type Culture Collection (Rockville, MD) were cultured as described previously (Partin et al., 1996). Cells were transiently transfected by the use of FuGene reagent (Boehringer Mannheim, Indianapolis, IN) with a combination of 90% glutamate receptor cDNA and 10% GFP cDNA (Chalfie et al., 1994), driven by the same cytomegalovirus promoter. Currents were recorded 24–72 hr after transfection on whole cells. Cells were voltage-clamped at −60 mV by the use of an Axopatch 200B (Axon Instruments). Thin-walled borosilicilate glass micropipets (catalog #TW150F; World Precision Instruments, Sarasota, FL) with a resistance of 2–5 MΩ were filled with (in mm): 135 CsCl, 10 CsF, 10 HEPES, 5 Cs-BAPTA, 1 MgCl2, and 0.5 CaCl2, pH7.2. After going into voltage clamp, each cell was lifted to a flow pipe constructed from θ tubing (catalog #BT150-10; Sutter Instrument Company, Novato, CA) and placed in the control solution stream close to the interface between continuously flowing control and drug-containing solutions. Extracellular solution contained (in mm): 145 NaCl, 5.4 KCl, 5 HEPES, 1 MgCl2, 1.8 CaCl2, and 0.1 mg/ml phenol red, pH 7.3. Solution flow was driven by a syringe pump (KD Scientific, New Hope, PA) at a rate of 0.2–0.6 ml/min. Cells were rapidly jumped into drug-containing solution for 10–500 msec with a 70 μm step controlled by a piezoelectric device (Burleigh Instruments, Fishers, NY). Responses were filtered at 5 kHz with a low-pass Bessel filter (Warner Instruments, Hamden, CT), digitized at 0.25–10 msec/point, and stored on a PowerMac computer, using an ITC-16 interface (Instrutech). Data acquisition and analysis were done using Synapse (Synergistic Research Systems).
Data analysis. The kinetics of desensitization was analyzed as described previously (Partin et al., 1996). Statistical ANOVAs were performed using Microsoft Excel software. Current traces were plotted using KaleidaGraph 3.5 (Synergy Software, Reading, PA). Visualization of three-dimensional protein structure was done on the GluR2 Protein Data Bank coordinates (Armstrong et al., 1999), using RasMac version 2.5-UCB (Berkley, CA) and MacLook version 2.1 (Molecular Applications Group, Palo Alto, CA). Three-dimensional images were constructed using Molscript version 2.0 (Kraulis, 1991) and Raster3D version 2.0 (Merritt and Bacon, 1997) on a Silicon Graphics Octane Workstation.
RESULTS
Rationale for the design of receptor chimeras
Previous studies had demonstrated that cyclothiazide is a selective, positive modulator of AMPA versus kainate non-NMDA receptors and, in fact, has an inhibitory effect on kainate receptors. GluR6 is a kainate receptor that is expressed robustly in both theXenopus oocyte and mammalian fibroblast heterologous systems. Therefore, to identify residues in AMPA receptors that contribute to drug modulation, small domains of GluR1 were swapped into GluR6 and analyzed for gain of function. Amino acids were investigated on the basis of their proximity to flip/flop as predicted from tertiary structure (Stern-Bach et al., 1994; Armstrong et al., 1999) (Fig.1). Initially, chimeras were screened for function and drug sensitivity in oocytes under slow solution perfusion. Because the kinetics of desensitization is poorly resolved under these circumstances, the ability of cyclothiazide to modulate desensitization was assayed by measuring the amplitude of the peak current in the absence or presence of cyclothiazide. Subsequently, desensitization kinetics of interesting chimeras was characterized by the use of rapid perfusion in transiently transfected HEK293 cells. Previous studies had shown that insertion into GluR6 of either the entire flip/flop domain of GluR1 (GluR6-Flip) (Partin and Mayer, 1996) or a single residue within flip/flop (GluR6-Q755S) (Partin et al., 1995) could confer a modest sensitivity to cyclothiazide.
Conferring increased cyclothiazide sensitivity on GluR6
As shown previously, cyclothiazide is a modulator causing a dramatic decrease in the desensitization of GluR1 (Fig.2b1) but not that of GluR6 (Fig. 2b2). Also in agreement with previous results, a point mutation in GluR6 that converts a glutamine residue to the serine as found at position 750 in GluR1(flip) results in a modest degree of cyclothiazide sensitivity (Fig. 2b3). Among the several GluR6/GluR1 chimeras that were designed and screened, only one was identified that further enhanced sensitivity to cyclothiazide, GluR6-BETA 7,8 + Q755S (Fig. 2b4). Glutamate responses of GluR6-BETA 7,8 + Q755S were potentiated 24.5 (± 9.5)-fold by cyclothiazide, compared with 2.2 (± 0.9)-fold for GluR6-Q755S; kainate responses of GluR6-BETA 7,8 + Q755S were potentiated 158.5 (± 82)-fold, versus 1.9 (± 0.5)-fold for GluR6-Q755S (Fig. 2c). The double chimera consisting of GluR6 containing both BETA 7,8 and the entire flip/flop domain of GluR1 was nonfunctional (data not shown). BETA 7,8 lies within the previously defined S1 domain (Stern-Bach et al., 1994) and coincides with a region that has structural homology with one of the connecting strands (crossover 1) between the two agonist-binding lobes of LAOBP (Figs. 1, 2a) (Oh et al., 1993; Armstrong et al., 1999). The data in Figure 2 suggest that Q755S and residues within BETA 7,8 act together to play an important role in cyclothiazide sensitivity.
Fig. 2.

Identification of a domain within S1 that enhances modulation by cyclothiazide. a, Schematic representation of the structure of the agonist-binding domain, focusing on β-strands 7 and 8 and α-helices J and K, also shown in Figure 1.b, Electrophysiological responses (bottom) of chimeric receptors expressed inXenopus oocytes, with schematic representations (top) of the chimeric constructs (GluR1 sequences shown in black; GluR6 sequences shown in gray). Control glutamate and kainate responses (hairline) are superimposed on responses in the presence of cyclothiazide (CTZ; bold). Closed arrowspoint to the peak current in the control; open arrowspoint to the peak current in the presence of cyclothiazide. Wild-type (wt) GluR1 currents were potentiated by cyclothiazide (b1), whereas wt GluR6 currents were consistently inhibited by cyclothiazide (b2). GluR6-Q755S currents were somewhat potentiated by cyclothiazide (Partin et al., 1995) (b3), but the double chimera GluR6-BETA 7,8 + Q755S was strongly potentiated by cyclothiazide (b4). Insets (b4), A higher magnification of control currents for GluR6-BETA 7,8 + Q755S. These experiments were done in the absence of Concanavalin A, a modulator of kainate receptor desensitization, and therefore the responses to agonists desensitized within 1–2 sec in oocytes. c, The mean potentiation by 100 μmCTZ of peak currents (IAGONIST+CTZ/IAGONIST) plotted for 300 μm glutamate (stippled bars) or 300 μm kainate (filled bars). Error bars represent SEM; the number of oocytes studied under each condition ranges from 5 to 10.
Identification of residues in the BETA 7,8 domain of GluR1 critical for cyclothiazide modulation
If the BETA 7,8 region is indeed involved with allosteric modulation of AMPA receptors by cyclothiazide, then mutation of individual residues within this domain in GluR1 that are divergent from GluR6 should produce a loss of modulation by cyclothiazide. GluR1 receptors with point mutations in BETA 7,8 (Fig.3a) were studied in HEK293 cells exposed to agonist by rapid perfusion. Thus, modulation of desensitization by cyclothiazide is reflected in the slowing of current decay in the continued presence of 10 mmglutamate, rather than as an increase in peak current amplitude. There was no effect on modulation by cyclothiazide compared with that of wt GluR1 when residues M499–K501 within BETA 7,8 were mutated to the residues found at the homologous positions in GluR6 (Fig. 3b,c). However, the mutation of S493T resulted in a dramatic reduction in the efficacy of cyclothiazide. Interestingly, a loss of modulation by cyclothiazide did not occur when S493 was mutated to alanine (Fig. 3b), which has a less bulky side chain than threonine. Testing the effect of increasing the size of the side chain by mutating it to glutamine or cysteine was not possible because these substitutions grossly impaired current amplitudes and prevented further quantitation, although qualitatively the effects of cyclothiazide appeared to be greatly diminished (data not shown). Inspection of the atomic coordinates of GluR2 indicated that of the four divergent residues in BETA 7,8, the S493 side chain is nearest to N750 of GluR2(flop) [7.79 Å, or 8.41 Å if that residue were the S750 found in GluR1(flip)]. That the size and/or shape of the side chain at position 493 is so critical for modulation (threonine adds an asymmetric Cβ-atom) suggests some type of an interaction between these residues. A functional interaction may be caused by a state-dependent, physical interaction between BETA 7,8 and flip/flop, a direct interaction between these residues and cyclothiazide, or the participation by these residues in the regulation of desensitization, thereby affecting modulation of desensitization in an allosteric manner. The GluR1 point mutations provide independent confirmation of the conclusions drawn from the GluR6-BETA 7,8 + Q755S chimera and implicate S493 in regulating GluR1 sensitivity to cyclothiazide. In addition to impacting cyclothiazide modulation, point mutations in BETA 7,8 affected control desensitization kinetics, which reached significance for I500Y (p = 0.00058; Fig. 3d) but not for S493T, which is consistent with the GluR6 “C1a” mutant described by Stern-Bach et al. (1998). The role of BETA 7,8 and the flip/flop region in control desensitization is addressed in further detail by the experiments described below.
Fig. 3.
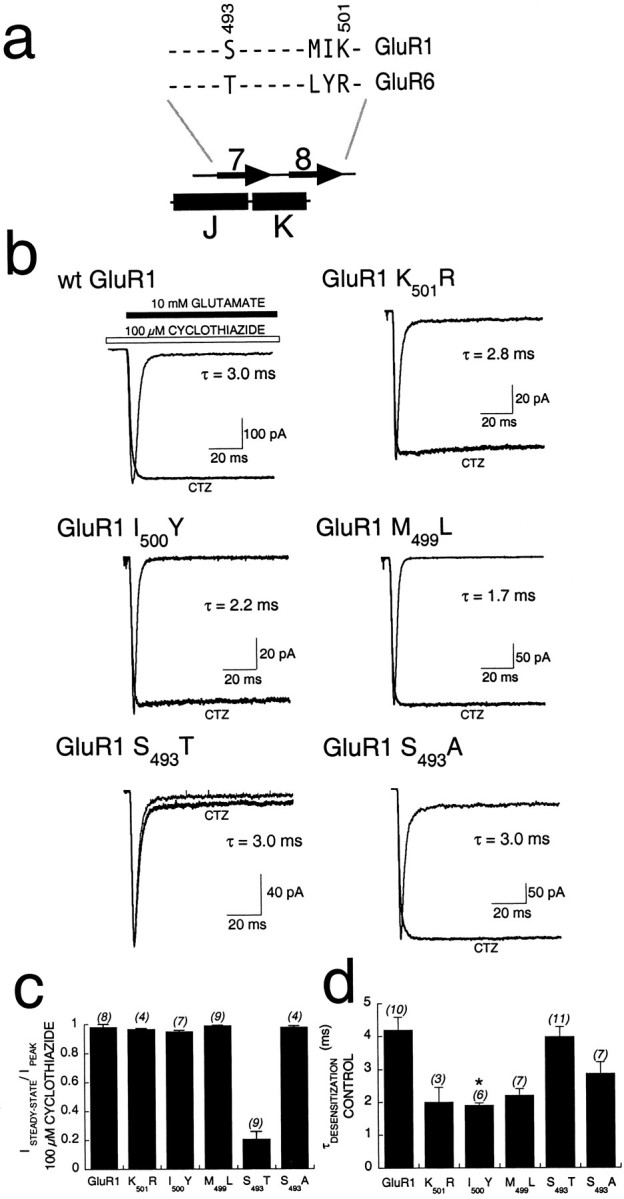
Effect of point mutations within the BETA 7,8 domain of GluR1 on modulation by cyclothiazide. a, An alignment of GluR1 and GluR6 sequences within the BETA 7,8 domain showing that there are 10 identical (dashes) and 4 nonidentical residues. Each of the divergent residues was mutated in GluR1 to the side chain found in GluR6 and analyzed for function and sensitivity to cyclothiazide. b, Individual, whole-cell responses in transiently transfected HEK293 cells rapidly perfused with 100 msec pulses of 10 mm glutamate in control conditions (hairline) or in the presence of 100 μmCTZ (bold line). The time constant for desensitization of control responses is indicated with eachtrace. Calibration bars for current are shown for the control responses; responses in the presence of cyclothiazide were scaled to the peak amplitude of the control response. The mutation S493T dramatically altered the ability of cyclothiazide to block desensitization, without altering control kinetics.c, Analysis of potentiation by cyclothiazide of point mutations, showing mean decay (ISTEADY-STATE/IPEAK) after 100 msec in the presence of 100 μm cyclothiazide and 10 mm glutamate. Cyclothiazide failed to block desensitization only for GluR1-S493T. Error bars represent SEM, and the number of cells studied is indicated inparenthesesabove eachcolumn. d, Mean time constants for desensitization of control responses for each point mutant. GluR1-I500Y desensitization kinetics was significantly faster than that of wt GluR1 (*p = 0.00058).
Functional interactions controlling desensitization kinetics
In agreement with our previous results (Partin et al., 1995), the Q755S point mutation in GluR6 not only introduced sensitivity to cyclothiazide but also slowed the kinetics of control desensitization in response to glutamate from τdesensitization = 3.8 ± 0.4 msec (n = 6) to τdesensitization = 16.4 ± 1.4 msec (n = 12; Fig.4a1,a2,b). Because Q755 and BETA 7,8 residues appeared to be important for cyclothiazide modulation of desensitization of GluR6-based chimeras (Fig. 2), it was important to determine whether mutation of these residues affected control desensitization kinetics. Control desensitization kinetics of GluR6/GluR1 chimeras was tested by fast perfusion. Introduction of the BETA 7,8 domain of GluR1 into GluR6-Q755S reversed the desensitization-slowing effect of the point mutation, restoring a time constant for desensitization similar to that of wt GluR6 (Fig.4a3). Introducing the BETA 7,8 domain alone into GluR6 caused a modest, but significant, increase in the rate of desensitization (τdesensitization = 2.6 ± 0.3 msec; n = 13; Fig. 4b). GluR1-BETA 7,8 contains a serine at position 493, which when mutated to threonine (the homologous residue is at position 504 of GluR6) abolished modulation by cyclothiazide (Fig. 3b). Thus, it was important to determine whether GluR6-T504S + Q755S had control desensitization kinetics like that of GluR6-BETA 7,8. The double-point mutant receptor demonstrated desensitization kinetics (τdesensitization = 5.5 ± 0.3 msec;n = 18) similar to that of wt GluR6 (Fig.4a4), indicating that T504 in BETA 7,8 is sufficient to correct the slowing of desensitization caused by introduction of GluR1-BETA 7,8 into GluR6. These data support the hypothesis that one residue in BETA 7,8 and one residue in flip/flop interact functionally to regulate desensitization. However, the data do not exclude an indirect, allosteric mechanism.
Fig. 4.
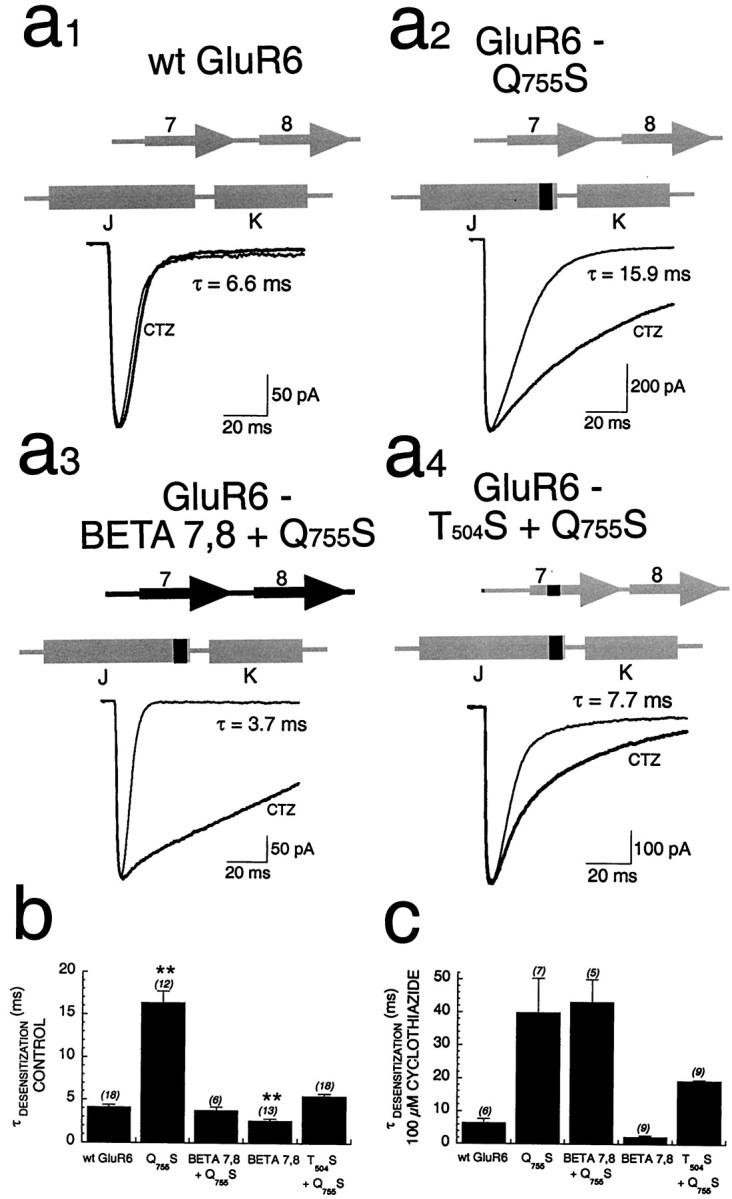
Amino acids regulating desensitization kinetics.a1–a4, Fast perfusion experiments performed in the whole-cell configuration on wt and chimeric GluR6 receptors transiently expressed in HEK293 cells, comparing control (hairline) andCTZ-modulated (bold) responses to 10 mm glutamate. Time constants for control desensitization for each trace are indicated. The structure of each chimera is shown above eachtrace (sequences derived from GluR1 are shown inblack; those derived from GluR6 are shown ingray). b, Summary of the mean control kinetics, showing that desensitization of GluR6-Q755S was significantly slower than that of wt GluR6 (**p < 0.0001), whereas desensitization of GluR6-BETA 7,8 is significantly faster than that of wt GluR6 (**p = 0.0001). However, the double chimera GluR6-BETA 7,8 + Q755S doesnot significantly differ from wt GluR6; thus, the presence of GluR1 sequence in both regions compensated for the defects induced by the presence of GluR1 sequence in only one of the two regions. c, Summary of the mean kinetics of desensitization in the presence of 100 μmcyclothiazide, showing that the actions of cyclothiazide were greatest on any chimera that contains the Q755S mutation (independent of the presence of the BETA 7,8 domain).
In the presence of cyclothiazide, desensitization kinetics was similar for GluR6-Q755S and GluR6-BETA 7,8 + Q755S (Fig. 4c). However, the ratio of the time constant of desensitization in the presence of cyclothiazide to the time constant of desensitization in control was much greater for GluR6-BETA 7,8 + Q755S (11.4-fold) than for GluR6-Q755S (2.3-fold) or GluR6-BETA 7,8 (0.9-fold), because of faster kinetics of control desensitization for the latter constructs. These data offer an explanation of the large effect that cyclothiazide had on potentiation of peak currents when tested in the oocyte system (Fig. 2).
Differential contributions of α-helices J and K to the modulation of GluR1 by cyclothiazide
The results presented thus far suggest that residues within BETA 7,8 and flip/flop affect both modulation by cyclothiazide and control desensitization kinetics and identify two residues (S493 and S750) that mediate these effects. Two prominent structural components within flip/flop are α-helices J and K (Fig. 1). Although it was clear from previous studies that the differential drug modulation of desensitization in flip versus flop isoforms of AMPA receptors was controlled by the S/N/Q site (i.e., at the position of Q755 of GluR6 or S750 of GluR1), the contribution of other residues within flip/flop had not been defined. Thus, in the present study GluR6/GluR1 chimeras of smaller domains within flip/flop were constructed and tested for sensitivity to cyclothiazide. Introduction of the entire flip/flop domain into GluR6 conferred cyclothiazide slowing of desensitization (Fig. 5a1) in agreement with previous studies in oocytes (Partin and Mayer, 1996). Cyclothiazide slowed desensitization for GluR6-Flip ∼38-fold, to 193 ± 17.5 msec (Fig. 5b), but not to the extend that occurred for wt GluR1. This effect was not seen when only the C-terminal 17 amino acids of flip/flop were inserted (GluR6-FlipC17τdesensitization = 3.4 ± 0.4 msec;n = 8; Fig. 5a2). Cyclothiazide also had little effect when α-helix K from GluR1 and Q755S were inserted into GluR6, with only a twofold slowing of desensitization (τdesensitization = 19.3 ± 1.9 msec;n = 9; Fig. 5a3). By contrast, for a chimera that included all of helix J, cyclothiazide slowed desensitization 18-fold (τdesensitization = 179.4 ± 12.7 msec;n = 5; Fig. 5a4), suggesting a dominant role for α-helix J in regulating drug modulation of GluR1. Both the GluR6-Helix J and GluR6-Helix K chimeras had slower kinetics of desensitization than either wt GluR1 or GluR6 had (Fig. 5c).
Fig. 5.
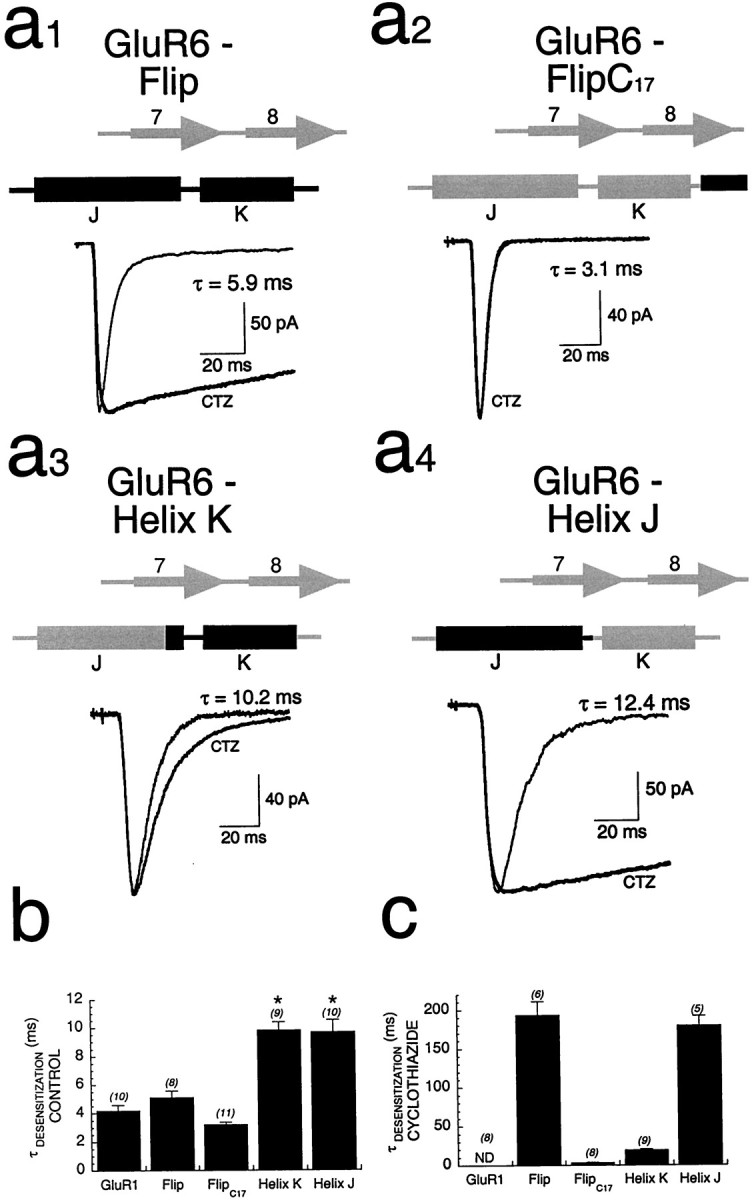
Dominance of α-helix J in determining cyclothiazide modulation of GluR1.a1–a4, Whole-cell responses of GluR6 chimeras transiently expressed in HEK293 cells show that inclusion of α-helix J (a4) appeared to be necessary and sufficient to match the efficacy of modulation by cyclothiazide seen with GluR6-Flip (a1).b, Quantitation of the efficacy of modulation of flip/flop domain chimeras as measured by comparing the mean time constants of desensitization in the presence of cyclothiazide is shown.c, The control kinetics of desensitization was significantly slower than that of wt GluR1 for both helix-swapping chimeras (*p < 0.001), whereas those of the GluR6-Flip and GluR6-FlipC17 chimeras were similar to those of wt GluR1. ND, Nondesensitizing.
The contributions of a residue homologous to one affecting desensitization of GluR3
Previously, Stern-Bach et al. (1998) identified a residue in GluR3 (L507) that, when mutated to the tyrosine found at the homologous position in GluR6 (L507Y), blocks desensitization. Their data suggest that this residue, together with sequences between M1 and M4, may participate in allosteric modulation by cyclothiazide. To probe this hypothesis further, a GluR6 chimera that includes helix J from GluR1, in conjunction with a mutation (Y490L in GluR6) that effectively inverts the mutation of Stern-Bach et al. (1998), was tested for allosteric modulation by cyclothiazide (Fig.6). The ability of cyclothiazide to block desensitization at the end of either a 100 or 500 msec application of glutamate was used to assess the contributions of these residues to allosteric modulation. Some desensitization was present at the end of the 500 msec pulse of agonist for all three chimeras. In particular, there was substantial desensitization (75–80%) by the end of the 500 msec pulse for the chimera replacing just helix J (as seen in Fig. 5) and the construct replacing helix J in conjunction with the Y490L mutation (Fig. 6b). However, when the T504S substitution was included (GluR6-Helix J + Y490L + T504S), desensitization was significantly reduced (p = 0.006) to ∼40% by the end of the pulse. Thus, the contributions of GluR1 S493 to allosteric modulation are evident even in a chimera that has a substitution at the nondesensitizing site. These data further support the hypothesis that S493 in GluR1 plays an important role in drug modulation.
Fig. 6.

Contributions of the nondesensitizing mutation to allosteric modulation. a, Whole-cell responses in transfected HEK293 cells to rapid applications of glutamate for 500 msec, in the presence of 100 μm cyclothiazide, are shown for wt GluR1 and three GluR6/GluR1 chimeras. None of these chimeras permitted the complete block of desensitization that is seen with wt GluR1. However, the GluR6 chimera containing the T504S mutation was more efficacious than were the two chimeras lacking T504S. b, Quantitation of the steady-state-to-peak ratio at the end of either a 100 msec (gray bars) or a 500 msec (black bars) application of glutamate is shown. Inclusion of T504S significantly (p = 0.006) increases the steady-state-to-peak ratio at the end of a 500 msec application compared with that seen with GluR6-Helix J + Y490L. pk, Peak; ss, steady-state.
Identification of residues in α-helices J and K critical for modulation by cyclothiazide
To finely map the role of amino acids in α-helices J and K, individual residues in GluR1 were mutated to alanine (or glycine in the case of A745), and the mutant receptors were assayed for sensitivity to cyclothiazide. Because of the large number of receptors studied, function was assayed in oocytes, where the ability of cyclothiazide to block desensitization was measured by determining the ratio of peak to steady-state current during a 60 sec application of glutamate. The effect of the mutations showed a periodicity (Fig. 7a), which may be the result of the tertiary structure in this region. Specifically, modulation by cyclothiazide was markedly impaired at two positions, V746(Iss/Ipk = 0.33 ± 0.06) and L755(Iss/Ipk = 0.40 ± 0.06) (Fig. 7b), compared with that of wt GluR1. A magnified portion of the GluR2(flop) crystal structure is shown in Figure7c, indicating the spatial relationship of four residues (S493, N750, V746, and L755) that are critical for allosteric modulation by cyclothiazide. A nearest-neighbor analysis (MacLook version 2.1) indicated that each of these four side chains is the nearest neighbor (≤5 Å) either of each other or of residues within BETA 7,8. Taken together, these data indicate that BETA 7, 8 and flip/flop form at least part of a functional domain, regulating sensitivity to cyclothiazide and affecting desensitization kinetics.
Fig. 7.
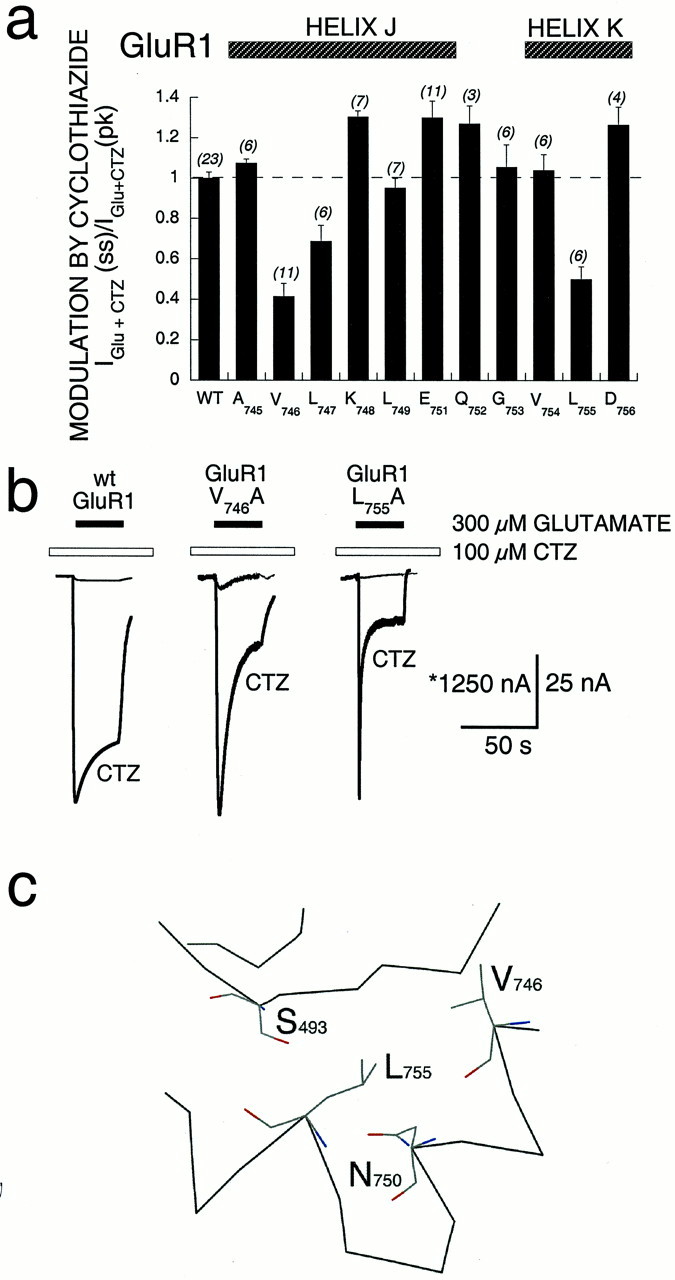
Effect of alanine substitution within flip/flop on modulation by cyclothiazide of GluR1. a, Individual residues within flip/flop were mutated, and modulation by cyclothiazide was characterized in Xenopus oocytes. A summary of the mean modulation by cyclothiazide [IGlu+CTZ(ss)/IGlu+CTZ (pk)] for all point mutants is shown. Ratios were normalized to that of wt GluR1 (WT). Each residue was mutated to alanine, except A745 that was mutated to glycine. Error bars represent SEM; the number of oocytes studied for each mutant is shown in parenthesesabove each column. b, Responses of wt GluR1 and two point mutations, V746A and L755A, show control responses (hairline) and responses in the presence of 100 μmCTZ(bold line). Both point mutations demonstrated less effective modulation by cyclothiazide. The asteriskrepresents calibration for wt GluR1. c, Molecular representation, using the atomic coordinates of GluR2 (Armstrong et al., 1999), shows the proximity of side chains that, when mutated, alter the efficacy of cyclothiazide, including S493, V746, L755, and N750.
DISCUSSION
The results reported here newly identify a residue within BETA 7,8 of the AMPA receptor GluR1 as functionally interacting with the α-helical J and K region (within the alternatively spliced flip/flop domain) to regulate desensitization and its modulation by cyclothiazide. In particular, introduction of BETA 7,8 of GluR1 into GluR6 with a point mutation in α-helix J (GluR6-Q755S) conferred greater sensitivity to cyclothiazide than did wt GluR6 or GluR6-Q755S. Moreover, a point mutation in β-sheet 7 of GluR1 (S493) affected cyclothiazide sensitivity in a manner that depended on the size of the side chain, in that increasing side chain size diminished the effects of cyclothiazide but decreasing the size had no effect. These findings are supportive of a functional interaction between α-helices J and K and β-sheets 7 and 8. Such an interpretation is further supported by the analysis of a GluR6 α-helical J and K point mutation that caused a defect in control desensitization kinetics, which could be compensated for by concomitant mutation at position T504 in BETA 7,8. Another indication of a functionally important domain–domain interaction was provided by scanning alanine mutagenesis across α-helices J and K, which identified the two additional residues (V746 and L755) as being important for modulation by cyclothiazide.
Examination of the crystal structure of the agonist-binding portion of GluR2(flop) reveals that α-helices J and K and BETA 7,8 both lie between the two lobes that form the agonist-binding site and are therefore in a position to play a role in the stabilization of lobe closure. The BETA 7,8 domain shares structural homology with one of the connecting strands (crossover 1) between the two lobes of the periplasmic amino acid-binding proteins LAOBP and QBP (Oh et al., 1993;Armstrong et al., 1999). Thus, BETA 7,8 might perform the same function for glutamate receptors, with specific residues undergoing large changes in torsion angles after ligand binding to stabilize the closed clamshell conformation. Glutamate receptors diverge from amino acid-binding proteins because of novel sequences C-terminal to helices J and K (homologous to helices VII and VIII of QBP) that permit the formation of a disulfide bond, allowing flip/flop to become a third connecting strand between the two lobes. It has been postulated that variable degrees of lobe closure reflect the different states of receptor activity: open, closed, and desensitized (Armstrong et al., 1999). That α-helices J and K and BETA 7,8 form connecting strands between the two lobes suggests that these two regions may act in concert to regulate the transition between open and closed clamshell conformations, thereby coupling agonist binding and channel gating.
Examination of the crystal structure also reveals that each of the two critical residues, S750 of α-helix J and S493 of BETA 7,8, is exposed on the surface of the protein, as illustrated in Figure 1, c and d. In a recent study, Stern-Bach et al. (1998) identified a residue in AMPA receptors (L507 of GluR3, homologous to L479 of GluR1) that, when mutated, completely blocks the onset of desensitization. As noted by Stern-Bach et al. (1998), this residue is also present on the surface of the protein, and the spatial relationship between it and the two residues identified here, S493 and S750, is notable (Fig. 1). It is clear that all three residues have a functional role in regulating desensitization, all lie within domain 1 (the large domain), and together they form a plane on the same exposed face of the protein. However, the residues appear to be too far apart to participate in a direct three-way interaction; L479 lies ∼15 Å from S493 and ∼20 Å from S750. In contrast, S493 and S750 are ∼8 Å from each other in this crystal structure, which probably represents the open, nondesensitized state of the intact receptor (Armstrong et al., 1999). The data presented here suggest that in the transition to the desensitized state, S493 and S750 either interact directly or allosterically.
The functional properties and spatial positioning of these three residues (L479, S493, and S750) suggest that together they form an interface that may be the site of a specific subunit–subunit interaction. Such an interface would thus contain one amino acid within α-helix D (directly involved with agonist binding, Fig. 1), one amino acid within a hinge region, and one amino acid within the alternatively spliced regulatory domain of flip/flop. Glutamate receptors are formed from four [or five (Premkumar and Auerbach, 1997)] individual subunits that assemble to make a receptor complex (Rosenmund et al., 1998). Each subunit can bind (at least) one molecule of agonist (Clements and Westbrook, 1991). There is evidence that binding of glutamate to a single subunit is sufficient for channel opening, although opening to larger conductance states requires the binding of agonist to more than one subunit (Rosenmund et al., 1998). Can a single subunit also undergo desensitization after opening? It has been widely postulated that desensitization occurs via an allosteric mechanism, whereby agonist binding induces a change in the conformation of one subunit that is extended to the subunit–subunit interface, allowing the entire protein to undergo a concerted shift in conformation to the desensitized state. Such a postulate predicts the existence of multiple interdomain contacts between subunits that regulate desensitization. The data shown here are consistent with L479, S493, and S750 serving just such a function but do not exclude the possibility that these residues participate in domain–domain interactions within a single subunit. Further structural and functional analyses will be required to resolve these issues.
Footnotes
This work was supported by American Heart Association Scientist Development Grant 0030100N. I thank Dr. Mark L. Mayer, in whose lab this project was initiated, Drs. Kurt Beam and Mike Tamkun for scientific discussions and critical reading of this manuscript, and Galen Pickard for his technical assistance.
Correspondence should be addressed to Dr. Kathryn M. Partin at the above address. E-mail: kpartin@lamar.colostate.edu.
REFERENCES
- 1.Armstrong N, Sun Y, Chen GQ, Gouaux E. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature. 1999;395:913–917. doi: 10.1038/27692. [DOI] [PubMed] [Google Scholar]
- 2.Bettler B, Egebjerg J, Sharma G, Pecht G, Hermans-Borgmeyer I, Moll C, Stevens CF, Heinemann S. Cloning of a putative glutamate receptor: a low affinity kainate-binding subunit. Neuron. 1992;8:257–265. doi: 10.1016/0896-6273(92)90292-l. [DOI] [PubMed] [Google Scholar]
- 3.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 4.Chen G-Q, Cui C, Mayer ML, Gouaux E. Functional characterization of a potassium-selective prokaryotic glutamate receptor. Nature. 1999;402:817–821. doi: 10.1038/45568. [DOI] [PubMed] [Google Scholar]
- 5.Clements JD, Westbrook GL. Activation kinetics reveal the number of glutamate and glycine binding sites on the N-methyl-d-aspartate receptor. Neuron. 1991;7:605–613. doi: 10.1016/0896-6273(91)90373-8. [DOI] [PubMed] [Google Scholar]
- 6.Cotton JLS, Partin KM. The contributions of Glu2 to allosteric modulation of AMPA receptors. Neuropharmacology. 2000;39:21–31. doi: 10.1016/s0028-3908(99)00105-7. [DOI] [PubMed] [Google Scholar]
- 7.Edmonds B, Gibb AJ, Colquhoun D. Mechanisms of activation of glutamate receptors and the time course of excitatory synaptic currents. Annu Rev Physiol. 1995;57:495–519. doi: 10.1146/annurev.ph.57.030195.002431. [DOI] [PubMed] [Google Scholar]
- 8.Everts I, Petroski R, Kizelsztein P, Teichberg VI, Heinemann SF, Hollmann M. Lectin-induced inhibition of desensitization of the kainate receptor GluR6 depends on the activation state and can be mediated by a single native or ectopic N-linked carbohydrate side chain. J Neurosci. 1999;19:916–927. doi: 10.1523/JNEUROSCI.19-03-00916.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsiao CD, Sun Y-J, Rose J, Wang B-C. The crystal structure of glutamine-binding protein from Escherichia coli. J Mol Biol. 1996;262:225–242. doi: 10.1006/jmbi.1996.0509. [DOI] [PubMed] [Google Scholar]
- 10.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structure. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 11.Mano I, Lamed Y, Teichberg VI. A Venus flytrap mechanism for activation and desensitization of AMPA receptors. J Biol Chem. 1996;271:15299–15302. doi: 10.1074/jbc.271.26.15299. [DOI] [PubMed] [Google Scholar]
- 12.Merritt EA, Bacon DJ. Raster3D: photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 13.Nakanishi N, Shneider NA, Axel R. A family of glutamate receptor genes: evidence for the formation of heteromultimeric receptors with distinct channel properties. Neuron. 1990;5:569–581. doi: 10.1016/0896-6273(90)90212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oh BH, Pandit J, Kang CH, Nikaido K, Gokcen S, Ames GF, Kim SH. Three-dimensional structures of the periplasmic lysine/arginine/ornithine-binding protein with and without a ligand. J Biol Chem. 1993;268:11348–11355. [PubMed] [Google Scholar]
- 15.O'Hara PJ, Sheppard PO, Thogersen H, Venezia D, Haldeman BA, McGrane V, Houamed KM, Thomsen C, Gilbert TL, Mulvihill ER. The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron. 1993;11:41–52. doi: 10.1016/0896-6273(93)90269-w. [DOI] [PubMed] [Google Scholar]
- 16.Paas Y. The macro- and microarchitectures of the ligand-binding domain of glutamate receptors. Trends Neurosci. 1998;21:117–125. doi: 10.1016/s0166-2236(97)01184-3. [DOI] [PubMed] [Google Scholar]
- 17.Partin KM, Mayer ML. Negative allosteric modulation of wild-type and mutant AMPA receptors by GYKI 53655. Mol Pharmacol. 1996;49:142–148. [PubMed] [Google Scholar]
- 18.Partin KM, Bowie D, Mayer ML. Structural determinants of allosteric regulation in alternatively spliced AMPA receptors. Neuron. 1995;14:833–843. doi: 10.1016/0896-6273(95)90227-9. [DOI] [PubMed] [Google Scholar]
- 19.Partin KM, Fleck MF, Mayer ML. AMPA receptor flip/flop mutants affecting deactivation, desensitization, and modulation by cyclothiazide, aniracetam, and thiocyanate. J Neurosci. 1996;16:6634–6647. doi: 10.1523/JNEUROSCI.16-21-06634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Premkumar LS, Auerbach A. Stoichiometry of recombinant N-methyl-d-aspartate receptor channels inferred from single-channel current patterns. J Gen Physiol. 1997;110:485–502. doi: 10.1085/jgp.110.5.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogawski MA. Therapeutic potential of excitatory amino acid antagonists: channel blockers and 2,3-benzodiazepines. Trends Pharmacol Sci. 1993;14:325–331. doi: 10.1016/0165-6147(93)90005-5. [DOI] [PubMed] [Google Scholar]
- 22.Rosenmund C, Stern-Bach Y, Stevens CF. The tetrameric structure of a glutamate receptor channel. Science. 1998;280:1596–1599. doi: 10.1126/science.280.5369.1596. [DOI] [PubMed] [Google Scholar]
- 23.Sommer B, Keinänen K, Verdoorn TA, Wisden W, Burnashev N, Herb A, Köhler M, Takagi T, Sakmann B, Seeburg PH. Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science. 1990;249:1580–1585. doi: 10.1126/science.1699275. [DOI] [PubMed] [Google Scholar]
- 24.Stern-Bach Y, Bettler B, Hartley M, Sheppard PO, O'Hara PJ, Heinemann SF. Agonist-selectivity of glutamate receptors is specified by two domains structurally related to bacterial amino acid binding proteins. Neuron. 1994;13:1345–1357. doi: 10.1016/0896-6273(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 25.Stern-Bach Y, Russo S, Neuman M, Rosenmund C. A point mutation in the glutamate binding site blocks desensitization of AMPA receptors. Neuron. 1998;21:907–918. doi: 10.1016/s0896-6273(00)80605-4. [DOI] [PubMed] [Google Scholar]
- 26.Sutcliffe M, Wo ZG, Oswald RE. Three-dimensional models of non-NMDA glutamate receptors. Biophys J. 1996;70:1575–1589. doi: 10.1016/S0006-3495(96)79724-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swanson GT, Gereau RW, Green T, Heinemann SF. Identification of amino acid residues that control functional behavior in GluR5 and GluR6 kainate receptors. Neuron. 1997;19:913–926. doi: 10.1016/s0896-6273(00)80972-1. [DOI] [PubMed] [Google Scholar]
- 28.Trussell LO, Otis TS. Physiology of AMPA receptors: biophysical characteristics that subserve integrative roles of synapses. In: Conti F, Hicks TP, editors. Excitatory amino acids and the cerebral cortex. MIT; Cambridge, MA: 1996. pp. 63–72. [Google Scholar]
- 29.Yamada K. Modulating excitatory synaptic transmission: potential treatment for neurologic disease? Neurobiol Dis. 1998;5:67–80. doi: 10.1006/nbdi.1998.0190. [DOI] [PubMed] [Google Scholar]