

Abstract
Impaired GABAergic inhibition may contribute to the development of hyperexcitability in epilepsy. We used the pilocarpine model of epilepsy to demonstrate that regulation of excitatory synaptic drive onto GABAergic interneurons is impaired during epileptogenesis. Synaptic input from granule cells (GCs), perforant path, and CA3 inputs onto hilar border interneurons of the dentate gyrus were examined in rat hippocampal slices during the latent period (1–8 d) after induction of status epilepticus (SE). Short-term depression (STD) of GC inputs to interneurons induced by brief (500–800 msec), repetitive (5–20 Hz) stimulation, as well as paired-pulse depression at both GC and CA3 inputs to interneurons, were significantly (p < 0.05) enhanced in SE-experienced rats. In contrast, we found no significant differences between SE-experienced and age-matched control rats in the properties of minimal EPSCs evoked at low frequency (0.3 Hz). Consistent with reduced GABAergic inhibition onto granule cells, paired-pulse depression of perforant path-evoked granule cell population spikes was lost in SE-experienced rats. Enhanced STD was partially mediated by group II metabotropic glutamate receptors, because the selective antagonist, 2S-2-amino-2-(1S,2S-2-carboxycyclopropyl-1-yl)-3-(xanth-9-yl)propanoic acid, attenuated STD in SE-experienced rats but had no effect on STD of GC inputs in the normal adult rat. The group II mGluR agonist, (2S′,1R′,2R′,3R′)-2-(2,3-dicarboxylcyclopropyl) glycine (1 μm), produced a greater depression of GC input to hilar border interneurons in SE-experienced rats than in controls. These results indicate that, in the SE-experienced rat, excitatory drive to hilar border inhibitory interneurons is weakened through a use-dependent mechanism involving group II metabotropic glutamate receptors.
Keywords: hippocampus, seizure, granule cell, short-term depression, metabotropic glutamate receptor, status epilepticus
GABAergic neurotransmission regulates cortical functions such as synaptic plasticity (Wigström and Gustafsson, 1983; Grover and Yan, 1999; Steele and Mauk, 1999), processing of sensory information (Dykes, 1997; Zheng and Knudsen, 1999), and learning and memory (Keverne and Brennan, 1996;Paulsen and Moser, 1998). A linkage between GABAergic neurotransmission and epilepsy has long been recognized (Olsen and Avoli, 1997). Inhibition of GABAA receptors triggers acute seizures (Prince, 1978), and drugs that potentiate GABAergic inhibition are typically anticonvulsant (George and Kulkarni, 1996; Olsen and Avoli, 1997; Bleck, 1999). However, it is not clear whether the anticonvulsant effect of GABA receptor potentiation reflects a general dampening of cortical excitability or a selective compensation for a specific deficit in GABAergic inhibition.
Disruptions in the strength of GABAergic inhibition occur in the epileptic hippocampus. However, both a loss or reduction (Sloviter, 1991; Bekenstein and Lothman, 1993; Merlin and Wong, 1993; Whittington and Jefferys, 1994; Rice et al., 1996; Hirsch et al., 1999; Williamson et al., 1999) or an increase (King et al., 1985; Haas et al., 1996; Swanson et al., 1998; Wilson et al., 1998) in synaptic inhibition have been reported. Depressed synaptic inhibition occurs in experimental epilepsy models, including the pilocarpine (Rice et al., 1996; Bausch and Chavkin, 1997), tetanus toxin (Empson and Jeffreys, 1993; Whittington and Jeffreys, 1994), and self-sustaining limbic status epilepticus (SE) (Mangan and Lothman, 1996) models. GABAA receptor antagonists also unmask epileptiform activity in chronic experimental models of epilepsy (Patrylo and Dudek, 1998; Smith et al., 1998).
Numerous alterations occur in hippocampal circuitry after SE, producing remodeled networks with properties significantly different from normal brain (Coulter, 1999; Sloviter, 1999). Many pathological alterations produced during epileptogenesis might disinhibit hippocampal circuitry, including changes in GABAA receptor subunit composition or properties (Bühl et al., 1996; Kapur and Macdonald, 1997; Brooks-Kayal et al., 1998; Macdonald and Kapur, 1999), death of GABAergic interneurons (Obenaus et al., 1993; Houser and Escaplez, 1996), changes in receptor-mediated regulation of GABAergic neurotransmission (Haas et al., 1996; Mangan and Lothman, 1996;Bausch and Chavkin, 1997), or loss of excitatory synaptic input onto GABAergic interneurons (Sloviter, 1991; Bekenstein and Lothman, 1993).
Excitatory synaptic inputs onto hilar border interneurons undergo short-term plasticity that is partially mediated by metabotropic glutamate receptors (mGluRs) (J. Doherty, D. Mott, S. Alagarsamy, P. J. Conn, and R. Dingledine, unpublished observations). Hippocampal GABAergic interneurons undergo both short-term (Galarreta and Hestrin, 1998) and long-term (McMahon and Kauer, 1997; Laezza et al., 1999) forms of synaptic plasticity. Small deficits in the regulation or strength of excitatory synaptic drive onto GABAergic interneurons during epileptogenesis can have significant consequences for local excitability, because individual interneurons typically synapse onto large numbers of principal neurons (Freund and Buzáski, 1996). Small reductions in the strength of inhibitory neurotransmission can trigger acute seizure-like activity (Miles and Wong, 1987; Chagnac-Amitai and Connors, 1989).
We demonstrated that short-term depression (STD) of excitatory afferents onto dentate hilar border interneurons is enhanced in SE-experienced rats. Diminished drive onto interneurons is partially mediated by enhanced mGluR-mediated depression of transmitter release.
Some of these data have appeared previously in preliminary form (Doherty and Dingledine, 1997b, 1998).
MATERIALS AND METHODS
Pilocarpine-induced status epilepticus. The pilocarpine-induced model of chronic epilepsy in rats (Turski et al., 1983, 1989; Cavalheiro et al., 1991; Mello et al., 1993) was chosen because it replicates several features of human temporal lobe epilepsy. Male Sprague Dawley rats (36–60 d old; 102–210 gm) were injected with methylscopolamine and terbutaline (2 mg/kg, i.p.). Thirty minutes later, experimental rats received a single injection of pilocarpine HCl, whereas sham-treated rats received saline injections. Although a range of pilocarpine doses (315–350 mg/kg, i.p.) was tested with the aim to produce the highest proportion of rats experiencing status epilepticus with the lowest mortality rate, a concentration of 335 mg/kg was used for >90% of the experiments. Seizure duration and frequency progressively increased until a state of SE characterized by rearing and falling was achieved. SE was allowed to proceed for 75–90 min and then was terminated with sodium pentobarbital (25–50 mg/kg, i.p.). Rats were allowed to recover for 1–8 d before hippocampal slices were prepared for electrophysiological recordings. A single intraperitoneal injection of pilocarpine evoked seizures in 94% (92 of 98) of treated rats. Typically, intermittent seizures characterized by head bobbing and forelimb clonus appeared 5–15 min after pilocarpine injection. Seizure severity and frequency increased progressively over 5–15 min, culminating in a state of SE in 84% (82 of 98) of treated rats. Slices taken from SE-experienced rats, defined as those undergoing >75 min of SE (n = 48), were examined for changes in synaptic inhibition in the dentate gyrus.
Interneuron recordings. All recordings from SE-experienced rats were made at 1–8 d after SE, before the appearance of extensive supragranular mossy fiber sprouting (Mello et al., 1993). The procedures for recording from interneurons in hippocampal slices were essentially as previously described (Doherty and Dingledine, 1997). Thin (225 μm) hippocampal slices were prepared from both pilocarpine- and sham-treated rats. Rats were first anesthetized with isoflurane, then brains were rapidly removed into ice-cold artificial CSF (ACSF) containing (in mm): 120 NaCl, 3.5 KCl, 0.75 CaCl2 · 2 H2O, 2.25 MgSO4 · 7 H2O, 24 NaHCO3, 1.25 NaH2PO4, 1 Na pyruvate, 10 glucose, pH 7.4 (295–305 mOsm). Slices from the dorsal half of the hippocampus were cut with a vibratome, incubated at room temperature (24–25°C) for 5–60 min, and finally transferred to a submerged recording chamber. Once in the recording chamber, slices were perfused with a room temperature ACSF containing (in mm):130 NaCl, 3.5 KCl, 1.5 CaCl2 · 2 H2O, 1.5 MgSO4 · 7 H2O, 24 NaHCO3, 1.25 NaH2PO4, 10 glucose, pH 7.4 (295–305 mOsm), at a rate of 2–3 ml/min. Individual interneurons located at the border of the hilus and the granule cell (GC) layer were visually selected for whole-cell patch-clamp recording using Hoffman modulation contrast optics (600×) and previously established criteria (Doherty and Dingledine, 1997, 1998). Interneurons in this study include basket cells, which synapse on the somata of granule cells, as well as interneurons that project into the molecular layer (Freund and Buzáski, 1996). No differences in the electrophysiological properties of excitatory synaptic inputs were detected between subsets of hilar border interneurons with different axonal projections; therefore results from all interneurons were considered together.
Field potential and whole-cell patch recordings were performed using an Axopatch 1D electrometer (Axon Instruments, Foster City, CA). Field potential responses were acquired with an ACSF-containing patch electrode placed in the granule cell layer. Sharpened tungsten microelectrodes placed in the outer molecular layer to a depth of 5–50 μm were used to evoke field potentials. Stimulus intensity was adjusted to produce a response at 40% of maximal population spike amplitude. All stimuli were triggered by Clampex protocols (Axon Instruments) and delivered through photoelectric stimulus isolation units (World Precision Instruments, Sarasota, FL). Seal formation and whole-cell configuration of patch-clamp recordings were achieved in current-clamp mode; passive membrane properties were measured before switching to voltage-clamp mode. Whole-cell responses were filtered at 3 kHz, digitized at 10–30 kHz, and collected directly to a computer using pClamp 7.0. Patch electrodes (5–7 MΩ) were pulled from borosilicate glass using a two-stage vertical puller and filled with solution containing (in mm) 130 CsOH, 140 methanesulfonic acid, 10 HEPES, 2 MgCl2, and biocytin (0.5–1%). Intracellular solution was buffered to pH 7.3 with CsOH and adjusted to 275–280 mOsm with H2O. Voltage-clamp experiments were performed at a holding potential of −70 mV in hilar border interneurons and 0 mV in granule cells. Series resistance, input resistance, and holding current were periodically monitored throughout experiments; only neurons with stable electrophysiological parameters were included. Glass micropipettes were used to deliver stimuli (0.3 Hz, 10–80 μA; 300–400 μsec) in the stratum granulosum (granule cells) 10–50 μm from the recording site or in the stratum pyramidale of the CA3b and CA3c regions. For minimal stimulation experiments, stimulus intensities were adjusted to the lowest level necessary to evoke visually identifiable EPSCs. Multiple sites were tested in each region to isolate a single reliable EPSC, as well as to prevent antidromic activation of interneurons in response to stimulation in the GC. Minimally evoked EPSCs were visually differentiated from synaptic failures as previously described (Doherty and Dingledine, 1997). Briefly, evoked events were required to meet several criteria to be accepted for analysis. These included a short mean latency (2–7 msec) from the stimulus artifact, characteristic fast rising and exponential decay phases, and peak amplitudes that exceeded 2 SD of the mean amplitude of the baseline electrical noise. Events chosen for analysis were confined to a 2 msec window centered around the mean latency to minimize the possibility of counting spontaneous EPSCs as evoked events. Stimulus trials that did not produce events meeting these criteria were designated as synaptic failures and were not included in the calculation of EPSC parameters. The failure rate in synaptic transmission for each excitatory input was defined as the percentage of failures over the total number of stimulus trials. Individual inputs were not considered for further analysis if the mean failure rate was >90% during control stimulation. Transmission rate was defined as 100% − failure rate.
Drugs. Pilocarpine HCl (315–350 mg/kg), methylscopolamine (10 mg/kg), terbutaline (10 mg/kg), and sodium pentobarbital (25 mg/kg) (Sigma, St. Louis, MO) were dissolved in 0.9% sterile saline. Bicuculline methobromide (10 μm), (+)-α-methyl-4-carboxyphenylglycine (500 μm), (2S′,1R′,2R′,3R′)-2-(2,3-dicarboxylcyclopropyl) glycine (DCG-IV; 1 μm) (Tocris Cookson, Ballwin, MO), and 2S-2-amino-2-(1S,2S-2-carboxycyclopropyl-1-yl)-3-(xanth-9-yl)propanoic acid (LY341495; 500 nm) were dissolved in ACSF and delivered by bath perfusion.
RESULTS
Depressed synaptic inhibition in the dentate gyrus
Field potential recordings of perforant path input to dentate granule cells were used to assess the strength of feedback and feedforward synaptic inhibition in the dentate gyrus from 1 to 28 d after pilocarpine-induced SE. Population spikes were recorded in the granule cell layer in response to paired stimuli delivered to the perforant path in hippocampal slices from both control and pilocarpine-treated rats (Fig.1A). In control rats, paired-pulse inhibition, determined by measuring the amplitude of the second population spike amplitude as a percentage of the first, was observed over an interstimulus interval ranging from 10 to 30 msec. Paired-pulse inhibition was replaced by facilitation in slices taken from SE-experienced rats when compared with control rats (Fig.1A,B), indicating a significant (p < 0.001, one-way ANOVA) loss of synaptic inhibition during paired stimulation. Paired-pulse facilitation of perforant path-evoked population spikes (224 ± 65%;n = 3) was observed within 1–2 d after induction of SE. However, the magnitude of the facilitation continued to increase for 4–5 d, reaching a peak at 305 ± 22% (Fig.1C; n = 6). The establishment of SE was required to produce the loss of paired-pulse inhibition in granule cells. In four pilocarpine-treated rats that experienced intermittent seizures, but not SE, paired-pulse inhibition remained normal, reducing the second population spike to 43 ± 13% of the first population spike amplitude at an interstimulus interval of 10 msec. This level of inhibition was not significantly different (p = 0.6, one-way ANOVA with post hocBonferroni test) from control slices (24 ± 9%; n= 9). Multiple population spikes were not observed in the dentate gyrus after either single or paired stimulation of the perforant path in slices taken from normal or SE-experienced rats.
Fig. 1.
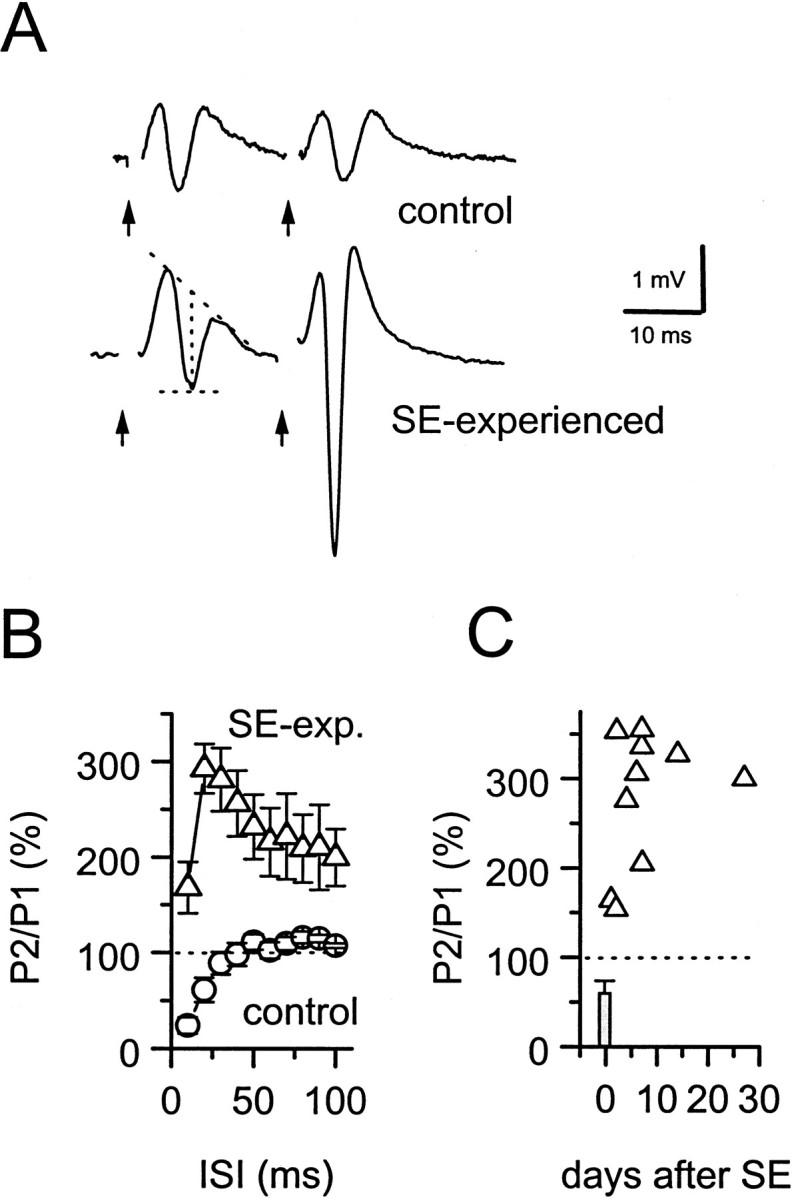
Synaptic inhibition in the dentate gyrus is impaired at 2–6 d after pilocarpine-induced status epilepticus.A, Field potential recordings evoked in the granule cell layer of the dentate gyrus after paired stimulation of the perforant path in untreated (top) or SE-experienced (bottom) rats. Population spike amplitudes were measured as the difference between the peak amplitude and the midpoint of the field EPSP (dotted lines). Stimulus artifacts (arrows) have been removed for clarity.B, Paired-pulse inhibition was seen at short (10–30 msec) interstimulus intervals (ISI) in untreated rats (○; n = 9 rats), but was replaced by facilitation in SE-experienced rats (▵;n = 7 rats). C, Paired-pulse facilitation (interpulse interval, 20 msec) peaks at 4–5 d after SE and persists for at least 30 d.
These data indicate an increase in granule cell excitability in SE-experienced rats; however, they alone do not indicate whether this results from a loss of inhibition or an increase in synaptic facilitation. Reduction of paired-pulse inhibition was not accompanied by noticeable deficits in monosynaptic IPSCs evoked at low frequency by stimulation within the granule cell layer in the presence of CNQX (3 μm) and d-APV (50 μm). The reliability of synaptic transmission at monosynaptic GABAergic synapses onto granule cells was high in both untreated (evoked IPSCs in 86 ± 4% of stimulus trials with minimal stimulation; n = 5 neurons) and SE-experienced (94 ± 6%; n = 3) rats. IPSC amplitudes in granule cells of SE-experienced rats produced by a minimally effective stimulus delivered to the granule cell layer (39 ± 10 pA; n = 3) were not significantly different from IPSCs in granule cells from control rats (27 ± 4 pA; n = 5; data not shown).
Excitatory synaptic input to hilar border interneurons in SE-experienced rats
To identify the mechanisms responsible for reduced synaptic inhibition in the dentate gyrus of SE-experienced rats, we studied the short-term regulation of excitatory synaptic inputs onto the heterogeneous group of interneurons located at the granule cell–hilar border in slices from SE-experienced rats. Whole-cell patch recordings were made from 10 granule cells and 57 visually identified hilar border interneurons in slices taken from SE-experienced rats. Synaptic responses from interneurons in sham-treated rats (n = 12) did not differ from untreated adult rats (n = 34); thus these recordings were grouped together in the control group. Spontaneous action potentials were observed at the resting membrane potential in a slightly larger proportion of hilar border interneurons from SE-experienced rats (71%) than from control rats (60%). However, there were no significant differences in resting membrane potential, action potential threshold, or input resistance in interneurons from SE-experienced rats when compared with interneurons from control rats (Table 1). This suggests that changes in interneuron membrane excitability were not sufficient to explain the observed loss of synaptic inhibition. Spontaneous EPSCs were observed in recordings from interneurons from both control and SE-experienced rats. Spontaneous EPSC rise and decay times were slower in interneurons from SE-experienced rats, but there were no changes in amplitude or frequency (Table 1).
Table 1.
Electrophysiological properties of hilar border dentate interneurons in pilocarpine-treated rats
| SE-experienced | Control | |
|---|---|---|
| Resting membrane potential (mV) | −52 ± 3 (25) | −52 ± 3 (19) |
| Action potential threshold (mV) | −50 ± 1 (7) | −50 ± 1 (7) |
| Input resistance (MΩ) | 258 ± 11 (26) | 264 ± 14 (19) |
| Spontaneous EPSCs (measured at −70 mV) | ||
| Amplitude (pA) | −13 ± 2 (16) | −11 ± 2 (13) |
| 10–90% rise time (msec) | 2.0 ± 0.1 (16)1-160 | 1.4 ± 0.1 (13) |
| Decay tau (msec) | 7.1 ± 0.5 (16)1-160 | 4.1 ± 0.5 (13) |
| Frequency (Hz) | 2.6 ± 0.6 (37) | 3.0 ± 0.5 (18) |
Parentheses denote the number of interneurons used for each measurement;
F1-160: p < 0.01.
To determine whether regulation of excitatory synaptic drive to hilar border interneurons is altered in SE-experienced rats, we isolated minimally evoked EPSCs from three different synaptic inputs as previously described (Doherty and Dingledine, 1997, 1998) (Doherty, Mott, Alagarsamy, Conn, and Dingledine, unpublished observations). Stimulation of dentate granule cells (Fig.2A), CA3 pyramidal cells (Fig. 2B), or perforant path axons evoked minimal inputs to hilar border interneurons from SE-experienced rats. There were no significant differences in either the reliability of synaptic transmission or the kinetics of evoked minimal EPSCs at excitatory inputs to hilar border interneurons in slices from normal adult and SE-experienced rats. These data, summarized in Table2 and the scattergrams in Figure 2, A and B, indicate that low-frequency synaptic excitation of hilar border interneurons appears normal in SE-experienced rats.
Fig. 2.
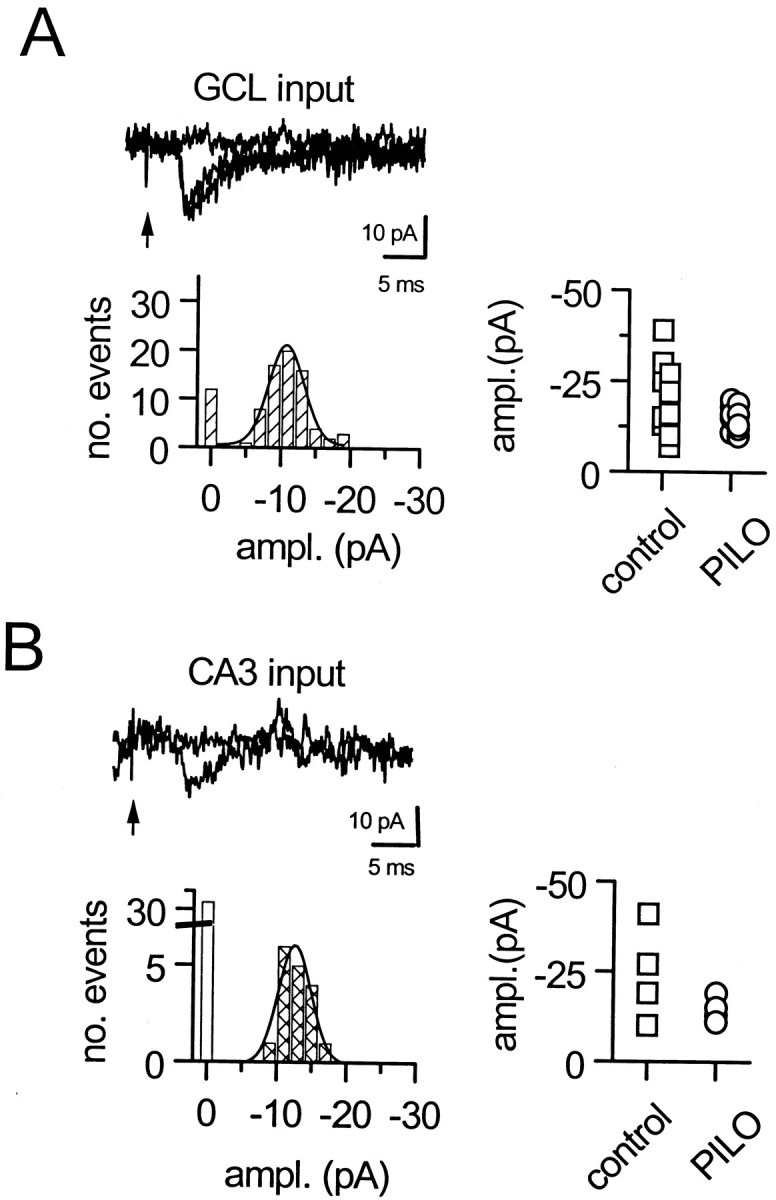
Properties of unitary EPSCs evoked by minimal stimulation onto hilar border interneurons. A, Dentate granule cell inputs to an interneuron from an SE-experienced adult rat.B, Input from stimulation of the CA3 pyramidal cell layer. In each case, the amplitude histogram is unimodally centered on −10 pA. The scattergrams to theright show the peak of the amplitude histogram for minimally evoked synaptic inputs to interneurons in control and SE-experienced rats. GCL, Granule cell layer;ampl., amplitude; PILO, pilocarpine.
Table 2.
Properties of evoked EPSCs onto hilar border interneurons (measured at −70 mV)
| SE-experienced | Control | |
|---|---|---|
| Granule cell layer inputs | ||
| Amplitude (pA) | −15 ± 1 (12) | −20 ± 3 (12) |
| Latency (msec) | 3.8 ± 0.2 (12) | 3.8 ± 0.4 (12) |
| Transmission rate (%) | 79 ± 6 (12) | 71 ± 6 (7) |
| CA3 inputs | ||
| Amplitude (pA) | −15 ± 2 (4) | −24 ± 7 (4) |
| Latency (msec) | 5.1 ± 0.7 (4) | 5.5 ± 1.0 (4) |
| Transmission rate (%) | 47 ± 4 (4) | 42 ± 12 (3) |
Parentheses denote the number of interneurons used for each measurement.
Enhanced short-term depression in SE-experienced rats
Excitatory inputs to hilar border interneurons in the juvenile and adult rat express synapse-specific forms of short-term plasticity (Doherty, Mott, Alagarsamy, Conn, and Dingledine, unpublished observations). Stimulation of granule cell synapses onto interneurons with brief high-frequency (20 Hz) stimulus trains produces STD in both control (Fig. 3A,top) and SE-experienced (Fig. 3, bottom) rats. STD of GC-evoked inputs to hilar border interneurons was significantly potentiated in SE-experienced rats (Fig. 3B). In SE-experienced rats, STD of GC-evoked EPSC amplitudes reached a plateau at 24 ± 2% of the first EPSC amplitude during 20 Hz trains (n = 16), a significantly (p < 0.001, one-way ANOVA) greater depression than was observed during 20 Hz trains in interneurons from control adult rats. The onset of STD was also more rapid at GC inputs to hilar border interneurons in SE-experienced rats. The time constant of the fitted exponential decay function in interneurons from SE-experienced rats (81 ± 6 msec;n = 16) was significantly (p < 0.001, unpaired t test) shorter than was observed for interneurons in control rats (120 ± 13 msec; n = 13). There were no significant changes in the kinetics of evoked EPSCs during repetitive stimulation in either control or SE-experienced rats (Fig. 3C).
Fig. 3.

Short-term depression of GC-evoked EPSC amplitudes during high-frequency (20 Hz) stimulation was more intense in hilar border interneurons from SE-experienced rats. A, Sample traces demonstrating short-term depression of EPSC amplitudes during high-frequency stimulation of GC inputs in interneurons from untreated (top) and SE-experienced (bottom) rats. The depicted EPSCs represent the average of 12–15 consecutive 20 Hz trains delivered at 3 sec intervals. B, Plot depicting the average depression of EPSC amplitudes produced by 20 Hz stimulation of GC inputs to interneurons from either untreated (●;n = 13 interneurons) or pilocarpine-treated (▵;n = 16 interneurons) rats. Each data set was well fit by a single exponential function (solid lines).C, Superimposed first and last EPSCs in a 20 Hz train delivered to GC inputs to interneurons from either an SE-experienced (bottom) or untreated (top) rat. For each pair, the amplitude of the last EPSC in the train was scaled to the amplitude of the first EPSC. The decay kinetics of EPSCs evoked during repetitive activation of GC inputs was not altered during the train in interneurons from either untreated or SE-experienced rats.
Changes in the magnitude of short-term plasticity induced by SE at each of the three excitatory inputs to hilar border interneurons that we examined are summarized in Figure 4. At granule cell inputs, STD was significantly enhanced at both 10 Hz (p = 0.03, unpaired t test) and 20 Hz (p < 0.0001, unpaired t test) but not at 5 Hz (Fig. 4A). Enhanced STD of granule cell input to hilar border interneurons occurs within 24 hr after induction of SE and persists for at least 8 d (Fig.4B). In contrast, STD of perforant path inputs to interneurons in SE-experienced rats was not different from STD in control adult rats at any frequency tested (Fig. 4C).
Fig. 4.

Short-term depression of evoked EPSCs is enhanced over a range of stimulus frequencies. A, Peak depression of GC-evoked EPSCs was measured at 0.3, 5, 10, and 20 Hz in untreated (○) and SE-experienced (●) rats. STD produced a significantly greater depression of EPSC amplitude at both 10 Hz (*p = 0.03, unpaired t test;n = 13) and 20 Hz (*p < 0.001, unpaired t test; n = 17).B, Enhancement of short-term depression at GC inputs to hilar border interneurons persists for at least 8 d after induction of SE. Each point represents the maximal STD achieved during 20 Hz stimulation of GC input for a single interneuron from normal adult (○; n = 11) and SE-experienced (●;n = 16) rats. Maximal depression was calculated to be the amplitude of the plateau current (500–800 msec latency) expressed as a percentage of the first EPSC amplitude.C, STD of perforant path inputs was not enhanced significantly in SE-experienced rats at any frequency tested.D, High-frequency stimulation of CA3 inputs produced a combination of facilitation and depression in the adult rat. In contrast, only HFS-induced depression was observed in SE-experienced rats.
CA3 inputs underwent short-term facilitation during repetitive stimulation at 5 Hz in control adult rats but showed no net plasticity at 10 Hz and depression at 20 Hz (Fig. 4D). In SE-experienced rats, however, repetitive stimulation of CA3 inputs produced depression at both frequencies tested (Fig.4D). Repetitive stimulation of the CA3 pyramidal cell layer often produced bursts of EPSCs during the trains in both control and SE-experienced rats, likely because of synchronized activation of CA3 pyramidal cells (Miles and Wong, 1987; Kneisler and Dingledine, 1995). Because this bursting behavior often obscured the short-term plasticity of evoked inputs, we also used a paired-pulse protocol to examine the effects of SE on activity-dependent plasticity at evoked inputs to interneurons. This paired stimulation protocol also demonstrated changes in short-term plasticity at both CA3 and GC inputs to interneurons in SE-experienced rats (Fig.5). For example, paired stimulation of GC input to interneurons, delivered at an interpulse interval of 50 msec, resulted in a depression of the second pulse to 77 ± 3% of the first EPSC amplitude in control rats (n = 19). In SE-experienced rats, paired stimulation of GC inputs resulted in a significantly greater (p = 0.002, unpairedt test) reduction of the second EPSC (58 ± 5% of the first EPSC amplitude; n = 10). In contrast to the synaptic depression observed at granule cell inputs, paired stimulation of CA3 inputs to hilar border interneurons at an interval of 50 msec produced facilitation to 225 ± 28% of the first EPSC amplitude in control rats (n = 14). In SE-experienced rats, however, pairing stimuli in CA3 resulted in a depression of evoked EPSCs onto interneurons (62 ± 21% of the first EPSC amplitude;n = 3). The bursting behavior produced by repetitive high-frequency stimulation (HFS) of CA3 inputs was not observed with paired stimulation.
Fig. 5.

Paired-pulse stimulation of evoked inputs to hilar border interneurons in normal adult and SE-experienced rats.A, Paired stimulation to GC inputs delivered at a 50 msec interpulse interval produced a depression of the second EPSC in normal adult rats (top left) that was potentiated in SE-experienced rats (top right). A summary of paired stimulation of GC inputs to interneurons from 19 normal adult (▵) and 10 SE-experienced (▴) rats is shown in the bottom panel. Paired-pulse depression was significantly (*p = 0.002, unpaired t test) greater at GC inputs to interneurons in SE-experienced rats.B, Paired stimulation (interpulse interval, 50 msec) at CA3 inputs produced a facilitation in EPSC amplitude in normal adult rats (top left). In contrast, paired stimulation of CA3 inputs in SE-experienced rats produced a depression (top right). Paired stimulation of CA3 inputs to interneurons from 15 normal adult (○) and three SE-experienced (●) rats is summarized in the bottom panel. The paired-pulse relationship at CA3 inputs to interneurons in SE-experienced rats was depressed significantly (*p < 0.019, unpairedt test), relative to the strong potentiation in normal adult rats.
Enhanced mGluR-mediated regulation of interneurons in SE-experienced rats
A near maximally effective concentration of the group II mGluR agonist, DCG-IV (1 μm), was significantly (p = 0.004) more effective in depressing GC input to hilar border interneurons in SE-experienced rats at 1–8 d after SE. A comparison of the time course of the effect of DCG-IV on GC-evoked inputs to hilar border interneurons is shown in Figure6A. In control adult rats, GC-evoked EPSCs were reduced by 27 ± 5% (n= 7 neurons). In contrast, DCG-IV produced a 68 ± 5% reduction in GC-evoked EPSCs in SE-experienced rats (Fig.6A,B; n = 11 neurons). Enhanced regulation of GC inputs by group II mGluRs was apparent as early as 1 d and persisted for at least 8 d after the induction of SE (Fig. 6C). The transmission rate of minimal GC inputs to hilar border interneurons was reduced by DCG-IV to 20 ± 17% of control (n = 3 neurons) in SE-experienced rats. However, as with GC inputs to hilar border interneurons in juvenile rats (Doherty and Dingledine, 1998), the amplitude of minimally evoked EPSCs was unchanged (Fig.6D). This is consistent with a presynaptic action of group II mGluRs at GC inputs to hilar border interneurons in adult control and SE-experienced rats, similar to that in untreated juvenile rats (Doherty and Dingledine, 1998).
Fig. 6.

Presynaptic inhibition of transmitter release at GC inputs to hilar border interneurons by the selective group II mGluR agonist, DCG-IV, was enhanced in the SE-experienced rat.A, Time course of the reversible depression of GC-evoked EPSC amplitude by DCG-IV (1 μm). GC inputs were more sensitive to DCG-IV in interneurons from SE-experienced rats (●;n = 4) than in interneurons from untreated adult rats (▵; n = 5). B, Maximal depression of GC-evoked EPSC amplitude induced by activation of group II mGluRs is significantly (*p = 0.005, unpairedt test) greater in SE-experienced rats (n = 11) than in untreated controls (n = 7). C, The sensitivity of GC inputs to depression by group II mGluRs is enhanced rapidly after induction of SE and persists for at least 8 d after induction. Each point in the scatterplot depicts the maximal effect of DCG-IV on evoked EPSC amplitudes at GC inputs to a single interneuron from either SE-experienced (●) or untreated adult (▵) rats. D, The mean amplitude of EPSCs evoked with minimal stimulation of a GC input to a hilar border interneuron from an SE-experienced rat was not depressed by DCG-IV (1 μm).
The mGluR antagonist, LY341495, antagonizes group II mGluRs with potencies in the low nanomolar range (Kingston et al., 1998; Schoepp et al., 1999). Thus, LY341495 at 500 nm provides a powerful inhibition of group II mGluRs; however, LY341495 is also a potent antagonist for mGluR8, and to a lesser degree mGluR7, at this concentration (Schoeppe et al., 1999). In SE-experienced rats, however, LY341495 (500 nm) attenuated STD induced by 20 Hz stimulation of GC inputs to hilar border interneurons (Fig.7A). The plateau reached during STD of GC-evoked EPSCs was significantly greater in the absence of LY341495 than in its presence (Fig. 7B). Thus, LY341495 produced a small but significant (p < 0.001, paired t test) attenuation of short-term plasticity at these synapses in SE-experienced rats, reducing the magnitude of STD by 13%. In contrast, LY341495 had no significant effect on the magnitude of STD in normal adult rats (Fig. 7B) (Doherty, Mott, Alagarsamy, Conn, and Dingledine, unpublished observations). These data together point to enhanced group II mGluR function at granule cell input to hilar border interneurons in the SE-experienced rat.
Fig. 7.

The selective group II mGluR antagonist, LY341495, attenuates STD of interneuron inputs in SE-experienced rats.A, Bath application of LY341495 (▵, 500 nm) significantly (p < 0.01, one-way ANOVA) attenuated the STD produced during 20 Hz stimulation of GC inputs to hilar border interneurons from SE-experienced rats (●).B, Group data illustrating the effect of LY341495 on the magnitude of STD at GC inputs to hilar border interneurons in the SE-experienced, but not control, rat. In control rats (n = 4), LY341495 had no effect on the plateau amplitude of GC-evoked EPSCs reached during 20 Hz trains. In SE-experienced rats (n = 4), LY341495 significantly (p < 0.01, paired t test) attenuated the plateau current reached during 20 Hz trains.
High-frequency stimulation evokes slow EPSCs in interneurons from SE-experienced rats
Stimulation of GC inputs at 5–20 Hz produced slow inward currents during the stimulus trains. Slow evoked currents are obvious in Figure3A and are shown in more detail in Figure8A. Slow currents were observed during high-frequency stimulation of GC inputs in 95% of hilar border interneurons in SE-experienced rats. In contrast, similar slow currents were observed in only 40% of hilar border interneurons from control adult rats. The mean amplitude of the slow current was −26 ± 11 pA (n = 20) in SE-experienced rats and −9 ± 5 pA in control adult rats (n = 20). The amplitude of this current was dependent on stimulus frequency, with higher stimulus frequencies producing larger currents (Fig. 8A,B). There was no correlation between the amplitude of the slow current and the magnitude of STD in SE-experienced rats (Fig. 8C). Thus, the slow current was unlikely to have had a systematic effect on the short-term depression of fast AMPA-mediated EPSCs. The current–voltage relationship for the slow current is depicted in Figure8D. Slow currents had an apparent reversal potential of >0 mV and showed significant rectification at negative membrane potentials. The selective NMDA receptor antagonist,d-APV (50 μm), significantly (p < 0.05, paired ttest) reduced the amplitude of the slow current (n = 4), suggesting that it was largely mediated by NMDA receptors activated during the train (Fig. 8E).
Fig. 8.

Properties of the slow current produced during HFS of afferent input to hilar border interneurons in SE-experienced rats.A, HFS of the GC input produced slow inward currents at a holding potential of −70 mV in a hilar border interneuron from an SE-experienced rat. The amplitude of the current was dependent on the frequency of stimulation. Each train was 800 msec in duration.B, Group data showing the relationship between STD and slow EPSC amplitude (n = 6 interneurons). Linear regression analysis indicated no significant correlation (r = 0.42; p = 0.91) between these two measurements. C, Plot showing the lack of correlation between the amplitude of the slow current and the amount of STD produced at that input. Each point represents data from a different interneuron from SE-experienced rats. Stimulation was delivered in all cases to GC inputs at a frequency of 20 Hz.D, Current–voltage relationship of the slow current. Note the pronounced rectification of the evoked current at hyperpolarized potentials. E, The selective NMDA receptor antagonist, d-APV (50 μm), significantly reduced the amplitude of the slow EPSC evoked during 20 Hz GC stimulation in four hilar border interneurons (Vh, −70 mV).
DISCUSSION
SE-induced alterations in short-term plasticity at inputs to inhibitory interneurons
Paired-pulse inhibition of granule cell population spikes was progressively impaired after pilocarpine-induced SE; it was evident at 24 hr after SE and peaked after 4–5 d. This result indicates that synaptic inhibition was rapidly impaired or excitation was enhanced after SE. We hypothesized that synaptic inhibition was rapidly reduced after SE because of a reduction of excitatory drive onto hilar border interneurons. Loss of paired-pulse inhibition may be partially attributable to a loss of inhibitory interneurons in the hilus after SE (Obenaus, 1993). In this study, we demonstrated that use-dependent depression at two different excitatory synapses onto surviving GABAergic hilar border interneurons is enhanced after pilocarpine-induced status epilepticus.
The magnitude of STD onto hilar border interneurons during repetitive stimulus trains at synapses from both dentate granule cell and CA3 pyramidal cell synapses is significantly enhanced in SE-experienced rats. However, enhanced STD after SE was synapse-specific because depression of perforant path synapses was not increased. Despite enhanced STD during high-frequency stimulation after SE, neither the probability of transmitter release nor the amplitude of minimal EPSCs was altered at these synapses during low-frequency stimulation. Thus, enhanced STD during repetitive activation represents a synapse- and context-dependent deficit in excitatory drive onto interneurons during epileptogenesis.
We demonstrated that depression of glutamate release at GC inputs by group II mGluRs is enhanced after pilocarpine-induced SE. This enhancement reverses a decrease in group II mGluR function at these synapses that occurs during development (Doherty, Mott, Alagarsamy, Conn, and Dingledine, unpublished observations). LY341495, a mGluR antagonist with selectivity for group II mGluRs, significantly attenuated STD at GC inputs in the SE-experienced rat but had no effect on STD at GC inputs in the normal adult. Thus, group II mGluR-mediated reduction of transmitter release contributes to STD in the SE-experienced adult rat.
Thus, some synaptic inputs onto hilar border interneurons are susceptible to a partial, reversible disconnection from their excitatory drive via an mGluR-mediated, activity-dependent process. This mGluR-mediated disconnection of inhibitory interneurons leads to a transient disinhibition of granule cell firing in juvenile rats (Doherty, Mott, Alagarsamy, Conn, and Dingledine, unpublished observations) and could therefore contribute to increased excitatory mossy fiber activation of CA3 pyramidal cells. We hypothesize that feedback inhibition of granule cell activity in the epileptic dentate gyrus is episodically reduced when hilar border interneurons experience a transient, presynaptic mGluR-mediated reduction in excitatory drive during repetitive synaptic activation. Additional work is needed to determine whether this shift in the balance between excitation and inhibition in the dentate gyrus during the latent period before spontaneous seizures appear enables further activity-dependent synaptic reorganization in the hippocampus during epileptogenesis and contributes to triggering seizure generation in the chronically epileptic brain.
Functional disconnection of interneuron inputs in SE-experienced rats
The dentate gyrus may act as a filter, resisting the transfer of epileptiform discharges from the entorhinal cortex to the hippocampus (Collins et al., 1983). Sufficiently intense stimulation can overwhelm this “dentate gate” (Stringer and Lothman, 1989). Synaptically mediated seizure activity can be propagated into the dentate gyrus through the entorhinal cortex (Barbarosie et al., 2000). Our hypothesis that a transient loss of feedback inhibition during high-frequency activation of the dentate gyrus, because of a functional disconnection of hilar border interneurons, could provide a physiological mechanism to overwhelm the dentate gate, facilitating the development of seizure discharges in downstream hippocampal circuitry.
Alterations in short-term plasticity were the only defects in excitatory neurotransmission onto interneurons observed after SE. We found no significant changes in either the properties of unitary EPSCs or the probability of transmitter release at these synapses after SE. Thus, functional disconnection of interneurons from excitatory inputs is subtle, manifesting only in response to an appropriate range of stimulus frequencies at affected synapses. Although these data are not consistent with a major deafferentation of hilar border interneurons in the SE-experienced dentate gyrus (Sloviter, 1991; Bekenstein and Lothman, 1993), we cannot rule out the possibility that some excitatory inputs to interneurons are lost after SE.
Changes in group II mGluR function in the SE-experienced dentate gyrus
Increased group II mGluR function during epileptogenesis may represent a return toward a juvenile phenotype. The group II mGluR agonist, DCG-IV, powerfully depresses synaptic transmission at GC inputs to hilar border interneurons in the juvenile rat (Doherty and Dingledine, 1998), but GC inputs in adult rats have a significantly reduced sensitivity to DCG-IV (Doherty, Mott, Alagarsamy, Conn, and Dingledine, unpublished observations). DCG-IV-mediated inhibition of GC inputs increases after pilocarpine-induced SE, matching the efficacy of DCG-IV observed in the juvenile hippocampus. Likewise, LY341495 reversibly attenuates STD of GC inputs in SE-experienced rats. In contrast, LY341495 has no effect on STD of GC inputs to hilar border interneurons in the normal adult rat, although it does attenuate STD significantly in the juvenile rat (Doherty, Mott, Alagarsamy, Conn, and Dingledine, unpublished observations). Group II mGluR-mediated function during epileptogenesis increases to the level seen in juvenile rats (Doherty and Dingledine, 1998), suggesting a recapitulation of a development program in the dentate gyrus during epileptogenesis. However, in contrast to the transient contribution of group II mGluRs to STD in normal juvenile rats (Doherty, Mott, Alagarsamy, Conn, and Dingledine, unpublished observations), a persistent mGluR component was revealed in SE-experienced rats (Fig. 7A).
Modulation of mGluR function in cortical circuitry is likely to be an important feature in the epileptogenic process. mGluR mRNA levels are rapidly modulated in the hippocampus after status epilepticus (Aronica et al., 1997). Although Klapstein et al. (1999) reported that perforant path sensitivity to group III agonists is reduced in kindled rats,Friedl et al. (1999) found no significant change in group III sensitivity at these synapses in the same model. In contrast, increased sensitivity to group II mGluR agonists in the SE-experienced dentate gyrus resembles enhanced group II sensitivity reported in the epileptic amygdala (Neugebauer et al., 1997). mGluR1a (Blumcke et al., 2000) and mGluR4 (Lie et al., 2000) immunoreactivities are increased in the epileptic human dentate gyrus. Group III mGluR function is reduced in hippocampal slices from surgically resected tissue (Dietrich et al., 1999).
We demonstrated enhanced mGluR-mediated inhibition of glutamatergic neurotransmission at GC synapses after SE contributes to enhanced STD in SE-experienced rats. Although these results are most consistent with a group II effect, 500 nm LY341495 partially antagonizes mGluR8 and mGluR7 (Schoeppe et al., 1999). Thus, an increase in group III mGluR function could also contribute to the enhanced STD at these synapses in SE-experienced rats. LY341495 did not completely block the enhanced STD in SE-experienced rats, indicating that other mechanisms must contribute to enhanced STD at these synapses. Perhaps activity-dependent increases in the rate of vesicle depletion, reduced vesicle pool replenishment (Stevens and Wesseling, 1999), or changes in the properties of postsynaptic AMPA receptors (Rozov and Burnashev, 1999) contribute to enhanced STD in SE-experienced rats. We cannot rule out a selective loss of a subpopulation of interneurons with weak STD in SE-experienced rats. However, the observation that the plateau of STD was typically greater in SE-experienced rats than in any of the control cells (Fig. 4B) argues against this possibility.
Functional implications
Although granule cells in vivo do not typically discharge at frequencies likely to activate presynaptic mGluRs (Jung and McNaughten, 1993), high-frequency granule cell discharges in the dentate gyrus can precede seizures in the chronically epileptic brain (Bragin et al., 1999; Finnerty and Jefferys, 2000). Moreover, granule cells in slices of human epileptic dentate gyrus can discharge at high frequencies when GABAergic inhibition is compromised (Franck et al., 1995). Transient reduction of inhibitory control during mGluR-mediated depression of interneuron input may contribute to the development of hyperexcitability in the latent period of epileptogenesis. Polysynaptic IPSCs onto granule cells undergo a progressive depression during high-frequency (30 Hz) stimulation of the perforant path in pilocarpine-treated rats at 8 weeks after SE (Isokawa, 1996).
Because hilar border interneurons synapse onto other inhibitory interneurons (Freund and Buzáski, 1996), enhanced STD at interneuron inputs might produce a paradoxical increase in feedback inhibition in the dentate gyrus. Indeed, augmentation of GABAergic inhibition in the hippocampus has also been reported in both human epilepsy (Swanson et al., 1998; Wilson et al., 1998) and chronic experimental models (Bühl et al., 1996; Haas et al., 1996). However, our data are most consistent with activity-dependent functional disinhibition of granule cells. Evidence for this includes impaired synaptic inhibition within the first week after SE observed in this study and the demonstration that a DCG-IV-mediated reduction in excitatory input onto hilar border interneurons results in impaired feedback inhibition of dentate granule cells (Doherty, Mott, Alagarsamy, Conn, and Dingledine, unpublished observations).
In conclusion, SE enhances mGluR-mediated depression of excitatory drive onto dentate hilar border interneurons, a potentially important regulatory mechanism to dynamically influence the strength of GABAergic inhibition in the dentate gyrus. This aberrant plasticity may play an important role in epileptogenesis and may trigger or amplify epileptiform activity in the epileptic hippocampus.
Footnotes
This work was supported by an American Epilepsy Society Research Training Fellowship (J.D.), the Charles E. Culpeper Foundation (J.D.), and the National Institutes of Health (R.D.). We thank Dr. Y. Ohfune and Tocris Cookson for DCG-IV, Dr. D. Schoepp for LY341495, Dr. V. Nadler for help with the pilocarpine model of chronic seizures, and Drs. S. Bausch and D. Mott for helpful comments on this manuscript.
Correspondence should be addressed to James Doherty, 5010 Rollins Research Center, Emory University Medical School, 1510 Clifton Road, Atlanta, GA 30322. E-mail:jdoherty@bimcore.emory.edu.
REFERENCES
- 1.Aronica EM, Gorter JA, Paupard M-C, Grooms SY, Bennett MVL, Zukin SR. Status epilepticus-induced alterations in metabotropic glutamate receptor expression in young and adult rats. J Neurosci. 1997;17:8588–8595. doi: 10.1523/JNEUROSCI.17-21-08588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ascády L, Kamondi A, Sík A, Freund TF, Buzáki G. GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci. 1998;18:3386–3404. doi: 10.1523/JNEUROSCI.18-09-03386.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbarosie M, Louvel J, Kurcewicz I, Avoli M. CA3-released entorhinal seizures disclose dentate gyrus epileptogenicity and unmask a temporoammonic pathway. J Neurophysiol. 2000;83:1115–1124. doi: 10.1152/jn.2000.83.3.1115. [DOI] [PubMed] [Google Scholar]
- 4.Bausch SB, Chavkin C. Changes in hippocampal circuitry after pilocarpine-induced seizures as revealed by opioid receptor distribution and activation. J Neurosci. 1997;17:477–492. doi: 10.1523/JNEUROSCI.17-01-00477.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bekenstein JW, Lothman EW. Dormancy of inhibitory interneurons in a model of temporal lobe epilepsy. Science. 1993;259:97–100. doi: 10.1126/science.8093417. [DOI] [PubMed] [Google Scholar]
- 6.Bleck TP. Management approaches to prolonged seizures and status epilepticus. Epilepsia. 1999;40 [Suppl 1]:S59–63. doi: 10.1111/j.1528-1157.1999.tb00880.x. [DOI] [PubMed] [Google Scholar]
- 7.Blumcke I, Becker AJ, Klein C, Scheiwe C, Lie AA, Beck H, Waha A, Friedl MG, Kuhn R, Emson P, Elger C, Wiestler OD. Temporal lobe epilepsy associated up-regulation of metabotropic glutamate receptors: correlated changes in mGluR1 mRNA and protein expression in experimental animals and human patients. J Neuropathol Exp Neurol. 2000;59:1–10. doi: 10.1093/jnen/59.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Bragin A, Engel J, Jr, Wilson CL, Fried I, Mathern GW. Hippocampal and entorhinal cortex high-frequency oscillations (100–500 Hz) in human epileptic brain and in kainic acid-treated rats with chronic seizures. Epilepsia. 1999;40:127–137. doi: 10.1111/j.1528-1157.1999.tb02065.x. [DOI] [PubMed] [Google Scholar]
- 9.Bragin AG, Jando G, Nadazdy Z, Hetke J, Wise K, Buzsaki G. Gamma (40–100 Hz) oscillations in the hippocampus of the behaving rat. J Neurosci. 1995;15:47–60. doi: 10.1523/JNEUROSCI.15-01-00047.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breakwell NA, Rowan MJ, Anwyl R. Metabotropic glutamate receptor dependent EPSP and EPSP-spike potentiation in area CA1 of the submerged rat hippocampal slice. J Neurophysiol. 1996;76:3126–3135. doi: 10.1152/jn.1996.76.5.3126. [DOI] [PubMed] [Google Scholar]
- 11.Brooks-Kayal A, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABAA receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- 12.Bühl EH, Otis TS, Mody I. Zinc-induced collapse of augmented inhibition by GABA in a temporal lobe epilepsy model. Science. 1996;271:369–373. doi: 10.1126/science.271.5247.369. [DOI] [PubMed] [Google Scholar]
- 13.Canepari M, Cherubini E. Dynamics of excitatory transmitter release: analysis of synaptic responses in CA3 hippocampal neurons after repetitive stimulation of afferent fibers. J Neurophysiol. 1998;79:1977–1988. doi: 10.1152/jn.1998.79.4.1977. [DOI] [PubMed] [Google Scholar]
- 14.Cavalheiro EA, Leite JP, Bortolotto ZA, Turski WA, Ikonomidou C, Turski L. Long-term effects of pilocarpine in rats: structural damage of the brain triggers kindling and spontaneous recurrent seizures. Epilepsia. 1991;32:778–782. doi: 10.1111/j.1528-1157.1991.tb05533.x. [DOI] [PubMed] [Google Scholar]
- 15.Chagnac-Amitai Y, Connors BW. Horizontal spread of synchronized activity in neocortex and its control by GABA-mediated inhibition. J Neurophysiol. 1989;61:747–758. doi: 10.1152/jn.1989.61.4.747. [DOI] [PubMed] [Google Scholar]
- 16.Collins RC, Tearse RG, Lothman EW. Functional anatomy of limbic seizures: focal discharges from medial entorhinal cortex in rat. Brain Res. 1983;280:25–40. doi: 10.1016/0006-8993(83)91170-8. [DOI] [PubMed] [Google Scholar]
- 17.Coulter D. Chronic epileptogenic cellular alterations in the limbic system after status epilepticus. Epilepsia. 1999;40 [Suppl 1]:S23–33. doi: 10.1111/j.1528-1157.1999.tb00875.x. [DOI] [PubMed] [Google Scholar]
- 18.Dietrich D, Kral T, Clusmann H, Friedl M, Schramm J. Reduced function of L-AP4-sensitive metabotropic glutamate receptors in human epileptic sclerotic hippocampus. Eur J Neurosci. 1999;11:1109–1113. doi: 10.1046/j.1460-9568.1999.00549.x. [DOI] [PubMed] [Google Scholar]
- 19.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 20.Doherty J, Dingledine R. The regulation of excitatory input to inhibitory interneurons of the dentate gyrus during hypoxia. J Neurophysiol. 1997a;77:393–404. doi: 10.1152/jn.1997.77.1.393. [DOI] [PubMed] [Google Scholar]
- 21.Doherty J, Dingledine R. Excitatory synaptic inputs to dentate interneurons in slices from pilocarpine-treated epileptic rats. Epilepsia. 1997b;38 [Suppl 8]:124. [Google Scholar]
- 22.Doherty J, Dingledine R. Differential regulation of excitatory synaptic inputs to hilar border interneurons in the dentate gyrus by metabotropic glutamate receptors. J Neurophysiol. 1998;79:2903–2910. doi: 10.1152/jn.1998.79.6.2903. [DOI] [PubMed] [Google Scholar]
- 23.Dykes RW. Mechanisms controlling neuronal plasticity in somatosensory cortex. Can J Physiol Pharmacol. 1997;75:535–545. [PubMed] [Google Scholar]
- 24.Empson RM, Jefferys JG. Synaptic inhibition in primary and secondary chronic foci by intrahippocampal tetanus toxin in the rat. J Physiol (Lond) 1993;465:595–614. doi: 10.1113/jphysiol.1993.sp019695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finnerty GT, Jefferys JG. 9–16 Hz oscillation precedes secondary generalization of seizures in the rat tetanus toxin model of epilepsy. J Neurophysiol. 2000;83:2217–2226. doi: 10.1152/jn.2000.83.4.2217. [DOI] [PubMed] [Google Scholar]
- 26.Franck JE, Pokorny J, Kunkel DD, Schwartzkroin PA. Physiologic and morphologic characteristics of granule cell circuitry in human epileptic hippocampus. Epilepsia. 1995;36:543–558. doi: 10.1111/j.1528-1157.1995.tb02566.x. [DOI] [PubMed] [Google Scholar]
- 27.Freund TF, Busáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 28.Friedl M, Clusmann H, Kral T, Dietrich D, Schramm J. Analysing metabotropic glutamate group III receptor mediated modulation of synaptic transmission in the amygdala-kindled dentate gyrus of the rat. Brain Res. 1999;821:117–123. doi: 10.1016/s0006-8993(99)01083-5. [DOI] [PubMed] [Google Scholar]
- 29.Galarreta M, Hestrin S. Frequency-dependent synaptic depression and the balance between excitation and inhibition in the neocortex. Nat Neurosci. 1998;7:587–594. doi: 10.1038/2822. [DOI] [PubMed] [Google Scholar]
- 30.George B, Kulkarni SK. Protective effects of GABAergic drugs and other anticonvulsants in lithium-pilocarpine-induced status epilepticus. Methods Find Exp Clin Pharmacol. 1996;18:335–340. [PubMed] [Google Scholar]
- 31.Grover LM, Yan C. Blockade of GABAA-receptors facilitates induction of NMDA receptor-independent long-term potentiation. J Neurophysiol. 1999;81:2814–2822. doi: 10.1152/jn.1999.81.6.2814. [DOI] [PubMed] [Google Scholar]
- 32.Haas KZ, Sperber EF, Moshé SL, Stanton PK. Kainic acid-induced seizures enhance dentate gyrus inhibition by downregulation of GABAB receptors. J Neurosci. 1996;16:4250–4260. doi: 10.1523/JNEUROSCI.16-13-04250.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirsch JC, Agassandian C, Merchán-Pérez A, Ben-Ari Y, DeFelipe J, Esclapez M, Bernard C. Deficit of quantal release of GABA in experimental models of temporal lobe epilepsy. Nat Neurosci. 1999;2:499–500. doi: 10.1038/9142. [DOI] [PubMed] [Google Scholar]
- 34.Houser CR, Escaplez M. Vulnerability and plasticity of the GABA system in the pilocarpine model of spontaneous recurrent seizures. J Neurosci. 1996;13:4470–4485. doi: 10.1016/s0920-1211(96)00054-x. [DOI] [PubMed] [Google Scholar]
- 35.Isokawa M. Decrement of GABAA receptor-mediated inhibitory postsynaptic currents in dentate granule cells in epileptic hippocampus. J Neurophysiol. 1996;75:1901–1908. doi: 10.1152/jn.1996.75.5.1901. [DOI] [PubMed] [Google Scholar]
- 36.Jung MW, McNaughten BL. Spatial selectivity of unit activity in the hippocampal granule cell layer. Hippocampus. 1993;3:165–182. doi: 10.1002/hipo.450030209. [DOI] [PubMed] [Google Scholar]
- 37.Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci. 1997;17:7532–7540. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keverne EB, Brennan PA. Olfactory recognition memory. J Physiol (Lond) 1996;90:399–401. doi: 10.1016/s0928-4257(97)87929-6. [DOI] [PubMed] [Google Scholar]
- 39.King GL, Dingledine R, Giacchino JL, McNamara JO. Abnormal neuronal excitability in hippocampal slices from kindled rats. J Neurophysiol. 1985;54:1295–1304. doi: 10.1152/jn.1985.54.5.1295. [DOI] [PubMed] [Google Scholar]
- 40.Kingston AE, Ornstein PL, Wright RA, Johnson BG, Mayne NG, Burnett JP, Belagaje R, Wu S, Schoepp DD. LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology. 1998;37:1–12. doi: 10.1016/s0028-3908(97)00191-3. [DOI] [PubMed] [Google Scholar]
- 41.Klapstein GJ, Meldrum BS, Mody I. Decreased sensitivity to group III mGluR agonists in the lateral perforant path following kindling. Neuropharmacology. 1999;38:927–933. doi: 10.1016/s0028-3908(99)00016-7. [DOI] [PubMed] [Google Scholar]
- 42.Kneisler TB, Dingledine R. Synaptic input from CA3 pyramidal cells to dentate basket cells in the rat hippocampus. J Physiol (Lond) 1995;487:125–146. doi: 10.1113/jphysiol.1995.sp020866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laezza F, Doherty J, Dingledine R. Long-term depression in hippocampal interneurons: joint requirement for pre- and postsynaptic events. Science. 1999;285:1411–1414. doi: 10.1126/science.285.5432.1411. [DOI] [PubMed] [Google Scholar]
- 44.Lie AA, Becker A, Behle K, Beck H, Malitschek B, Conn PJ, Kuhn R, Nitsch R, Plaschke M, Schramm J, Elger CE, Wiestler OD, Blümcke I. Up-regulation of the metabotropic glutamate receptor mGluR4 in hippocampal neurons with reduced seizure vulnerability. Ann Neurol. 2000;47:26–35. [PubMed] [Google Scholar]
- 45.MacDonald RL, Kapur J. Acute cellular alterations in the hippocampus after status epilepticus. Epilepsia. 1999;40 [Suppl 1]:S9–20. doi: 10.1111/j.1528-1157.1999.tb00873.x. [DOI] [PubMed] [Google Scholar]
- 46.Mangan PS, Lothman EW. Profound disturbances of pre- and postsynaptic GABAB-receptor-mediated processes in region CA1 in a chronic model of temporal lobe epilepsy. J Neurophysiol. 1996;76:1282–1296. doi: 10.1152/jn.1996.76.2.1282. [DOI] [PubMed] [Google Scholar]
- 47.McMahon LL, Kauer JA. Hippocampal interneurons express a novel form of synaptic plasticity. Neuron. 1997;18:295–305. doi: 10.1016/s0896-6273(00)80269-x. [DOI] [PubMed] [Google Scholar]
- 48.Mello LEAM, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- 49.Merlin LR, Wong RKS. Synaptic modifications accompanying epileptogenesis in vitro: long-term depression of GABA-mediated inhibition. Brain Res. 1993;627:330–340. doi: 10.1016/0006-8993(93)90338-n. [DOI] [PubMed] [Google Scholar]
- 50.Miles R, Wong RK. Inhibitory control of local excitatory circuits in the guinea-pig hippocampus. J Physiol (Lond) 1987;388:611–629. doi: 10.1113/jphysiol.1987.sp016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neugebauer V, Keele NB, Shinnick-Gallagher P. Epileptogenesis in vivo enhances the sensitivity of inhibitory metabotropic glutamate receptors in basolateral amygdala neurons in vitro. J Neurosci. 1997;17:983–995. doi: 10.1523/JNEUROSCI.17-03-00983.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Obenaus A, Esclapez M, Houser CR. Loss of glutamate decarboxylase mRNA-containing neurons in the rat dentate gyrus following pilocarpine-induced seizures. J Neurosci. 1993;13:4470–4485. doi: 10.1523/JNEUROSCI.13-10-04470.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohfune Y, Shimamoto K, Ishida M, Shinozaki H. Synthesis of L-2-(2,3-dicarboxycyclopropyl)glycine: a novel conformationally restricted glutamate analog. Bioorg Med Chem. 1993;3:15–18. [Google Scholar]
- 54.Okazaki MM, Molnár P, Nadler V. Recurrent mossy fiber pathway in rat dentate gyrus: synaptic currents in presence and absence of seizure-induced growth. J Neurophysiol. 1999;81:1645–1660. doi: 10.1152/jn.1999.81.4.1645. [DOI] [PubMed] [Google Scholar]
- 55.Olsen RW, Avoli M. GABA and epileptogenesis. Epilepsia. 1997;38:399–407. doi: 10.1111/j.1528-1157.1997.tb01728.x. [DOI] [PubMed] [Google Scholar]
- 56.Patrylo PR, Dudek FE. Physiological unmasking of new glutamatergic pathways in the dentate gyrus of hippocampal slices from kainate-induced epileptic rats. J Neurophysiol. 1998;79:418–429. doi: 10.1152/jn.1998.79.1.418. [DOI] [PubMed] [Google Scholar]
- 57.Paulsen O, Moser EI. A model of hippocampal memory encoding and retrieval: GABAergic control of synaptic plasticity. Trends Neurosci. 1998;21:273–278. doi: 10.1016/s0166-2236(97)01205-8. [DOI] [PubMed] [Google Scholar]
- 58.Prince DA. Neurophysiology of epilepsy. Annu Rev Neurosci. 1978;1:395–415. doi: 10.1146/annurev.ne.01.030178.002143. [DOI] [PubMed] [Google Scholar]
- 59.Psarropoulou C, Matsokis N, Angelatou F, Kostopoulos G. Pentylenetetrazol-induced seizures decrease γ-aminobutyric acid-mediated recurrent inhibition and enhance adenosine-mediated depression. Epilepsia. 1994;35:12–19. doi: 10.1111/j.1528-1157.1994.tb02906.x. [DOI] [PubMed] [Google Scholar]
- 60.Rice A, Rafiq A, Shapiro SM, Jakoi ER, Coulter DA, DeLorenzo RJ. Long-lasting reduction of inhibitory function and gamma-aminobutyric acid type A receptor subunit mRNA expression in a model of temporal lobe epilepsy. Proc Natl Acad Sci USA. 1996;93:9665–9669. doi: 10.1073/pnas.93.18.9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rozov A, Burnashev N. Polyamine-dependent facilitation of postsynaptic AMPA receptors counteracts paired-pulse depression. Nature. 1999;401:594–598. doi: 10.1038/44151. [DOI] [PubMed] [Google Scholar]
- 62.Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 63.Sloviter RS. Permanently altered hippocampal structure, excitability, and inhibition after experimental status epilepticus in the rat: the “dormant basket cell” hypothesis and its possible relevance to temporal lobe epilepsy. Hippocampus. 1991;1:41–66. doi: 10.1002/hipo.450010106. [DOI] [PubMed] [Google Scholar]
- 64.Sloviter RS. Status epilepticus-induced neuronal injury and network reorganization. Epilepsia. 1999;40 [Suppl 1]:S34–39. doi: 10.1111/j.1528-1157.1999.tb00876.x. [DOI] [PubMed] [Google Scholar]
- 65.Smith KL, Lee CL, Swann JW. Local circuit abnormalities in chronically epileptic rats after intrahippocampal tetanus toxin injection in infancy. J Neurophysiol. 1998;79:106–116. doi: 10.1152/jn.1998.79.1.106. [DOI] [PubMed] [Google Scholar]
- 66.Steele PM, Mauk MD. Inhibitory control of LTP and LTD: stability of synaptic strength. J Neurophysiol. 1999;81:1559–1566. doi: 10.1152/jn.1999.81.4.1559. [DOI] [PubMed] [Google Scholar]
- 67.Stevens CF, Wesseling JF. Identification of a novel process limiting the rate of synaptic vesicle cycling at hippocampal synapses. Neuron. 1999;24:1017–1028. doi: 10.1016/s0896-6273(00)81047-8. [DOI] [PubMed] [Google Scholar]
- 68.Stringer JL, Lothman EW. Maximal dentate activation: characteristics and alterations after repeated seizures. J Neurophysiol. 1989;62:136–143. doi: 10.1152/jn.1989.62.1.136. [DOI] [PubMed] [Google Scholar]
- 69.Swanson TH, Sperling MR, O'Connor MJ. Strong paired pulse depression of dentate granule cells in slices from patients with temporal lobe epilepsy. J Neural Transm. 1998;105:613–625. doi: 10.1007/s007020050083. [DOI] [PubMed] [Google Scholar]
- 70.Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA. Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse. 1989;3:154–171. doi: 10.1002/syn.890030207. [DOI] [PubMed] [Google Scholar]
- 71.Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: a behavioural electroencephalographic and neuropathological study. Behav Brain Res. 1983;9:315–336. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- 72.Wasterlain CG, Shirasaka Y, Mazarati AM, Spigelman I. Chronic epilepsy with damage restricted to the hippocampus: possible mechanisms. Epilepsy Res. 1996;26:255–265. doi: 10.1016/s0920-1211(96)00058-7. [DOI] [PubMed] [Google Scholar]
- 73.Whittington MA, Jefferys JG. Epileptic activity outlasts disinhibition after intrahippocampal tetanus toxin in the rat. J Physiol (Lond) 1994;481:593–604. doi: 10.1113/jphysiol.1994.sp020466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wigström H, Gustafsson B. Facilitated induction of hippocampal long-lasting potentiation during blockade of inhibition. Nature. 1983;301:603–604. doi: 10.1038/301603a0. [DOI] [PubMed] [Google Scholar]
- 75.Williamson A, Patrylo PR, Spencer DD. Decrease in inhibition in dentate granule cells from patients with medial temporal lobe epilepsy. Ann Neurol. 1999;45:92–99. doi: 10.1002/1531-8249(199901)45:1<92::aid-art15>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 76.Wilson CL, Khan SU, Engel J, Jr, Isokawa M, Babb TL, Behnke EJ. Paired pulse suppression and facilitation in human epileptogenic hippocampal formation. Epilepsy Res. 1998;31:211–230. doi: 10.1016/s0920-1211(98)00063-1. [DOI] [PubMed] [Google Scholar]
- 77.Zheng W, Knudsen EI. Functional selection of adaptive auditory space map by GABAA-mediated inhibition. Science. 1999;284:962–965. doi: 10.1126/science.284.5416.962. [DOI] [PubMed] [Google Scholar]