

Abstract
Brain injury in newborns can cause deficits in motor and sensory function. In most models of neonatal brain injury, thalamic damage often occurs. Using the Rice–Vannucci model of neonatal hypoxic–ischemic brain injury, we have shown that neuronal degeneration in somatosensory thalamus is delayed in onset (∼24 hr) compared with cortical and striatal injury and exhibits prominent structural features of apoptosis. In the present study, we examined whether cell death in the thalamus has molecular features of apoptosis. Fas death receptor protein expression increased rapidly after neonatal hypoxia–ischemia, in concert with cleavage of procaspase 8 to its active form. Concurrently, the levels of Bax in mitochondrial-enriched cell fractions increase, and cytochrome c accumulates in the soluble fraction. Mitochondria accumulate in a perinuclear distribution by 6 hr after hypoxia–ischemia. Cytochrome oxidase subunit 1 protein levels also increase at 6 hr after hypoxia–ischemia. Increased levels of Fas death receptor, Bax, and cytochrome c, activation of caspase 8, and abnormalities in mitochondria in the thalamus significantly precede the activation of caspase 3 and the appearance of neuronal apoptosis at 24 hr. We conclude that the delayed neurodegeneration in neonatal rat ventral basal thalamus after hypoxic–ischemic injury is apoptosis mediated by death receptor activation.
Keywords: mitochondria, neonatal brain injury, Bax, Fas death receptor, caspase, cytochrome oxidase
A large amount of investigation has focused on cytokine- and hypoxia–ischemia-mediated injury to the developing cortex and periventricular white matter as the cause of the neurodevelopmental handicaps suffered by infants who have experienced perinatal brain injury. Energy failure, free radical, cytokine, and excitatory amino acid release, and caspase-dependant cell death are known to contribute to injury in the neocortex, striatum, and periventricular white matter (McDonald et al., 1988; Barks and Silverstein, 1992; Hagan et al., 1996; Liu et al., 1996; Martin et al., 1997a, b; Back et al., 1998; Cheng et al., 1998). However, the degeneration of thalamus and other nonforebrain structures after hypoxia–ischemia is studied less frequently. Injury to somatosensory thalamus has been described in human newborns after hypoxia–ischemia (Barkovich, 1995; Roland et al., 1998) and may contribute to sensorimotor deficits in infants with perinatal brain injury and cerebral palsy. Sensorimotor deficits have been detected in neonatal rats subjected to the Rice–Vannucci model (Rice et al., 1981) of hypoxic–ischemic injury (Bona et al., 1997). A few detailed neuropathological studies of animal models have revealed injury to the developing thalamus after neonatal hypoxia–ischemia (Towfighi et al., 1991). In addition, we have demonstrated recently that injury to the thalamus occurs in a delayed manner and exhibits prominent structural features of apoptosis when compared with the early necrotic cell death seen in the forebrain after hypoxia–ischemia (Northington et al., 2001).
The mechanisms of apoptotic neurodegeneration in the thalamus and other brain regions remote from the forebrain after neonatal hypoxia–ischemia are completely unknown. However, death receptor-activated pathways, altered mitochondrial function, and changes in expression of mitochondrial-related bcl-2 family proteins are likely important effectors of programmed cell death in the present model of neonatal brain injury (Nelson and Silverstein, 1994;Silverstein, 1998; Felderhoff-Mueser et al., 2000). Fas death receptor protein expression is increased in hippocampus bilaterally during the 24 hr immediately after neonatal hypoxia–ischemia (Felderhoff-Mueser et al., 2000), and administration of cytokine antagonists afford neuroprotection in the present model (Liu et al., 1996). In adult models of apoptosis, mitochondrial accumulation occurs during the critical chromatolytic phase before apoptosis simultaneously with increased expression of the proapoptosis proteins Bax and Bak (Martin, 1999; Martin et al., 1999).
In this study, we investigate whether a cascade of events (including death receptor activation, alteration in the ratio of proapoptosis- and antiapoptosis-regulating proteins in mitochondria, altered mitochondrial activity and location, cytochrome c accumulation, and cleavage of caspases into active subunits) leads to apoptotic neurodegeneration in the thalamus after neonatal hypoxia–ischemia.
MATERIALS AND METHODS
Hypoxia–ischemia in 7-d-old rats. The Rice–Vannucci (Rice et al., 1981) neonatal adaptation of the Levine procedure (Levine, 1960) was used to cause hypoxic–ischemic brain injury in 7-d-old (p7) rats. In brief, rat pups were anesthetized with 2.5% halothane and 15% nitrous oxide in O2. The right common carotid artery was permanently ligated (in sham controls the ligature was passed around the artery and removed). After the wound was sutured, the pups recovered from anesthesia and were returned to the dam. Two hours later, pups were placed in an airtight container in a 37°C water bath through which humidified 8% O2and balance nitrogen flowed for 150 min. After hypoxia, pups were returned to the dam until death.
The animals were killed, and the brains were retrieved at 0, 1.5, 3, 6, and 24 hr after the end of hypoxia for histological analysis (n = 6 for each time point) or at 3, 6, and 24 hr after the end of hypoxia for Western blotting. Because of the small size of the immature rat thalamus, three hemithalami per pooled sample were required to generate adequate tissue homogenate for immunoblot analysis. Thus tissue from12 to 15 animals per time point were used to generate four to five pooled samples at each time point for immunoblotting. The multiple early time points were chosen because our data, as well as that of others, showed a faster progression of injury after hypoxia–ischemia in neonates as compared with adults (Towfighi et al., 1995; Martin et al., 1997a; Northington et al., 2001). Control groups consisted of sham-operated littermates (n = 4–6 for histological analysis; n= 4 pooled thalami for Western blotting). Because the entire thalamus could be used to create tissue homogenates in sham controls, only eight animals were required to generate adequate tissue samples for immunoblotting. Sham controls were killed on postnatal day 7 to provide age-matched tissue for comparison of expression of apoptosis-related proteins and levels of cytochrome oxidase activity.
All animal studies received previous approval from the Animal Care and Use Committee of Johns Hopkins University School of Medicine and were performed in accordance with the National Institutes of HealthGuide for the Care and Use of Laboratory Animals (United States Department of Health and Human Services 85-23, 1985).
Tissue preparation. Animals were killed with an overdose of pentobarbital (65 mg/kg, i.p.) and exsanguinated with cold 0.1m PBS, pH 7.4, via intracardiac perfusion. Brains were perfusion fixed with 4% paraformaldehyde in PBS for 30 min at 4 ml/min. The brains were removed, post-fixed in 4% paraformaldehyde overnight, cryoprotected in 30% sucrose, and frozen in isopentane (−30°C). Sixty micrometer coronal sections were cut on a sliding microtome for cresyl violet and immunohistochemical staining.
Immunoblotting. Samples of thalamus were rapidly microdissected under a surgical microscope, immediately after death, and frozen on dry ice. Pooled samples (150–200 mg) were homogenized in cold 20 mm Tris-HCl, pH 7.4, containing 10% (w/v) sucrose, 20 μg/ml aprotinin (Trayslol), 20 μg/ml leupeptin, 20 μg/ml antipain, 20 μg/ml pepstatin A, 20 μg/ml chymostatin, 0.1 mm phenylmethylsulfonyl fluoride, 10 mmbenzamidine, 1 mm EDTA, and 5 mm EGTA. Crude homogenates were centrifuged at 1000 × g for 10 min. The supernatant (S1 fraction) was then centrifuged at 114,000 ×g for 20 min, and the resulting supernatant (S2 soluble fraction) was collected. The pellet (P2, mitochondrial-enriched, membrane fraction) was washed in homogenization buffer (without sucrose) three times by resuspension and centrifugation at 114,000 × g for 20 min. The P2 fraction was then resuspended fully in homogenization buffer supplemented with 20% (w/v) glycerol. This subcellular fractionation protocol has been verified to be enriched in mitochondria but also contains other organelle constituents (i.e., endoplasmic reticulum and Golgi apparatus) (Martin et al., 2000). Protein concentrations were measured by a Bio-Rad (Hercules, CA) protein assay with bovine serum albumin as a standard.
Samples of membrane or soluble proteins were subjected to SDS-PAGE and electroeluted onto nitrocellulose membranes. The reliability of sample loading and protein transfer was evaluated by staining nitrocellulose membranes with Ponceau S before immunoblotting and by quantification of Coomassie-stained gels and Ponceau-stained blots with OD. Blots were blocked with 2.5% nonfat dry milk with 0.1%Tween 20 in 50 mm Tris-buffered saline, pH 7.4, and incubated overnight at 4°C with antibody. After primary incubation, blots were washed, incubated with peroxidase-conjugated secondary antibodies (0.2 μg/ml), and developed with enhanced chemiluminescence. To quantify cell death protein immunoreactivity, films were scanned using Adobe Photoshop, and OD was performed with IP Lab Gel H software. The OD of the corresponding lanes in Coomassie-stained gels or Ponceau-stained blots was used to correct the OD of the cell death protein immunoreactivity for differences in protein loading. Protein levels are thus expressed as relative OD measurements compared with control lanes in the same blot.
Antibodies. Anti-Fas death receptor antibody (Santa Cruz Biotechnology, Santa Cruz, CA) is a rabbit polyclonal antibody generated against an epitope mapping to the C terminal of human Fas and is noncross-reactive with other tumor necrosis factor receptor (TNFR) type-1 receptors. It recognizes a single band at 45 kDa and higher molecular weight bands after SDS-PAGE. Jurkat cells known to express high levels of the Fas death receptor were used as a positive control. Anti-caspase 8 antibody (Santa Cruz Biotechnology) is a rabbit polyclonal antibody raised against a recombinant protein corresponding to amino acids 217–350 of human caspase 8. Anti-Bax (Upstate Biotechnology, Lake Placid, NY) and anti-Bcl-2 antibodies (Santa Cruz Biotechnology) are affinity-purified rabbit polyclonal antibodies generated against a peptide corresponding to amino acids 1–21 of human Bax and a peptide mapping at the N terminal of human Bcl-2, respectively. Anti-Bax antibody recognizes a single 23 kDa band, a faint band at 18.5 kDa representing the cleaved form of Bax, and several less intense high-molecular weight bands in P2 protein fractions. Anti-Bcl-2 antibody recognizes a band at 26 kDa and several less intense high-molecular weight bands in P2 protein fractions. A second rabbit polyclonal anti-Bax antibody (Santa Cruz Biotechnology) generated against a peptide homologous to amino acids 11–30 of human Bax gave results identical to those of the Upstate Biotechnology antibody. Because these proteins were fractionated in denaturing gels, it is unlikely that the high-molecular weight bands represent various Bax and Bcl-2 homodimers and heterodimers. Anti-cytochrome c antibody (Santa Cruz Biotechnology) is a rabbit polyclonal antibody directed against amino acids 1–104 of full-length horse cytochrome c. It recognizes a band at 12–15 kDa. Anti-cytochrome oxidase subunit 1 (anti-COX1) antibody (Molecular Probes, Eugene, OR) is a mouse monoclonal antibody generated against subunit 1 of human cytochrome oxidase. The antibody recognizes only a single band of ∼35 kDa in nonboiled P2 protein samples subjected to SDS-PAGE. Anti-caspase 3 antibody (Santa Cruz Biotechnology) is a rabbit polyclonal antibody generated against amino acids 1–277 of full-length human caspase-3, and it recognizes the 32 kDa proenzyme with an intense band of immunoreactivity and the 12, 17, and 20 kDa active fragments with less intense single bands. Cleaved caspase 3 antibody (Cell Signaling Technology, Beverly, MA) is a rabbit polyclonal antibody generated against a synthetic peptide corresponding to residues surrounding the cleavage site of human caspase 3.
Cytochrome oxidase histochemistry. To identify the levels of oxidative metabolism and intracellular distribution of oxidatively active mitochondria, we used the cytochrome oxidase histochemical method of Wong-Riley (1979, 1989) as described previously for our laboratory (Martin et al., 1997a). Briefly, brain sections from control and hypoxic–ischemic rat pups were exposed to the assay simultaneously. The enzymatic reaction medium was prepared immediately before each experiment and consisted of 100 mmphosphate buffer, pH 7.4, 0.1% horse heart cytochrome c (Sigma, St. Louis, MO), 117 mm sucrose, and 1.4 mmdiaminobenzidine tetrahydrochloride. In this histochemical reaction,in situ cytochrome oxidase catalyzes the transfer of electrons (donated by diaminobenzidine) from cytochrome c, provided as substrate, to O2 to form H2O. The donation of electrons from diaminobenzidine is a chromogenic reaction that yields the formation of an insoluble precipitate in the vicinity of cytochrome oxidase activity, thus revealing both the location of the mitochondria and the relative metabolic activity of cytochrome oxidase (Wong-Riley, 1979). Sections were incubated for 2.5 hr at 37°C in a Dubnoff metabolic shaker incubator. After the reaction, sections were rinsed in phosphate buffer, mounted on glass slides, and coverslipped.
Statistical analysis. To quantify changes in the expression of Fas death receptor protein, mitochondrial apoptosis-regulating proteins, COX1 protein, and caspase 8 and 3 active fragments, protein expression was corrected for loading differences and then expressed as a percent of control. In some blots, two different controls were used, and the average optical density of the two bands was used for percent control calculations. Mean and SD for protein expression in tissue obtained at similar time points were calculated, and ANOVA was used to compare differences in protein expression over time. Post hoc testing for differences in expression at 3, 6, and 24 hr was performed with Fisher's analysis. A p value of <0.05 was used to determine significance.
RESULTS
In the ipsilateral thalamus at 24 hr after neonatal hypoxia–ischemia, many degenerating neurons display light microscopic features of apoptosis (Fig.1B, arrows) (Northington et al., 2001). This contrasts with the normal morphology of thalamic neurons from sham controls (Fig. 1A).
Fig. 1.
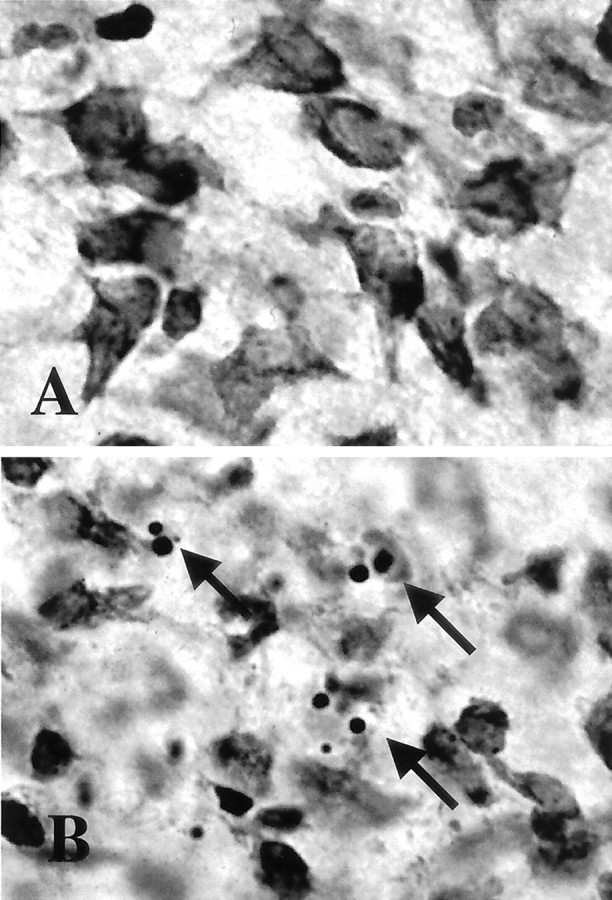
Thalamic neurons die by apoptosis after neonatal hypoxia–ischemia. A, B, Compared with sham controls (A), neurons at 24 hr after hypoxia–ischemia (B) contain many apoptotic profiles as seen in cresyl violet-stained sections of the ventral basal thalamus (arrows).
Fas receptor expression is induced in thalamus rapidly after neonatal hypoxia–ischemia
By immunoblotting, Fas death receptor protein expression is increased in the diencephalon after neonatal hypoxia–ischemia (Fig.2). In membrane protein fractions of newborn rat thalamus, Fas was detected at 45 kDa, corresponding to the known molecular weight of Fas death receptor protein in Jurkat cells. Jurkat cells were chosen as a positive control because they are known to express high levels of Fas death receptor (Felderhoff-Mueser et al., 2000). A comparison of samples from sham-operated controls and p7 rat pups at 3, 6, and 24 hr after hypoxia–ischemia revealed a significant (p < 0.05) increase in Fas levels after hypoxia–ischemia (Fig. 2) in the rat thalamus. This increase occurred as early as 3 hr after hypoxia–ischemia. The increase in Fas death receptor was not caused by a generalized increase in similar cytokine receptor expression because expression of the closely related TNF receptor 1 did not change in response to neonatal hypoxia–ischemia (Fig. 2).
Fig. 2.
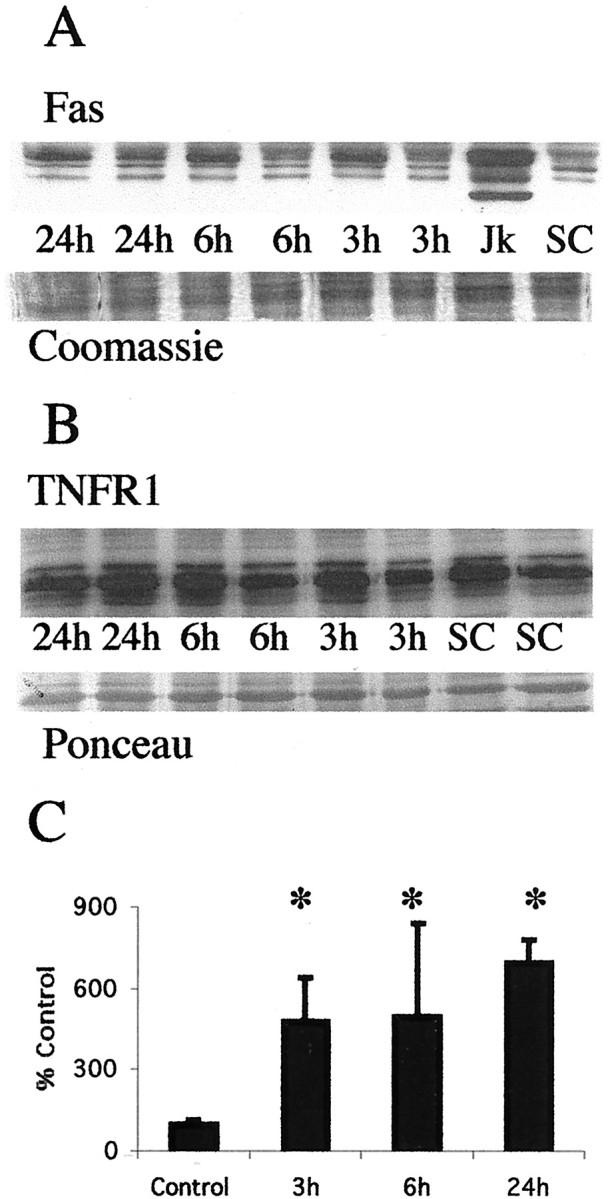
Fas death receptor protein levels increase in the thalamus after neonatal hypoxia–ischemia. A, Top, Immunoblot showing increased Fas death receptor protein in membrane fractions from thalamic homogenates obtained 3, 6, and 24 hr after neonatal hypoxia–ischemia compared with noninjured control samples. Jurkat cell lysates were used as a positive control, because they express high levels of Fas as detected at 45 kDa. The corresponding 45 kDa band in controls and injured thalamic samples was used for quantification. Bottom, The corresponding Coomassie-stained gel. B, Top, Immunoblot showing no change in TNFR1 death receptor protein in membrane fractions from thalamic homogenates obtained 3, 6, and 24 hr after neonatal hypoxia–ischemia compared with noninjured control samples.Bottom, The corresponding Ponceau-stained blot. ForA and B each lanerepresents a pooled sample of thalamus from three animals at the indicated time point (i.e., sham control and 3, 6, or 24 hr after hypoxia–ischemia). C, Graph representing changes in Fas death receptor protein levels in the thalamus over time after neonatal hypoxia–ischemia. Results are shown as the mean ± SD of four to five pooled samples per time point (*p < 0.05 compared with control). Jk, Jurkat cell lysates;SC, sham control.
Caspase 8 is cleaved to its active subunits after neonatal hypoxia–ischemia
Procaspase 8 undergoes cleavage and activation in cell culture model systems of apoptosis (Dragovich et al., 1998; Nagata, 1999a,b). Procaspase 8 (54–55 kDa) is cleaved to its 30 and 18 kDa subunits in soluble protein from ipsilateral thalamus after neonatal hypoxia–ischemia (Fig. 3). The levels of procaspase 8 in soluble fractions decrease as the expression of cleaved subunits increases after hypoxia–ischemia (Fig. 3). Jurkat cells, which are highly sensitive to Fas-mediated apoptosis, express both procaspase 8 and the active subunits (Fig. 3).
Fig. 3.
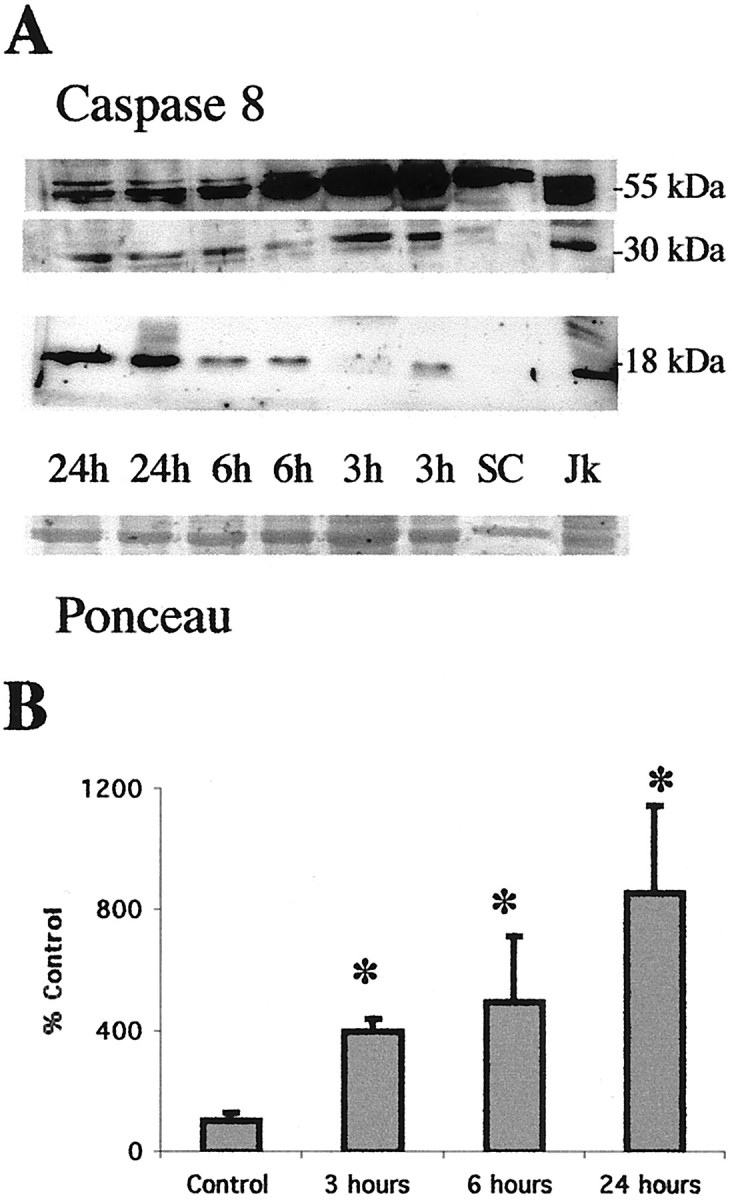
Procaspase 8 is cleaved to active fragments in the thalamus after neonatal hypoxia–ischemia. A, Immunoblot showing that procaspase 8 (54–55 kDa) levels decrease concurrently with an increase in the levels of the 30 and 18 kDa active fragments of caspase 8 in cytosolic fractions from the thalamus at 3, 6, and 24 hr after neonatal hypoxia–ischemia. There is a progressive increase in expression of the active subunits during the first 24 hr after hypoxia–ischemia compared with controls (SC). Anti-caspase 8 antibody also reacts strongly with a 55, 30, and 18 kDa protein in Jk cells consistent with the proenzyme and active fragment forms of caspase 8. Bottom, The corresponding Ponceau-stained blot. Each lane represents a pooled sample of thalamus from three animals at the indicated time point (i.e., sham control and 3, 6, or 24 hr after hypoxia–ischemia).B, Graph representing changes in abundance of the 18 kDa active subunit of caspase 8 in the thalamus over time after neonatal hypoxia–ischemia. After correcting for protein-loading differences and comparing with control, results are shown as the mean ± SD of four to five pooled samples per time point (*p < 0.05 compared with control).
A potential target of Fas- and caspase 8-mediated apoptosis is the Bcl-2 family protein Bid that has been shown to cause cytochrome c release and subsequent apoptosis (Li et al., 1998). We found no evidence of Bid cleavage in either soluble or mitochondrial-enriched fractions (data not shown).
Ratio of mitochondrial apoptosis-regulating proteins is altered rapidly after hypoxia–ischemia in the thalamus before the appearance of prominent apoptosis
After neonatal hypoxia–ischemia, levels of the proapoptosis protein Bax rapidly increase in mitochondrial-enriched fractions of thalamus (Fig. 4A). Additionally, at 24 hr there is faint immunoreactivity of an 18 kDa Bax band (Fig. 4A) consistent with a cleaved fragment of Bax reported also to have potent proapoptosis activity (Wood and Newcomb, 2000). This increase in Bax protein, within the mitochondrial fraction, occurs without a concomitant change in the level of the antiapoptosis Bcl-2 protein in the mitochondrial fraction within the same time period (Fig. 4B). The increase in expression of Bax and stable expression of Bcl-2 proteins result in a marked shift in protein abundance in favor of proapoptosis Bax (Fig.4C). A significant change in the relative abundance of Bax and Bcl-2 is evident as early as 3 hr after neonatal hypoxia–ischemia.
Fig. 4.
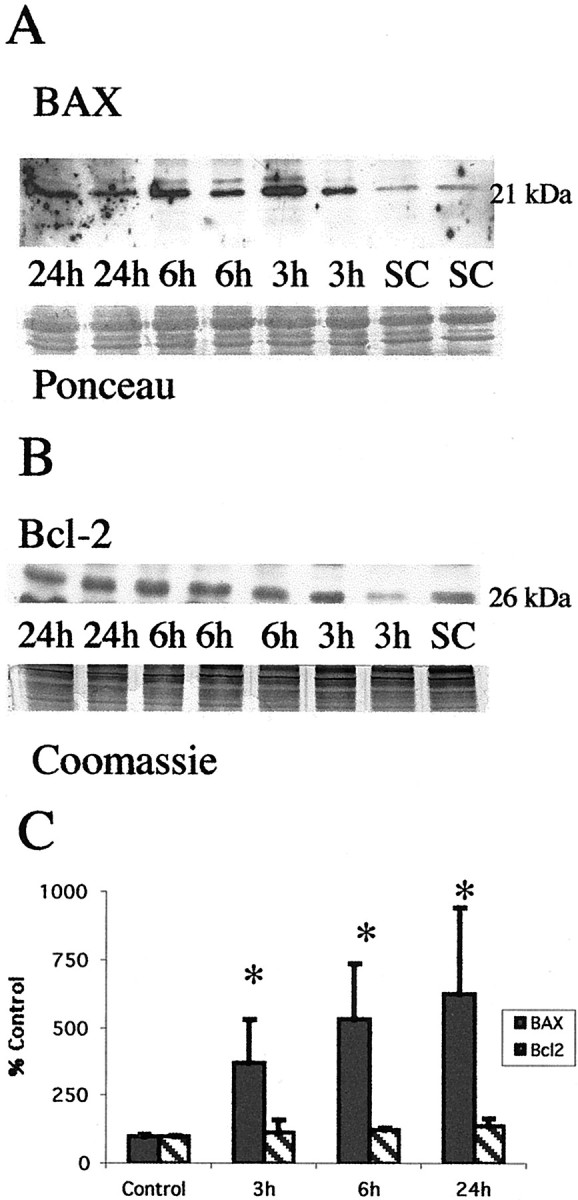
Neonatal hypoxia–ischemia causes an elevation in proapoptosis Bax protein levels in the mitochondrial fraction but does not alter levels of antiapoptosis Bcl-2. A, Top, Immunoblot shows increased Bax protein levels in mitochondrial membrane fractions from the thalamus from homogenates obtained 3, 6, and 24 hr after neonatal hypoxia–ischemia compared with noninjured control samples (SC). Anti-Bax antibody recognizes the expected 21 kDa band corresponding to Bax protein. Bottom, The corresponding Ponceau-stained blot is shown. Each lanerepresents a pooled sample of thalamus from three animals at the indicated time point (i.e., sham control and 3, 6, or 24 hr after hypoxia–ischemia). B, In comparison Bcl-2 protein expression is not changed in mitochondrial membrane fractions from thalamus homogenates obtained 3, 6, and 24 hr after neonatal hypoxia–ischemia compared with noninjured control samples (SC). Anti-Bcl-2 antibody recognizes the expected 26 kDa band corresponding to Bcl-2 protein. Bottom, The corresponding Coomassie-stained gel is shown. C, Graph represents alteration in relative amounts of Bax and Bcl-2 protein in the mitochondrial membrane fraction in the thalamus over time after neonatal hypoxia–ischemia. By 3 hr, there is a significant shift in the Bax/Bcl-2 ratio favoring proapoptosis Bax. After correcting for protein-loading differences and comparing with control, results are shown as the mean ± SD of four to five pooled samples per time point (*p < 0.05 compared with control).
Cytochrome c rapidly accumulates in the soluble fraction of thalamus after neonatal hypoxia–ischemia
Cytochrome c levels increase in the soluble fraction as early as 3 hr after neonatal hypoxia–ischemia (Fig.5A). This increase is coincident with the appearance of cleaved caspase-8 and before the increase in expression of the 12 kDa subunit of caspase 3. The initial increase in the amount of cytochrome c in the soluble fraction at 3 hr is sustained at 6 and 24 hr after neonatal hypoxia–ischemia (Fig.5B).
Fig. 5.

Cytochrome c accumulates in the soluble fraction in the thalamus after neonatal hypoxia–ischemia. A, Top, Immunoblot showing increased cytochrome c protein in soluble fractions from thalamic homogenates obtained 3, 6, and 24 hr after neonatal hypoxia–ischemia compared with noninjured control samples (SC). Bottom, The corresponding Ponceau-stained blot. Each lane represents a pooled sample of thalamus from three animals at the indicated time point (i.e., sham control and 3, 6, or 24 hr after hypoxia–ischemia).B, Graph showing the accumulation of cytochrome c in the thalamus over time after neonatal hypoxia–ischemia. After correcting for protein-loading differences and comparing with control, results are shown as the mean ± SD of four to five pooled samples per time point (*p < 0.05 compared with control).
Caspase-3 is cleaved into its active subunits at 24 hr after neonatal hypoxia–ischemia in the thalamus
Caspases 3 and 8 are primary targets of death receptor-mediated apoptosis cascades (Nagata, 1999b; Yamada et al., 1999). When cleaved from its 32 kDa proenzyme form, the active 12, 17, and 20 kDa fragments have a central role in activating DNA fragmentation and other irreversible steps in apoptosis (Du et al., 1997). Caspase 3 is cleaved to its active forms in the thalamus after neonatal hypoxia–ischemia (Fig. 6A). Interestingly this activation is found only at 24 hr after hypoxia–ischemia (Fig. 6B). This result was confirmed using both the Santa Cruz Biotechnology anti-caspase 3 antibody that recognizes procaspase 3 and the active fragments and an antibody against the 17 kDa cleaved caspase 3 fragment.
Fig. 6.
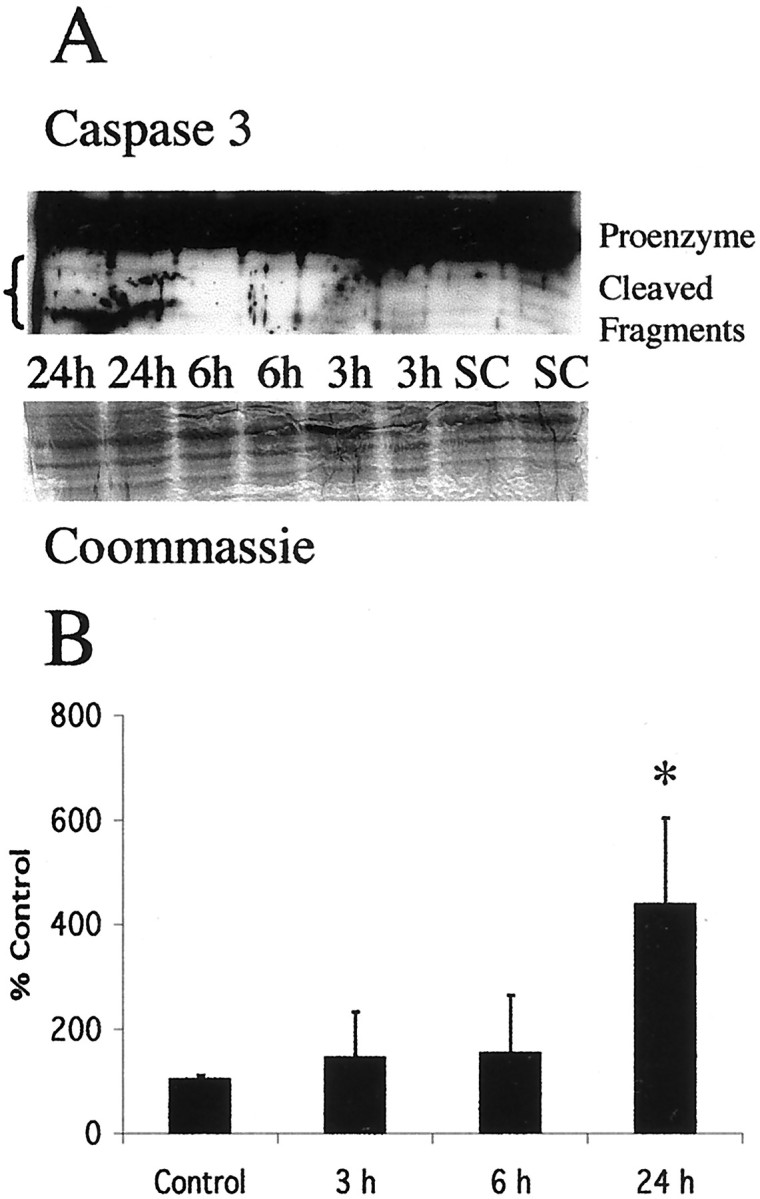
Cleavage of procaspase 3 to active fragments occurs at 24 hr after neonatal hypoxia–ischemia. A, Top, Immunoblot shows levels of 32 kDa procaspase 3 and its lower molecular weight cleavage products in thalamus cytosolic fractions from homogenates obtained 3, 6, and 24 hr after neonatal hypoxia–ischemia compared with noninjured control samples (SC). Bottom, The corresponding Coomassie-stained blot is shown. Each lane represents a pooled sample of thalamus from three animals at the indicated time point (i.e., sham control and 3, 6, or 24 hr after hypoxia–ischemia).B, Graph represents change in expression of the12 kDa cleavage product in the thalamus over time after neonatal hypoxia–ischemia. Not until 24 hr is there a significant increase in the expression of the 12 kDa fragment. After correcting for protein-loading differences and comparing with control, results are shown as the mean ± SD of four to five pooled samples per time point (*p < 0.05 compared with control).
Mitochondria accumulate at perinuclear locations in thalamic neurons after neonatal hypoxia–ischemia
Using a histochemical detection method for cytochrome oxidase activity to identify mitochondria, we found that neurons in the ventral basal thalamus, after neonatal hypoxia–ischemia, accumulate mitochondria (Fig. 7). These changes occur before the appearance of a significant number of apoptotic profiles at 24 hr (Northington et al., 2001). In normal neurons, cytochrome oxidase activity is evenly distributed throughout the cell soma in ventral basal thalamic neurons (Fig. 7A). In contrast, by 3–6 hr after neonatal hypoxia–ischemia, mitochondria within neurons in the ipsilateral ventral basal thalamus exhibit intense cytochrome oxidase activity and form prominent perinuclear aggregates of mitochondria (Fig. 7B). These changes are most prominent in neurons in the chromatolytic stage of early neuronal apoptosis (Fig. 7C, cytochrome oxidase histochemistry and cresyl violet counterstaining) according to a recently proposed staging scheme for apoptosis of neurons (Al-Abdulla and Martin, 1998;Al-Abdulla et al., 1998; Martin et al., 1999). In these cells, dense cytochrome oxidase-positive aggregates cluster adjacent to peripherally displaced nuclei. By 24 hr, cells in late phases of apoptosis with tightly condensed chromatin aggregates have lost or are losing immunoreactivity for cytochrome oxidase (Fig. 7D, cytochrome oxidase histochemistry and cresyl violet counterstaining)
Fig. 7.
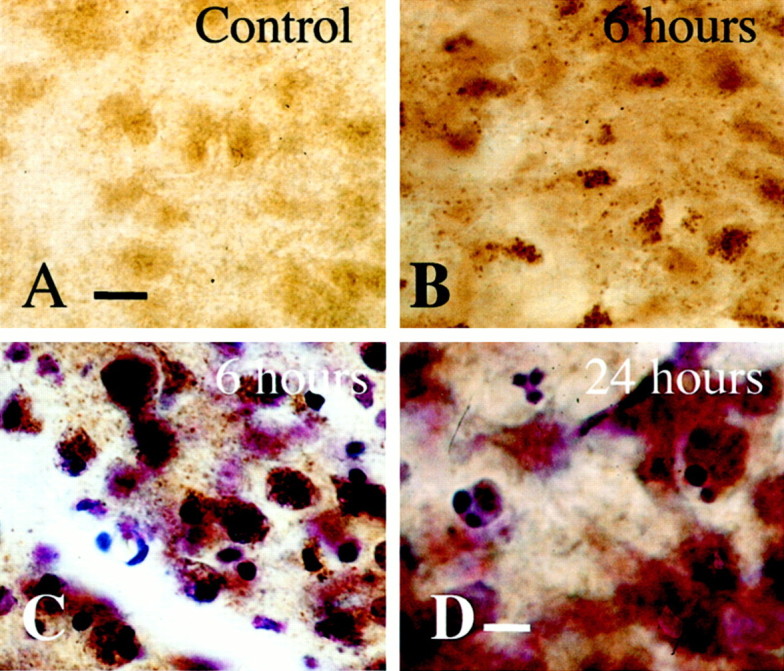
Mitochondria accumulate in a perinuclear location in ventral basal thalamic neurons after neonatal hypoxia–ischemia. Histochemistry for cytochrome oxidase activity in the thalamus after neonatal hypoxia–ischemia shows intense cytochrome oxidase activity in mitochondria and alteration in appearance and intracellular location of mitochondria after neonatal hypoxia–ischemia. At 6 hr, mitochondria are densely immunoreactive for cytochrome oxidase, assuming a punctate appearance and clustering near nuclei that are in the chromatolytic phase of apoptosis (B, C, cytochrome oxidase histochemistry and cytochrome oxidase histochemistry counterstained with cresyl violet, respectfully). By 24 hr (D), cytochrome oxidase immunoreactivity is dissipating in thalamic neurons undergoing late stages of apoptotic degeneration. Three cells with densely condensed chromatin are shown with variable but decreasing levels of cytochrome oxidase activity. Scale bars: A–C, 12 μm; D, 6 μm.
The accumulation of mitochondria in the thalamus after neonatal hypoxia–ischemia was confirmed indirectly by immunoblotting. By immunoblotting, levels of COX1 increased after neonatal hypoxia–ischemia (Fig. 8). Expression of this inner mitochondrial membrane protein increases in the thalamus (Fig. 8A). In the thalamus, a significant increase in expression of COX1 is evident at 6 hr after hypoxia–ischemia (Fig.8B) coincident with the peak of cytochrome oxidase activity in neurons in early phases of apoptotic neurodegeneration (Fig. 7C).
Fig. 8.

COX1 protein increases in thalamic mitochondrial protein fractions coincident with mitochondrial accumulation after neonatal hypoxia–ischemia. A, Top, Immunoblot shows an increase in COX1 protein in mitochondria-enriched membrane fractions from the thalamus from homogenates obtained 3, 6, and 24 hr after neonatal hypoxia–ischemia compared with noninjured control samples (SC). Bottom, The corresponding Ponceau-stained blot is shown. Each lane represents a pooled sample of thalamus from three animals at the indicated time point (i.e., sham control and 3, 6, or 24 hr after hypoxia–ischemia).B, Graph represents changes in COX1 protein levels in the thalamus over time after neonatal hypoxia–ischemia. The maximal increase at 6 hr corresponds nicely with the peak in mitochondrial accumulation seen at 6 hr in Figure 7C. After correcting for protein-loading differences and comparing with control, results are shown as the mean ± SD of four to five pooled samples per time point (*p < 0.05 compared with control).
DISCUSSION
We found previously that neuronal degeneration in the neonatal thalamus after hypoxia–ischemia is apoptosis (Northington et al., 2001). In this study, we explored the mechanisms and conclude that delayed neuronal degeneration in neonatal rat thalamus after hypoxia–ischemia has biochemical and molecular features of apoptosis. The findings that support this conclusion are (1) increased Fas death receptor levels, (2) an alteration in the levels of Bax and Bcl-2 favoring apoptosis-promoting Bax, (3) intracellular redistribution of active mitochondria, (4) accumulation of soluble cytochrome c, and (5) cleavage of caspases 8 and 3 to active forms. This cascade of biochemical and molecular events is consistent with Fas-mediated, mitochondrial-amplified, apoptosis as a major mechanism of delayed neurodegeneration after neonatal hypoxia–ischemia.
Thalamic damage after hypoxia–ischemia in newborns has long been recognized and is particularly important in infants with extrapyramidal cerebral palsy (Malamud, 1950; Volpe, 1995; Roland et al., 1998). Human neuroimaging and neuropathological studies have revealed that the thalamus is among the selectively vulnerable brain regions in the human newborn (Yokochi et al., 1991; Barkovich, 1995; Roland et al., 1998). Despite recognition of injury to the thalamus in children with cerebral palsy, the contribution of delayed injury and injury to nonforebrain regions to the development of cerebral palsy has been less well studied. The ventral basal thalamus contains afferent and efferent connections to the ipsilateral cortex and functions prominently in sensorimotor integration (Erzurumlu and Jhaveri, 1990). Injury to these circuits interrupts cortical sensory function and the integration of sensory information into volitional movement (White, 1989). Damage to motor relay nuclei in the thalamus may lead to movement disorders such as ataxia or dystonia (White, 1989) and when this injury occurs in the immature brain may contribute to the motor and sensory handicaps suffered by children with cerebral palsy.
In the model of neonatal brain injury used in these studies, the ipsilateral forebrain is the most vulnerable region and clearly represents the ischemic core of the lesion (Rice et al., 1981). However, the thalamus is also damaged in this and most other models of neonatal brain injury (present data) (Myers, 1975; Towfighi et al., 1991, 1995; Martin et al., 1997a; Northington et al., 2001). We postulated that this thalamic injury is apoptosis. We identified increased Fas death receptor protein levels in the ventral basal thalamus after neonatal hypoxia–ischemia. This observation suggests an additional link between cytokine- and hypoxia–ischemia-mediated neonatal brain injury. Previous studies have demonstrated activation of interleukin (IL) (Szaflarski et al., 1995) neuroprotection with cytokine antagonists and in the absence of IL-1β-converting enzyme (Liu et al., 1996, 1999). The mechanism of hypoxia–ischemia-induced, cytokine-mediated cell death in the developing brain has not been clearly defined. The present study demonstrates that hypoxia–ischemia induces a cytokine death receptor that functions in apoptosis (Nagata, 1999b). Although we have not shown directly that Fas is induced in thalamic neurons, the rapid induction (by 3 hr) and the sustained elevation over the relevant time preceding thalamic neuronal apoptosis make Fas death receptor induction unlikely to be the result of inflammation.
An elevation in Fas death receptor is a link to multiple pathways of cell death. Fas is a member of the TNFR family. Oligomerization of Fas death receptor and recruitment of FADD/MORT1 and procaspase 8 create the death-inducing signal complex (DISC). DISC is a potent apoptosis stimulus in multiple cell culture models. When procaspase 8 is bound by the DISC, autocleavage to its active fragments occurs. Cleaved caspase 8 then acts directly and indirectly to cleave caspase 3, depending on cell type and injury stimulus (Dragovich et al., 1998; Nagata, 1999a,b). Cleavage of caspase 3 is generally considered one of the irreversible steps immediately responsible for the execution of apoptosis in which the cell develops the morphological features recognizable as late-stage apoptosis (Petit et al., 1996). Our data demonstrate early Fas death receptor elevation and early and progressive cleavage of caspase 8 to its active form, followed by delayed caspase 3 cleavage just at the time of appearance of significant apoptosis in the thalamus after neonatal hypoxia–ischemia. This change is selective for Fas because TNFR1 was not changed. We have not yet shown activation of caspase 8 and caspase 3 specifically in thalamic neurons because of the lack of specific reagents. Fas death receptor protein and downstream caspase 3 cleavage likely bracket an important signaling cascade for apoptotic neurodegeneration in the thalamus.
Direct caspase 8 cleavage of caspase 3 is the original model of Fas-mediated apoptosis signaling (Dragovich et al., 1998); however mitochondria and mitochondrial apoptosis-regulating proteins of thebcl-2 family are now known to amplify cell death signals greatly (Susin et al., 1997). Bid, a cytosolic protein with homology to the BH3 domain of the Bcl-2 family, is cleaved in several models of Fas-mediated apoptosis and causes cytochrome c release from the mitochondria (Susin et al., 1997; Nagata, 1999a). We did not find evidence of Bid cleavage in the present model. However, we did find cytochrome c accumulation in the soluble fraction. Most of thebcl-2 family of proteins that regulate the rate of programmed cell death are normally present within mitochondrial membranes. Several in vivo and in vitro systems have shown the ratio of the proapoptosis protein Bax and the antiapoptosis proteins Bcl-2 and Bcl-x(l) to be critical in determining cell survival (Krajewski et al., 1995; Vekrellis et al., 1997; Isenmann et al., 1998; Antonawich et al., 1999; Martin, 1999; Shimizu et al., 1999; Almeida et al., 2000).
We find a marked alteration in the balance of proapoptosis and antiapoptosis bcl-2 proteins in the P2 fraction of thalamus after hypoxia–ischemia. This accumulation of Bax in the mitochondrial-enriched fraction occurs before caspase 3 cleavage and the appearance of large numbers of apoptotic profiles (present data) (Northington et al., 2001). A fourfold increase in the amount of proapoptosis protein Bax in the mitochondrial-enriched fraction was detected by 3 hr after injury, whereas the amount of Bcl-2 in the mitochondria is not changed during the first 24 hr after hypoxia–ischemia. These changes in the relative ratio of Bax to Bcl-2 favors the formation of Bax homodimers, the configuration in which Bax exerts its proapoptosis activity (Gross et al., 1998). Bax normally exists as a cytosolic protein and is translocated to the mitochondria after ligation of Fas receptor. This translocation is inhibited by Bcl-2 (Murphy et al., 2000). The present data suggest active mitochondrial translocation of Bax greatly in excess of the steady-state amount of Bcl-2. This is consistent with a strong death signal and may participate in mitochondrial amplification of Fas-mediated apoptosis. Cleavage of Bax to an 18 kDa isoform also enhances its cell death potency (Wood and Newcomb, 2000). Although neither antibody used in the present study was designed to detect the 18 kDa isoform, there is weak expression of an 18 kDa Bax band in the diencephalon at 24 hr after neonatal hypoxia–ischemia. The late appearance of this isoform in mitochondrial-enriched fractions is consistent with published reports of calpain-mediated Bax cleavage after translocation of Bax to the mitochondria (Wood and Newcomb, 1999).
Release of cytochrome c and changes in mitochondrial morphology and membrane potential and function precede caspase 3 activation inin vitro model systems (Zamzami et al., 1996; Vander Heiden et al., 1997). Cytochrome c has been identified as APAF2, and cytochrome c release is the immediate upstream event preceding caspase 3 cleavage and the execution phase of apoptosis (Susin et al., 1999). Our data show that cytochrome c accumulates in the soluble fraction of thalamus, in concert with caspase 8 cleavage and an altered Bax/Bcl-2 ratio and before the appearance of large amounts of caspase 3 cleavage. In axotomy models of neuronal apoptosis, mitochondrial trafficking is altered with perinuclear accumulation of mitochondria in neurons as they undergo apoptosis (Al-Abdulla and Martin, 1998; Martin et al., 1999), a finding very similar to that of the present study. These previous in vitro and in vivo observations support our conclusions described here. Within 3–6 hr after neonatal hypoxia–ischemia, neurons in the ventral basal thalamus exhibit marked accumulation of mitochondria (as revealed by intense cytochrome oxidase activity) and simultaneously display altered morphology. The mitochondria assume a prominent punctate appearance and concentrate in a perinuclear location. As thalamic neurons enter late stages of apoptosis, the cytoplasm becomes progressively devoid of COX activity. These data are consistent with immunoblotting for cytochrome oxidase subunit 1 protein, which shows maximal levels at 6 hr after neonatal hypoxia–ischemia in the thalamus. Taken together these data provide evidence of the participation of mitochondria in thalamic neurodegeneration after neonatal hypoxia–ischemia.
In summary, these experiments provide the first evidence of the presence of crucial components of Fas-mediated, mitochondrial-amplified, apoptosis in the thalamus after neonatal brain injury. These findings are important for the further understanding of the mechanisms of neuronal apoptosis in the immature brain. Eventually, these studies may be important for development of appropriately timed and targeted therapies for the protection of the neonatal brain and rescue of neurons after hypoxic–ischemic insults.
Footnotes
These studies were supported by Johns Hopkins Internal Research Grant funds (F.J.N.), a United Cerebral Palsy Grant (F.J.N.), United States Army Department of Defense Grant DAMD17-99-1-9553, and National Institutes of Health Grants AG 16282 (L.J.M.) and NS 35902 (D.M.F.). We gratefully acknowledge the expert technical assistance of Ann Sheldon, Ann Price, and George Kuck III.
Correspondence should be addressed to Dr. Frances J. Northington, Eudowood Neonatal Pulmonary Division, Department of Pediatrics, CMSC 210, Johns Hopkins Hospital, 600 North Wolfe Street, Baltimore, MD 21287. E-mail: fnorthin@welchlink.welch.jhu.edu.
REFERENCES
- 1.Al-Abdulla NA, Martin LJ. Apoptosis of retrogradely degenerating neurons occurs in association with the accumulation of perkaryal mitochondria and oxidative damage to the nucleus. Am J Pathol. 1998;153:447–456. doi: 10.1016/S0002-9440(10)65588-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Abdulla NA, Portera-Cailliau C, Martin LJ. Occipital cortex ablation in adult rat causes retrograde neuronal death in the lateral geniculate nucleus that resembles apoptosis. Neuroscience. 1998;86:191–209. doi: 10.1016/s0306-4522(98)00014-1. [DOI] [PubMed] [Google Scholar]
- 3.Almeida OF, Conde GL, Crochemore C, Demeneix BA, Fischer D, Hassan AH, Meyer M, Holsboer F, Michaelidis TM. Subtle shifts in the ratio between pro- and antiapoptotic molecules after activation of corticosteroid receptors decide neuronal fate. FASEB J. 2000;14:779–790. doi: 10.1096/fasebj.14.5.779. [DOI] [PubMed] [Google Scholar]
- 4.Antonawich FJ, Federoff HJ, Davis JN. BCL-2 transduction, using a herpes simplex virus amplicon, protects hippocampal neurons from transient global ischemia. Exp Neurol. 1999;156:130–137. doi: 10.1006/exnr.1998.7004. [DOI] [PubMed] [Google Scholar]
- 5.Back SA, Li Y, Gan X, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barkovich A. Profound asphyxia in the premature infant: image findings. AJNR Am J Neuroradiol. 1995;95:1837–1846. [PMC free article] [PubMed] [Google Scholar]
- 7.Barks JD, Silverstein FS. Excitatory amino acids contribute to the pathogenesis of perinatal hypoxic-ischemic brain injury. Brain Pathol. 1992;2:235–243. doi: 10.1111/j.1750-3639.1992.tb00697.x. [DOI] [PubMed] [Google Scholar]
- 8.Bona E, Johansson BB, Hagberg H. Sensorimotor function and neuropathology five to six weeks after hypoxia-ischemia in seven-day-old rats. Pediatr Res. 1997;42:678–683. doi: 10.1203/00006450-199711000-00021. [DOI] [PubMed] [Google Scholar]
- 9.Cheng Y, Deshmukh M, D'Costa A, Demaro JA, Gidday JM, Shah A, Sun Y, Jacquin MF, Johnson EM, Jr, Holtzman DM. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J Clin Invest. 1998;101:1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dragovich T, Rudin CM, Thompson CB. Signal transduction pathways that regulate cell survival and cell death. Oncogene. 1998;17:3207–3213. doi: 10.1038/sj.onc.1202587. [DOI] [PubMed] [Google Scholar]
- 11.Du Y, Bales KR, Dodel RC, Hamilton-Byrd E, Horn JW, Czilli DL, Simmons LK, Ni B, Paul SM. Activation of a caspase 3-related cysteine protease is required for glutamate-mediated apoptosis of cultured cerebellar granule neurons. Proc Natl Acad Sci USA. 1997;94:11657–11662. doi: 10.1073/pnas.94.21.11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erzurumlu RS, Jhaveri S. Thalamic axons confer a blueprint of the sensory periphery onto the developing rat somatosensory cortex. Brain Res Dev Brain Res. 1990;56:229–234. doi: 10.1016/0165-3806(90)90087-f. [DOI] [PubMed] [Google Scholar]
- 13.Felderhoff-Mueser U, Taylor DL, Greenwood K, Kozma M, Stibenz D, Joashi UC, Edwards AD, Mehmet H. Fas/CD95/APO-1 can function as a death receptor for neuronal cells in vitro and in vivo and is upregulated following cerebral hypoxic-ischemic injury to the developing rat brain. Brain Pathol. 2000;10:17–29. doi: 10.1111/j.1750-3639.2000.tb00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 1998;17:3878–3885. doi: 10.1093/emboj/17.14.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hagan P, Barks JD, Yabut M, Davidson BL, Roessler B, Silverstein FS. Adenovirus-mediated over-expression of interleukin-1 receptor antagonist reduces susceptibility to excitotoxic brain injury in perinatal rats. Neuroscience. 1996;75:1033–1045. doi: 10.1016/0306-4522(96)00225-4. [DOI] [PubMed] [Google Scholar]
- 16. Isenmann S, Stoll G, Schroeter M, Krajewski S, Reed JC, Bahr M. Differential regulation of Bax, Bcl-2, and Bcl-X proteins in focal cortical ischemia in the rat. Brain Pathol 8 1998. 49 62, discussion 62–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krajewski S, Mai JK, Krajewska M, Sikorska M, Mossakowski MJ, Reed JC. Upregulation of bax protein levels in neurons following cerebral ischemia. J Neurosci. 1995;15:6364–6376. doi: 10.1523/JNEUROSCI.15-10-06364.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levine S. Anoxic-ischemic encephalopathy in rats. Am J Pathol. 1960;36:1–17. [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 20.Liu XH, Eun BL, Silverstein FS, Barks JD. The platelet-activating factor antagonist BN 52021 attenuates hypoxic-ischemic brain injury in the immature rat. Pediatr Res. 1996;40:797–803. doi: 10.1203/00006450-199612000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Liu XH, Kwon D, Schielke GP, Yang GY, Silverstein FS, Barks JD. Mice deficient in interleukin-1 converting enzyme are resistant to neonatal hypoxic-ischemic brain damage. J Cereb Blood Flow Metab. 1999;19:1099–1108. doi: 10.1097/00004647-199910000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Malamud N. Status marmoratus: a form of birth injury following either birth injury or inflammation of the central nervous system. J Pediatr. 1950;37:610. doi: 10.1016/s0022-3476(50)80272-x. [DOI] [PubMed] [Google Scholar]
- 23.Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. 1999;58:459–471. doi: 10.1097/00005072-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 24.Martin LJ, Brambrink AM, Koehler RC, Traystman RJ. Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia-ischemia. J Comp Neurol. 1997a;377:262–285. doi: 10.1002/(sici)1096-9861(19970113)377:2<262::aid-cne8>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 25.Martin LJ, Brambrink AM, Lehmann C, Portera-Cailliau C, Koehler RC, Rothstein J, Traystman RJ. Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann Neurol. 1997b;42:335–348. doi: 10.1002/ana.410420310. [DOI] [PubMed] [Google Scholar]
- 26.Martin LJ, Kaiser A, Price AC. Motor neuron degeneration after sciatic nerve avulsion in adult rat evolves with oxidative stress and is apoptosis. J Neurobiol. 1999;40:185–201. [PubMed] [Google Scholar]
- 27.Martin LJ, Price AC, Kaiser A, Shaikh AY, Liu Z. Mechanisms for neuronal degeneration in amyotrophic lateral sclerosis and in models of motor neuron death (review). Int J Mol Med. 2000;5:3–13. doi: 10.3892/ijmm.5.1.3. [DOI] [PubMed] [Google Scholar]
- 28.McDonald JW, Silverstein FS, Johnston MV. Neurotoxicity of N-methyl-d-aspartate is markedly enhanced in developing rat central nervous system. Brain Res. 1988;459:200–203. doi: 10.1016/0006-8993(88)90306-x. [DOI] [PubMed] [Google Scholar]
- 29.Murphy KM, Streips UN, Lock RB. Bcl-2 inhibits a Fas-induced conformational change in the Bax N-terminus and Bax mitochondrial translocation. J Biol Chem. 2000;275:17255–17258. doi: 10.1074/jbc.C900590199. [DOI] [PubMed] [Google Scholar]
- 30.Myers RE. Four patterns of perinatal brain damage and their conditions of occurrence in primates. Adv Neurol. 1975;10:223–234. [PubMed] [Google Scholar]
- 31.Nagata S. Biddable death. Nat Cell Biol. 1999a;1:E143–E145. doi: 10.1038/14094. [DOI] [PubMed] [Google Scholar]
- 32.Nagata S. Fas ligand-induced apoptosis. Annu Rev Genet. 1999b;33:29–55. doi: 10.1146/annurev.genet.33.1.29. [DOI] [PubMed] [Google Scholar]
- 33.Nelson C, Silverstein FS. Acute disruption of cytochrome oxidase activity in brain in a perinatal rat stroke model. Pediatr Res. 1994;36:12–19. doi: 10.1203/00006450-199407001-00003. [DOI] [PubMed] [Google Scholar]
- 34.Northington F, Ferriero D, Graham E, Traystman R, Martin L (2001) Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol Dis, in press. [DOI] [PubMed]
- 35.Petit PX, Susin SA, Zamzami N, Mignotte B, Kroemer G. Mitochondria and programmed cell death: back to the future. FEBS Lett. 1996;396:7–13. doi: 10.1016/0014-5793(96)00988-x. [DOI] [PubMed] [Google Scholar]
- 36.Rice JE, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- 37.Roland EH, Poskitt K, Rodriguez E, Lupton BA, Hill A. Perinatal hypoxic-ischemic thalamic injury: clinical features and neuroimaging [see comments]. Ann Neurol. 1998;44:161–166. doi: 10.1002/ana.410440205. [DOI] [PubMed] [Google Scholar]
- 38.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC [see comments]. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 39.Silverstein FS. Can inhibition of apoptosis rescue ischemic brain? J Clin Invest. 1998;101:1809–1810. doi: 10.1172/JCI3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Susin SA, Zamzami N, Castedo M, Daugas E, Wang HG, Geley S, Fassy F, Reed JC, Kroemer G. The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J Exp Med. 1997;186:25–37. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Brenner C, Larochette N, Prevost MC, Alzari PM, Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J Exp Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szaflarski J, Burtrum D, Silverstein FS. Cerebral hypoxia-ischemia stimulates cytokine gene expression in perinatal rats. Stoke. 1995;26:1093–1100. doi: 10.1161/01.str.26.6.1093. [DOI] [PubMed] [Google Scholar]
- 43.Towfighi J, Yager JY, Housman C, Vannucci RC. Neuropathology of remote hypoxic-ischemic damage in the immature rat. Acta Neuropathol. 1991;81:578–587. doi: 10.1007/BF00310141. [DOI] [PubMed] [Google Scholar]
- 44.Towfighi J, Zec N, Yager J, Housman C, Vannucci RC. Temporal evolution of neuropathologic changes in an immature rat model of cerebral hypoxia: a light microscopic study. Acta Neuropathol. 1995;90:375–386. doi: 10.1007/BF00315011. [DOI] [PubMed] [Google Scholar]
- 45.Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria [see comments]. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- 46.Vekrellis K, McCarthy MJ, Watson A, Whitfield J, Rubin LL, Ham J. Bax promotes neuronal cell death and is downregulated during the development of the nervous system. Development. 1997;124:1239–1249. doi: 10.1242/dev.124.6.1239. [DOI] [PubMed] [Google Scholar]
- 47.Volpe JJ. Neurology of the newborn, pp 314–369. Saunders; Philadelphia: 1995. [Google Scholar]
- 48.White E. Cortical circuits: synaptic organization of the cereb cortex: structure, function, and theory. Birkhauser; Boston: 1989. [Google Scholar]
- 49.Wong-Riley M. Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res. 1979;171:11–28. doi: 10.1016/0006-8993(79)90728-5. [DOI] [PubMed] [Google Scholar]
- 50.Wong-Riley MT. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]
- 51.Wood D, Newcomb EW. Cleavage of bax enhances its cell death function. Exp Cell Res. 2000;256:375–382. doi: 10.1006/excr.2000.4859. [DOI] [PubMed] [Google Scholar]
- 52.Wood DE, Newcomb EW. Caspase-dependent activation of calpain during drug-induced apoptosis. J Biol Chem. 1999;274:8309–8315. doi: 10.1074/jbc.274.12.8309. [DOI] [PubMed] [Google Scholar]
- 53.Yamada H, Tada-Oikawa S, Uchida A, Kawanishi S. TRAIL causes cleavage of bid by caspase-8 and loss of mitochondrial membrane potential resulting in apoptosis in BJAB cells. Biochem Biophys Res Commun. 1999;265:130–133. doi: 10.1006/bbrc.1999.1641. [DOI] [PubMed] [Google Scholar]
- 54.Yokochi K, Aiba K, Kodama M, Fujimoto S. Magnetic resonance imaging in athetotic cerebral palsied children. Acta Paediatr Scand. 1991;80:818–823. doi: 10.1111/j.1651-2227.1991.tb11955.x. [DOI] [PubMed] [Google Scholar]
- 55.Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis [see comments]. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]