

Abstract
Transmission at the end-bulb synapse formed by auditory nerve terminals onto the soma of neurons in the avian nucleus magnocellularis is characterized by high transmitter release probability and strong synaptic depression. Activation of presynaptic GABABreceptors minimizes depression at this synapse and significantly enhances synaptic strength during high-frequency activity. Here we investigate synaptic mechanisms underlying this phenomenon. EPSC amplitudes evoked by 200 Hz trains increased more than twofold when release probability was reduced with Cd2+ or baclofen. This effect was not exhibited by a transmitter depletion model of presynaptic depression, which predicts that EPSC amplitudes reach a common steady-state amplitude during high-frequency trains, despite alterations of initial release probability. However, an additional source of postsynaptic depression was sufficient to explain our findings. Aniracetam, a modulator of AMPA receptors that reduces desensitization, decreased the amount of synaptic depression during trains, indicating that desensitization occurred during trains of stimuli. However, this effect of aniracetam was absent when release probability was lowered with baclofen or Cd2+. No effect of aniracetam on the NMDA component of the EPSC was seen, confirming a postsynaptic site of action of aniracetam. When desensitization was reduced with aniracetam, steady-state EPSC amplitudes during trains were found to converge over a wide range of release probabilities, as predicted by the depletion model. Additional evidence of AMPA receptor desensitization was provided by direct measurement of quantal amplitudes immediately after stimulus trains. Thus, presynaptic modulation by GABAB receptors regulates the extent of AMPA receptor desensitization and controls synaptic strength, thereby modulating the flow of information at an auditory synapse.
Keywords: short-term depression, AMPA receptors, desensitization, cochlear nucleus, GABAB receptors, end-bulb synapse, auditory
Synaptic strength varies with the frequency of synaptic activity as a consequence of several forms of short- and long-term plasticity (Feng, 1940; Lundberg and Quilisch, 1953; DelCastillo and Katz, 1954; Kusano and Landau, 1975). Synaptic depression is the predominant form of short-term plasticity at synapses with high probability of transmitter release (Zucker, 1989) and has generally been attributed to depletion of a pool of readily releasable transmitter vesicles (Takeuchi, 1958; Thies, 1965; Betz, 1970; Dobrunz and Stevens, 1997; Wu and Borst, 1999). Depression is especially pronounced in large synapses of the auditory pathway. In the avian nucleus magnocellularis (nMag), action potentials in the auditory nerve evoked at low frequency (<1 Hz) generate large AMPA-mediated EPSCs (Zhou and Parks, 1992), resulting from release of 100–200 transmitter quanta onto soma of the postsynaptic nMag neurons (Zhang and Trussell, 1994a). However, mean firing rates of auditory nerve fibers in vivo range from 86 to 327 Hz (Warchol and Dallos, 1990; Salvi et al., 1992). At these frequencies, synaptic responses exhibit pronounced depression sufficient to reduce single-fiber EPSPs below action potential threshold, eventually interrupting the relay of timing information required for sound localization (Zhang and Trussell, 1994b; Brenowitz et al., 1998).
GABAB receptors located on end-bulb terminals of auditory nerve fibers modulate synaptic strength in nMag in a frequency-dependent manner. Activation of presynaptic GABAB receptors reduces glutamate release by 85% during low-frequency auditory nerve activity (Otis and Trussell, 1996). However, at high rates of auditory nerve activity (up to 500 Hz), GABAB receptor activation increases the steady-state amplitudes of synaptic responses up to fivefold relative to control, by lowering initial transmitter release and slowing onset of depression during stimulus trains (Brenowitz et al., 1998). Because the enhancement of synaptic strength by GABABreceptor activation allowed suprathreshold transmission to persist longer during high-frequency trains, this mechanism may play an important role in allowing faithful relaying of ongoing auditory stimuli. This finding was unexpected, because presynaptic depletion models of depression indicate that, during high-frequency stimulation, response amplitudes reach a steady state determined by the rates of transmitter release and vesicle recycling but not by the initial transmitter release probability (PR) (O'Donovan and Rinzel, 1997; Tsodyks and Markram, 1997). Thus, alterations in PR are not expected to affect steady-state EPSC amplitudes (EPSCSS) during high-frequency trains. Convergence of steady-state EPSC amplitudes evoked at high frequency, despite changes in PR, has been confirmed in cortical (Markram and Tsodyks, 1996; Abbott et al., 1997) and cerebellar (Kreitzer and Regehr, 2000) synapses.
Previous studies of nMag have characterized AMPA receptor desensitization to applied glutamate or to single synaptic stimuli (Trussell et al., 1993; Raman and Trussell, 1995a; Otis et al., 1996b). Here we describe a component of synaptic depression that persists during repetitive stimulation of the end-bulb synapse and was attributed to receptor desensitization. DecreasingPR by activation of presynaptic GABAB receptors or with Cd2+ reduced or eliminated the contribution of desensitization. After reduction of desensitization with aniracetam, lowering PR no longer caused enhancement of steady-state EPSC amplitudes during high-frequency trains. Instead, EPSCs reached the same steady-state amplitude despite large changes in release probability. These findings suggest that, during periods of high-frequency activity, synaptic depression was enhanced under high but not lowPR conditions. Thus, activation of presynaptic GABAB receptors may control synaptic strength by regulating the extent of AMPA receptor desensitization.
MATERIALS AND METHODS
Physiology. Brainstem slices (300 μm) were prepared from embryonic day 17–20 chicks (Zhang and Trussell, 1994a; Turecek and Trussell, 2000). During dissection, storage, and recording, slices were maintained in warmed, oxygenated saline containing (in mm): 140 NaCl, 20 glucose, 10 HEPES, 5 KCl, 3 CaCl2, and 1 MgCl2, pH 7.35. During recordings (34–37οC), slices were perfused at 3–5 ml/min. Neurons were viewed with a Zeiss(Oberkochen, Germany) Axioskop and Olympus Optical (Tokyo, Japan) 60× water immersion lens using differential interference contrast optics and infrared illumination. For measurement of AMPA-mediated EPSCs, saline was supplemented with (in μm): 100 dl-APV, 10 7-Cl-kynurenate, 10 SR-95531, and 2 strychnine. In other experiments, NMDA-mediated EPSCs were pharmacologically isolated by supplementing saline with (in μm): 20 GYKI-52466, 20 6,7-dinitro-7-quinoxaline-2,3-dione (DNQX), 20 SR-95531, 20 glycine, and 2 strychnine. Neurons were voltage clamped with an Axopatch 200A or 200B amplifier (Axon Instruments, Foster City, CA) at −30 mV (for recording AMPA receptor-mediated EPSCs), +50 mV (for recording NMDA receptor-mediated EPSCs), or −60 mV [for recording miniature synaptic currents (mEPSCs)]. Electrode series resistance (2–8 MΩ) was compensated 80–95%. Pipettes were filled with an intracellular solution containing (in mm): 125 CH3O3SCs (Cs-methanesulfonate), 15 CsCl, 10 HEPES, 5 BAPTA, and 1 MgCl2, pH 7.25. For measurement of NMDA responses, 2 Na2-ATP was added to the pipette solution. Synaptic responses were obtained by positioning a stimulus electrode (2–4 MΩ) onto nearby myelinated fibers 20–100 μm from the postsynaptic cell body. Individual afferent auditory nerve axons were stimulated by 100–200 μsec, 5–50 V pulses delivered via an isolated stimulus unit (Iso-flex; A.M.P.I., Jerusalem, Israel). Currents were filtered at 5–10 kHz and sampled at 20 kHz. Aniracetam stocks (0.5 m, 100×) were prepared in DMSO and added to extracellular solutions immediately before use. The final working concentration of aniracetam was 5 mm and aniracetam-containing solutions included 1% (v/v) DMSO. For all experiments using aniracetam, control extracellular solutions were also supplemented with 1% DMSO. Baclofen and Cd2+ were either added to extracellular solutions or pressure applied with a puffer pipette (2–4 μm tip diameter). Means are reported ± SE. Chemicals and drugs were obtained from Sigma (St. Louis, MO), Research Biochemicals (Natick, MA), and Tocris Cookson (Ballwin, MO).
mEPSC analysis. Frequency of spontaneous mEPSCs was enhanced by addition of SrCl2 (2–4 mm) to extracellular solutions. Whole-cell currents were digitally sampled on a second channel using a Cygnus (Medina, OH) FLA-01 signal conditioner to increase gain 10×. mEPSCs were detected using derivative or template detection algorithms implemented in Axograph software (Axon Instruments).
Modeling synaptic depression. For simulations of synaptic depression, the model consisted of a synapse withN0 release sites, each of which releases a vesicle with probability PRafter a presynaptic action potential. Immediately after release, sites become refractory and subsequently recover with a single-exponential time course, τD. This value is assumed to be Ca2+-independent. Before the first stimulus,
This notation was used by Weis et al. (1999), whereN1− refers to the releasable pool size immediately before stimulus 1, andN1+ refers to the releasable pool size after stimulus 1. Thus,
After recovery during the interval between the first and second stimuli,
where int is the interval between stimuli andtrec is the exponential time constant for the transition of release sites from a refractory to an active state. This model was iteratively calculated for stimulus trains of arbitrary lengths.
Desensitization is modeled using a coefficient, β, that scales quantal amplitudes uniformly at all release sites. The amount of desensitization induced by each EPSC is modeled as having an exponential relationship to the quantal content:
where β is the fraction of nondesensitized receptors,mi is the quantal content of EPSCi, and parameters A andB define the function relating release and desensitization.β recovers with a single-exponential time course between stimuli (τβ). Before the first stimulus,
Immediately after the first stimulus,
where m1 is the quantal content of the first EPSC. Before the second stimulus,
After the second stimulus, receptor availability is expressed as:
The value of β was calculated iteratively throughout the train. The amplitude of an EPSC on the ith stimulus of a train using the desensitization model is:
where q is the quantal amplitude. To obtain values for parameters in the model, simulations were compared with data in Figure 4B. First, the purely presynaptic depression model was fit to data obtained in aniracetam. This yielded values of PR = 0.65 and τrec = 75 msec. Parameters affecting desensitization were then determined by fitting the model to the control data in Figure 4B, yielding valuesA = 0.90, B = 1.5, and τβ = 100 msec.
Fig. 4.

Effect of aniracetam on synaptic depression.Ai, A single EPSC in 5 mm aniracetam (gray trace). Control peak (black trace) is indicated with an arrow. In this cell, aniracetam increased the peak amplitude by 60% (from −6.19 to −10.24 nA). Aii, A 200 Hz train in control (black trace) and aniracetam (gray trace) is shown. Traces have been normalized to the amplitude of the first peak in each train. Averages of five traces are shown. Bi, Normalized peak amplitudes during 200 Hz stimulus trains (control,black circles; aniracetam, gray circles). Data from 13 cells. Depression is significantly reduced by aniracetam for stimuli 2–10 of trains (p < 0.01; paired t test). Bii, Relative enhancement of EPSC amplitudes by aniracetam throughout the train, calculated as 100% · (EPSCANI − EPSCCON)/EPSCCON. C, Effect of aniracetam on synaptic depression in the continuous presence of 20 μm Cd2+. Ci, EPSC trains at 200 Hz in 20 μm Cd2+(black trace) and 20 μmCd2+ plus 5 mm aniracetam (gray trace). In this example, aniracetam increased the peak of the first EPSC by 55.8% (from 0.84 to 1.30 nA).Cii, Responses during trains in the presence of Cd2+ (black circles) and Cd2+ plus aniracetam (gray circles) were normalized to the first response amplitude and plotted versus stimulus number. No effect of aniracetam on synaptic depression was seen when PR was lowered with 20 μm Cd2+ (n = 12).
RESULTS
Enhancement of synaptic strength by reducing release probability
Stimulation of auditory nerve fibers at 200 Hz evoked large inward currents in nMag neurons voltage clamped at a holding potential of −30 mV. During stimulus trains, the average amplitude of the first EPSC was −8.11 ± 0.62 nA and depressed to 7.1 ± 2.2% of this value during trains of 10 stimuli (n = 6) (Fig.1A,B; see Fig. 4B). As shown previously (Brenowitz et al., 1998), activation of presynaptic GABAB receptors by bath application of baclofen (50 μm) reduced initial response amplitudes to 16 ± 3% of control (n = 6). However, depression during 200 Hz trains was minimal in baclofen, so that after two to three stimuli, absolute EPSC amplitudes in baclofen were enhanced 215 ± 35% relative to controls (n = 6) (Fig.1Aii,C, filled circles).
Fig. 1.

Enhancement of synaptic strength by lowering release probability. Ai, Trains of 10 EPSCs were evoked at 200 Hz under control conditions (bottom trace) or in the presence of 50 μm baclofen (top trace). The first peak in baclofen was reduced to 19.3% of control. Vm = −30 mV. Averages of 5–10 trials are shown. Stimulus artifacts have been removed.Aii, EPSCs 8–10 have been enlarged to illustrate the increase in EPSC amplitudes when initial release probability was reduced with baclofen (control, black trace; baclofen,gray trace). Bi, Same as inA, but in the presence of Cd2+ (20 μm) (top trace). Peak of EPSC1in Cd2+ was reduced to 20.1% of control.Bii, EPSCs 8–10 have been enlarged to illustrate enhancement of EPSCSS in the presence of Cd2+ (control, black trace; Cd2+, gray trace). C,Filled circles show amplitude ratio of EPSCs in baclofen to controls for each stimulus. As the control EPSCs depress during the train, this value increases. Average enhancement of EPSCs for stimuli 8–10 in this cell was 223%. Open circles show ratios of EPSCs in Cd2+ relative to controls. Average steady-state enhancement of EPSCs in this cell was 186%. Data inA–C are from two different neurons. D, Bar graph comparing results for Cd2+(n = 6) and baclofen (n = 6).Black bars show ratios of EPSC1 amplitude in 50 μm baclofen or 20 μmCd2+ relative to control (EPSC1, BAC (or Cd)/EPSC1, CON). Gray barsshow ratios of steady-state EPSC amplitude (average of EPSCs 8–10 of 200 Hz trains) in baclofen or Cd2+ relative to control (EPSCSS, BAC (or Cd)/EPSCSS, CON).
Because activation of presynaptic GABAB receptors with baclofen reduces Ca2+ currents (Bean, 1989; Dittman and Regehr, 1996; Wu and Saggau, 1997), we examined whether block of Ca2+ currents with Cd2+ could produce a similar enhancement of EPSCSS (average of EPSCs 8–10). Bath application of 20 μm Cd2+reduced amplitudes of single EPSCs to 14 ± 2% of controls, statistically indistinguishable from the effect of 50 μmbaclofen (Fig. 1B,D, black bars). Stimulus trains delivered at 200 Hz in the presence of 20 μm Cd2+ caused enhancement of EPSCs to 180 ± 10% relative to control (n = 6) (Fig. 1B–D, gray bars). With equivalent levels of block of the first EPSC, the enhancement of EPSCs by Cd2+ and baclofen during 200 Hz trains was similar, suggesting that effects of baclofen result solely from a reduction of the initial probability of transmitter release (Kreitzer and Regehr, 2000). When trains of 30–50 stimuli were delivered (n = 6; data not shown), EPSC enhancement persisted under conditions of loweredPR, indicating that this enhancement is not a transient phenomenon attributable to a failure of EPSC amplitudes to reach steady-state.
Synaptic strength is maximized at intermediate values of initial release probability
To explore further the effect of changing initialPR on EPSCSS, we delivered 200 Hz stimulus trains while varyingPR with Cd2+. A high concentration of Cd2+ (100 μm), sufficient to block transmission in nMag completely, was applied by bath perfusion or local pressure ejection near the cell. Cd2+ levels were varied by gradual reperfusion of control bath solution (containing 0 μm Cd2+) or by repositioning the puffer pipette various distances from the cell (Fig.2Ai). Trains of EPSCs were recorded at 15 sec intervals in concentrations of Cd2+ that ranged from 0 to 100 μm, producing a wide range of amplitudes of the first EPSC (EPSC1) in each train (Fig.2Ai). Enhancement of EPSCSS was seen to accompany reduction of PR(Fig. 2Aii). Figure 2B illustrates the relationship between EPSCSS andPR. Figure 2, A andB, shows an example in which maximal enhancement of EPSCSS was 411–422% when EPSC1 was reduced to between 7 and 12% of its control value. Further reductions ofPR caused EPSCSSto decline and approach zero, as expected with nearly complete block of Ca2+ channels. Similar observations were made in 11 neurons. Data were pooled from six neurons in which a large number of responses (25–135) were recorded (Fig. 2C). In this group, average enhancement of EPSCSS was 232% when EPSC1 was blocked with Cd2+ to between −1 and −2 nA. These experiments demonstrate that EPSCSS amplitudes were enhanced under conditions of reducedPR.
Fig. 2.

Relationship between EPSCSSand PR. Ai, Trains (200 Hz) of 10 stimuli were recorded in levels of Cd2+ranging from 0 to 100 μm. The first EPSC in each train is shown.Aii, Superimposed traces in high Cd2+(gray trace) and in 0 Cd2+(black trace). B, EPSCSS(averages of EPSCs 8–10) are plotted versus amplitude of the first EPSC for each train. C, Pooled results from six neurons. Data from each neuron were grouped in bins associated with EPSC1 values from 0–1, 1–2 nA, etc. Means and SEs were calculated for each bin. A linear fit to the data points (excluding the leftmost point associated with the smallest value of EPSC1) indicated a significant negative correlation between EPSC1 and EPSCSS (r2 = 0.968;p < 0.0001).
Because modulation of PRin vivo may occur via activation of presynaptic GABAB receptors, we used the same experimental approach to record EPSC trains in the presence of baclofen concentrations ranging from 0 to 100 μm (Fig.3). At 100 μm, the effect of baclofen is expected to be saturating, given an IC50 of 9 μm for baclofen of inhibition of the initial EPSC (data not shown). As noted earlier, baclofen enhanced EPSCSS by more than twofold during 200 Hz trains, but unlike Cd2+, no suppression of EPSCSS was obtained even at the highest concentrations of baclofen. Figure 3, A andB, shows data from a neuron in which EPSCSS was enhanced 3.6-fold when the first EPSC in the train was reduced to 6.8% of its control value. As release during the first stimulus of the train was progressively blocked with saturating baclofen, the value of EPSCSSincreased and reached a plateau (Fig. 3B). Similar observations were made in 13 neurons. Data were pooled from six neurons in which numerous responses (43–96) were obtained (Fig.3C). In this group, EPSCSS was enhanced on average by 218% when EPSC1 was reduced to less than −3 nA with baclofen. Thus, at high stimulus rates, full activation of GABAB receptors appears to maximize synaptic strength.
Fig. 3.

Effect of baclofen on EPSCSS.Ai, Trains (200 Hz) were delivered in baclofen concentrations ranging from 0 to 100 μm. The first EPSC of the train is shown. Aii, Responses in 0 and 100 μm baclofen are shown. After the second stimulus of the train, EPSCs in baclofen are larger than controls. B, EPSCSS is plotted against EPSC1 of each train. As PR is lowered with baclofen, steady-state EPSCs increase in size, reaching a plateau as saturating levels of baclofen are reached. In this cell, enhancement of EPSCSS relative to control was 361% when EPSC1 was reduced with baclofen to between 5.1 and 8.1% of its control value. C, Pooled data from six neurons. Data were binned as in Figure 2C. A linear fit to theeight rightmost data points was highly significant (r2 = 0.903;p < 0.001).
Contribution of AMPA receptor desensitization to depression during high-frequency trains
One hypothesis to account for the changes we observed in EPSCSS that accompany changes inPR is that AMPA receptors undergo desensitization during stimulus trains delivered only whenPR is high. To investigate the contribution of AMPA receptor desensitization to synaptic depression, we evoked trains of stimuli at 200 Hz in the presence of 5 mm aniracetam, a drug that reduces AMPA receptor desensitization (Vyklicky et al., 1991, Raman and Trussell, 1995b;Partin et al., 1996). Although desensitization will occur to high concentrations of glutamate in the presence of aniracetam, its onset is slowed dramatically in nMag, and should be minimal with stimulus intervals of 5 msec (Raman and Trussell, 1995b; J. Lawrence and L. O. Trussell, unpublished observations). Effects of aniracetam on single EPSCs are shown in Fig.4Ai. Control EPSC amplitudes were −7.60 ± 0.65 versus −10.52 ± 0.97 in 5 mm aniracetam (an increase of 37.2 ± 4.3%;n = 16). Half-decay times were 0.80 ± 0.04 for controls versus 2.33 ± 0.12 msec in aniracetam (an increase of 197 ± 16%; n = 16) (Fig. 4Ai). Effects of aniracetam on peak amplitude and decay rate of AMPA-mediated EPSCs are attributed to modulation of receptor kinetics, which include an increase in the open time of the channel and a slowing of entry into desensitized states (Raman and Trussell, 1995b; Partin et al., 1996).
To determine the effect of aniracetam on synaptic depression during 200 Hz stimulation, we normalized peaks to the amplitude of the first EPSC in each train (Fig. 4Aii). By comparing relative amounts of depression after normalization, effects of aniracetam on desensitization during stimulus trains can be explored. Because EPSCs recorded at high frequency in aniracetam exhibited summation, peaks were measured from a baseline obtained by extrapolating the decay of the preceding EPSC using a single-exponential function. Synaptic depression during 200 Hz trains was reduced by aniracetam (Fig.4Bi). After normalization of EPSC1, EPSCs 2–10 were significantly larger in aniracetam (p < 0.002 for each stimulus; pairedt test; n = 13). Enhancement by aniracetam of EPSCs 2–10 of the train was calculated after normalization of EPSC1 as 100% · (EPSCANI− EPSCCON)/EPSCCON (Fig.4Bii). Maximal enhancement of the EPSC by aniracetam was seen during the second stimulus (131% larger than control) and declined slightly during trains (110% enhancement of the 10th stimulus). Thus, over 50% of synaptic AMPA receptors remained desensitized throughout 200 Hz trains of 10 stimuli.
In contrast, when release was blocked by ∼85% with 20 μm Cd2+ (Fig.4Ci,Cii) or 50 μmbaclofen (data not shown), no effect of aniracetam on normalized EPSC amplitudes was observed (Fig. 4Cii). This suggests that desensitization contributes to synaptic depression under control release conditions but not when PR is reduced by baclofen or Cd2+. Notably, the approximately twofold enhancement of EPSCs during 200 Hz trains by relief of desensitization (Fig. 4Bii) was similar to the enhancement of EPSCs observed when release was blocked with Cd2+ or baclofen (Figs.1D, 2C, 3B). This similarity suggests that relief of desensitization may contribute to the enhancement of EPSCSS observed whenPR was lowered with Cd2+or baclofen.
Convergence of EPSC amplitudes with relief of desensitization
Because desensitization caused a decrease in EPSCSS under high-release conditions, we predicted that EPSCSS would remain constant over a wide range of initial release probabilities if desensitization was reduced by aniracetam. To test this hypothesis, experiments similar to those presented in Figure 2 were conducted in the continuous presence of 5 mm aniracetam. Trains were recorded at 15 sec intervals while PR was varied by bath application and subsequent washout of 100 μmCd2+ (Fig.5Ai). Under these conditions, EPSCSS amplitudes converged on the same value during 200 Hz trains despite large changes inPR (Fig.5Aii,B). Figure 5B indicates that, whereas initial EPSC amplitudes varied fourfold (from −2 to −8 nA), steady-state EPSCs remained constant at ∼1.5 nA. Similar results were obtained in eight neurons. Figure 5C (filled circles) shows pooled data from seven neurons from which a large number of responses (21–60) were recorded, as described in Figure 2. The slope of a linear regression of the eight rightmost data points in Figure 5C (filled circles) was not significantly different from zero (slope of −0.029;p = 0.198). For comparison, the control data from Figure 2C were scaled up by 37% to account for the effect of aniracetam on the initial EPSC amplitude and were plotted in Figure5C as open circles.
Fig. 5.

Convergence of EPSCSS in the presence of aniracetam A, In the continuous presence of 5 mm aniracetam, 200 Hz trains of 10 stimuli were delivered in various concentrations of Cd2+ ranging from 0 to 100 μm. Ai, The first EPSC of each train is shown. Aii, Superimposed traces in high Cd2+ (gray trace) and low Cd2+ (black trace) are shown.B, EPSCSS is plotted versus EPSC1 for each train. C, Data from seven cells in aniracetam (filled circles). As in Figure 2, data from each neuron were grouped in bins associated with EPSC1 values from 0–1, 1–2 nA, etc. Means and SEs were calculated for each bin. The slope of a linear fit to the data points (excluding the 2 leftmost pointsassociated with EPSC1 values from 0 to 2 nA) was not significantly different from 0 (r2 = 0.26;p = 0.20). Control data from Figure2C have been scaled up by a factor of 1.37 to account for the effect of aniracetam on the amplitude of a single EPSC (see Fig. 4A) and are shown for comparison (open circles).
The experiments presented in Figure 5 demonstrate that, in the presence of aniracetam, EPSCs approach the same amplitude during trains despite large variations in the transmitter release probability, in agreement with predictions of purely presynaptic depletion models of synaptic depression (see below). These data indicate that the progressive decline in EPSCSS seen asPR increased (Figs. 2C,3B) results from AMPA receptor desensitization under high-release conditions. Thus, relief of desensitization can account for the enhancement of EPSCSS that results from lowering PR by GABAB receptor activation or Cd2+ application.
Alternative actions of aniracetam
Control experiments were performed to determine whether presynaptic effects of aniracetam might have influenced the above conclusions, although previous studies found no effect of aniracetam on quantal content of EPSCs (Vyklicky et al., 1991). Effects of aniracetam on transmitter release were expected to alter paired-pulse ratios (EPSC2/EPSC1) of NMDA responses, as reported previously for cyclothiazide (Bellingham and Walmsley, 1999). NMDA-mediated EPSCs were recorded at +50 mV in the presence of AMPA receptor blockers. Paired stimuli were delivered at 20 msec intervals under control conditions (Fig.6Ai) or in the presence of 5 mm aniracetam (Fig. 6Aii). Paired-pulse ratios for the NMDA component of the EPSC were compared in nine neurons (Fig. 6B). For controls, average paired-pulse ratio was 0.27 ± 0.01; in the presence of 5 mm aniracetam, the average value was 0.26 ± 0.01. These values were not significantly different (p = 0.480; paired t test). Paired-pulse ratios of AMPA-mediated EPSCs were significantly larger (paired-pulse ratio of 0.49 ± 0.02; p < 0.01; paired t test) (Fig. 6C). Based on the higher affinity of the NMDA receptor for glutamate, we attribute this finding to greater occupancy of NMDA receptors at the onset of the second stimulus. Receptor desensitization may also contribute to paired-pulse depression (PPD) of NMDA responses. Additional evidence for a purely postsynaptic site of action for aniracetam is indicated by the absence of an effect of aniracetam on the relative amplitudes of AMPA-mediated EPSCs during stimulus trains under lowPR conditions (Fig.4Cii).
Fig. 6.

NMDA component of EPSC is not affected by aniracetam. A, Pairs of EPSCs at 20 msec intervals were recorded at +50 mV in GYKI-52466 and DNQX under control conditions (Ai) and in 5 mm aniracetam (Aii). Average of five traces. Peak 2 (bottom traces) in Ai and Aii was obtained by subtraction of the averaged single EPSC (data not shown).B, Paired-pulse ratios (EPSC2/EPSC1) for responses in control solutions and in 5 mm aniracetam (n = 9). C, Average values of EPSC2/EPSC1 for NMDA component of EPSC (control and aniracetam) and AMPA component (control;n = 9).
As transmitter release declines during high-frequency trains, it is possible that postsynaptic sites are exposed to progressively lower concentrations of glutamate. If this were true, then EPSC enhancement by aniracetam during trains might simply be an outcome of the drug increasing the affinity of AMPA receptors, apart from preventing desensitization. We determined whether the effectiveness of aniracetam changed with quantal content by comparing the effects of the drug on mEPSCs and evoked EPSCs. If receptors were farther from saturation during mEPSCs than during EPSCs, then the effects of aniracetam on peak current should be greatest for mEPSCs. However, this was not the case. In a group of six neurons, mEPSCs enhancement by aniracetam was 14.5 ± 5.2%, slightly less than enhancement of evoked EPSCs, which was 25.7 ± 6.9%. These values were not significantly different (p = 0.12; pairedt test). Thus, effects of aniracetam on affinity of AMPA receptors for glutamate cannot account for the enhancement of EPSC amplitudes during trains.
Quantal amplitudes are depressed after single stimuli and after trains
It has been shown previously in nMag that desensitization of AMPA receptors causes a decrease in the size of the mEPSC immediately after a single evoked EPSC (Otis et al., 1996b). Others, however, have not observed a decrease in postsynaptic responsiveness to transmitter after a single EPSC (Silver et al., 1998; Bellingham and Walmsley, 1999) or during trains (Silver et al., 1998). We therefore reexamined this effect, in particular contrasting changes in quantal size with single or multiple conditioning EPSCs. After EPSCs evoked in the presence of Sr2+ (2–4 mm), an elevated frequency of asynchronous quantal release is observed in nMag (Otis et al., 1996b) and other preparations (Rahamimoff and Yaari, 1973; Xu-Friedman and Regehr, 1999). Therefore, we recorded single EPSCs and trains of five EPSCs at 200 Hz, in the presence of Sr2+. Amplitude and latency of quantal events were measured after the stimulus (Fig.7Ai,Bi). Between 50 and 100 consecutive traces were recorded to obtain sufficient quantal events for analysis (Fig. 7Aii,Bii). Quantal amplitudes were sorted by latency and binned into groups of 30 successive events. For each bin, mean amplitude was plotted against mean latency from the peak of the preceding EPSC (Fig.7Aiii,Biii). After one and five stimuli, quantal amplitudes were depressed by 36.5 ± 3.9 (n = 5) and 36.3 ± 3.5% (n = 5), respectively. Amplitudes recovered with a single-exponential time constant of 21.1 ± 2.9 and 10.6 ± 2.0 msec, respectively. The overall extent of depression after one or five stimuli was not statistically different; however, the time course of recovery was significantly faster after five stimuli (p < 0.01;t test). In these experiments, a maximum train length of five stimuli was used because longer trains resulted in a high frequency of asynchronous quantal release whose superposition prevented resolution of individual events. These results are in agreement with the results obtained using aniracetam (Fig. 4), indicating the persistent desensitization of AMPA receptors during high-frequency stimulus trains.
Fig. 7.

Depression of quantal size during stimulus trains.Ai, Averaged EPSC recorded in 3 mmSr2+. Aii, Twenty superimposed traces after a single stimulus. Asynchronously released quanta are visible arising from the decay of the preceding EPSC. Dotted line indicates zero-current baseline. Aiii, Means and SE of amplitude and latency for quantal events binned in groups of 30. For this synapse, the amplitude of the first bin was depressed by 49% relative to the steady-state amplitude at longer latencies. A single-exponential fit to the data gave a recovery time course of 27 msec. Bi, Averaged traces from a train of five stimuli at 200 Hz in 3 mm Sr2+.Bii, Twenty-five superimposed traces after the fifth EPSC of the train shown in Bi. Biii, Mean amplitude and latency for quantal events binned in groups of 30. In this synapse, the amplitude of the first bin was depressed by 38% relative to steady-state amplitude at longer latencies. Recovery time course of quantal amplitudes was 11 msec.
Modeling depression and desensitization
Simulations were performed to determine whether the experimental results presented in Figures 2 and 5 could be explained by incorporating receptor desensitization into a depletion model of synaptic depression (Fig. 8). The model consists of a synapse with N0 release sites that release transmitter with probabilityPR after a presynaptic action potential. Sites becomes refractory immediately after release and subsequently recover with a single-exponential time course (see Materials and Methods). Although alternative presynaptic models have been developed (Wu and Borst, 1999; Kraushaar and Jonas, 2000; Matveev and Wang, 2000), a depletion model was used here because it accurately reproduced our synaptic data obtained in the presence of aniracetam. Postsynaptic desensitization was modeled by introducing a scaling factor in the model that reduced quantal size uniformly at all sites and was dependent on the amount of release. Desensitization had an exponential relationship to the amount of release and recovered with a single-exponential time course. Parameters of the model were determined by fitting the simulated results to the data shown in Figure4B. The presynaptic component of the model is similar in many respects to previously published models (O'Donovan and Rinzel, 1997, Dittman and Regehr, 1998, Weis et al., 1999; Lu and Trussell, 2000).
Fig. 8.
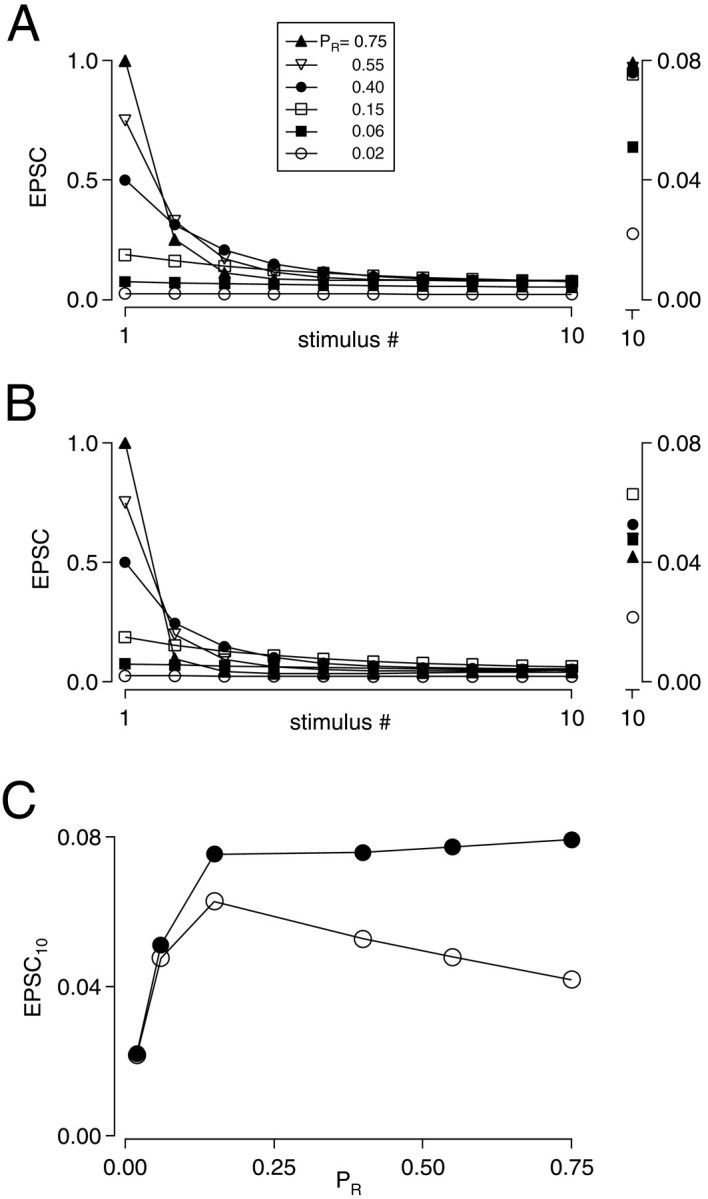
Depletion and desensitization model of synaptic depression. A, Simulations based on a presynaptic depletion model that does not include receptor desensitization. Trains of 10 stimuli at 200 Hz with PR values ranging from 0.02 to 0.75. Smaller responses to stimulus #1 correspond to lower PR values. Time constant of recovery from depression was 100 msec. EPSCs reached similar steady-state values when PR ranged from 0.15 to 0.75. Inset on right shows the 10th response on an expanded vertical scale. Note that data points are superimposed from trains withPR of 0.15 to 0.75. Y values (normalized to EPSC1) for these four data points ranged from 0.075 to 0.079. B, Desensitization was included in the model as described in Materials and Methods. PR was varied as inA. Steady-state EPSCs were maximal atPR = 0.15. Inset shows 10th response on an expanded vertical scale. C, EPSCSS is plotted versus EPSC1. Results of the presynaptic model are shown by filled circles; results of simulations incorporating postsynaptic desensitization are shown with open circles.
Figure 8A shows results of simulations of 200 Hz stimulus trains using a presynaptic depletion model in whichPR ranged from 0.02 to 0.75. At steady state, each presynaptic action potential evoked release from ∼8% of the total population of release sites when initial release probability is greater than 0.15 (Fig. 8A,C). AsPR decreased below 0.15, the amount of release was no longer limited by interstimulus recovery but instead was limited by extremely low fusion probability. As shown in Figure8C (filled circles), a purely presynaptic model of depression predicted that steady-state EPSC amplitudes evoked at high frequency (i.e., the stimulus interval is much faster than the time constant of recovery from depression) converge on the same value despite large changes in initial release probability. However, when the model incorporated postsynaptic receptor desensitization (see Materials and Methods), we observed a progressive reduction of EPSCSS as PRincreased (Fig. 8B,C, open circles), in agreement with our experimental results (Figs.2B,C, 3, 5C). The ability of this model to reproduce these key features of our experimental results strongly suggests that desensitization can account for the decline in EPSCSS with highPR.
DISCUSSION
This work demonstrates that desensitization of AMPA receptors contributes strongly to synaptic depression during high-frequency activity at avian end-bulb synapses in the auditory brainstem. Desensitization was dependent on the amount of transmitter release at the onset of a stimulus train. With high initial transmitter release probability, approximately half of the postsynaptic AMPA receptors were desensitized at the end of a 50 msec, 200 Hz stimulus train. However, desensitization decreased as PR was reduced, thereby enhancing amplitudes of steady-state EPSCs during stimulus trains. Thus, relief of AMPA receptor desensitization can account for the enhancement of steady-state EPSC amplitudes that accompany reduction of release probability with baclofen or Cd2+.
During stimulus trains, transmitter release approaches a steady-state that occurs when vesicle depletion after each stimulus is equal to recovery between stimuli. Depression models that consider only transmitter depletion predict that, during high-frequency stimulation, the steady-state EPSC amplitude is independent ofPR (except for extremely lowPR) because of an inverse relationship between PR and the size of the steady-state vesicle pool. With highPR, steady-state pool size is small and a large fraction of the pool is released with each stimulus, but with low PR, the steady-state pool size is large and only a small proportion of available release sites undergo exocytosis with each stimulus. This ideal relationship is an approximation that holds at stimulus frequencies for which the interval between stimuli is much shorter than the time constant of recovery from depression. In our experiments, block of desensitization resulted in convergence of EPSC amplitudes during high-frequency trains, indicating that release approaches a steady-state during the stimulus trains. If Ca2+-dependent processes cause acceleration of recovery from synaptic depression, as described at other synapses (Dittman and Regehr, 1998; Stevens and Wesseling, 1998;Wang and Kaczmarek, 1998; Wang and Zucker, 1998) (but see Weis et al., 1999; Wu and Borst, 1999), EPSCSS should have declined with reductions in presynaptic Ca2+ influx, i.e., as EPSC1 was lowered. However, EPSCSS was independent of EPSC1 over a wide range of release probabilities after block of postsynaptic depression. This suggests that, over the range of Ca2+ influx associated with this range of release probabilities, Ca2+-dependent processes act at a constant level, and perhaps under our experimental conditions Ca2+-dependent recovery processes are saturated by Ca2+ influx during trains of stimuli.
Changes in release probability have presynaptic and postsynaptic effects on transmission at the end-bulb synapse. Under low-release conditions, release of relatively few vesicles results in a rapidly decaying glutamate transient at a receptor cluster opposite an active release site. With higher release probability, transmitter will be released synchronously from many sites, generating a slower phase of decay of the glutamate transient in the synaptic cleft (Otis et al., 1996a). We observed a current plateau between EPSCs during 200 Hz trains, which has been attributed to accumulation of glutamate in the synaptic cleft (Turecek and Trussell, 2000). Modeling studies have suggested that the glutamate concentration in the synaptic cleft will remain above 10 μm for tens of milliseconds after release under high PR conditions. AMPA receptors undergo desensitization in the presence of micromolar concentrations of agonist insufficient to cause channel openings (Raman and Trussell, 1992). Experiments in which glutamate was applied to membrane patches excised from nMag neurons indicate that 10 μm glutamate will induce ∼70% steady-state desensitization of AMPA receptors (Raman and Trussell, 1992). Thus, low concentrations of glutamate that persist in the synaptic cleft during trains may contribute to desensitization that we observed during periods of high-frequency synaptic activation.
In addition, brief exposure of AMPA receptors to high concentrations of agonists can induce a form of desensitization that accompanies channel opening. One millisecond application of 1 mm glutamate to membrane patches at room temperature caused 50% desensitization of AMPA receptors, which subsequently recovered with a 16 msec exponential time constant (Raman and Trussell, 1995a). During trains, the relative contribution to desensitization of prolonged exposure to low glutamate concentrations and brief exposure to high concentrations is not known.
At room temperature, a single evoked EPSC desensitized 35–40% of synaptic AMPA receptors, recovering with a time constant of 68 msec (Otis et al., 1996b). At near-physiological temperatures, we observed a greater extent of desensitization, which recovered with a time constant of 21 msec. Faster recovery may be attributable to more rapid clearance of glutamate from the synaptic cleft and faster gating kinetics of AMPA receptors. Desensitization of AMPA receptors during synaptic activation was also shown to be dependent on the total amount of transmitter release (Trussell et al., 1993). PPD of EPSCs in low Ca2+ showed little sensitivity to cyclothiazide, but in higher Ca2+concentrations, relief of PPD by cyclothiazide was observed. Moreover, synaptic depression during 200 Hz trains was reduced by cyclothiazide (Zhang and Trussell, 1994a) and by a glutamate scavenging enzyme (Turecek and Trussell, 2000), indicating persistent AMPA receptor desensitization during periods of repetitive stimulation. These results are consistent with our observation that desensitization persists during stimulus trains delivered under conditions of highPR but not under conditions of lowPR.
Because we demonstrate that desensitization depends on the amount of evoked release during trains, it is predicted that presynaptic depression will allow receptors to recover from desensitization during periods of prolonged activity. During 200 Hz stimulation, <10% of release sites undergo exocytosis after each stimulus so that, on average, several stimuli will elapse between successive events of vesicle fusion at a particular release site. However, we saw little decline in the extent of receptor desensitization during 50 msec trains (Fig. 4Bii), as estimated by the enhancing effect of aniracetam. The persistence of AMPA receptor desensitization despite >90% reduction in transmitter release may be explained by accumulation and slow clearance of transmitter in the synaptic cleft.
Although a large reduction of PR by Cd2+ ultimately caused a decline in EPSCSS from its plateau value (Fig.2C), reduction of PR by GABAB receptor activation using a saturating concentration of agonist always raised EPSCSS. Based on a 3rd or 4th power relationship between calcium influx and transmitter release (Dodge and Rahamimoff, 1967; Augustine and Charlton, 1986; Borst and Sakmann, 1996), an 85% reduction of a single EPSC by saturating levels of baclofen indicates that maximal activation of GABAB receptors reduced presynaptic calcium influx by 38–47%. This observation suggests precise regulation of the coupling between GABAB receptors and Ca2+ channels involved in transmitter release, so that release is never actually inhibited during intense activity. Sites of such regulation could include the number of GABAB receptors, levels of expression of G-proteins, extent of modulation of particular Ca2+ channels subtypes, and the degree of coupling of different Ca2+ channel types to release.
Synaptic depression has been proposed to play an important role in promoting network stability in the cerebral cortex (Galarreta and Hestrin, 1998) and has been described as a mechanism that enables neurons to maintain responsiveness to the firing patterns of a large number of afferents (Abbott et al., 1997). However, at end-bulb synapses in the cochlear nucleus, a role for synaptic depression is less clear. Bushy cells serve as relays in a timing pathway enabling sound localization (Carr and Konishi, 1990; Overholt et al., 1992). In the avian brainstem, GABAergic neurons from the superior olivary nucleus are activated by sounds and project to nMag, in which they may activate GABAB receptors on auditory nerve terminals (Lachica et al., 1994; Monsivais et al., 2000). The feed-forward nature of this pathway should cause GABA release by superior olivary neurons to increase approximately in parallel with end-bulb synaptic activity. Because desensitization is likely to result from in vivo firing rates of the auditory nerve, activation of presynaptic GABAB receptors could serve to minimize synaptic depression by relieving desensitization, thereby allowing suprathreshold transmission to persist at higher rates of synaptic activity. Such an enhancement of gain may help widen the dynamic range of sensory signaling.
Footnotes
This work was supported by National Institutes of Health Grants NS28901 (L.O.T.) and GM07507 (S.B.). We thank Drs. V. Alvarez, J. Diamond, R. Fettiplace, T. Lu, I. Raman, and R. Turecek for helpful discussions and comments on this manuscript.
Correspondence should be addressed to Stephan Brenowitz, Auditory Neuroscience L-335A, Oregon Health Sciences University, 3181 SW Sam Jackson Park Road, Portland, OR 97201. E-mail: brenowit@ohsu.edu.
REFERENCES
- 1.Abbott LF, Varela JA, Sen K, Nelson SB. Synaptic depression and cortical gain control. Science. 1997;275:220–224. doi: 10.1126/science.275.5297.221. [DOI] [PubMed] [Google Scholar]
- 2.Augustine GJ, Charlton MP. Calcium dependence of presynaptic calcium current and post-synaptic response at the squid giant synapse. J Physiol (Lond) 1986;381:619–640. doi: 10.1113/jphysiol.1986.sp016347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- 4.Bellingham MC, Walmsley B. A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron. 1999;23:159–170. doi: 10.1016/s0896-6273(00)80762-x. [DOI] [PubMed] [Google Scholar]
- 5.Betz WJ. Depression of transmitter release at the neuromuscular junction of the frog. J Physiol (Lond) 1970;206:629–644. doi: 10.1113/jphysiol.1970.sp009034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borst JG, Sakmann B. Calcium influx and transmitter release in a fast CNS synapse. Nature. 1996;383:431–434. doi: 10.1038/383431a0. [DOI] [PubMed] [Google Scholar]
- 7.Brenowitz S, David J, Trussell L. Enhancement of synaptic efficacy by presynaptic GABA(B) receptors. Neuron. 1998;20:135–141. doi: 10.1016/s0896-6273(00)80441-9. [DOI] [PubMed] [Google Scholar]
- 8.Carr CE, Konishi M. A circuit for detection of interaural time differences in the brain stem of the barn owl. J Neurosci. 1990;10:3227–3246. doi: 10.1523/JNEUROSCI.10-10-03227.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DelCastillo J, Katz B. Statistical factors involved in neuromuscular facilitation and depression. J Physiol (Lond) 1954;124:574–585. doi: 10.1113/jphysiol.1954.sp005130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dittman JS, Regehr WG. Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J Neurosci. 1996;16:1623–1633. doi: 10.1523/JNEUROSCI.16-05-01623.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 13.Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol (Lond) 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng TP. Studies on the neuromuscular junction XVIII. The local potentials around n–m junctions induced by single and multiple volleys. Chin J Physiol. 1940;15:367–404. [Google Scholar]
- 15.Galarreta M, Hestrin S. Frequency-dependent synaptic depression and the balance of excitation and inhibition in the neocortex. Nat Neurosci. 1998;1:587–594. doi: 10.1038/2822. [DOI] [PubMed] [Google Scholar]
- 16.Kraushaar U, Jonas P. Efficacy and stability of quantal GABA release at a hippocampal interneuron-principal neuron synapse. J Neurosci. 2000;20:5594–5607. doi: 10.1523/JNEUROSCI.20-15-05594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kreitzer AC, Regehr WG. Modulation of transmission during trains at a cerebellar synapse. J Neurosci. 2000;20:1348–1357. doi: 10.1523/JNEUROSCI.20-04-01348.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kusano K, Landau EM. Depression and recovery of transmission at the squid giant synapse. J Physiol (Lond) 1975;245:13–22. doi: 10.1113/jphysiol.1975.sp010832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lachica EA, Rubsamen R, Rubel EW. GABAergic terminals in nucleus magnocellularis and laminaris originate from the superior olivary nucleus. J Comp Neurol. 1994;348:403–418. doi: 10.1002/cne.903480307. [DOI] [PubMed] [Google Scholar]
- 20.Lu T, Trussell LO. Inhibitory transmission mediated by asynchronous transmitter release. Neuron. 2000;26:683–694. doi: 10.1016/s0896-6273(00)81204-0. [DOI] [PubMed] [Google Scholar]
- 21.Lundberg A, Quilisch H. Presynaptic potentiation and depression of neuromuscular transmission in frog and rat. Acta Physiol Scand. 1953;[Suppl iii] 30:111–120. [PubMed] [Google Scholar]
- 22.Markram H, Tsodyks M. Redistribution of synaptic efficacy between neocortical pyramidal neurons. Nature. 1996;382:807–810. doi: 10.1038/382807a0. [DOI] [PubMed] [Google Scholar]
- 23.Matveev V, Wang X. Implications of all-or-none synaptic transmission and short-term depression beyond vesicle depletion: a computational study. J Neurosci. 2000;20:1575–1588. doi: 10.1523/JNEUROSCI.20-04-01575.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monsivais P, Yang L, Rubel EW. GABAergic inhibition in nucleus magnocellularis: implications for phase locking in the avian auditory brainstem. J Neurosci. 2000;20:2954–2963. doi: 10.1523/JNEUROSCI.20-08-02954.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Donovan MJ, Rinzel J. Synaptic depression: a dynamic regulator of synaptic communication with varied functional roles. Trends Neurosci. 1997;20:431–433. doi: 10.1016/s0166-2236(97)01124-7. [DOI] [PubMed] [Google Scholar]
- 26.Otis TS, Trussell LO. Inhibition of transmitter release shortens the duration of the excitatory synaptic current at a calyceal synapse. J Neurophysiol. 1996;76:3584–3588. doi: 10.1152/jn.1996.76.5.3584. [DOI] [PubMed] [Google Scholar]
- 27.Otis TS, Wu YC, Trussell LO. Delayed clearance of transmitter and the role of glutamate transporters at synapses with multiple release sites. J Neurosci. 1996a;16:1634–1644. doi: 10.1523/JNEUROSCI.16-05-01634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otis TS, Zhang S, Trussell LO. Direct measurement of AMPA receptor desensitization induced by glutamatergic synaptic transmission. J Neurosci. 1996b;16:7496–7504. doi: 10.1523/JNEUROSCI.16-23-07496.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Overholt EM, Rubel EW, Hyson RL. A circuit for coding interaural time differences in the chick brainstem. J Neurosci. 1992;12:1698–1708. doi: 10.1523/JNEUROSCI.12-05-01698.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Partin KM, Fleck MW, Mayer ML. AMPA receptor flip/flop mutants affecting deactivation, desensitization, and modulation by cyclothiazide, aniracetam, and thiocyanate. J Neurosci. 1996;16:6634–6647. doi: 10.1523/JNEUROSCI.16-21-06634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rahamimoff R, Yaari Y. Delayed release of transmitter at the frog neuromuscular junction. J Physiol (Lond) 1973;228:241–257. doi: 10.1113/jphysiol.1973.sp010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raman IM, Trussell LO. The kinetics of the response to glutamate and kainate in neurons of the avian cochlear nucleus. Neuron. 1992;9:173–186. doi: 10.1016/0896-6273(92)90232-3. [DOI] [PubMed] [Google Scholar]
- 33.Raman IM, Trussell LO. The mechanism of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor desensitization after removal of glutamate. Biophys J. 1995a;68:137–146. doi: 10.1016/S0006-3495(95)80168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raman IM, Trussell LO. Concentration-jump analysis of voltage-dependent conductances activated by glutamate and kainate in neurons of the avian cochlear nucleus. Biophys J. 1995b;69:1868–1879. doi: 10.1016/S0006-3495(95)80057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salvi RJ, Saunders SS, Powers NL, Boettcher FA. Discharge patterns of cochlear ganglion neurons in the chicken. J Comp Physiol [A] 1992;170:227–241. doi: 10.1007/BF00196905. [DOI] [PubMed] [Google Scholar]
- 36.Silver RA, Momiyama A, Cull-Candy SG. Locus of frequency-dependent depression identified with multiple-probability fluctuation analysis at rat climbing fibre-Purkinje cell synapses. J Physiol (Lond) 1998;510:881–902. doi: 10.1111/j.1469-7793.1998.881bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron. 1998;21:415–424. doi: 10.1016/s0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- 38.Takeuchi A. The long-lasting depression in neuromuscular transmission of frog. Jpn J Physiol. 1958;8:102–113. doi: 10.2170/jjphysiol.8.102. [DOI] [PubMed] [Google Scholar]
- 39.Thies R. Neuromuscular depression and the apparent depletion of transmitter in mammalian muscle. J Neurophysiol. 1965;28:427–442. doi: 10.1152/jn.1965.28.3.427. [DOI] [PubMed] [Google Scholar]
- 40.Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-z. [DOI] [PubMed] [Google Scholar]
- 41.Tsodyks MV, Markram H. The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc Natl Acad Sci USA. 1997;94:719–723. doi: 10.1073/pnas.94.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turecek R, Trussell LO. Control of synaptic depression by glutamate transporters. J Neurosci. 2000;20:2054–2063. doi: 10.1523/JNEUROSCI.20-05-02054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vyklicky L, Jr, Patneau DK, Mayer ML. Modulation of excitatory synaptic transmission by drugs that reduce desensitization at AMPA/kainate receptors. Neuron. 1991;7:971–984. doi: 10.1016/0896-6273(91)90342-w. [DOI] [PubMed] [Google Scholar]
- 44.Wang C, Zucker RS. Regulation of synaptic vesicle recycling by calcium and serotonin. Neuron. 1998;21:155–167. doi: 10.1016/s0896-6273(00)80523-1. [DOI] [PubMed] [Google Scholar]
- 45.Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- 46.Warchol ME, Dallos P. Neural coding in the chick cochlear nucleus. J Comp Physiol [A] 1990;166:721–734. doi: 10.1007/BF00240021. [DOI] [PubMed] [Google Scholar]
- 47.Weis S, Schneggenburger R, Neher E. Properties of a model of Ca2+-dependent vesicle pool dynamics and short term synaptic depression. Biophys J. 1999;77:2418–2429. doi: 10.1016/S0006-3495(99)77079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu LG, Borst JG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/s0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- 49.Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- 50.Xu-Friedman MA, Regehr WG. Presynaptic strontium dynamics and synaptic transmission. Biophys J. 1999;76:2029–2042. doi: 10.1016/S0006-3495(99)77360-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang S, Trussell LO. Voltage clamp analysis of excitatory synaptic transmission in the avian nucleus magnocellularis. J Physiol (Lond) 1994a;480:123–136. doi: 10.1113/jphysiol.1994.sp020346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang S, Trussell LO. A characterization of excitatory postsynaptic potentials in the avian nucleus magnocellularis. J Neurophysiol. 1994b;72:705–718. doi: 10.1152/jn.1994.72.2.705. [DOI] [PubMed] [Google Scholar]
- 53.Zhou N, Parks TN. Developmental changes in the effects of drugs acting at NMDA or non-NMDA receptors on synaptic transmission in the chick cochlear nucleus (nuc. magnocellularis). Brain Res Dev Brain Res. 1992;67:145–152. doi: 10.1016/0165-3806(92)90215-i. [DOI] [PubMed] [Google Scholar]
- 54.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]