

Abstract
Tumor-necrosis-factor-α (TNF-α) prevented secondary death of retinal ganglion cells (RGCs) after axotomy of the optic nervein vivo. This RGC rescue was confirmed in vitro in a mixed retinal culture model. In accordance with our previous findings, TNF-α decreased outward potassium currents in RGCs. Antagonism of the TNF-α-induced decrease in outward potassium currents with the potassium channel opener minoxidilsulfate (as verified by electrophysiology) abolished neuroprotection. Western blot analysis revealed an upregulation of phospho-Akt as a consequence of TNF-α-induced potassium current reduction. Inhibition of the phosphatidylinositol 3-kinase–Akt pathway with wortmannin decreased TNF-α-promoted RGC survival. These data point to a functionally relevant cytokine-dependent neuroprotective signaling cascade in adult CNS neurons.
Keywords: tumor necrosis factor-α, retinal ganglion cells, neuroprotection, outward potassium current, PKB/Akt, PI3-K, retrograde cell death
Tumor-necrosis-factor-α (TNF-α) was discovered as a serum protein released after systemic treatment of rodents with “bacille Calmette-Guérin” and lipopolysaccharide (Carswell et al., 1975). The biological responses to TNF-α are mediated by two types of TNF receptors, which can be differentiated by their molecular weight of ∼55 kDa (TNFRI) and 75 kDa (TNFRII). Whereas TNFRI is ubiquitously expressed, TNFRII appears to be restricted to cells of hematopoetic origin (Fiers, 1991) and does not possess a death domain (Rath and Aggarwal, 1999). A multitude of studies over the last decade have revealed that TNF-α can exert opposing effects on the cellular level: induction of apoptosis via TNFRI-death domains and adapter proteins that couple the receptor complex to the activation of caspase-8 on the one hand, increased cell-survival via activation of NF-κB-dependent genes on the other (Ashkenazi and Dixit, 1998). TNF-α has been shown to potentiate the induction of neuronal apoptosis by HIV-1 Tat in primary human neurons (Shi et al., 1998), and has neurotoxic effects on primary human astrocytes because of its ability to inhibit glutamate uptake (Fine et al., 1996). In vivo studies have demonstrated damaging effects in cerebral ischemia (Dawson et al., 1996; Barone et al., 1997; Meistrell et al., 1997; Nawashiro et al., 1997), experimental autoimmune encephalomyelitis (Klinkert et al., 1997) and head injury models (Shohami et al., 1996, 1997). Furthermore, TNF-α contributes to apoptosis in rat hippocampal neurons during experimental meningitis (Bogdan et al., 1997). On the other hand, TNF-α seems to protect primary hippocampal neurons against hypoxia or nitric oxide-induced injury (Tamatani et al., 1998) or cultured mesencephalic neurons against glutamate neurotoxicity (Shinpo et al., 1999). Neuronal damage after focal cerebral ischemia or administration of kainic acid was increased in mice lacking TNFRI (Gary et al., 1998).
Only few in vitro studies have related TNF-α-induced changes of ion balance or electrophysiological properties to neuroprotection. TNF-α maintains calcium homeostasis in cultured neurons after glucose deprivation and excitatory amino acid toxicity via induction of calbindin (Cheng et al., 1994). In addition, TNF-α may protect cultured rat cortical neurons against NMDA neurotoxicity by increasing transient outward potassium currents (Houzen et al., 1997).
In recent studies, Akt has been identified as a part of the TNF-α signaling pathway that leads to inflammatory responses and inhibition of apoptosis in vitro (Nidai Ozes et al., 1999; Pastorino et al., 1999; Reddy et al., 2000). A connection between membrane depolarization and phosphatidylinositol 3-kinase (PI3-K)-dependent Akt phosphorylation was found in cultured rat sympathetic neurons (Vaillant et al., 1999).
Using the patch-clamp technique, we have recently demonstrated that an acute application of TNF-α resulted in a decrease in outward potassium currents in dissociated retinal ganglion cells (RGCs) (R. Meyer, R. Diem, U. Wagner, M. Labes, and M. Bähr, unpublished observations). In the present study, we injected TNF-α intraocularly after transection of the optic nerve (ON) and assessed RGC viability 14 d after the lesion. Now we relate TNF-α-promoted cell survival in vivo to its electrophysiological effects observed on an in vitro model. Moreover, we investigated the downstream signaling cascade that leads to phosphorylation of Akt and neuroprotection in vivo.
MATERIALS AND METHODS
Unilateral ON transection. Surgical procedures have been described elsewhere (Klöcker et al., 1997; Kermer et al., 1998). Briefly, adult female Sprague Dawley rats (200–250 gm) purchased from Charles River Wiga (Sulzfeld, Germany) were anesthetized by intraperitoneal injection of chloral hydrate (0.42 gm/kg body weight). A skin incision close to the superior orbital rim was performed, and the right orbita was opened. The lachrymal gland was resected subtotally. After spreading of the superior extraocular muscles the ON was exposed by longitudinal incision of the eye retractor muscle and the perineurium. ON transection was performed ∼2 mm from the posterior pole of the eye without damaging retinal blood supply. This was verified funduscopically after surgery.
In vivo labeling and counting of axotomized RGCs. The procedure has been described in detail previously (Kermer et al., 1999a). Retrograde labeling of RGCs was achieved by placing a small sponge soaked in 5% FluoroGold (FG; Fluorochrome, Denver, CO) at the ocular stump of the transected ON. Fourteen days later the retinas were processed as described (Kermer et al., 1998), blinded, and counted by two independent investigators. The counting was performed under a fluorescence microscope (Axiovert 35, Zeiss, Oberkochen, Germany) using a DAPI filter (365/420 nm). The FG-positive RGCs were counted in 12 distinct areas of 62,500 μm2 each (three areas per retinal quadrant at three different eccentricities of 1/6, 1/2, and 5/6 of the retinal radius). Overestimation of RGC density by retinal shrinkage caused by intraocular injection was excluded (Klöcker et al., 1998). Data are given as mean ± SEM. Statistical significance was assessed using one-way ANOVA followed by the Duncan's test.
Retrograde labeling of retinal ganglion cells for mixed retinal cell culture. Rat pups were anesthetized by diethylether at postnatal day 5, when their superior colliculi offer good surgical access, because they are not yet overgrown by the visual cortex. The skull cartilage was opened dorsal to the lambda fissure, and FG (5% in normal saline) was applied to both colliculi using a micropipette (Leifer et al., 1984).
Intravitreal drug administration. For intraocular injection of recombinant human TNF-α (5 μg/ml; Roche, Basel, Switzerland), anti-mouse TNF-RI antibodies (MAB 225, 0.1 mg/ml; AF-425-PB, 0.1 mg/ml; R & D Systems GmbH, Wiesbaden, Germany) or vehicle (PBS), animals were anesthetized with diethylether. By means of a glass microelectrode with a tip diameter of 30 μm, 2 μl of drug or vehicle was injected into the vitreous space, puncturing the eye at the cornea–sclera junction. The injections were done at days 0, 4, 7, and 10 after lesion.
In another set of experiments, intraocular application of the potassium channel opener minoxidilsulfate (Mnxs; 400 μm, Sigma, Deisenhofen, Germany) or the PI3-K-inhibitor wortmannin (WM; 0.1 mm; dissolved in 15% dimethylsulfoxide; Sigma) alone and simultaneously with TNF-α or tetraetylammonium (TEA; 300 mm; Sigma) alone was performed following the same protocol.
Cell culture techniques. The culturing procedure of RGCs was performed as described elsewhere (Guenther et al., 1994). Both eyes of one decapitated Sprague Dawley rat in the age of 6–16 d (Charles River Wiga, Sulzfeld, Germany) were enucleated. The retinas were incubated for 20 min at 37°C in 2.0 ml of calcium- and magnesium-free PBS, containing bovine serum albumin (0.3%),dl-cysteine (0.18 mg/ml) and papain (15 U/ml). After mechanical dissociation by tituration with a glass pipette, the suspension was centrifuged at 900 × g for 10 min. For electrophysiological measurements, cells were plated onto poly-l-lysine (PLL)-coated glass coverslips in 35 mm tissue culture dishes (Greiner, Frickenhausen, Germany) that contained 2 ml of modified Eagles's medium (MEM) supplemented with 10% fetal calf serum, 33 mm glutamine, 24 mm NaHCO3, and 10 μg/ml antibiotic solution.
For counting of FG-positive RGCs, 100 μl aliquots of a 4 ml cell suspension extracted from four retinas were plated onto 9 mm PLL-coated glass coverslips. Four coverslips per group were counted under a fluorescence microscope (Axiovert 35; Zeiss) using a DAPI filter (365/420 nm) 2, 22, and 46 hr after plating. TNF-α (5 ng/ml), Mnxs (100 μm, alone or together with TNF-α), WM (0.1 mm, alone or together with TNF-α), and TEA (100 μm) were added to the medium before and 20 hr after plating. Data are given as mean ± SEM. Statistical significance was assessed using Student's t test.
Electrophysiology. Measurements were performed using the whole-cell configuration of the patch-clamp technique (Hamill et al., 1981). For current recording, an EPC-9 patch-clamp amplifier and the Pulse software (Heka, Lambrecht, Germany) were used. A routine correction for leak currents and capacitive transients was performed using a P/n method. Only experiments with series resistances <30 MΩ were used. Series resistance errors were compensated in the range of 50–90% with a routine of the Pulse software. Data analysis was performed with the programs PulseFit and IGOR Pro (Wavemetrics, Lake Oswego, OR). Patch pipettes were pulled from borosilicate glass capillaries (Science Products, Hofheim, Germany) with a resistance range of 2–6 MΩ and polished using a DMZ puller (Zeitz Instruments, Augsburg, Germany). The experiments were performed with RGCs maintained in culture dishes for 4–36 hr. The coverslips were placed in 35 mm dishes on the stage of an inverted microscope (Axiovert 135; Zeiss). TNF-α (5 ng/ml) alone or together with Mnxs (100 μm) was applied by perfusion with a tube placed in front of the cell.
RGCs were identified by their large size, the presence of a large sodium current of at least 0.5 nA (Lipton and Tauck, 1987), and by retrograde labeling with FG.
The standard external solution contained the following ion concentrations (in mm): 130 NaCl, 5 KCl, 2 CaCl2, 2 MgCl, and 10 HEPES, pH 7.4. The intracellular solution contained (in mm): 10 NaCl, 130 KCl, 2 MgCl, 10 HEPES, and 10 EGTA, pH 7.4.
Results are presented as mean ± SEM. Statistical significance was assessed using one-way ANOVA followed by the Duncan's test.
Western blotting. The Western blot analysis was performed as described elsewhere (Kermer et al., 2000). Retinas were homogenized and lysed (150 mm NaCl, 50 mmTris, pH 8.0, 2 mm EDTA, and 1% Triton X-100, containing 0.1 mm PMSF and 2 μg/ml pepstatin, leupeptin, and aprotinin) on ice for 20 min. Cell debris were pelleted at 13,000 × g for 15 min, and protein concentration of the supernatant was determined using the BCA reagent (Pierce, Rockford, IL). The lysates (20 μg of protein per lane) were separated by reducing SDS-PAGE, and proteins were transferred to a polyvinylidene difluoride membrane and blocked with 5% skim milk in 0.1% Tween 20 in PBS (PBS-T). After incubation with the primary antibody against phospho-Akt (New England Biolabs GmbH, Schwalbach, Germany; 1:1000 in 1% skim milk in PBS-T), membranes were washed in PBS-T and incubated with HRP-conjugated secondary antibodies against rabbit IgG (Dianova, Hamburg, Germany; 1:2000 in PBS-T). Labeled proteins were detected using the ECL-plus reagent (Amersham, Arlington Heights, IL).
For Western blot analysis of BDNF levels, the primary antibody (Promega, Madison, WI) was diluted 1:500 in 1% skim milk in PBS-T; for protein detection, a HRP-conjugated secondary antibody against chicken IgY was used (Promega; 1:1000 in PBS-T).
RESULTS
TNF-α prevents axotomy-induced retrograde death of RGCs in vivo via activation of the TNFRI
In unlesioned control retinas retrogradely labeled from the superior colliculi with the fluorescent tracer DiI, the mean RGC density was 2068 ± 82 cells/mm2(n = 4; see Fig. 2a) in accordance with earlier reports (Klöcker et al., 1997). Comparable cell densities have been observed by other investigators (Eschweiler and Bähr, 1993; Mansour-Robaey et al., 1994). In vehicle (PBS)-treated rats, no more than 405.5 ± 39.72 (mean ± SEM; n = 4) RGCs per square millimeter were detectable within 14 d after ON transection, as determined by retrograde FG labeling from the axon stump (Figs. 1,2a–c). To examine a potential positive or negative effect of TNF-α on the survival of axotomized RGCs, 2 μl of recombinant human TNF-α (5 μg/ml) were injected intravitreously on days 0, 4, 7, and 10 after the lesion. Human recombinant TNF-α was selected because it has been described to have less toxic side effects in rodents when given systemically (Fiers, 1991) and this was confirmed by our own observations. Compared with vehicle treatment, TNF-α increased survival of RGCs to 1131.6 ± 131.95 cells per square millimeter (n = 5; Figs. 1, 2a–c). An antibody to the TNFRI that has agonist activity (according to supplier's information; 0.1 mg/ml, 2 μl injections) had a similar effect (1245.9 ± 214.53 cells per square millimeter; n = 3, Fig. 2a). Cell rescue was completely abolished under combined treatment with TNF-α (5 μg/ml) and an antibody against the TNFRI with inhibitory properties (Matthews and Neale, 1987) (0.1 mg/ml; 395.7 ± 22.89 cells per square millimeter; n = 5; Fig.2a). To test whether endogenous TNF-α has any neuroprotective effect, the inhibitory antibody (0.1 mg/ml, 2 μl injections) was given as a monotherapy. The result was very similar to the lesioned retinas with vehicle treatment (409.9 ± 48.58 cells per square millimeter; n = 6; Fig. 2a).
Fig. 2.
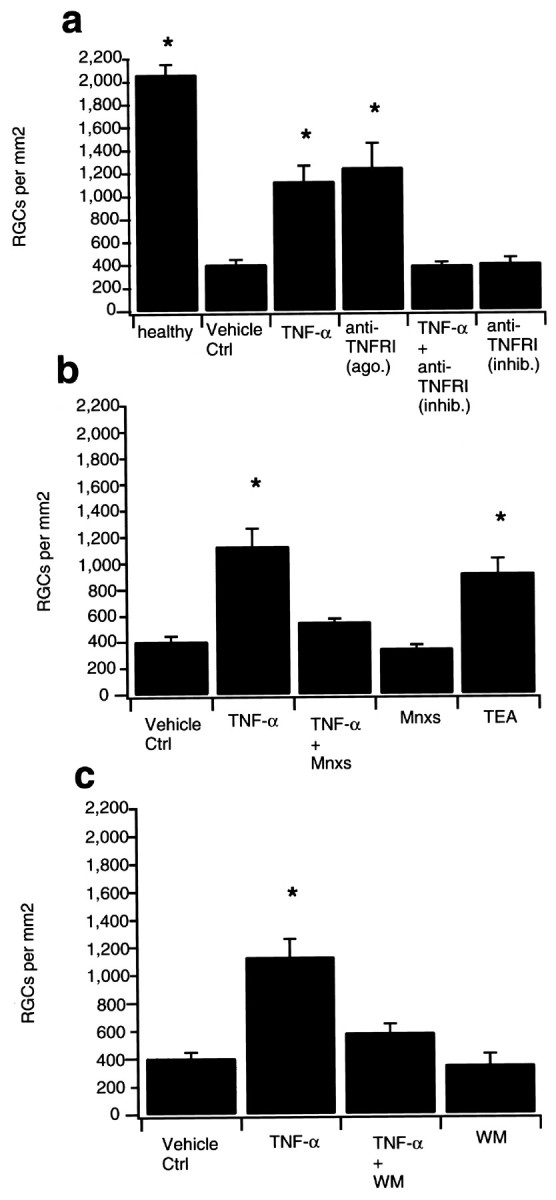
a, TNF-α protects axotomized RGCs from retrograde cell death via activation of the TNFRI. Data are given as the mean (± SEM) of retrogradely labeled RGCs per square millimeter 14 d after ON transection. The left bar shows RGC counts of unoperated healthy controls retrogradely labeled from the superior colliculus (healthy). Vehicle treatment (Vehicle Ctrl) with intraocular injection of 2 μl PBS on postlesional days 0, 4, 7, and 10 served as controls for the different treatment groups. Injection of TNF-α (TNF-α; 5 μg/ml) following the same protocol significantly increased the number of surviving RGCs. An antibody directed against the TNFRI with agonist activity [anti-TNFRI (ago.); 0.1 mg/ml; 2 μl per injection] had a similar rescue effect. TNF-α-induced RGC survival was completely blocked under a combined therapy with TNF-α and an antibody against the TNFRI with inhibitory properties [anti-TNFRI (inhib.); 0.1 mg/ml; 2 μl per injection]. Application of the
Fig. 1.
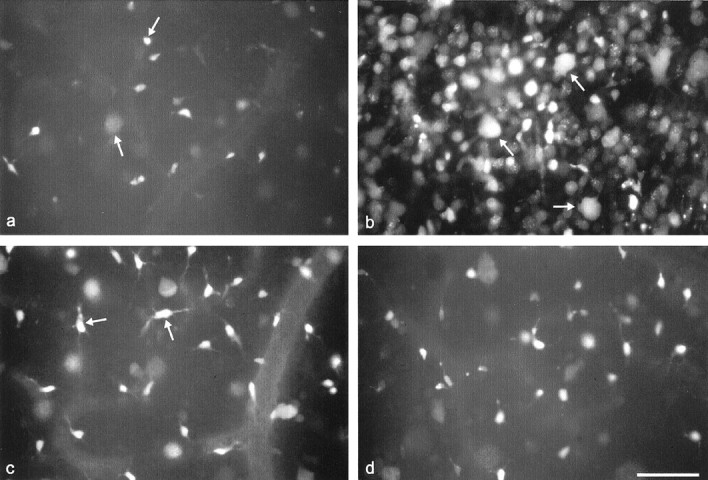
Retrogradely labeled RGCs. a,Axotomy without treatment. Note appearance of only a few FG-labeled RGCs that look pale or shrunken and hardly display any dendritic processes (arrows). b, Retinal whole-mounts intraocularly treated with 2 μl injections of TNF-α (5 μg/ml) on days 0, 4, 7, and 10 after ON transection. RGC counts are significantly increased with many regularly shaped somata (arrows). c, Combined treatment with TNF-α and Mnxs (400 μm; 2 μl per injection) on days 0, 4, 7, and 10 after axotomy. Mnxs blocks TNF-α-induced neuroprotection. Note the predominance of cells with irregularly sized and ramificated dendritic processes corresponding to microglia (arrows). d, Combined treatment with TNF-α and WM (0.1 mm; 2 μl per injection). WM abolishes TNF-α promoted RGC survival. Scale bar, 100 μm.
Potassium channel opening and inhibition of PI3-K block TNF-α-induced RGC rescue in vivo
Using the whole-cell patch-clamp technique, we have recently shown that acute application of TNF-α decreased potassium currents with delayed rectifying and A-type like characteristics in dissociated RGCs (Meyer, Diem, Wagner, Labes, and Bähr, unpublished observations). It has been demonstrated that attenuating outward potassium currents with conventional blockers, such as TEA reduced neuronal apoptosis in other cell culture models (Yu et al., 1997). To demonstrate the significance of the electrophysiological properties of TNF-α in ourin vivo model, we assessed RGC survival under a combined treatment of TNF-α (5 μg/ml) and Mnxs (400 μm; 2 μl injections each; n = 6). The additional intravitreal injection of the potassium channel opener significantly reduced the neuroprotective effect of TNF-α on RGCs 14 d after ON transection (549.9 ± 22.4 vs 1131.6 ± 131.9; p < 0.05; Fig. 2b). RGC counts under treatment with Mnxs alone (n = 5) did not differ significantly from control experiments (347.1 ± 26.3 vs 405.5 ± 39.72; Fig. 2b). To mimic the electrophysiological effect of TNF-α on potassium channels, TEA (300 mm) was injected intravitreously following the same treatment protocol (n = 4). TEA-induced rescue was not significantly different from RGC counts after treatment with TNF-α (920.5 ± 108.8 vs 1131.6 ± 131.9; Fig.2b).
In sympathetic neurons, it has been shown that membrane depolarization by enhanced concentrations of KCl was sufficient to activate the cell-protective RAS-PI3-K-Akt pathway (Vaillant et al., 1999). To test whether TNF-α-induced potassium current reduction leads to a downstream activation of this pathway in RGCs, we injected WM (0.1 mm; 2 μl injections) immediately after TNF-α on days 0, 4, 7, and 10 after the lesion. WM is a naturally occurring inhibitor of PI3-K and abolishes Akt phosphorylation and antiapoptotic effects of this signaling pathway in vitro (Alessi et al., 1996; Franke et al., 1997). We could recently demonstrate that intraocular injection of various doses of WM decreased Akt phosphorylation in untreated controls in vivo (Kermer et al., 2000). The additional injection of WM resulted in a significantly reduced RGC rescue compared with TNF-α treatment alone (n = 5; 585.5 ± 61.8 vs 1131.6 ± 131.9; p < 0.05; Fig. 2c) whereas intraocular injection of WM alone did not alter RGC counts after axotomy (n = 3; 353 ± 80; Fig.2c).
Minoxidilsulfate abolishes TNF-α-induced potassium current reduction in dissociated RGCs
The results described so far indicate that Mnxs blocks RGC rescue caused by TNF-α treatment after axotomy of the optic nerve in vivo. Extending our recent patch-clamp results, which showed a TNF-α-evoked reduction of potassium currents in dissociated RGCs to 49.7 ± 5.3% (Meyer, Diem, Wagner, Labes, and Bähr, unpublished observations), we now demonstrate that Mnxs abolishes this electrophysiological effect, suggesting a strong correlation between potassium current modification and cell viability. Voltage-gated outward potassium currents were analyzed at a depolarizing potential of +50 mV using the whole-cell configuration of the patch-clamp technique. Mean currents at the end of 20 msec pulses were used for analysis. Acute application of Mnxs (100 μm) together with TNF-α (5 ng/ml) led to a significantly smaller decrease in potassium currents than TNF-α application alone (85.9 ± 2.7%;n = 4; Figs.3a–d,4). This small reduction in amplitude was close to control values, in which superfusion with physiological salt solution alone caused a current decrease to 88.0 ± 6.4% (n = 8; Figs. 3g,h, 4). Application of Mnxs alone induced an increase in potassium currents to 119.3 ± 1.7% (n = 4; Figs.3e,f, 4). No changes in sodium peak amplitudes were observed in any experimental setting. Voltage dependence of the potassium channel activation as well as the activation kinetics were unchanged (data not shown).
Fig. 3.
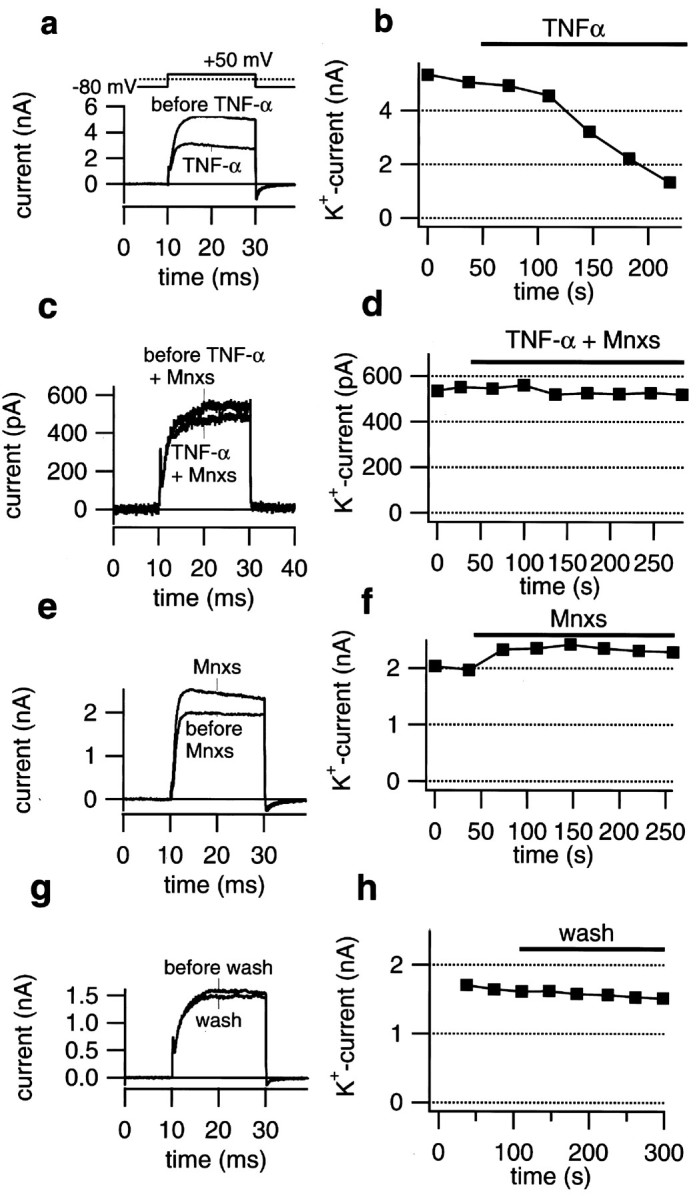
Minoxidilsulfate abolished TNF-α-evoked potassium current reduction in dissociated RGCs. a,Whole-cell current traces recorded at +50 mV before and during application of TNF-α (5 ng/ml). Application of TNF-α significantly decreased outward potassium currents. b, Mean outward potassium currents (K+current), measured at the end of the depolarizing pulses, are plotted versus time. Treatment with TNF-α resulted in an acute decrease of potassium currents beginning within the first 100 sec of the application. c, Whole-cell current traces recorded at +50 mV before and during simultaneous application of TNF-α (5 ng/ml) and minoxidilsulfate (Mnxs; 100 μm). Mnxs blocked TNF-α-induced reduction of outward potassium currents. d, Mean outward potassium currents are plotted versus time. Under a combined treatment of TNF-α and Mnxs, no significant changes in potassium current amplitudes were observed. e, Whole-cell current traces recorded at +50 mV before and during application of 100 μm Mnxs. Treatment with Mnxs resulted in a significant increase of outward potassium currents when compared with control values. f,Mean outward potassium currents, measured at the end of the depolarizing pulses, are plotted versus time. Treatment with Mnxs increased potassium current amplitudes immediately after beginning of the application. g, Whole-cell current traces recorded at +50 mV before and during superfusion with bath solution (Wash) served as control experiments. Under control conditions a small run down in potassium currents could be observed.h, Mean outward potassium currents are plotted versus time. Superfusion with bath solution resulted in a small decrease in potassium current amplitudes.
Fig. 4.

Quantification of outward potassium current changes. Data are given as the mean (± SEM) of alterations in potassium current amplitudes 150 sec after addition of compounds using the whole-cell patch-clamp technique. Absolute values are normalized so that the last value obtained before application is considered 100%. Acute application of TNF-α (TNF-α; 5 ng/ml) resulted in a significant decrease of outward potassium currents when compared with control values. Superfusion with bath solution (Wash) served as control experiments that showed a small run down of potassium current amplitude over the whole time period. Simultaneous application of 5 ng/ml TNF-α and 100 μmminoxidilsulfate (TNF-α+ Mnxs)abolished TNF-α-induced current reduction. Mnxs application alone (Mnxs; 100 μm) resulted in a significant increase of potassium current amplitudes compared with control experiments. *Statistically significant when compared withWash experiments (p < 0.05; one-way ANOVA followed by Duncan's test). The number of experiments are given in parentheses.
TNF-α induces Akt phosphorylation
Axotomy without treatment led to a decreased Akt phosphorylation compared with the contralateral control on postlesional day 4, as previously shown in our experimental paradigm by Western blot analysis (Kermer et al., 2000). Thereby it could be excluded that these results are caused by decreased levels of the inactive, unphosphorylated form of the Akt protein. On day 7 after axotomy, phospho-Akt concentrations of the lesioned side have reached the level of the corresponding contralateral control again. Western blot analysis of phospho-Akt was performed on days 4 and 7 after ON transection in axotomized retinas treated with 2 μl injections of TNF-α (5 μg/ml) on days 0 and 4 or 0, 4, and 7, respectively, as well as in the corresponding contralateral control retinas. As revealed by densitometric analysis of three paired samples corresponding three different animals in each experimental group, TNF-α treatment resulted in an increase in phospho-Akt in the lesioned eye on day 7 after axotomy (138.4 ± 9.1; mean ± SEM) compared with the contralateral control (75.0 ± 10.9; Fig. 5b). This effect was not seen after vehicle injection (data not shown). Cotreatment with 2 μl injections of Mnxs (400 μm) decreased Akt phosphorylation down to control levels (55.2 ± 0.7 vs 53.9 ± 8.8), as shown in Figure 5c, indicating that potassium current reduction was sufficient to induce phosphorylation of the Akt protein. Simultaneous application of the PI3-K inhibitor WM (0.1 mm; 2 μl per injection) together with TNF-α blocked the TNF-α-induced phosphorylation of Akt or even decreased phospho-Akt below control levels (50.0 ± 25.5 vs 79.7 ± 2.7; Fig. 5d).
Fig. 5.
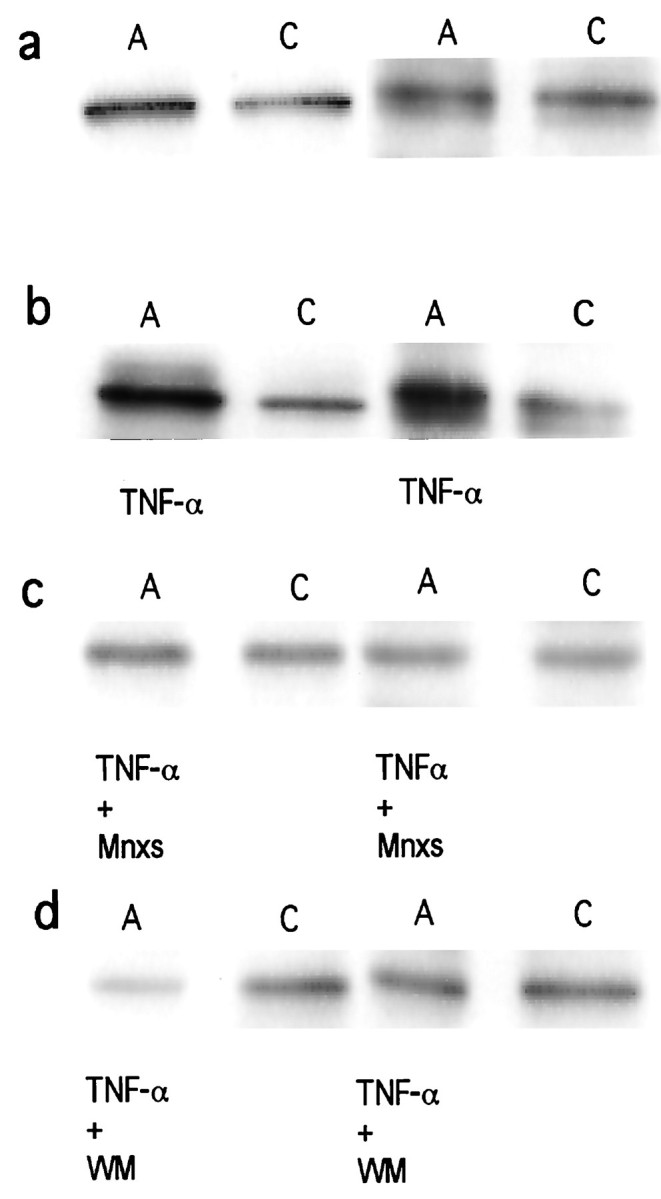
Western blot analysis of phospho-Akt.a–d, Each panel shows axotomized (A) and corresponding contralateral control (C) retinas of two independent animals on postlesional day 7. a, Axotomy without treatment (A) compared with the corresponding contralateral control (C). On day 7 after ON transection no differences in phospho-Akt levels could be detected. b,Axotomy treated with 2 μl injections of TNF-α (5 μg/ml) on days 0, 4, and 7 after axotomy (A; TNF-αbelow) compared with the corresponding contralateral control retina (C). Note the strong increase of phospho-Akt in axotomized retinas under treatment with TNF-α. c,Axotomized retinas treated with a combined therapy of TNF-α (5 μg/ml) and minoxidilsulfate (400 μm) on postlesional days 0, 4, and 7 (A; TNF-α+ Mnxsbelow) compared with the corresponding contralateral control (C). Mnxs abolishes TNF-α-induced Akt phosphorylation. d, Axotomy treated with TNF-α (5 μg/ml) and wortmannin (0.1 mm) on postlesional days 0, 4, and 7 (A; TNF-α+ WM below) compared with the corresponding contralateral control (C). Note the inhibition of Akt phosphorylation down to or even below control levels.
Phospho-Akt levels of retinas treated with TNF-α on postlesional day 4 reached control values, but did not show a further increase of Akt phosphorylation (data not shown).
TNF-α does not induce upregulation of BDNF
Western blot analysis of brain-derived neurotrophic factor (BDNF) was performed on days 4 and 7 after ON transection in axotomized retinas treated with 2 μl injections of TNF-α (5 μg/ml) on days 0 and 4 or 0, 4, and 7, respectively, as well as in the corresponding contralateral control retinas. In another set of experiments, unlesioned retinas treated with TNF-α following the same protocol were tested on days 4 and 7 including the contralateral controls. As revealed by densitometric analysis of three paired samples corresponding to three different animals in each experimental group, treatment with TNF-α did not induce upregulation of BDNF, neither after axotomy nor in unlesioned retinas, when compared with the corresponding contralateral control (data not shown).
TNF-α rescues RGCs from retrograde cell death in mixed retinal culture via potassium current reduction and Akt phosphorylation
It has been described that electrophysiological properties and ion channel expression of retinal ganglion cells or Müller glia can be affected by different culturing procedures and surrounding conditions of in vitro systems (Barres et al., 1988; Ishii et al., 1997). To investigate whether TNF-α-induced neuroprotection and its mechanisms depend on the in vivo pattern of connections among retinal cell types, we focused on postnatal day 6–16 retinal ganglion cells in a mixed retinal culture as an in vitro system. Retinal ganglion cells were identified by retrograde FG staining 2–3 d after injection of both superior colliculi and counted 2, 22, and 46 hr after plating. When mixed retinal culture cells were maintained in serum- and glutamine-supplemented medium, but in the absence of protective growth factors, >50% of ganglion cells degenerated during the first 22 hr, and >75% ganglion cell loss occurred during 46 hr incubation time. To perform these experiments, mixed retinal culture cells were treated with 5 ng/ml TNF-α over the whole culturing period, a concentration sufficient to obtain at least a 50% potassium current reduction in RGCs in the whole-cell patch-clamp mode (Meyer, Diem, Wagner, Labes, and Bähr, unpublished observations). For presentation in the text, absolute survival rates are normalized; the value obtained 2 hr after the culturing procedure is considered 100% survival for each group. TNF-α led to 96.2 ± 7.1% RGC survival 22 hr and 36.0 ± 2.2% survival 46 hr after plating, whereas vehicle treatment with PBS produced only 46.7 ± 7.7% RGC survival at 22 hr and 18.5 ± 2.3% survival at 46 hr (n = 4; Fig.6a). Combined treatment with TNF-α and Mnxs (100 μm) over the same time period blocked the TNF-α-induced neuroprotection (40.7 ± 2.8% survival after 22 hr and 22.4 ± 0.9% after 46 hr incubation time; n = 4; Fig. 6b), whereas Mnxs alone did not reduce RGC counts below control values (data not shown). TNF-α treatment in combination with WM (0.1 mm) reduced RGC rescue to 52.3 ± 5.6% after 22 hr and 28.0 ± 4.2% after 46 hr incubation time as shown in Figure 6d, whereas again results from WM application alone did not significantly differ from control experiments (data not shown). Potassium current reduction by TEA (100 μm) produced 71.0 ± 3.1% RGC survival after 22 hr and 33.5 ± 4.0% survival after 46 hr incubation time (n = 4; Fig. 6c).
Fig. 6.
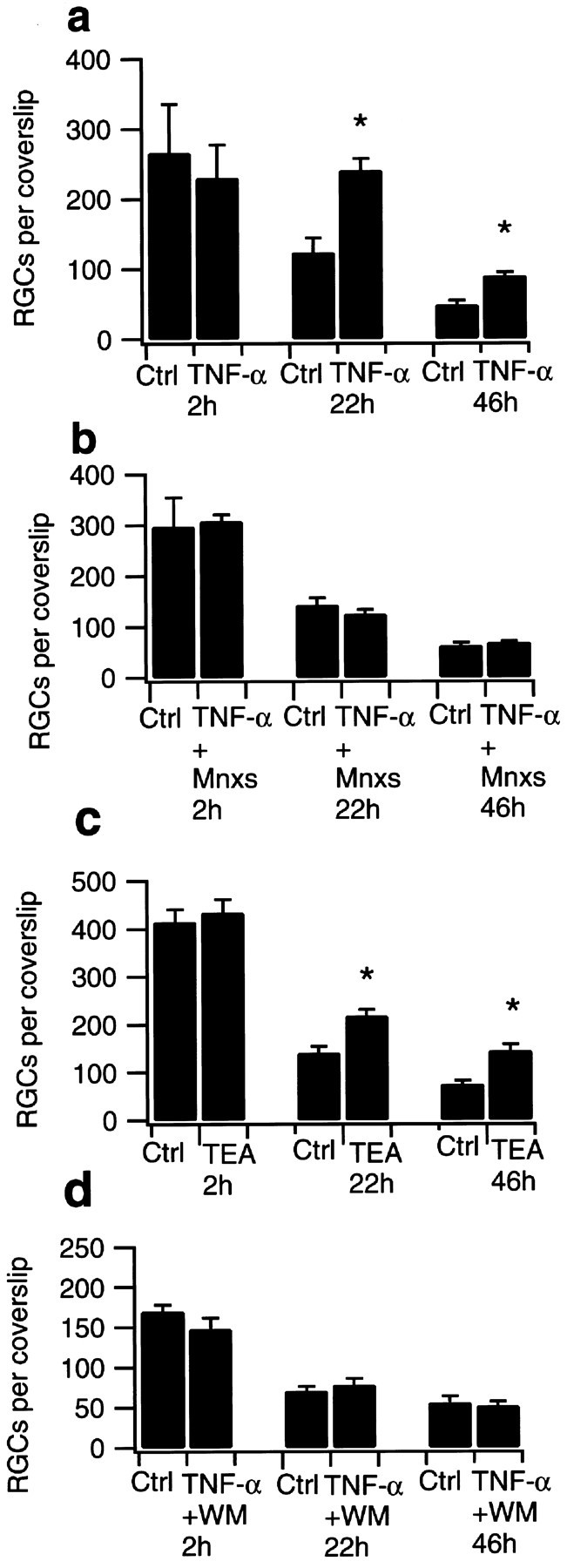
RGC survival in mixed retinal culture. Data are given as the mean (± SEM) of retrogradely labeled RGCs per 9 mm coverslip counted 2, 22, and 46 hr after plating. For each treatment group and time, four coverslips were counted. Tested substances were added to the medium before and 20 hr after plating. a–drepresent independent experiments. Differences in absolute cell counts result from variability of RGC numbers in different animals and fluctuations in culturing procedure. a, RGC counts under TNF-α treatment (5 ng/ml) did not show significant differences 2 hr after plating procedure when compared with control values. Treatment with TNF-α significantly increased the number of surviving RGCs after 22 and 46 hr. RGCs cultured in normal medium derived from the same preparation served as controls (Ctrl).b, Simultaneous treatment with TNF-α (5 ng/ml) and minoxidilsulfate (Mnxs; 100 μm) abolished TNF-α-induced RGC rescue after 22 and 46 hr. Application of Mnxs (100 μm) alone did not show significant differences when compared with control values (data not shown). c,Culturing in tetraethylammonium (TEA; 100 μm) resulted in significantly increased RGC survival after 22 and 46 hr when compared with control values. No differences in RGC counts were detected 2 hr after plating. RGCs cultured under standard conditions derived from the same preparation served as controls (Ctrl). d, Simultaneous treatment with TNF-α (5 ng/ml) and wortmannin (WM; 0.1 mm) blocked TNF-α-induced RGC rescue after 22 and 46 hr. RGCs cultured in WM (0.1 mm) alone did not show significant differences in counts when compared with control values (data not shown). *Statistically significant when compared with control counts (p < 0.05; Student's ttest).
DISCUSSION
Recently we have demonstrated that TNF-α inhibits outward potassium currents in dissociated RGCs via activation of the TNFRI by using the patch-clamp technique (Meyer, Diem, Wagner, Labes, and Bähr, unpublished observations). In the present study, we relate its electrophysiological properties to neuroprotection. We show that TNF-α rescues RGCs from retrograde cell death after axonal lesion in an in vivo paradigm. For the first time, we demonstrate that the neuroprotective action of TNF-α in vivo is most likely mediated by potassium current reduction and activation of the antiapoptotic PI3-K–Akt pathway.
Fiber tract lesion of the retinocollicular projection in the rat is an established model to investigate secondary neuronal loss in degenerative processes of the mammalian CNS because of its good surgical accessibility and well known kinetics of cell death (Villegas-Perez et al., 1988; Mey and Thanos, 1993; Bähr and Bonhoeffer, 1994; Bähr and Wizenmann, 1996; Klöcker et al., 1997, 1998; Kermer et al., 1999a, 2000). Transection of the ON induces a delayed death of 80–90% of RGCs within 14 d, starting around day 4 and reaching a maximum on day 7 after axotomy (Eschweiler and Bähr, 1993; Mansour-Robaey et al., 1994). It has been shown that this retrograde cell death can be ascribed to apoptosis (Garcia-Valenzuela et al., 1994; Rabacchi et al., 1994; Isenmann et al., 1997). Caspase-3 (CPP32)-like protease has been identified as an important mediator of apoptotic RGC death in our experimental paradigm (Kermer et al., 1998, 1999b). Morphological changes common to apoptotic cells include cell shrinkage. These alterations in cell volume are typically mediated by changes in the level of potassium as the predominant intracellular ion (Barbiero et al., 1995; Beauvais et al., 1995; Bortner and Cidlowski, 1996; Benson et al., 1996). Loss of intracellular potassium has been proposed as an early event in programmed cell death. Elevation of extracellular potassium as well as attenuation of outward potassium currents prevents this death (Galli et al., 1995; De Luca et al., 1996; Yu et al., 1997). The hypothesis that TNF-α might have a neuroprotective effect on RGCs was based on the electrophysiological observation that acute application of TNF-α reduced both types of potassium currents present in RGCs, a delayed rectifying current that inactivates slowly, and a fast inactivating A-type current (Meyer, Diem, Wagner, Labes, and Bähr, unpublished observations). When mimicking the TNF-α-induced potassium current reduction by using TEA, a similar rescue effect could be achieved in our in vivo as well as our in vitro model. Counteracting the electrophysiological effect of TNF-α with Mnxs, an ATP-sensitive potassium channel opener (Schwanstecher et al., 1998), abolished RGC rescue in both experimental settings. In contrast to our findings, survival promoting effects of potassium channel activators have been described for cultured hippocampal neurons that were protected against oxidative injury and amyloid β-peptide toxicity under therapy with diazoxide, levocromakalim, and pinacidil (Goodman and Mattson, 1996). Treatment with cromakalim and diazoxide also abolished fluctuations in intracellular calcium levels and associated cell death of hippocampal pyramidal neurons (Abele et al., 1990). In these model systems, potassium channel activators are expected to act neuroprotective by hyperpolarizing the membrane and thereby raising the threshold for induction of excitotoxicity. In our experimental setting, treatment with the NMDA antagonist memantine did not result in significant rescue of axotomized RGCs in vivo, indicating that excitotoxicity does not play a major role in retrograde cell death after axotomy of the optic nerve (Klöcker et al., 1999). This is in good agreement with results demonstrating even adverse effects of the NMDA antagonist MK-801 on the survival of axotomized RGCs (Schmitt and Sabel, 1996). For similar reasons, TNF-α-induced decrease of NMDA- and AMPA-dependent currents described in hippocampal neurons (Furukawa and Mattson, 1998) appears to be an alternative neuroprotective signaling pathway but should not account for neuronal rescue under our experimental conditions.
For the present findings, cell rescue caused by other cytokines or soluble factors stimulated by TNF-α under in vivoconditions seems unlikely, because neuroprotection was completely blocked after simultaneous injection of TNF-α and an antibody against the TNFRI with inhibitory properties. This does not completely exclude upregulation of soluble factors via de novo protein synthesis after TNFRI activation. However, the time course with immediate onset of the electrophysiological changes after TNF-α application seen in our patch-clamp experiments renders this mechanism unlikely. One the other hand, this would not exclude the possibility that the electrophysiological changes ascribed to TNF-α contribute to synthesis of other survival-promoting factors such as BDNF. Recently, it has been described that the neuroprotective actions of interleukin-6, another proinflammatory cytokine that can influence neurotransmitter-operated ion currents (Qiu et al., 1995), on embryonic sensory neurons are mediated through a mechanism requiring endogenous BDNF (Murphy et al., 2000). Exogenously applied BDNF has been shown previously to prevent secondary RGC loss in our experimental paradigm (Klöcker et al., 1998). To test a possible upregulation of BDNF under treatment with TNF-α, Western blots were performed on postlesional days 4 and 7, which did not detect a TNF-α-induced increase of BDNF levels. However, in spite of unchanged BDNF levels, peptide trophic factor action may play an additional role in depolarization-induced neuronal survival. For cultured RGCs, it has been shown that depolarization with high extracellular potassium could increase surface levels of the neurotrophin receptor TrkB (Meyer-Franke et al., 1998).
Application of the inhibitory antibody directed against the TNFRI alone did not reduce RGC counts under control values, indicating that endogenous TNF-α does not play an active part in RGC survival after ON transection. This observation could be explained by much higher concentrations achieved under treatment with exogenous TNF-α. TNF-α concentrations applied in our in vivo setting were higher than those electrophysiologically tested or those used for mixed retinal culture experiments because, under in vivoconditions, biological half life of an relatively small protein as well as dilution effects were difficult to predict.
An alternative explanation would be the species specificity of TNF-α. It has been described that the TNFRI in rodents binds both rodent and human TNF-α, whereas the TNFRII only binds rodent TNF-α (Fiers, 1991). In our experimental setting, recombinant human TNF-α was chosen because it had less toxic systemic side effects after intraocular injection (R. Diem, unpublished observations). Although the presence of the TNFRII on RGCs could be excluded by RT-PCR (Meyer, Diem, Wagner, Labes, and Bähr, unpublished observations), we cannot exclude differences in TNFRI binding affinity or activation of signaling pathways between both types of TNF-α.
The neuroprotective effect of membrane depolarization caused by elevated extracellular potassium or attenuated outward potassium currents has often been ascribed to increased calcium influx through voltage-dependent calcium channels. It has been shown that the L-type calcium channel blocker nifedipine was able to inhibit the survival effect of high extracellular potassium on cerebellar granule cells, whereas application of an L-type agonist enhanced cell rescue (Galli et al., 1995). Depolarization induced by high extracellular potassium elevates cAMP levels in RGCs by activating a calcium-dependent type-1 adenylyl cyclase and thereby enhances their trophic responsiveness (Meyer-Franke et al., 1995). Activation of the antiapoptotic PI3-K-dependent Akt-pathway may provide another downstream mechanism for mediating the survival effects of membrane depolarization-induced elevated intracellular calcium as shown for cultured sympathetic neurons, which require nerve growth factor and neural activity for survival (Vaillant et al., 1999). Whereas the role of calcium in our lesion model is under current investigation, we here examined the phosphorylation of Akt as a consequence of TNF-α-induced potassium current reduction. Recent in vitro data indicate that TNF-α can activate the PI3-K-Akt pathway and that this mechanism is involved in the activation of the nuclear transcription factor NFκB (Nidai Ozes et al., 1999). Phospho-Akt in turn can phosphorylate and thereby inactivate the proapoptotic protein Bad (Datta et al., 1997) as well as unprocessed or active caspase-9 (Cardone et al., 1998), which leads to decreased levels of the downstream effector caspase-3 (Li et al., 1997). In our experimental setting, Western blot analysis of axotomized retinas revealed a phosphorylation of Akt after TNF-α treatment when compared with the corresponding contralateral control. The hypothesis that TNF-α-induced potassium current reduction and phosphorylation of the Akt protein are associated in a linear order was supported by the observation that combined treatment with TNF-α and the potassium channel opener Mnxs or the PI3-K inhibitor WM, respectively, decreased TNF-α promoted RGC survival to almost identical counts in our in vivo setting. This theory was confirmed by Western blot analysis of axotomized retinas treated with a combined therapy of TNF-α and Mnxs, which showed a decrease of phospho-Akt to control levels.
Calcium influx after KCl-induced membrane depolarization has been demonstrated to activate Ras via the neuronal exchange factor Ras-GRF (Farnsworth et al., 1995). Ras may be the link between calcium influx and PI3-K/Akt as shown for cultured sympathetic neurons (Vaillant et al., 1999). We are aware that we provide only indirect evidence for membrane depolarization, and the involvement of calcium influx as well as the activation of Ras remains speculative. An additional direct interaction between the TNFRI and the PI3-K-Akt pathway as well as the presence of not yet characterized interrelated signal transduction steps cannot be excluded at present.
Experimental evidence indicates that the level of intracellular potassium regulates the apoptotic process by controlling the activity of two key apoptotic enzymes, caspase-3 and the internucleosomal DNA cleavage nuclease (Hughes et al., 1997). It is not clear how potassium levels inhibit caspase-3 activity. Proteases have been demonstrated to be sensitive to ionic strength in many aspects including their activity and conformational state (Polgar and Patthy, 1992; Polgar, 1995). However, in the context of the above results, it seems reasonable to also consider other multistep pathways.
Taken together, our present results demonstrate that TNF-α is a survival factor for axotomized RGCs in an in vivo model. Moreover, our study relates TNF-α-induced electrophysiological changes in outward potassium currents to Akt phosphorylation and neuroprotection in vivo. Future studies are necessary to examine the role of calcium as a connection between membrane-related potassium current changes and intracellular signal transduction pathways in our lesion model.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant Ba 949/12-2 and the Herman-and-Lilly-Schilly Foundation (M.B.).We thank B. Kramer and S. Thomsen for excellent technical assistance. We also thank U. Förstermann and G. Dietz for critically reading this manuscript.
Correspondence should be addressed to Ricarda Diem, Neurologische Universitätsklinik, Verfügungsgebäude, Auf der Morgenstelle 15, D-72076 Tübingen, Germany. E-mail:ricarda.diem@uni-tuebingen.de.
REFERENCES
- 1.Abele AE, Miller RJ. Potassium channel activators abolish excitotoxicity in cultured hippocampal pyramidal neurons. Neurosci Lett. 1990;115:195–200. doi: 10.1016/0304-3940(90)90454-h. [DOI] [PubMed] [Google Scholar]
- 2.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanisms of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 3.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 4.Bähr M, Bonhoeffer F. Perspectives on axonal regeneration in the mammalian CNS. Trends Neurosci. 1994;17:473–479. doi: 10.1016/0166-2236(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 5.Bähr M, Wizenmann A. Retinal ganglion cell axons recognize specific guidance cues present in the deafferented adult rat superior colliculus. J Neurosci. 1996;16:5106–5116. doi: 10.1523/JNEUROSCI.16-16-05106.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbiero G, Duranti F, Bonelli G, Amenta JS, Baccino FM. Intracellular ionic variations in the apoptotic death of L cells by inhibitors of cell cycle progression. Exp Cell Res. 1995;217:410–418. doi: 10.1006/excr.1995.1104. [DOI] [PubMed] [Google Scholar]
- 7.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 8.Barres BA, Silverstein BE, Corey DP, Chun LL. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron. 1988;1:791–803. doi: 10.1016/0896-6273(88)90127-4. [DOI] [PubMed] [Google Scholar]
- 9.Beauvais F, Michel L, Dubertret L. Human eosinophils in culture undergo a striking and rapid shrinkage during apoptosis. Role of K+ channels. J Leukoc Biol. 1995;57:851–855. doi: 10.1002/jlb.57.6.851. [DOI] [PubMed] [Google Scholar]
- 10.Benson RS, Heer S, Dive C, Watson AJ. Characterization of cell volume loss in CEM-c7A cells during dexamethasone-induced apoptosis. Am J Physiol. 1996;270:C1190–C1203. doi: 10.1152/ajpcell.1996.270.4.C1190. [DOI] [PubMed] [Google Scholar]
- 11.Bogdan I, Leib SL, Bergeron M, Chow L, Täuber MG. Tumor necrosis factor-α contributes to apoptosis in hippocampal neurons during experimental group B streptococcal meningitis. J Infect Dis. 1997;176:693–697. doi: 10.1086/514092. [DOI] [PubMed] [Google Scholar]
- 12.Bortner CD, Cidlowski JA. Absence of volume regulatory mechanisms contributes to the rapid activation of apoptosis in thymocytes. Am J Physiol. 1996;271:C950–C961. doi: 10.1152/ajpcell.1996.271.3.C950. [DOI] [PubMed] [Google Scholar]
- 13.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 14.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 16.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 17.Dawson DA, Martin D, Hallenbeck JM. Inhibition of tumor necrosis factor-alpha reduces focal cerebral ischemic injury in the spontaneously hypertensive rat. Neurosci Lett. 1996;218:41–44. doi: 10.1016/0304-3940(96)13116-5. [DOI] [PubMed] [Google Scholar]
- 18.De Luca A, Weller M, Fontana A. TGF-beta-induced apoptosis of cerebellar granule neurons is prevented by depolarization. J Neurosci. 1996;16:4174–4185. doi: 10.1523/JNEUROSCI.16-13-04174.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eschweiler GW, Bähr M. Flunarizine enhances rat retinal ganglion cell survival after axotomy. J Neurol Sci. 1993;116:34–40. doi: 10.1016/0022-510x(93)90086-e. [DOI] [PubMed] [Google Scholar]
- 20.Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature. 1995;376:524–527. doi: 10.1038/376524a0. [DOI] [PubMed] [Google Scholar]
- 21.Fiers W. Tumor necrosis factor. Characterization at the molecular, cellular and in vivo level. FEBS Lett. 1991;285:199–212. doi: 10.1016/0014-5793(91)80803-b. [DOI] [PubMed] [Google Scholar]
- 22.Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, Gelbard HA. Tumor necrosis factor α inhibits glutamate uptake by primary human astrocytes. J Biol Chem. 1996;271:15303–15306. doi: 10.1074/jbc.271.26.15303. [DOI] [PubMed] [Google Scholar]
- 23.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKT ion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 24.Furukawa K, Mattson MP. The transcription factor NF-κB mediates increases in calcium currents and decreases in NMDA- and AMPA/kainate-induced currents induced by tumor necrosis factor-α in hippocampal neurons. J Neurochem. 1998;70:1876–1886. doi: 10.1046/j.1471-4159.1998.70051876.x. [DOI] [PubMed] [Google Scholar]
- 25.Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Valenzuela E, Gorczyca W, Darzynkiewicz Z, Sharma SC. Apoptosis in adult retinal ganglion cells after axotomy. J Neurobiol. 1994;25:431–138. doi: 10.1002/neu.480250408. [DOI] [PubMed] [Google Scholar]
- 27.Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab. 1998;18:1283–1287. doi: 10.1097/00004647-199812000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Goodman Y, Mattson MP. K+ channel openers protect hippocampal neurons against oxidative injury and amyloid β-peptide toxicity. Brain Res. 1996;706:328–332. doi: 10.1016/0006-8993(95)01367-9. [DOI] [PubMed] [Google Scholar]
- 29.Guenther E, Schmid S, Grantyn R, Zrenner E. In vitro identification of retinal ganglion cells in culture without the need of dye labeling. J Neurosci Methods. 1994;51:177–181. doi: 10.1016/0165-0270(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 30.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 31.Houzen H, Kikuchi S, Kanno M, Shinpo K, Tashiro K. Tumor necrosis factor enhancement of transient outward potassium currents in cultured rat cortical neurons. J Neurosci Res. 1997;50:990–999. doi: 10.1002/(SICI)1097-4547(19971215)50:6<990::AID-JNR9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 32.Hughes FM, Bortner CD, Purdy GD, Cidlowski JA. Intracellular K+ suppresses the activation of apoptosis in lymphocytes. J Biol Chem. 1997;272:30567–30576. doi: 10.1074/jbc.272.48.30567. [DOI] [PubMed] [Google Scholar]
- 33.Isenmann S, Wahl C, Krajewski S, Reed JC, Bähr M. Upregulation of Bax protein in degenerating retinal ganglion cells precedes apoptotic cell death after optic nerve lesion in the rat. Eur J Neurosci. 1997;9:1763–1772. doi: 10.1111/j.1460-9568.1997.tb01534.x. [DOI] [PubMed] [Google Scholar]
- 34.Ishii M, Horio Y, Tada Y, Hibino H, Inanobe A, Ito M, Yamada M, Gotow T, Uchiyama Y, Kurachi Y. Expression and clustered distribution of an inwardly rectifying potassium channel, KAB-2/Kir4.1, on mammalian retinal Müller cell membrane: their regulation by insulin and laminin signals. J Neurosci. 1997;17:7725–7735. doi: 10.1523/JNEUROSCI.17-20-07725.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kermer P, Klöcker N, Labes M, Bähr M. Inhibition of CPP32-like proteases rescues axotomized retinal ganglion cells from secondary cell death in vivo. J Neurosci. 1998;18:4656–4662. doi: 10.1523/JNEUROSCI.18-12-04656.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kermer P, Klöcker N, Bähr M. Long-term effect of inhibition of ced 3-like caspases on the survival of axotomized retinal ganglion cells in vivo. Exp Neurol. 1999a;158:202–205. doi: 10.1006/exnr.1999.7094. [DOI] [PubMed] [Google Scholar]
- 37.Kermer P, Klöcker N, Labes M, Thomsen S, Srinivasan A, Bähr M. Activation of caspase-3 in axotomized rat retinal ganglion cells in vivo. FEBS Lett. 1999b;453:361–364. doi: 10.1016/s0014-5793(99)00747-4. [DOI] [PubMed] [Google Scholar]
- 38.Kermer P, Klöcker N, Labes M, Bähr M. Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent Akt phosphorylation and inhibition of caspase-3 in vivo. J Neurosci. 2000;20:722–728. [PubMed] [Google Scholar]
- 39.Klinkert WE, Kojima K, Lesslauer W, Rinner W, Lassmann H, Wekerle H. TNF-alpha receptor fusion protein prevents experimental auto-immune encephalomyelitis and demyelination in Lewis rats: an overview. J Neuroimmunol. 1997;72:163–168. doi: 10.1016/s0165-5728(96)00183-x. [DOI] [PubMed] [Google Scholar]
- 40.Klöcker N, Bräunling F, Isenmann S, Bähr M. In vivo neurotrophic effects of GDNF on axotomized retinal ganglion cells. NeuroReport. 1997;8:3439–3442. doi: 10.1097/00001756-199711100-00005. [DOI] [PubMed] [Google Scholar]
- 41.Klöcker N, Cellerino A, Bähr M. Free radical scavenging and inhibition of nitric oxide synthase potentiates the neurotrophic effects of brain-derived neurotrophic factor on axotomized retinal ganglion cells in vivo. J Neurosci. 1998;18:1038–1046. doi: 10.1523/JNEUROSCI.18-03-01038.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klöcker N, Kermer P, Gleichmann M, Weller M, Bähr M. Both the neuronal and inducible isoforms contribute to upregulation of retinal nitric oxide synthase activity by brain-derived neurotrophic factor. J Neurosci. 1999;19:8517–8527. doi: 10.1523/JNEUROSCI.19-19-08517.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leifer D, Lipton SA, Barnstable CJ, Masland RH. Monoclonal antibody to Thy-1 enhances regeneration of processes by rat retinal ganglion cells in culture. Science. 1984;224:303–306. doi: 10.1126/science.6143400. [DOI] [PubMed] [Google Scholar]
- 44.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 45.Lipton SA, Tauck DL. Voltage-dependent conductances of solitary ganglion cells dissociated from the rat retina. J Physiol (Lond) 1987;385:361–391. doi: 10.1113/jphysiol.1987.sp016497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mansour-Robaey S, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc Natl Acad Sci USA. 1994;91:1632–1636. doi: 10.1073/pnas.91.5.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matthews N, Neale ML. Lymphokines and interferons, a practical approach. (Clemens MJ, Morris AG, Gearing AJH, eds), p 296. IRL; Oxford: 1987. [Google Scholar]
- 48.Meistrell ME, Botchkina GI, Wang H, Di Santo E, Cockroft KM, Bloom O, Vishnubhakat JM, Ghezzi P, Tracey KJ. Tumor necrosis factor is a brain damaging cytokine in cerebral ischemia. Shock. 1997;8:341–348. [PubMed] [Google Scholar]
- 49.Mey J, Thanos S. Intravitreal injections of neurotrophic factors support the survival of axotomized retinal ganglion cells in adult rats in vivo. Brain Res. 1993;602:304–317. doi: 10.1016/0006-8993(93)90695-j. [DOI] [PubMed] [Google Scholar]
- 50.Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- 51.Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG, Reichhardt LF, Barres BA. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–693. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy PG, Borthwick LA, Altares M, Gauldie J, Kaplan D, Richardson PM. Reciprocal actions of interleukin-6 and brain-derived neurotrophic factor on rat and mouse primary sensory neurons. Eur J Neurosci. 2000;12:1891–1899. doi: 10.1046/j.1460-9568.2000.00074.x. [DOI] [PubMed] [Google Scholar]
- 53.Nawashiro H, Martin D, Hallenbeck JM. Inhibition of tumor necrosis factor and amelioration of brain infarction in mice. J Cereb Blood Flow Metab. 1997;17:229–232. doi: 10.1097/00004647-199702000-00013. [DOI] [PubMed] [Google Scholar]
- 54.Nidai Ozes O, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-κB activation by tumor necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–89. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 55.Pastorino JG, Tafani M, Farber JL. Tumor necrosis factor induces phosphorylation and translocation of BAD through a phosphatidylinositide-3-OH kinase-dependent pathway. J Biol Chem. 1999;274:19411–19416. doi: 10.1074/jbc.274.27.19411. [DOI] [PubMed] [Google Scholar]
- 56.Polgar L. Effects of ionic strength on the catalysis and stability of prolyl oligopeptidase. Biochem J. 1995;312:267–271. doi: 10.1042/bj3120267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Polgar L, Patthy A. Cleavage of the Lys 196-Ser197 bond prolyl oligopeptidase: enhanced catalytic activity for one of the two active enzyme forms. Biochemistry. 1992;31:10769–10773. doi: 10.1021/bi00159a018. [DOI] [PubMed] [Google Scholar]
- 58.Qiu Z, Parsons KL, Gruol DL. Interleukin-6 selectively enhances the intracellular calcium response to NMDA in developing CNS neurons. J Neurosci. 1995;15:6688–6699. doi: 10.1523/JNEUROSCI.15-10-06688.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rabacchi SA, Bonfanti L, Liu XH, Maffei L. Apoptotic cell death induced by optic nerve lesion in the neonatal rat. J Neurosci. 1994;14:5292–5301. doi: 10.1523/JNEUROSCI.14-09-05292.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rath PC, Aggarwal BB. TNF-induced signaling in apoptosis. J Clin Immunol. 1999;19:350–364. doi: 10.1023/a:1020546615229. [DOI] [PubMed] [Google Scholar]
- 61.Reddy SA, Huang JH, Liao WS. Phosphatidylinositol 3-kinase as a mediator of TNF-induced NF-kappa B activation. J Immunol. 2000;164:1355–1363. doi: 10.4049/jimmunol.164.3.1355. [DOI] [PubMed] [Google Scholar]
- 62.Schmitt U, Sabel BA. MK-801 reduces retinal ganglion cell survival but improves visual performance after controlled optic nerve crush. J Neurotrauma. 1996;13:791–800. doi: 10.1089/neu.1996.13.791. [DOI] [PubMed] [Google Scholar]
- 63.Schwanstecher M, Sieverding C, Dorschner H, Gross I, Aguilar-Bryan L, Schwanstecher C, Bryan J. Potassium channel opener require ATP to bind to and act through sulfonylurea receptors. EMBO J. 1998;17:5529–5535. doi: 10.1093/emboj/17.19.5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shi B, Raina J, Lorenzo A, Busciglio J, Gabuzda D. Neuronal apoptosis induced by HIV-1 Tat protein and TNF-alpha: potentiation of neurotoxicity mediated by oxidative stress and implications for HIV-1 dementia. J Neurovirol. 1998;4:281–290. doi: 10.3109/13550289809114529. [DOI] [PubMed] [Google Scholar]
- 65.Shinpo K, Kikuchi S, Moriwaka F, Tashiro K. Protective effects of the TNF-ceramide pathway against glutamate neurotoxicity on cultured mesencephalic neurons. Brain Res. 1999;819:170–173. doi: 10.1016/s0006-8993(98)01354-7. [DOI] [PubMed] [Google Scholar]
- 66.Shohami E, Bass R, Wallach D, Yamin A, Gallily R. Inhibition of tumor necrosis factor alpha (TNFalpha) activity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab. 1996;16:378–384. doi: 10.1097/00004647-199605000-00004. [DOI] [PubMed] [Google Scholar]
- 67.Shohami E, Gallily R, Mechoulam R, Bass R, Ben Hur T. Cytokine production in the brain following closed head injury: dexabinol (HU-211) is a novel TNF-alpha inhibitor and an effective neuroprotectant. J Neuroimmunol. 1997;72:169–177. doi: 10.1016/s0165-5728(96)00181-6. [DOI] [PubMed] [Google Scholar]
- 68.Tamatani M, Che YC, Matsuzaki H, Ogawa S, Okado H, Miyake S, Mizuno T, Tohyama M. Tumor necrosis factor induces bcl-2 and bcl-x expression through NFκB activation in primary hippocampal neurons. J Biol Chem. 1998;274:8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- 69.Vaillant AR, Mazzoni I, Tudan C, Boudreau M, Kaplan DR, Miller FD. Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-Akt pathway to synergistically regulate neuronal survival. J Cell Biol. 1999;146:955–966. doi: 10.1083/jcb.146.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Villegas-Perez MP, Vidal Sanz M, Bray GM, Aguayo AJ. Influences of peripheral nerve grafts on the survival and regrowth of axotomized retinal ganglion cells in adult rats. J Neurosci. 1988;8:265–280. doi: 10.1523/JNEUROSCI.08-01-00265.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]