

Abstract
It has been found that insulin-like growth factor I (IGF-I) exerts cytoprotection against Aβ amyloid-induced neuronal cell death. Deposits of Aβ amyloid are one of the pathological hallmarks of Alzheimer's disease (AD). Here, we examined whether IGF-I exerts protective activity against cell death induced by a familial AD (FAD)-linked mutant of amyloid precursor protein (APP), and we found that IGF-I protected cells from toxicity of FAD-associated V642I mutant of APP in multiple cell systems. IGFBP-3 blocked this action of IGF-I, but not of des(1-3)IGF-I, which was as active as IGF-I in the presence of IGFBP-3. The data also demonstrated that the IGF-I receptor (IGF-IR) mediates the protective activity of IGF-I. The antagonizing function of the IGF-I/IGF-IR system against V642I-APP, which is further antagonized by IGFBP-3, provides a molecular clue to the understanding of AD pathophysiology and to the establishment of potential therapy for AD.
Keywords: IGF-I, IGFBP, des(1-3)IGF-I, amyloid precursor protein, Alzheimer's disease, neuroprotection
Alzheimer's disease (AD) is the most prevalent neurodegenerative disease pathologically characterized by extracellular Aβ amyloid plaques, intracellular neurofibrillary tangles, and extensive neuronal death. Amyloid precursor protein (APP) is the precursor of Aβ with a single transmembrane structure. V642I/F/G mutations in APP695 (the numbering is based onKang et al., 1987) were the first established causes of familial AD (FAD) (Hardy, 1992). It has been found that any of the three V642I/F/G mutants of APP causes cell death, when the cognate cDNAs are expressed in NK1 cells, a neuron-like transformant of COS cells (Yamatsuji et al., 1996a; Giambarella et al., 1997a,b), in neuron/neuroblastoma hybrid F11 cells (Yamatsuji et al., 1996b; Hashimoto et al., 2000a), in PC12 cells (Wolozin et al., 1996; Zhao et al., 1997), and in primary cultured central neurons (Luo et al., 1999). Subsequent studies (Yamatsuji et al., 1996a,b; Wolozin et al., 1996;Giambarella et al., 1997a; Hashimoto et al., 2000a) have clarified that V642I-APP, K595N/M596L-APP, and N141I-presenilin (PS)-2, the genes causing FAD, induce neuronal cell death through pertussis toxin (PTX)-sensitive manner, suggesting that intracellular signal transducers mediate the cytotoxicity of these FAD gene products. Although one potential mechanism underlying the neurotoxicity of the FAD genes could be that neuronal death occurs by cytotoxic Aβ (Loo et al., 1993), particularly Aβ1-42, whose secretion is stimulated by the FAD genes (Citron et al., 1992; Cai et al., 1993; Suzuki et al., 1994; Yamatsuji et al., 1996b), even Aβ triggers intracellular signaling mechanisms (Mark et al., 1997; Pike et al., 1997; Lee et al., 2000; Miranda et al., 2000; Nakagawa et al., 2000) to exert its neurotoxicity. Therefore, effective countermeasures against neuronal cell death in these types of FAD might be feasible even after Aβ deposition if the countermeasures could suppress death signals inside the neurons expressing the FAD genes.
A few years ago, Dore et al. (1997) found that insulin-like growth factor-I (IGF-I) exerts protective activity against Aβ-induced neurotoxicity and suggested that IGF-I could be a new therapeutic reagent for AD. However, no information has become available regarding whether IGF-I is effective in protection against cell death induced by FAD genes. Also, although those authors assumed the involvement of the IGF-I receptor (IGF-IR) in neuroprotection by IGF-I, based mainly on the lower potencies of IGF-II and insulin, no direct evidence was provided. Identifying the target receptor for the action of IGF-I in antagonizing AD-related insults is extremely important not only to understand the protection mechanism but also in creating small-molecular weight IGF-I mimetics with anti-AD activity. In this study, we examined whether IGF-I can antagonize cell death caused by V642I-APP in cultures, found that it does antagonize against V642I-APP-induced cell death in an IGF-binding protein (IGFBP)-sensitive manner, and demonstrated that IGF-IR mediates this action of IGF-I.
MATERIALS AND METHODS
Wild-type APP695 (wtAPP) and V642I mutant of APP695 (V642I-APP) cDNAs were described previously (Yamatsuji et al., 1996a). Wild-type human IGF-IR (wtIGF-IR) and IGF-IR mutants were described previously (Takahashi et al., 1995). Recombinant human IGF-I and des(1-3)IGF-I were from Fujisawa Pharmaceutical (Osaka, Japan) and Peninsula Laboratories (San Carlos, CA), respectively. We also used IGF-I from Boehringer Mannheim (Mannheim, Germany) and Becton-Dickinson (Franklin Lakes, NJ), each of which yielded similar results. Insulin and EGF were from Sigma (St. Louis, MO); PDGF and IGFBP-3 were from Upstate Biotechnology (Lake Placid, NY); and NGF and IGF-II were from Becton-Dickinson.
For Northern blot analysis, total RNA was extracted from NK1 or F11 cells, and 10 μg of total RNA was subjected to agarose gel electrophoresis followed by the transfer to Hybond-N nylon membrane. cDNA fragments used as probes were as follows: EGF receptor (EGFR) cDNA, BamHI fragment (∼1 kb); insulin receptor (IR) cDNA,XhoI fragment (∼1 kb); IGF-I receptor (IGF-IR) cDNA,NcoI fragment (∼1.8 kb); IGF-II/mannose-6-phosphate receptor cDNA, HindIII fragment (∼1.5 kb); p75 low-affinity NGF receptor (LNGFR) cDNA,HindIII–XbaI fragment (∼1.6 kb); TrkA cDNA,BamHI–NheI fragment (∼1.2 kb); PDGF receptor (PDGFR) β cDNA, sense (5′-GAT GGA AGG TGA TTG AGT CTG TGA GCT CTG ACG GCC ATG AGT ACA TCT-3′) and antisense (3′-CTA CCT TCC ACT AAC TCA GAC ACT CGA GAC TGC CGG TAC TCA TGT AGA-5′) (annealed and filled with Klenow fragment of DNA polymerase I in the presence of [α-32P] dCTP). Hybridization was performed at 65°C overnight for cDNA probes and at 37°C overnight for the oligonucleotide probe. TrkA cDNA (Meakin et al., 1992) was kindly provided by Dr. Andy A. Welcher, and EGFR cDNA, originally deposited by Dr. T. Yamamoto (The Institute of Medical Science, University of Tokyo, Tokyo, Japan), was kindly provided by RIKEN Gene Bank (Tsukuba, Ibaraki, Japan). IR cDNA and IGF-II/mannose-6-phosphate receptor cDNA used were described previously (Yonezawa et al., 1992; Ikezu et al., 1995).
For RT-PCR, total RNA (1 μg) of NK1 or F11 cells was subjected to one step RT-PCR, which was performed using rTth DNA polymerase (RT-PCR high Plus, TOYOBO). Reverse transcription was performed at 60°C for 30 min followed by 40 cycles of PCR (at 94°C for 1 min and 60°C for 1.5 min). Primers used are as follows: IR, sense primer, 5′-GCG ATA TGG TGA TGA GGA GCT GCA-3′ and antisense primer, 5′-GGT CTC TGC CTC ACC CTT GAT GAT-3′; IGF-IR, sense primer, 5′-ACA GAG TAC CCT TTC TTT GAG AGC-3′, antisense primer, 5′-AAG AAC ACA GGA TCT GTC CAC GAC-3′; GAPDH, sense primer, 5′-TCC ACC ACC CTG TTG CTG TA-3′, antisense primer, 5′-ACC ACA GTC CAT GCC ATC AC-3′.
For transfection experiments with NK1 cells, as described previously (Yamatsuji et al., 1996a), cells were seeded at 4 × 104 cells per well in a 24 well plate or at 106 cells per dish in a 100 mm dish and cultured for 24 hr in DME plus 10% calf serum (CS) and penicillin–streptomycin. Cells were transfected with cDNA and Lipofectamine (Life Technologies, Gaithersburg, MD) (cDNA and Lipofectamine, 0.5 μg and 1 μl in a 24 well plate for ELISA, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL), and immunohistochemical assay; 10 μg and 20 μl in a 100 mm dish for immunoblotting) in serum-free DME. This protocol yielded similar expression of cDNAs in these two settings. Media were changed to DME plus 1% CS after 24 hr serum-free culture. Various growth factors were then added to these media. After 24 hr of incubation, cells were fixed; the APP-positive cells and their nuclei were differentially stained with anti-APP monoclonal antibody 22C11 and acridine orange; and percentages of apoptotic cells with nuclear condensation, fragmentation, and compaction among APP-positive cells were measured, according to the method described previously (Yamatsuji et al., 1996a). Nucleosomally fragmented DNA was visualized in situ by TUNEL or quantitated by ELISA, as described previously (Yamatsuji et al., 1996a).
For transfection experiments with F11 cells, which were grown in Ham's F-12 plus 18% fetal bovine serum (FBS) and antibiotics, the protocol was as follows. F11 cells (7 × 104 cells per well in a 6 well plate cultured in Ham's F-12 plus 18% FBS for 12–16 hr) were transfected with V642I-APP by lipofection [V642I-APP cDNA 0.5 μg, Lipofectamine 1 μl, Plus reagent (Life Technologies) 2 μl] in the absence of serum for 3 hr, and were incubated with Ham's F-12 plus 18% FBS for 2 hr. Then, culture media were changed to Ham's F-12 plus 10% FBS and IGF-I with or without inhibitors or αIR3 (Oncogene Science, Cambridge MA), and cells were cultured for an additional 67 hr. Seventy-two hours after transfection, cell mortality was measured by Trypan blue exclusion assay. Trypan blue exclusion assay was performed as follows. Cells were suspended in cultured medium by pipetting gently, and 50 μl of 0.4% Trypan blue solution (Sigma, St. Louis, MO) was mixed with 200 μl of the cell suspension (final concentration 0.08%). Stained cells were counted within 3 min after the mixture with Trypan blue solution to determine cell mortality. The cell mortality assessed by this method thus represents the population of dead cells in total cells, including both adhesive and floating cells, at the termination of experiments. The cell mortality assessed by this method was in a reciprocally linear relationship to the cell viability assessed by WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt], using Cell Counting kit-8 (Wako Chemicals, Tokyo, Japan), as described previously (Hashimoto et al., 2000b). TUNEL experiments in F11 cells were performed using the protocol described previously (Yamatsuji et al., 1996b). In brief, 24 hr after transfection with V642I-APP and culture in the absence of serum, cells were treated with IGF-I or des(1-3)IGF-I. TUNEL was performed 24 hr after the onset of the treatment. F11/ecdysone receptor (EcR)/V642I cells were described previously (Niikura et al., 2000). In brief, F11/EcR/V642I cells were seeded and cultured in the presence of 10% FBS for 24 hr. Then cells were treated with 40 μm ecdysone (EcD) in the presence of 10% FBS for 48 hr. IGF-I, IGF-I plus anti-IGF-I antibody, or des(1-3)IGF-I, was incubated with EcD. Anti-IGF-I antibody was IGF-I (Ab-2), which was goat polyclonal antibody neutralizing human IGF-I, purchased from Oncogene.
The catalytically negative procaspase-3 cDNA was constructed from intact procaspase-3 cDNA, provided by Dr. Masayuki Miura (Osaka University, Osaka, Japan), using a kit (Clontech, Palo Alto, CA) and 5′-ATT ATT CAG GCC TCA CGT GGT ACA GAA CTG-3′ (the underlined TCA corresponds to the mutated residue). F11 cells were transfected with catalytically negative procaspase-3 mutant (CN-procaspase-3) cDNA with pcDNA or V642I-APP cDNA by Lipofectamine Plus (0.4 μg of CN-procaspase-3 cDNA, 0.6 μg of V642I-APP cDNA, Lipofectamine 2 μl, Plus reagent 4 μl) in the presence or absence of 10 nm IGF-I or 100 μm Ac-DEVD-CHO (Peptide Institute, Minoh, Osaka, Japan). Forty-eight hours after the onset of transfection, cell lysates were submitted to immunoblot analysis with anti-caspase-3 antibody, anti-APP antibody, and anti-actin antibody. The acetyl-l-aspartyl-l-glutamyl-l-valyl-l-aspart-1-al dependent cleavage of CN-procaspase-3 was then assessed. While in this system, we were unable to detect active caspase-3 fragment generated by V642I-APP, this failure was attributed to slow expression of V642I-APP over 1 d and relatively rapid disappearance of the active caspase-3 fragment.
IGF-IR was precipitated by monoclonal anti-IGF-IR antibody αIR3. After 12 hr serum starvation, NK1 cells (106 cells per well) were treated with 10 nm IGF-I or insulin at 37°C for 2 min. Cells were lysed with 300 μl of ice-cold buffer (50 mm Tris–HCl, pH 7.4, 1 mm EDTA, 1% NP-40, 1 mm DTT, 1 mm PMSF, and 1 mmNa3 VO4). After centrifugation, the supernatant was incubated with 2 μg of αIR3 bound to Protein G-Sepharose (Amersham Pharmacia Biotech, Uppsala, Sweden). The precipitate was applied to 7.5% SDS-PAGE, blotted onto a PVDF sheet, blocked with 10 mm Tris–HCl, pH 7.4, 2% skim milk, and 1% BSA, incubated with RC20H [HRP-conjugated anti-phosphotyrosine antibody; 1∶5000 dilution; Transduction Laboratories (Lexington, KY)] for 1 hr at room temperature, and visualized by ECL.
For the experiments to investigate the antagonism of mutationally activated IGF-IR against V642I-APP-induced apoptosis, cells were transfected with 0.5 μg of V642I-APP cDNA with V922E-IGF-IR, V922I-IGF-IR, or wtIGF-IR cDNA (0.5 μg each) by lipofection, according to the method described previously (Takahashi et al., 1995), and apoptotic populations in V642I-APP-expressing cells were measured. V922E-IGF-IR is mutationally activated IGF-IR with augmented tyrosine kinase, IRS-1 phosphorylation, and PI 3-kinase activation (Takahashi et al., 1995). V922I-IGF-IR is a demonstrated negative control mutant.
RESULTS
Antagonizing effect of IGF-I against V642I-APP-induced cell death
Three FAD-linked V642 mutants of APP transiently expressed in NK1 cells cause apoptotic cell death (Yamatsuji et al., 1996a; Giambarella et al., 1997a). In this system, the FAD mutants yield the highest incidence of apoptosis among all possible nineteen V642 mutants, and wtAPP exhibits the lowest toxicity, indicating that this apoptosis is linked to the FAD trait. Using V642I-APP and this system, we searched for an antagonizing factor among the known growth factors that have so far been reported to protect against death of cells in various systems: EGF, PDGF, IGF-I, IGF-II, insulin, and NGF. Apoptosis observed in a number of systems has been protected by specific growth factors: for instance, PDGF in glial cells of rat optic nerves (Barres et al., 1992); NGF in rat sympathetic neurons and PC12 cells (Levi-Montalcini, 1987; Batistatou and Greene, 1991; Mesner et al., 1992); FGF in vascular endothelial cells (Araki et al., 1990); EGF in embryonal cells (Rawson et al., 1991); and IGF-I and -II and PDGF in c-myc-induced apoptosis of Rat-1 cells (Harrington et al., 1994).
Figure 1 indicates that the IGF-I family protected V642I-APP-expressing NK1 cells from apoptosis. One nanomolar of IGF-I, but not that of insulin, significantly suppressed V642I-APP-induced apoptosis, which was strongly antagonized by both IGF-I and insulin at 10 nm, whereas IGF-II showed a much smaller effect. The other factors were without effect up to 100 nm. Both IGF-I and insulin antagonized V642I-APP-induced apoptosis in a dose-dependent manner. The IC50 values were ∼0.5 nm for IGF-I and ∼5 nm for insulin. The expression of V642I-APP was not affected by IGF-I (data not shown), indicating that expression of V642I-APP is not a target of this IGF-I action.
Fig. 1.
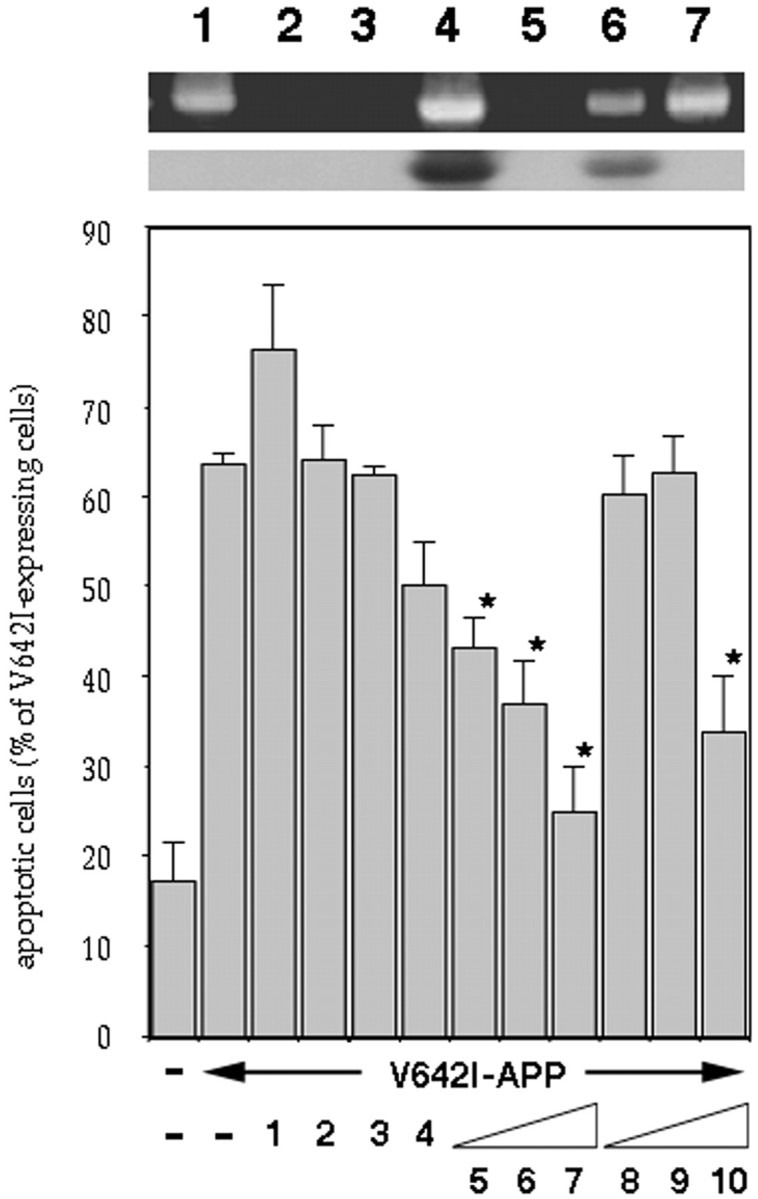
Effects of growth factors on V642I-APP-induced apoptosis. The effects of various growth factors on apoptosis in NK1 cells caused by transient expression of V642I-APP. Twenty-four hours after transfection of 0.5 μg of V642I-APP cDNA, cells were treated with or without various growth factors (lane 1, 100 ng/ml PDGF; lane 2, 10 nm EGF; lane 3, 10 nm NGF; lane 4, 10 nm IGF-II;lanes 5–7, 0.1, 1, 10 nm IGF-I; lanes 8–10; 0.1, 1, 10 nm insulin) for 24 hr, and then the percentage of apoptotic cells in V642I-APP-expressing cells was measured. Transfections were done three times independently, and the data indicate means ± SE of the three transfections. *Statistically significant versus V642I-APP transfection withp < 0.01 by Student's t test.Insets, RT-PCR clarifying the presence or absence of IGF-IR and IR in NK1 cells. In the top panel, total RNA samples (lanes 5–7), IGF-IR cDNA (lanes 3, 4), or IR cDNA (lanes 1, 2) were amplified by RT-PCR with IGF-IR primers (lanes 2, 4, 6) or IR primers (lanes 1, 3, 5), as described in Materials and Methods. Lane 7 indicates the amplified band for GAPDH as a marker of 450 bp. All amplified bands were with sizes corresponding to the expected sizes, which were 490 bp for IR and 448 bp for IGF-IR. In the bottom panel, the bands in the top panel were transferred onto nylon membrane and hybridized with IGF-IR cDNA probe.
Northern blot analysis revealed that NK1 cells expressed mRNA of 5.8 kb EGF receptor, 9.0 kb IGF-II/mannose-6-phosphate receptor, and 11 kb IGF-I receptor (IGF-IR), but no hybridizable mRNA of TrkA (the expected size, 3.2 kb), p75 low-affinity NGF receptor (LNGFR) (3.8 kb), the PDGFβ receptor (5.6 kb), or the insulin receptor (IR) (5.5, 6.0, and 7.0 kb) (data not shown). The positive and negative expressions of IGF-IR and IR, respectively, in these cells were confirmed by RT-PCR (Fig. 1, top inset). The identity of the amplified band to IGF-IR was further confirmed by the positive hybridization of the band with IGF-IR cDNA (Fig. 1, bottom inset). Consistent with other lines of evidence (see below), these data suggested that the action of insulin was mediated by IGF-IR in these cells.
Blockade of the IGF-I action by IGFBP-3
Serum contains IGFBPs that modulate IGFs-I and -II effects (Baxter, 1991). Among at least six subtypes of IGFBPs, IGFBP-3 is the major IGF binding activity in serum and is known to affect the pleiotropic actions of IGF-I (Clemmons, 1992). We thus examined whether IGFBP-3 affects the anti-apoptotic function of IGF-I. IGFBP-3 at 10 nm blocked the anti-V642I-APP effect of IGF-I, whereas IGFBP-3 itself had no effect on V642I-APP-induced apoptosis (Fig.2A). At similar concentrations, IGFBP-3 blocks the prevention by IGF-I of the spontaneous onset of apoptosis in cultured preovulatory follicles (Chun et al., 1994). Buckbinder et al. (1995) have reported that (1) IGFBP-3 is an autocrine inhibitor of growth that mediates the action of p53; and (2) 10 nmIGFBP-3 is sufficient to completely block mitogenesis by IGF-I. Therefore, the observed inhibition of IGF-I action by IGFBP-3 was reasonable. Combined with the fact that IGFBP-3 does not act on insulin, these results indicate that the observed action of IGF-I is not nonspecific and that IGFBP-3 can nullify the protective action of IGF-I against V642I-APP-induced apoptosis.
Fig. 2.

Anti-apoptotic function of des(1-3)IGF-I and effects of IGFBP-3. A, B, During the latter 24 hr incubation, wtAPP- or V642I-APP-transfected cells were treated with 10 nm IGF-I or des(1-3)IGF-I in the presence (+) or absence (−) of 10 nm IGFBP-3 (A), various concentrations of des(1-3)IGF-I (B), or 10 nm des(1-3)IGF-I (C). Otherwise, exactly the same experiments were done as in Figure 1. In this figure, three independent transfections were done, and the data are shown as means ± SE of the three transfections. In A andB, apoptotic cells were measured. In C, nucleosomally fragmented DNA was measured by ELISA. Mock means transfection with an empty vector alone. IGFBP-3 alone exerted no toxicity (data not shown). D, Cells were transfected with or without V642I-APP cDNA and then treated with or without 10 nm des(1-3)IGF-I with the same protocol. Nucleosomally fragmented DNA was visualized in situ and stained by TUNEL. The results are representative of three independent experiments, each of which yielded similar results.
The cytoprotective action of des(1-3)IGF-I
The latter finding may be important in planning IGF-I administration in FAD patients carrying the V642I mutation. Because IGFBP-3 is abundantly present in the plasma, exogenously provided IGF-I might be nullified. Considering the fact that the N-terminal 3 residues of IGF-I are essential for the binding to IGFBP-3 (Ross et al., 1989), we next examined the anti-apoptotic function of des(1-3)IGF-I, which lacks the N-terminal three residues of IGF-I, and its modification by IGFBP-3. Again, 10 nm des(1-3)IGF-I almost completely inhibited V642I-APP-induced apoptosis. The potency was similar to that of IGF-I (Fig. 2B). The anti-apoptotic function of des(1-3)IGF-I was not affected by IGFBP-3 (Fig. 2A). This suggests that IGFBP-3 blocks the anti-apoptotic function of IGF-I through direct interaction.
With two different assays for nucleosomal DNA fragmentation, we confirmed the anti-apoptotic function of des(1-3)IGF-I against V642I-APP. Consistent with the nuclear morphology assay for apoptosis, the ELISA assay of fragmented DNA revealed that des(1-3)IGF-I blocked oligonucleosomal DNA fragmentation induced by V642I-APP (Fig.2C). We also examined the TUNEL assay. Expression of V642I-APP drastically augmented the in situ labeling of the internucleosomally fragmented DNA in NK1 cells, as reported previously (Yamatsuji et al., 1996a). Figure 2Dindicates that 10 nm des(1-3)IGF-I completely protected cells from V642I-APP-induced DNA fragmentation.
Anti-apoptotic actions of IGF-I in V642I-APP-transfected neuronal systems
We confirmed the protective action of IGF-I and des(1-3)IGF-I in two different neuronal systems. One was a simple transfection system using F11 neuronal cells (Yamatsuji et al., 1996b). F11 cells are hybrid cells of a rat embryonic day 13 primary cultured neuron and a mouse neuroblastoma NTG18 cell, and as one of the best immortalized cell lines for primary cultured neurons, they carry a number of neuronal traits, such as maintenance of neuronal gangliosides and generation of action potentials without any differentiation factor treatment (Platika et al., 1985). As reported previously (Yamatsuji et al., 1996b), when F11 cells were transfected with V642I-APP cDNA, significant amounts of DNA fragmentation, detected by TUNEL, were induced (Fig.3A). When 10 nmIGF-I (data not shown) or 10 nm des(1-3)IGF-I was present in the cultured medium, DNA fragmentation of V642I-APP-transfected F11 cells was drastically suppressed. We also examined whether IGF-I protects against caspase-3 cleavage stimulated by V642I-APP transfection, using catalytically negative procaspase-3 (CN-procaspase-3). When F11 cells transfected with CN-procaspase-3 were cotransfected with V642I-APP cDNA, but not an empty vector, 67.8 ± 8.7% (mean ± SD; n = 3) of CN-procaspase-3 was cleaved in an Ac-DEVD-CHO-inhibitable manner. Because Ac-DEVD-CHO is a specific inhibitor of caspases, the Ac-DEVD-CHO-sensitive cleavage of CN-procaspase-3, which has no self-cleaving activity, reflects intracellular activation of caspase-3. When CN-procaspase-3-transfected F11 cells were cotransfected with V642I-APP cDNA and treated with 10 nm IGF-I, only 8.4 ± 5.8% of CN-procaspase-3 was cleaved. These data indicate that IGF-I also suppresses the cleavage of procaspase-3 stimulated by V642I-APP, a process essential for induction of apoptosis.
Fig. 3.
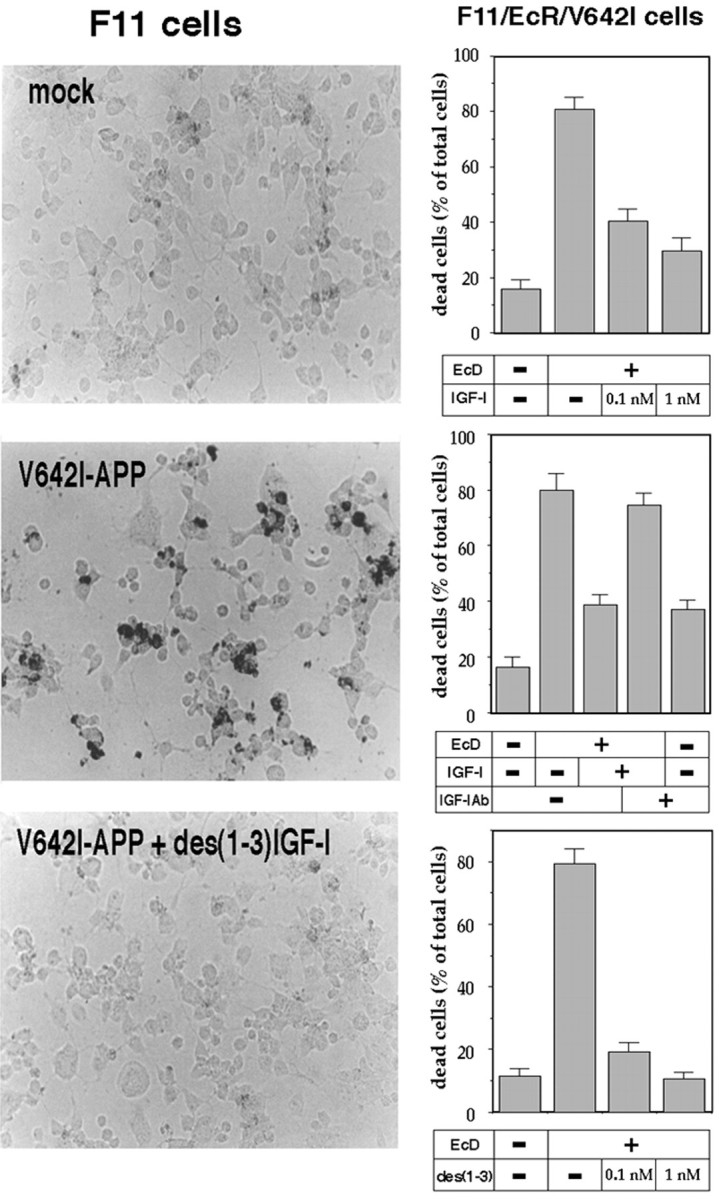
Effect of IGF-I and des(1-3)IGF-I on V642I-APP-induced neurotoxicity. In the left panels, F11 neuronal cells were transfected with or without V642I-APP cDNA and cultured in the presence or absence of 10 nm des(1-3)IGF-I. Twenty-four hours after the onset of treatment, intracellular fragmented DNA was stained in situ by TUNEL. The results indicate the representative fields. Similar experiments were repeated three times. In the right panels, F11/EcR/V642I cells were treated with or without 40 μm EcD for 48 hr in the presence or absence of IGF-I or des(1-3)IGF-I. Cell mortality was measured by Trypan blue exclusion assay. In the experiments shown in the right middle panel, cells were cultured with or without EcD in the presence or absence of 1 nm IGF-I or in the presence or absence of 5 μg/ml anti-IGF-I antibody. Values indicate means ± SD of six independent treatments.
Another system used for confirmation was F11/EcR/V642I cells, which were established by stably transfecting F11/EcR cells with EcD-inducible V642I-APP cDNA. F11/EcR cells are the stable F11 cells that express both the EcD receptor (EcR) and the retinoid X receptor (Hashimoto et al., 2000a). When F11/EcR/V642I cells are treated with EcD, they undergo apoptotic cell death of a clonal nature (Niikura et al., 2000). In this system, it has been demonstrated that EcD-induced toxicity is attributable to expressed V642I-APP and through apoptosis (Niikura et al., 2000). In F11/EcR/V642I cells, high picomolar to low nanomolar of IGF-I dose-dependently inhibited death of these cells induced by V642I-APP expression. This action of IGF-I was inhibited by neutralizing anti-IGF-I antibody (Fig. 3, right middle). This was also the case with des(1-3)IGF-I. One nanomolar of des(1-3)IGF-I completely recovered cell viability impaired by EcD induction of V642I-APP (Fig.3, right bottom) and abolished virtually all apoptotic morphological changes, including rounding, shrinking, and detaching from plates (data not shown). Collecting these data, it was shown that both low nanomolar IGF-I and des(1-3)IGF-I block V642I-APP-induced apoptosis in neuronal cells.
Involvement of IGF-IR in the action of IGF-I
The observed high potency of IGF-I as well as negative expression of IR in NK1 cells (see above) and F11 cells (data not shown) pointed to an intermediary role for IGF-IR. We thus examined the involvement of IGF-IR by three different approaches: (1) whether the signal inhibitors of IGF-IR block this anti-apoptotic action of IGF-I; (2) whether anti-IGF-IR blocking antibody influences it; and (3) whether mutationally activated IGF-IR reproduces it.
Effects of signal inhibitors of IGF-IR
Figure 4A shows that either 100 μm genistein or 10 nmwortmannin, which had no effect on basal death rates (data not shown), significantly inhibited the anti-apoptotic function of IGF-I against V642I-APP in NK-1 cells. This was also the case in F11 cells. In these cells, either 100 μm genistein or 10 nmwortmannin had no effect on basal cell death rates or V642I-APP-induced cytotoxicity (Fig. 4B). Under the conditions used, the transfection efficiency was estimated to be 60–70% (Hashimoto et al., 2000a). It thus followed that 70–80% of V642I-APP-transfected cells underwent death for 72 hr. Treatment with 10 nm IGF-I completely protected cells from V642I-APP-induced cytotoxicity. The suppressing effect of IGF-I against V642I-APP-induced cell death was significantly attenuated by 10 nm wortmannin and by 100 μm genistein (Fig.4B), although inhibition by genistein was not complete. These compounds are inhibitors designed to block tyrosine kinases and PI 3-kinases, which IGF-IR activates ligand-dependently. Although their effects may not be exclusive for these targets, the effective concentrations of genistein and wortmannin were reasonably low in each case to selectively suppress tyrosine kinases (Akiyama et al., 1987) and PI 3-kinase (Nakanishi et al., 1992), respectively. Considering the facts that (1) genistein inhibits the tyrosine kinase activity of IGF-IR (Taghon and Sadler, 1994) and (2) genistein does not inhibit the MEK/MAP kinases (Cox et al., 1996), it was highly likely that to exert its suppressing action against IGF-I cytoprotection, genistein directly inhibited IGF-IR kinase, but not further downstream MEK–MAP kinases. In support of this idea, the genistein-sensitive rescue action of IGF-I was not blocked by PD98059, a specific MEK inhibitor (Fig. 4B).
Fig. 4.
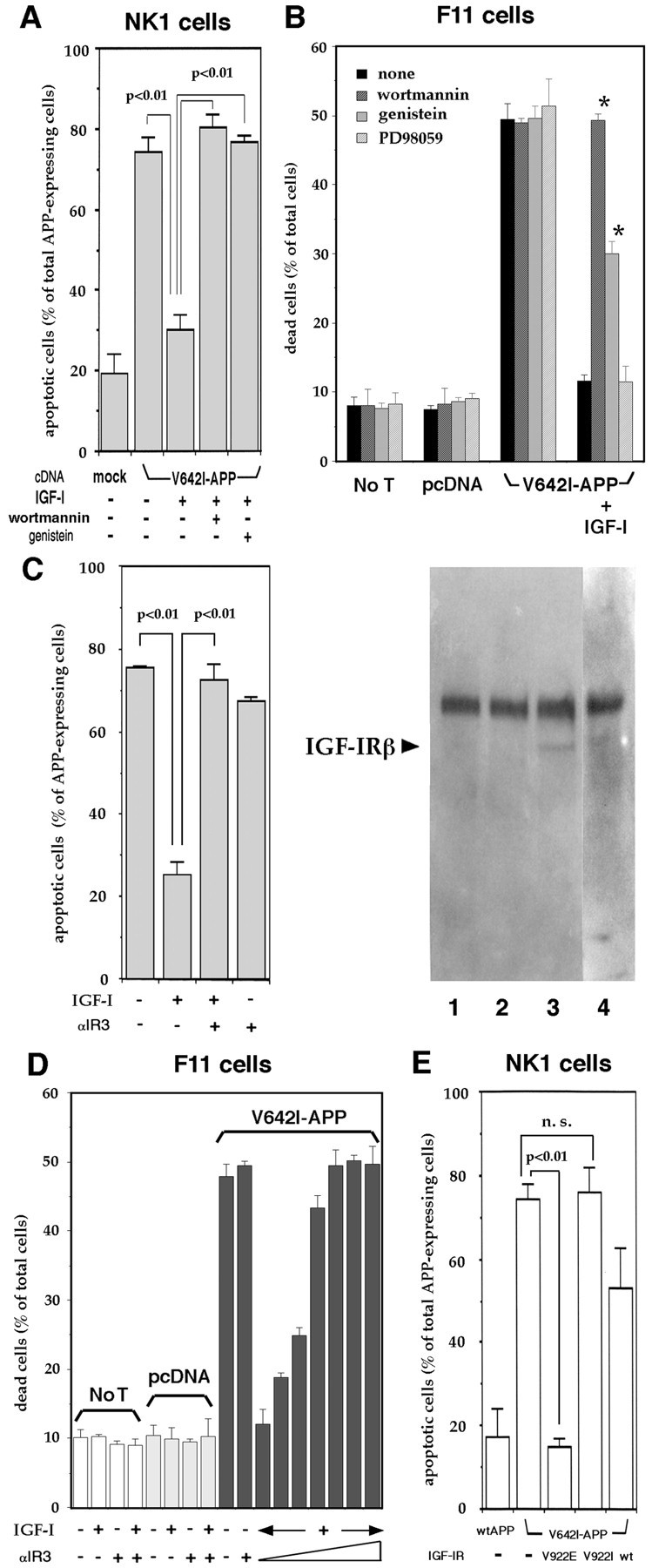
Involvement of IGF-IR in the anti-V642I-APP action of IGF-I. A, NK1 cells were transfected with V642I-APP cDNA or an empty plasmid (mock), and 24 hr after transfection, cells were treated with or without 10 nm IGF-I in the presence or absence of 10 nm wortmannin or 100 μm genistein, and apoptotic cells were measured as described in the legend of Figure 1. In this figure, three independent transfections were done, and the data are shown as means ± SE of the three transfections. p < 0.01 by Student'st test. B, F11 cells were transfected with or without (No T) V642I-APP cDNA or pcDNA in the presence or absence of 10 nm IGF-I with or without either 10 nm wortmannin, 100 μm genistein, or 50 μm PD98059. Cell mortality was measured by Trypan blue exclusion assay 72 hr after the onset of transfection. Values indicate means ± SD of three independent experiments. *Statistically significant versus V642I-APP plus IGF-I with p < 0.01 by Student's t test. C, Blocking effect of αIR3 on the anti-apoptotic function of IGF-I. In the left panel, 24 hr after transfection, V642I-APP-transfected NK1 cells were treated with 10 nm IGF-I in the presence or absence of 10 nm αIR3. Otherwise, exactly the same conditions as in Figure 1. Values indicate means ± SE of three independent experiments. In the right panel, phosphotyrosine immunoblot of the αIR3 precipitate from NK1 cells is indicated. Cells treated with vehicle (lane 1), 10 nm IGF-I (lane 3), or 10 nm insulin (lane 4) were lysed and precipitated by αIR3. In lane 2, the lysate of NK1 cells treated with 10 nm IGF-I was precipitated by nonspecific mouse IgG. These samples were blotted onto a sheet and probed with RC20, recombinant anti-phosphotyrosine antibody. The results are representative of three similar experiments. Thearrow indicates the IGF-IR β chain, corresponding to ∼100 kDa. D, Effects of αIR3 on the basal death rates, V642I-APP-stimulated cell death, and the inhibitory effect of IGF-I. In the left 10 columns, F11 cells were transfected with or without (No T) V642I-APP cDNA (V642I-APP) or pcDNA (pcDNA) and cultured in the presence or absence of 10 nm IGF-I and/or 10 nm αIR3. In theright seven columns, F11 cells were transfected with V642I-APP cDNA and cultured in the presence of 10 nm IGF-I with increasing concentrations (0, 1, 3, 10, 20, 50, and 100 nm from left to right) of αIR3. Cell mortality was measured by Trypan blue exclusion assay 72 hr after the onset of transfection. Values indicate means ± SD of three independent experiments. In IGF-I-treated, V642I-APP-transfected cells, the statistical differences of the death rates in the presence of ≥1 nm αIR3 are significant against the death rate in the absence of αIR3 (p < 0.01 by Student'st test). E, NK1 cells were transfected with wtAPP or V642I-APP cDNA with or without V922E-IGF-IR (V922E), V922I-IGF-IR (V922I), or wtIGF-IR (wt) cDNA. Forty-eight hours after transfection, the incidence of apoptotic nuclear changes in APP-expressing cells was measured. V922E-IGF-IR is demonstrated to be constitutively active in receptor tyrosine kinase activity, IRS-1 phosphorylation, and PI 3-kinase activation. V922I-IGF-IR is a negative control mutant. The same experiments were repeated three times independently, and the data are presented as means ± SE. p < 0.01 by Student's t test. n.s., Not significant.
Effect of anti-IGF-IR antibody
We investigated the effect of αIR3, an anti-IGF-IR monoclonal antibody that blocks binding of IGF-I to IGF-IR but has little affinity for IR (Steele-Perkins and Roth, 1990; Kato et al., 1993). Figure 4C shows that a low concentration of αIR3, which has been shown to totally antagonize the mitogenic action of IGF-I (Steele-Perkins and Roth, 1990), nullified the anti-apoptotic action of IGF-I in NK-1 cells, whereas normal IgG had no effect (data not shown). αIR3 per se did not affect V642I-APP-induced apoptosis. Because it had been thought that this antibody recognized only human and rodent IGF-IR, we confirmed that αIR3 recognizes monkey IGF-IR in NK1 cells. The data showed that αIR3, not normal IgG, precipitated an ∼100 kDa phosphotyrosine protein from NK1 cell lysates only after stimulation by 10 nm IGF-I (Fig. 4C, right panel), suggesting that this protein was IGF-IR β chain precipitated with its α chain by αIR3. These provide evidence that αIR3 recognizes monkey IGF-IR.
We performed similar experiments in which the IGF-I-antagonizing effect of αIR3 was examined in F11 cells transfected with 0.5 μg of V642I-APP cDNA. In this system, V642I-APP-induced cytotoxicity was totally suppressed by 10 nm coexisting IGF-I. The death-suppressing action of IGF-I was dose-dependently nullified by αIR3: the antagonizing effect of αIR3 reached complete at ∼10 nm, a low concentration similar to that of employed IGF-I. Although it has been reported that IGF-I-mimetic action was observed for growth promotion by high concentrations (∼100 nm) of αIR3 (Kato et al., 1993), no such effect was observed at high concentrations of this antibody as far as IGF-I protection against cell death was concerned. Consistent with the Northern blot analysis data that F11 cells expressed mRNA of IGF-IR, but little of IR (data not shown), these data suggest that IGF-I protects against V642I-APP-induced death in F11 cells through IGF-IR.
Effect of mutationally activated IGF-IR
We next examined the function of V922E-IGF-IR in the absence of IGF-I. This IGF-IR mutant, carrying a point mutation V922E in its transmembrane region, exhibits constitutively augmented tyrosine kinase activity in its β chain and acts as a constitutively active IGF-IR (Takahashi et al., 1995). By cotransfecting this mutationally activated IGF-IR cDNA, V642I-APP-induced apoptosis was drastically blocked (Fig. 4E). In contrast, cotransfection of a control mutant V922I-IGF-IR, whose tyrosine kinase is not constitutively active (Takahashi et al., 1995), had no antagonizing function. Cotransfection of wtIGF-IR cDNA seemed to slightly suppress apoptosis. However, this effect of wtIGF-IR was only weakly significant against apoptosis without IGF-IR (p ≈ 0.03 by Student's t test), whereas this might suggest the presence of a basally active fraction in wtIGF-IR (and its absence in V922I-IGF-IR). In these cotransfection experiments, there was no difference in the expression of V642I-APP (data not shown). These data indicate that IGF-IR could sufficiently protect cells from death induced by V642I-APP.
DISCUSSION
We have herein shown that IGF-I action protects cells from apoptotic toxicity by the FAD-associated V642I mutant of APP in NK1 cells, F11 cells, and F11/EcR/V642I cells. IGF-I has so far been shown to serve as an anti-cell death factor in a number of systems (D'Mello et al., 1993; Harrington et al., 1994; Sell et al., 1995); but this is the first report that defines the antagonizing action of IGF-I on FAD gene-caused cell death. This study also shows that IGF-IR acts as the target of IGF-I cytoprotection against AD-relevant insults. Based mainly on the relatively higher potency of IGF-I compared with IGF-II, Dore et al. (1997) inferred that IGF-IR is involved in the action of IGF-I against Aβ-induced neuronal death. As the first report demonstrating anti-cell death function of IGF-IR, Sell et al. (1995) assigned IGF-IR in the IGF-I antagonism against etoposide-induced apoptosis in fibroblasts. In the present study, the actions of inhibitors further suggest that the downstream target of IGF-I could be the intracellular signaling pathways of IGF-IR involving tyrosine kinases and PI 3-kinases. Their involvement was also supported by the action of V922E-IGF-IR. This mutant IGF-IR exhibits constitutively augmented receptor tyrosine kinase activity and activates the IRS-1–PI 3-kinase cascade and glucose uptake without binding to IGF-I, whereas it does not turn on the Ras–MAP kinase pathway or thereby could not stimulate cell proliferation at all in the absence of IGF-I (Takahashi et al., 1995). Therefore, considering the versatile functions of Akt, the downstream Ser–Thr kinase of PI-3 kinase, including inhibition of cytochrome c release (Kennedy et al., 1999), inhibition of BAD, caspase-9, or a Forkhead transcription factor by phosphorylation (Zha et al., 1996; Datta et al., 1997; Cardone et al., 1998; Brunet et al., 1999), posttranslational modification of a cytosolic factor downstream of cytochrome c and upstream of caspase-9 (Zhou et al., 2000), inhibition of the transcriptional activity of p53 (Yamaguchi et al., 2001), or transcriptional activation of Bcl-x (Suzuki et al., 1998; Leverrier et al., 1999), the observed cytoprotection by V922E-IGF-IR suggests that activation of the PI 3-kinase–Akt cascade would be essential for IGF-IR to execute anti-apoptosis against V642I-APP through a Ras-independent mechanism. This idea is not only consistent with multiple reports (Yao and Cooper, 1995; Mockridge et al., 2000) that activation of PI 3-kinase, not of the Ras/Raf/MEK/MAP kinase cascade, is essential for receptor tyrosine kinases to antagonize apoptosis in various kinds of cells, but consistent with the study of Kulik et al. (1997) showing that anti-apoptotic signaling by IGF-IR is mediated by PI-3 kinase and Akt.
The pathogenesis of AD has so far been discussed mainly from the viewpoint of offending factors, insults that could account for the pathophysiology of AD, such as Aβ or excitotoxins. However, recent studies have begun to specify the protective actions of neurotrophic factors, including IGF-I (Dore et al., 1997; this study), activity-dependent neurotrophic factor (Gozes and Brenneman, 1996; Guo et al., 1999), and basic FGF (Guo et al., 1999), and suggest that (1) the human body is equipped with a defense mechanism against the processes of AD; and (2) the pathology of AD may not be a simple result of offending processes but a result of confrontation between offending and defending mechanisms. IGF-I is a polypeptide normally circulating in plasma, mainly produced by the liver but also produced by a number of tissues in a paracrine manner. The major form of IGF-I produced by the brain seems to be des(1-3)IGF-I (Sara et al., 1986). IGF-IR is expressed in a variety of tissues, including the brain. Therefore, IGF-I or des(1-3)IGF-I can act as an in vivo defense mechanism through IGF-IR. There is a basic question of why the onset of AD in patients with V642 mutations in APP occurs around the fifth decade of life (Rossor et al., 1993), despite the fact that these mutations, present in APP throughout life, induce rapid neurotoxicity in vitro (Yamatsuji et al., 1996a,b; Giambarella et al., 1997a;Hashimoto et al., 2000a; Niikura et al., 2000). A simple interpretation, based on the antagonizing action of IGF-I and IGF-IR, is that during the period when the defense system is functional, no neurotoxicity occurs, but that after the function of IGF-I is impaired in the fifth decade of life, the clinical onset of AD may occur in individuals expressing the V642 mutant of APP. In support, plasma IGF-I bioactivity continuously decreases with aging (Johanson and Blizzard, 1981; Rudman et al., 1981; Florini et al., 1985; Zapf et al., 1990). Also, abnormal decrease in IGF-I production may facilitate or increase the risk of the onset of FAD. Consistent with this idea, Mustafa et al. (1999) reported that decreased IGF-I production is associated with FAD caused by K595N/M596L mutant APP.
The present study also suggests that IGF-I could be envisaged as a basis for developing therapies for AD, as Dores et al. (2000) have so far argued. Yet the observed inhibition by IGFBP-3 of the protective action of IGF-I suggests that the potential usefulness of IGF-I itself may be limited. Some IGFBPs, chief among them IGFBP-3, circulate in the plasma. IGFBPs are also produced locally. Therefore, intravenously administered IGF-I may be captured by IGFBPs, potentially resulting in attenuation of its cytoprotective activity. However, the present study further notes that IGFBP-nonbinding forms of IGF-I could be effective alternates for therapeutic administration. Although this idea seems attractive, it should be stressed that IGFBP-nonbinding forms of IGF-I have only a short half-life in the body (Gillespie et al., 1996). Another therapeutic intervention that circumvents the IGFBP inhibition could be virus-mediated transfer of V922E-IGF-IR, which functions independently from IGF-I and IGFBP. In addition, V922E-IGF-IR has an additional advantage over IGF-I. As the major action, IGF-I stimulates cell growth in various tissues. Overproduction of IGF-I causes gigantism or acromegaly caused by overgrowth of skeletal tissues. Furthermore, IGF-I has growth-stimulating effects on neural progenitor cells (Aberg et al., 2000) and astrocytic brain tumors (Glick et al., 1997) in the adult nervous system. In contrast, V922E-IGF-IR does not stimulate cell proliferation but exerts glucose uptake and cytoprotection (Takahashi et al., 1995; this study). Therefore, gene transfer of V922E-IGF-IR may be more suitable in aiming at selectively suppressing cell death with minimal unwanted effects of IGF-I. The present study thus provides a molecular clue not only to the understanding of the pathophysiology of AD but also to the development of its potential treatments.
Footnotes
T. N., Y. H., and T. O. contributed equally to this study.
This work was supported in part by grants from Naito Foundation, Brain Science Foundation, Takeda Medical Research Foundation, Takeda Science Foundation, the Japan Medical Association, the Ministry of Health and Welfare of Japan, the Ministry of Education, Science, and Culture of Japan and the Organization for Pharmaceutical Safety and Research, and supported by Keio University Grant-in-Aid for Encouragement of Young Medical Scientists (M. K.), Keio University Medical Science Fund (K. K.), and Keio University Special Grant-in-Aid for Innovative Collaborative Research Projects (I. N.). We especially thank J. Avruch for critical reading of this manuscript and helping us with this study, N. Rosenthal and J. C. Engert for critical reading of this manuscript, D. Cooper for stimulating discussion, A. A. Welcher for TrkA cDNA, K. Yonezawa for IR cDNA and cooperation, J. T. Potts Jr, Y. and Y. Tamai, and E. Ogata for enthusiastic encouragement, and D. Wylie for expert technical assistance. We are also indebted to Tomo Yoshida for excellent assistance and generous encouragement and Kazumi Nishihara for indispensable assistance.
Correspondence should be addressed to Dr. Ikuo Nishimoto, Department of Pharmacology and Neurosciences, Keio University School of Medicine, Shinanomachi, Shinjuku-ku, Tokyo 160–8582, Japan. E-mail:nisimoto@mc.med.keio.ac.jp.
REFERENCES
- 1.Aberg MA, Aberg ND, Hedbacker H, Oscarsson J, Eriksson PS. Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J Neurosci. 2000;20:2896–2903. doi: 10.1523/JNEUROSCI.20-08-02896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem. 1987;262:5592–5595. [PubMed] [Google Scholar]
- 3.Araki S, Shimada Y, Kaji K, Hayashi H. Apoptosis of vascular endothelial cells by fibroblast growth factor deprivation. Biochem Biophys Res Commun. 1990;168:1194–1200. doi: 10.1016/0006-291x(90)91155-l. [DOI] [PubMed] [Google Scholar]
- 4.Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- 5.Batistatou A, Greene LA. Aurintricarboxylic acid rescues PC12 cells and sympathetic neurons from cell death caused by nerve growth factor deprivation: correlation with suppression of endonuclease activity. J Cell Biol. 1991;115:461–471. doi: 10.1083/jcb.115.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baxter RC. Physiological roles of IGF binding proteins. In: Spencer EM, editor. Modern concepts of insulin-like growth factors. Elsevier; New York: 1991. pp. 371–380. [Google Scholar]
- 7.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 8.Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, Kley N. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature. 1995;377:646–649. doi: 10.1038/377646a0. [DOI] [PubMed] [Google Scholar]
- 9.Cai X-D, Golde TE, Younkin SG. Release of excess amyloid β protein from a mutant amyloid β protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- 10.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 11.Chun SY, Billig H, Tilly JL, Furuta I, Tsafriri A, Hsueh AJ. Gonadotropin suppression of apoptosis in cultured preovulatory follicles: mediatory role of endogenous insulin-like growth factor I. Endocrinology. 1994;135:1845–1853. doi: 10.1210/endo.135.5.7525255. [DOI] [PubMed] [Google Scholar]
- 12.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the β-amyloid precursor protein in familial Alzheimer's disease increases β-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 13.Clemmons DR. IGF binding proteins: regulation of cellular actions. Growth Regul. 1992;2:80–87. [PubMed] [Google Scholar]
- 14.Cox ME, Ely CM, Catling AD, Weber MJ, Parsons SJ. Tyrosine kinases are required for catecholamine secretion and mitogen-activated protein kinase activation in bovine adrenal chromaffin cells. J Neurochem. 1996;66:1103–1112. doi: 10.1046/j.1471-4159.1996.66031103.x. [DOI] [PubMed] [Google Scholar]
- 15.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 16.D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dore S, Kar S, Quirion R. Insulin-like growth factor I protects and rescues hippocampal neurons against β-amyloid- and human amylin-induced toxicity. Proc Natl Acad Sci USA. 1997;94:4772–4777. doi: 10.1073/pnas.94.9.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dore S, Kar S, Zheng WH, Quirion R. Rediscovering good old friend IGF-I in the new millennium: possible usefulness in Alzheimer's disease and stroke. Pharm Acta Helv. 2000;74:273–280. doi: 10.1016/s0031-6865(99)00037-0. [DOI] [PubMed] [Google Scholar]
- 19.Florini JR, Prinz PN, Vitiello MV, Hintz RL. Somatomedin-C levels in healthy young and old men: relationship to peak and 24-hour integrated levels of growth hormone. J Gerontol. 1985;40:2–7. doi: 10.1093/geronj/40.1.2. [DOI] [PubMed] [Google Scholar]
- 20.Giambarella U, Yamatsuji T, Okamoto T, Matsui T, Ikezu T, Murayama Y, Levine MA, Katz A, Gautam N, Nishimoto I. G protein βγ complex-mediated apoptosis by familial Alzheimer's disease mutant of APP. EMBO J. 1997a;16:4897–4907. doi: 10.1093/emboj/16.16.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giambarella U, Murayama Y, Ikezu T, Fujita T, Nishimoto I. Potential CRE suppression by familial Alzheimer's mutants of APP independent of adenylyl cyclase regulation. FEBS Lett. 1997b;412:97–101. doi: 10.1016/s0014-5793(97)00753-9. [DOI] [PubMed] [Google Scholar]
- 22.Gillespie CM, Hazel SJ, Walton PE, Martin AA. Effects of chronic renal failure on plasma clearance of insulin-like growth factor I, des-(1-3)IGF-I, and LR3IGF-I. Am J Physiol. 1996;271:E649–E657. doi: 10.1152/ajpendo.1996.271.4.E649. [DOI] [PubMed] [Google Scholar]
- 23.Glick RP, Lichtor T, Unterman TG. Insulin-like growth factors in central nervous system tumors. J Neurooncol. 1997;35:315–325. doi: 10.1023/a:1005876819455. [DOI] [PubMed] [Google Scholar]
- 24.Gozes I, Brenneman DE. Activity-dependent neurotrophic factor (ADNF). An extracellular neuroprotective chaperonin? J Mol Neurosci. 1996;7:235–244. doi: 10.1007/BF02737061. [DOI] [PubMed] [Google Scholar]
- 25.Guo Q, Sebastian L, Sopher BL, Miller MW, Glazner GW, Ware CB, Martin GM, Mattson MP. Neurotrophic factors [activity-dependent neurotrophic factor (ADNF) and basic fibroblast growth factor (bFGF)] interrupt excitotoxic neurodegenerative cascades promoted by a PS1 mutation. Proc Natl Acad Sci USA. 1999;96:4125–4130. doi: 10.1073/pnas.96.7.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardy J. Framing β-amyloid. Nat Genet. 1992;1:233–234. doi: 10.1038/ng0792-233. [DOI] [PubMed] [Google Scholar]
- 27.Harrington EA, Bennett MR, Fanidi A, Evan GI. c-Myc-induced apoptosis in fibroblasts is inhibited by specific cytokines. EMBO J. 1994;13:3286–3295. doi: 10.1002/j.1460-2075.1994.tb06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hashimoto Y, Niikura T, Ito Y, Nishimoto I. Multiple mechanisms underlie neurotoxicity by different type Alzheimer's disease mutations of amyloid precursor protein. J Biol Chem. 2000a;275:34541–34551. doi: 10.1074/jbc.M005332200. [DOI] [PubMed] [Google Scholar]
- 29.Hashimoto Y, Jiang H, Niikura T, Ito Y, Hagiwara A, Umezawa K, Abe Y, Murayama Y, Nishimoto I. Neuronal apoptosis by apolipoprotein E4 through low-density lipoprotein receptor-related protein and heterotrimeric GTPases. J Neurosci. 2000b;20:8401–8409. doi: 10.1523/JNEUROSCI.20-22-08401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikezu T, Okamoto T, Giambarella U, Yokota T, Nishimoto I. In vivo coupling of insulin-like growth factor II/mannose 6-phosphate receptor to heteromeric G proteins. Distinct roles of cytoplasmic domains and signal sequestration by the receptor. J Biol Chem. 1995;270:29224–29228. doi: 10.1074/jbc.270.49.29224. [DOI] [PubMed] [Google Scholar]
- 31.Johanson AJ, Blizzard RM. Low somatomedin-C levels in older men rise in response to growth hormone administration. Johns Hopkins Med J. 1981;149:115–117. [PubMed] [Google Scholar]
- 32.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 33.Kato H, Faria TN, Stannard B, Roberts CT, Jr, LeRoith D. Role of tyrosine kinase activity in signal transduction by the insulin-like growth factor-I (IGF-I) receptor. Characterization of kinase-deficient IGF-I receptors and the action of an IGF-I-mimetic antibody (alpha IR-3). J Biol Chem. 1993;268:2655–2661. [PubMed] [Google Scholar]
- 34.Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19:5800–5810. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kulik G, Klippel A, Weber MJ. Antiapoptotic signaling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 37.Leverrier Y, Thomas J, Mathieu AL, Low W, Blanquier B, Marvel J. Role of PI3-kinase in Bcl-X induction and apoptosis inhibition mediated by IL-3 or IGF-1 in Baf-2 cells. Cell Death Diff. 1999;6:290–296. doi: 10.1038/sj.cdd.4400492. [DOI] [PubMed] [Google Scholar]
- 38.Levi-Montalcini R. The nerve growth factor: thirty-five years later. EMBO J. 1987;6:1145–1154. doi: 10.1002/j.1460-2075.1987.tb02347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo JJ, Wallace W, Riccioni T, Ingram DK, Roth GS, Kusiak JW. Death of PC12 cells and hippocampal neurons induced by adenoviral-mediated FAD human amyloid precursor protein gene expression. J Neurosci Res. 1999;55:629–642. doi: 10.1002/(SICI)1097-4547(19990301)55:5<629::AID-JNR10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 41.Mark RJ, Keller JN, Kruman I, Mattson MP. Basic FGF attenuates amyloid β-peptide-induced oxidative stress, mitochondrial dysfunction, and impairment of Na+/K+-ATPase activity in hippocampal neurons. Brain Res. 1997;756:205–214. doi: 10.1016/s0006-8993(97)00196-0. [DOI] [PubMed] [Google Scholar]
- 42.Meakin SO, Suter U, Drinkwater CC, Welcher AA, Shooter EM. The rat trk protooncogene product exhibits properties characteristic of the slow nerve growth factor receptor. Proc Natl Acad Sci USA. 1992;89:2374–2378. doi: 10.1073/pnas.89.6.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mesner PW, Winters TR, Green SH. Nerve growth factor withdrawal-induced cell death in neuronal PC12 cells resembles that in sympathetic neurons. J Cell Biol. 1992;119:1669–1680. doi: 10.1083/jcb.119.6.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miranda S, Opazo C, Larrondo LF, Munoz FJ, Ruiz F, Leighton F, Inestrosa NC. The role of oxidative stress in the toxicity induced by amyloid β-peptide in Alzheimer's disease. Prog Neurobiol. 2000;62:633–648. doi: 10.1016/s0301-0082(00)00015-0. [DOI] [PubMed] [Google Scholar]
- 45.Mockridge JW, Benton EC, Andreeva LV, Latchman DS, Marber MS, Heads RJ. IGF-I regulates cardiac fibroblast apoptosis induced by osmotic stress. Biochem Biophys Res Commun. 2000;273:322–327. doi: 10.1006/bbrc.2000.2934. [DOI] [PubMed] [Google Scholar]
- 46.Mustafa A, Lannfelt L, Lilius L, Islam A, Winblad B, Adem A. Dement Decreased plasma insulin-like growth factor-I level in familial Alzheimer's disease patients carrying the Swedish APP 670/671 mutation. Geriatr Cogn Disord. 1999;10:446–451. doi: 10.1159/000017188. [DOI] [PubMed] [Google Scholar]
- 47.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 48.Nakanishi S, Kakita S, Takahashi I, Kawahara K, Tsukuda E, Sano T, Yamada K, Yoshida M, Kase H, Matsuda Y. Wortmannin, a microbial product inhibitor of myosin light chain kinase. J Biol Chem. 1992;267:2157–2163. [PubMed] [Google Scholar]
- 49.Niikura T, Murayama N, Hashimoto Y, Ito Y, Yamagishi Y, Matsuoka M, Takeuchi Y, Aiso S, Nishimoto I. V642I APP-inducible neuronal cells: A model system for investigating Alzheimer's disorders. Biochem Biophys Res Commun. 2000;274:445–454. doi: 10.1006/bbrc.2000.3143. [DOI] [PubMed] [Google Scholar]
- 50.Pike CJ, Ramezan-Arab N, Cotman CW. β-Amyloid neurotoxicity in vitro: evidence of oxidative stress but not protection by antioxidants. J Neurochem. 1997;69:1601–1611. doi: 10.1046/j.1471-4159.1997.69041601.x. [DOI] [PubMed] [Google Scholar]
- 51.Platika D, Boulos MH, Braizer L, Fishman MC. Neuronal traits of clonal cell lines derived by fusion of dorsal root ganglia neurons with neuroblastoma cells. Proc Natl Acad Sci USA. 1985;82:3499–3503. doi: 10.1073/pnas.82.10.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rawson CL, Loo DT, Duimstra JR, Hedstrom OR, Schmidt EE, Barnes DW. Death of serum-free mouse embryo cells caused by epidermal growth factor deprivation. J Cell Biol. 1991;113:671–680. doi: 10.1083/jcb.113.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ross M, Francis GL, Szabo L, Wallace JC, Ballard FJ. Insulin-like growth factor (IGF)-binding proteins inhibit the biological activities of IGF-1 and IGF-2 but not des-(1-3)-IGF-1. Biochem J. 1989;258:267–272. doi: 10.1042/bj2580267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rossor MN, Newman S, Frackowiak RS, Lantos P, Kennedy AM. Alzheimer's disease families with amyloid precursor protein mutations. Ann NY Acad Sci. 1993;695:198–202. doi: 10.1111/j.1749-6632.1993.tb23052.x. [DOI] [PubMed] [Google Scholar]
- 55.Rudman D, Kutner MH, Rogers CM, Lubin MF, Fleming GA, Bain RP. Impaired growth hormone secretion in the adult population: relation to age and adiposity. J Clin Invest. 1981;67:1361–1369. doi: 10.1172/JCI110164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sara VR, Carlsson-Skwirut C, Andersson C, Hall E, Sjogren B, Holmgren A, Jornvall H. Characterization of somatomedins from human fetal brain: identification of a variant form of insulin-like growth factor I. Proc Natl Acad Sci USA. 1986;83:4904–4907. doi: 10.1073/pnas.83.13.4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sell C, Baserga R, Rubin R. Insulin-like growth factor I (IGF-I) and the IGF-I receptor prevent etoposide-induced apoptosis. Cancer Res. 1995;55:303–306. [PubMed] [Google Scholar]
- 58.Steele-Perkins G, Roth RA. Insulin-mimetic anti-insulin receptor monoclonal antibodies stimulate receptor kinase activity in intact cells. J Biol Chem. 1990;265:9458–9463. [PubMed] [Google Scholar]
- 59.Suzuki J, Kaziro Y, Koide H. Synergistic action of R-Ras and IGF-1 on Bcl-xL expression and caspase-3 inhibition in BaF3 cells: R-Ras and IGF-1 control distinct anti-apoptotic kinase pathways. FEBS Lett. 1998;437:112–116. doi: 10.1016/s0014-5793(98)01213-7. [DOI] [PubMed] [Google Scholar]
- 60.Suzuki N, Cheung TT, Cai X-D, Odaka A, Otvos L, Eckman C, Golde TE, Younkin SG. An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (βAPP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 61.Taghon MS, Sadler SE. Insulin-like growth factor 1 receptor-mediated endocytosis in Xenopus laevis oocytes. A role for receptor tyrosine kinase activity. Dev Biol. 1994;163:66–74. doi: 10.1006/dbio.1994.1123. [DOI] [PubMed] [Google Scholar]
- 62.Takahashi K, Yonezawa K, Nishimoto I. Insulin-like growth factor I receptor activated by a transmembrane mutation. J Biol Chem. 1995;270:19041–19045. doi: 10.1074/jbc.270.32.19041. [DOI] [PubMed] [Google Scholar]
- 63.Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacana E, Sunderland T, Zhao B, Kusiak JW, Wasco W, D'Adamio L. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- 64.Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M (2001) Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem, in press. [DOI] [PubMed]
- 65.Yamatsuji T, Okamoto T, Takeda S, Murayama Y, Tanaka N, Nishimoto I. Expression of V642 APP mutant causes cellular apoptosis as Alzheimer trait-linked phenotype. EMBO J. 1996a;15:498–509. [PMC free article] [PubMed] [Google Scholar]
- 66.Yamatsuji T, Okamoto T, Takeda S, Fukumoto H, Iwatsubo T, Suzuki N, Asami-Odaka A, Ireland S, Kinane TB, Nishimoto I. Neuronal DNA fragmentation by familial Alzheimer's V642 mutants of APP via heteromeric G proteins. Science. 1996b;272:1349–1352. doi: 10.1126/science.272.5266.1349. [DOI] [PubMed] [Google Scholar]
- 67.Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- 68.Yonezawa K, Ueda H, Hara K, Nishida K, Ando A, Chavanieu A, Matsuba H, Shii K, Yokono K, Fukui Y, Calas B, Grigorescu F, Dhand R, Gout I, Otsu M, Waterfield MD, Kasuga M. Insulin-dependent formation of a complex containing an 85-kDa subunit of phosphatidylinositol 3-kinase and tyrosine-phosphorylated insulin receptor substrate 1. J Biol Chem. 1992;267:25958–25965. [PubMed] [Google Scholar]
- 69.Zapf J, Schmid C, Guler HP, Waldvogel M, Hauri C, Futo E, Hossenlopp P, Binoux M, Froesch ER. Regulation of binding proteins for insulin-like growth factors (IGF) in humans. Increased expression of IGF binding protein 2 during IGF I treatment of healthy adults and in patients with extrapancreatic tumor hypoglycemia. J Clin Invest. 1990;86:952–961. doi: 10.1172/JCI114797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 71.Zhao B, Chrest FJ, Horton WE, Jr, Sisodia SS, Kusiak JW. Expression of mutant amyloid precursor proteins induces apoptosis in PC12 cells. J Neurosci Res. 1997;47:253–263. [PubMed] [Google Scholar]
- 72.Zhou H, Li XM, Meinkoth J, Pittman RN. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol. 2000;151:483–494. doi: 10.1083/jcb.151.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]