

Abstract
Sympathetic neurons die by apoptosis when they are deprived of nerve growth factor (NGF). Activation of caspases by cytochrome c released from mitochondria is central to this death. In this report we present evidence that cellular redox state regulates cytochrome c redistribution in these neurons. An increase of mitochondrial-produced reactive oxygen species (ROS) occurred in rat sympathetic neurons in cell culture within 3 hr of NGF withdrawal. Caspase inhibitors blocked this ROS burst. By 6 hr after NGF deprivation, glutathione (GSH) levels had increased, neutralizing elevated hydrogen peroxide levels and returning cellular redox state to basal levels. By 12 hr after deprivation, ROS levels had again increased and remained elevated during the rest of the apoptotic process. The later ROS burst appeared to have both caspase-dependent and caspase-independent components and was coincident with the period of cytochrome c release. Inhibition of protein synthesis with cycloheximide (CHX) and treatment with the antioxidant compound, N-acetyl-l-cysteine (l-NAC), blocked both the early and late ROS bursts by increasing cellular GSH levels (Ratan et al., 1994; Tan et al., 1998). Both compounds, and a membrane-permeant form of GSH, also inhibited cytochrome c release and death. Treatment of NGF, CHX-,l-NAC-, and GSH-saved cells with hydrogen peroxide caused rapid cytochrome c release. These data suggest a role for cellular redox state in regulating cytochrome c release during apoptosis induced by NGF withdrawal.
Keywords: reactive oxygen, mitochondria, caspase, cytochrome c, apoptosis, redox
Approximately 50% of neurons produced during embryogenesis die by apoptosis (Oppenheim, 1991). This programmed death is characterized by atrophy, internucleosomal DNA fragmentation, dependence on caspase activity (Alnemri et al., 1996) and, in many cases, dependence on protein synthesis (Martin et al., 1988; Deckwerth and Johnson, 1993; Deshmukh et al., 1996). Availability of a sufficient quantity of a required neurotrophic factor provided by target, or other tissues, is a primary determinant of which neurons survive the period of developmental apoptosis (Oppenheim, 1991). Only those cells that obtain sufficient quantities of the neurotrophin live.
The prototypical model for investigating the cellular and molecular events underlying apoptosis during neurogenesis is embryonic sympathetic neurons in cell culture (Martin et al., 1988). These neurons undergo apoptotic death, either in vivo or in vitro, when they do not obtain enough of their required neurotrophic factor, nerve growth factor (NGF). Release of cytochrome c from the intermembrane space of mitochondria into the cytoplasm is central to the apoptotic death of many cell types, including NGF-deprived sympathetic neurons (Reed, 1997; Deshmukh and Johnson, 1998; Martinou et al., 1999). Once in the cytoplasm, cytochrome c activates caspase proteases (Alnemri et al., 1996), presumably by binding onto the caspase regulatory protein, apoptosis protease activating factor-1 (Apaf-1), or a related protein (Li et al., 1997). Caspases, in turn, cleave many important substrates and are directly responsible for cellular demise.
Suppression of protein synthesis has a potent anti-apoptotic effect on NGF-deprived sympathetic neurons (Martin et al., 1988). Recent evidence shows this effect is entirely mediated via block of cytochrome c release from mitochondria (Deshmukh and Johnson, 1998; Neame et al., 1998), implying that protein synthesis suppression blocks production of one or more proteins that are required for cytochrome c redistribution. In this report, we present evidence suggesting an alternate mode of protection by protein synthesis blockade. Withdrawal of NGF from rat sympathetic neurons caused elevated levels of mitochondrial-produced reactive oxygen species (ROS). These ROS caused a cellular pro-oxidant state that appeared to be necessary for cytochrome c release. Inhibition of protein synthesis increased cellular glutathione (GSH) concentration (Ratan et al., 1994) and, thereby, inhibited ROS accumulation (Tan et al., 1998). Thus, much of the anti-apoptotic effect of blocking protein synthesis in NGF-deprived sympathetic neurons may be caused by antioxidant inhibition of cytochrome c discharge from mitochondria rather than by block of pro-apoptotic protein production.
MATERIALS AND METHODS
Reagents. Monochlorobimane, Mitotracker Red CMXRos, 2′, 7′-bis(2-carboxyethyl)carboxyfluorescein (BCECF), and 5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) were purchased from Molecular Probes (Eugene, OR). Caspase inhibitor, boc-aspartyl(OMe)-fluoromethylketone, was purchased from Enzyme Systems Products (Livermore, CA), and nerve growth factor 2.5S was purchased from Harlan Bioproducts (Indianapolis, IN). Radioactive amino acids were purchased from ICN (Costa Mesa, CA). U74500A was a gift from Pharmacia and Upjohn (Kalamazoo, MI). All other reagents were purchased from Sigma (St. Louis, MO) unless otherwise stated.
Cell culture and survival assay. Superior cervical ganglia were dissected from embryonic day 20 or 21 Sprague Dawley (Harlan Bioproducts) rat fetuses. Neurons were dissociated from the ganglia as described (Johnson and Argiro, 1983; Franklin et al., 1995). Cells for survival and protein synthesis assays were plated on a collagen substrate in 24 well Costar tissue culture dishes (Corning, Corning, NY). Cells to be used for fluorescent or confocal microscopy experiments were plated on a collagen substrate coated on number 1 glass coverslips glued with SILASTIC medical adhesive (Dow Corning, Midland, MI) over holes cut in the bottoms of 35 mm Falcon tissue culture dishes (Beckton Dickinson, Franklin Lakes, NJ). Approximately one-half ganglion was plated per culture well for all experiments except for whole-culture GSH assays in which three to five ganglia were plated per culture (in 35 mm culture dishes). Cultures were maintained in medium containing Eagle's minimum essential medium with Earle's salts (Life Technologies, Gaithersburg, MD) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 20 μm fluorodeoxyuridine, 20 μm uridine, 1.4 mml-glutamine, and 50 ng/ml 2.5S NGF.
Cultures were deprived of NGF by incubation in the standard culture medium lacking NGF and containing a polyclonal NGF-neutralizing antibody (Harlan Bioproducts; Franklin and Johnson, 1998). At the end of all survival experiments, treatment media was replaced with standard culture medium containing NGF. After 5–7 d in this medium, cultures were fixed by exposing them for 30 min at 4° C to 4% paraformaldehyde in PBS, pH 7.2. They were then stained with 0.1% crystal violet to enhance visualization (Franklin et al., 1995). Counting by a naive observer determined number of surviving neurons. All counts were normalized to the number of neurons in sibling cultures maintained since the time of plating in standard culture medium containing NGF. Adding NGF back to the culture medium after survival experiments resulted in significant hypertrophy of living neurons while dead ones degenerated, allowing unambiguous determination of cells saved from death by the various experimental treatments. Therefore, the moment of death in this rescue paradigm was the time at which readdition of NGF no longer promoted long-term survival. All experiments were begun when cells had been in culture for 6–9 d.
Confocal and fluorescent microscopy. For confocal microscopy, neurons were visualized with a 60× plan oil immersion lens (numerical aperture 1.4) on an inverted microscope (Nikon Diaphot 200). Confocal imaging was accomplished by a Bio-Rad (Hercules, CA) MRC 1024 confocal laser-scanning microscope equipped with a mixed gas argon–krypton laser. The scan head of the confocal microscope was mounted transversely onto the inverted microscope. The laser emitted three strong lines in exact coalignment at 488, 568, and 647 nm. A motorized wheel with neutral density filters was controlled by computer software to vary laser intensity. The microscope and its settings were controlled by 24 bit MRC-1024 Laser Sharp software (version 3.0; Bio-Rad) running on a Compac Prosignia 300 computer.
Neurons, chosen at random with phase-contrast microscopy, were scanned by the confocal microscope at 512 × 512 pixel resolution. Digitized images were stored on computer hard drives or zip drives (Iomega, Roy, UT) for later analysis. Generally, eight separate fields of view were scanned per plate. Image quantification was accomplished by measuring raw pixel intensity of stained neuronal somas with Sigmagel software (SPSS, Chicago, IL) set to square spot operation mode. In all cases, the quantified spot covered a 60 μm2 area of the soma. Because we were interested in cytoplasmic changes, nuclei were excluded from measurements. The measured intensity of each neuron was normalized to that of control neurons maintained in the standard culture medium containing NGF and receiving the same concentration of dye for the same period as did the experimental cells. Control neurons were always from the same plating as the experimental cells. Presentation versions of saved images were prepared with Adobe Photoshop 5.0 (Adobe Systems, San Jose, CA).
All fluorescence microscopy was accomplished with a Nikon TE300 inverted microscope. Light was provided by a mercury lamp, and images were captured by a cooled CCD camera (MicroMAX; Princeton Instruments, Trenton, NJ) using Metamorph software (Universal Imaging Corporation, West Chester, PA) running on a Compaq 400 MHz computer. Filter cubes were changed manually. The 380 ± 15, 440 ± 15, and 490 ± 15 nm excitation filters were controlled by a Lambda 10–2 optical filter changer (Sutter Instruments, Novato, CA). Fluorescence intensity was measured in a 33 μm2 area of the soma of each cell with the Metamorph elliptical region tool. All microscopy was done at room temperature.
ROS measurement. We used the redox-sensitive dye CM-H2 DCFDA. This dye is nonfluorescent in reduced form and is readily membrane-permeant. Once in a cell, esterases cleave its acetate groups. The thiol-reactive chloromethyl group then binds to cellular thiols trapping the dye inside the cell where oxidation converts it to the fluorescent form. CM-H2 DCFDA is oxidized by cellular hydrogen peroxide, hydroxyl radicals, and various free radical products lying downstream from hydrogen peroxide. It is relatively insensitive to oxidation by superoxide (Royall and Ischiropoulis, 1993). However, because hydrogen peroxide is produced by dismutation of superoxide (Halliwell and Gutteridge, 1999), CM-H2 DCFDA serves as an indirect indicator of superoxide production. Therefore, in this report, when we refer to ROS detected by this dye we denote mostly hydrogen peroxide, hydroxyl radicals, or their downstream free radical products.
Most cultures were incubated for 20–25 min at 35°C in the appropriate experimental medium containing CM-H2DCFDA (10 μm). They were then washed two or three times with Leibovitz's L-15 medium containing the experimental treatments and left in the last wash for confocal microscopy. To determine how well the dye was retained, cells loaded with CM-H2 DCFDA were treated with 10 mmH2O2 to oxidize the dye. After 20 min of this treatment, CM-H2 DCFDA intensity was determined, and the H2O2 was washed out. The average fluorescence intensity of these neurons was about the same 30 min after washout as it was 20 min after the beginning of the treatment (4.8 ± 0.41-fold and 5.0 ± 0.29-fold increases before and after washout respectively; p > 0.55 by ttest; n = 28–32 neurons). Thus, there appeared to be no significant leakage of dye from cells. We also found that CM-H2 DCFDA was relatively insensitive to photo-oxidation when laser intensity was kept at low levels, also making it superior to earlier versions of the dye (data not shown). For the experiments described here, CM-H2 DCFDA was also superior to the redox-sensitive dye dihydrorhodamine. Staining with the latter dye, unlike CM-H2 DCFDA, depends on mitochondrial membrane potential, which changes during the apoptotic process (Neame et al., 1998). Cultures treated withN-acetyl-l-cysteine (l-NAC) were loaded with CM-H2DCFDA in medium lackingl-NAC out of concern that interaction of CM-H2DCFDA with l-NAC thiols might lower loading concentration. After treatment with the dye, cultures were returned to L-15 medium containing the appropriatel-NAC concentrations. The CM-H2DCFDA was excited with the 488 nm line of the confocal laser. The FITC photomultiplier of the confocal scope was used for image acquisition.
Immunocytochemistry. Neurons were immunostained for cytochrome c by a modification of the techniques described by Easton et al. (1997), Deshmukh and Johnson (1998), and Putcha et al. (1999). At the end of experiments, cells were fixed for 30 min at 4°C with 4% freshly made paraformaldehyde in PBS, pH 7.2. Cultures were then washed three times with Tris-buffered saline containing 100 mm Tris-HCl, pH 7.6, and 0.9% NaCl. Washing was followed by incubation for at least 30 min at room temperature in blocking buffer (Tris-buffered saline containing 0.3% Triton X-100 and 5% normal goat serum). Cultures were then incubated overnight at 4°C in the same solution containing the anti-cytochrome c primary antibody (200 ng/ml; PharMingen, San Diego, CA). After washing three times with blocking buffer, cultures were incubated for 2–4 hr at 4°C in blocking buffer containing an FITC-conjugated anti-mouse secondary antibody (1.7 μg/ml; PharMingen). Cultures were then washed two times with Tris-buffered saline, left in this medium, and viewed by fluorescence or confocal microscopy.
Glutathione assays. Reduced GSH concentrations were estimated by fluorescence microscopic imaging of single cells stained with monochlorobimane (MCB), a dye that is enzymatically bound to GSH (Fernandez-Checa and Kaplowitz, 1990). In most MCB experiments, cultures were stained for 30 min at room temperature in L-15 medium containing MCB (5 μm). They were then washed two times in the same medium lacking MCB and left in the second wash for recording the fluorescence intensity of incorporated MCB. During the last 30 min of some experiments, cultures were incubated at 35° in a 5% CO2 atmosphere in the appropriate experimental medium containing MCB (5 μm). No difference in results was noted with the two staining methods. MCB was excited at 380 ± 15 nm. Emission filter was 510 ± 20 nm. This technique allowed determination of relative GSH levels in the somas of individual neurons without interference from GSH in non-neuronal cells.
Total GSH and oxidized (GSSG) glutathione concentrations were determined in whole cultures by a modification of the method of Tietze (1969). In this assay, the sulfhydryl group of GSH reacts with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) to produce yellow 5-thio-2-nitrobenzoic acid (TNB). The mixed disulfide, GSTNB (GSH and TNB) that is produced is subsequently reduced by glutathione reductase to recycle the GSH and produce more TNB. The rate of TNB production is proportional to the recycling reaction which was, in turn, proportional to GSH concentration. Because of the use of glutathione reductase, which reduces GSSG to GSH, this assay measured both GSH and GSSG and, therefore, gave total glutathione concentration. Cells (from three to five ganglia) were lysed in 10 mm HCl by freezing (−70°C) and thawing at room temperature three times. The resultant lysates were deproteinated to eliminate interference from protein sulfhydryl groups. Deproteination was accomplished by precipitating proteins with 5% sulfosalycilic acid followed by centrifugation. The supernatant was removed, and pH was corrected to 7.0 with 4.0 N triethanolamine. Absorbance was read at 414 nm with a Bio-Rad model 550 microplate reader. Total GSH plus GSSG concentration was determined from standard curves.
Effects of compounds on protein synthesis. Protein synthesis was assayed by incorporation of radiolabeled amino acids as described (Franklin and Johnson, 1998). Labeling medium contained the appropriate concentrations of compounds and 10 μCi/ml TRAN35S-label (70%l-methionine, 15%l-cysteine) or 10 μCi/ml [3H]leucine.
pH measurement. Intracellular pH was measured by fluorescence microscopy of cells loaded with the pH-sensitive dye BCECF. Cultures were exposed for 15 min to medium appropriate for the experimental treatments and containing 8 μmBCECF. After loading, and 2× washout of the dye with L-15 medium, cultures were exposed to L-15 medium containing the appropriate experimental treatments. The BCECF was alternately excited at 440 ± 10 and 490 ± 10 nm. Emission filter was 515 ± 10 nm. The ratios of emission intensities at the two different excitation wavelengths was determined in a 33 μm2area of individual cells. As a positive control, intracellular pH was altered by exposing cells to L-15 medium, pH 6.0–8.0, containing the protonophore nigericin (5 μm; Molecular Probes).
Statistical analysis. Statistical analysis was done with SigmaPlot 5.0 (SPSS) or by SigmaStat 2.0 (SPSS). As appropriate, statistical comparisons were made by t test or by Kruskal–Wallis one-way ANOVA on Ranks with Dunn's multiple comparisons test. Other types of statistical comparisons are noted in the text or figure legends. Means in all figures are shown as ±SEM.
RESULTS
Increased ROS levels in NGF-deprived sympathetic neurons were blocked by protein synthesis inhibition and attenuated by caspase inhibition
Elevated levels of ROS occur in rat sympathetic neurons in cell culture within 3 hr of NGF deprivation (Greenlund et al., 1995;Dugan et al., 1997). This ROS increase appears transient, with levels declining to baseline by 8 hr after NGF withdrawal. Because none of these cells commit to apoptotic death until ∼18 hr after NGF removal (Deckwerth and Johnson, 1993), it was of interest to determine ROS levels at later periods. For these experiments we used the redox-sensitive dye 5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate (CM-H2 DCFDA), which fluoresces upon oxidation. Consistent with the reports of a transient increase in ROS levels after NGF deprivation, average CM-H2 DCFDA intensity was 1.22 ± 0.18-fold above average control intensity by 3 hr after deprivation but had returned to baseline levels by 6 hr after withdrawal (Fig. 1A). By 12 hr after NGF deprivation, average CM-H2DCFDA intensity was again well above baseline and continued to increase with time, reaching a peak average of threefold to fourfold above control intensity by 24–30 hr after withdrawal. Many individual cells showed much larger increases, with CM-H2DCFDA intensity rising by as much as 22-fold (Fig. 1B). Because intensity of fluorescein-based dyes can be affected by pH (Nedergaard et al., 1990; Reynolds and Hastings, 1995), we used the pH-sensitive dye BCECF to determine whether cellular pH changed after NGF deprivation. Table1 shows there were no alterations of pH after withdrawal (p < 0.001 compared with pH of cells maintained in NGF). Treatment of CM-H2DCFDA-loaded cells with H2O2 (10 mm) for 30 min caused an average 7.4 ± 0.9-fold increase of dye intensity. Therefore, the elevated CM-H2 DCFDA fluorescence after NGF deprivation was not caused by alterations of cellular pH but was, likely, attributable solely to dye oxidation caused by increased ROS levels.
Fig. 1.
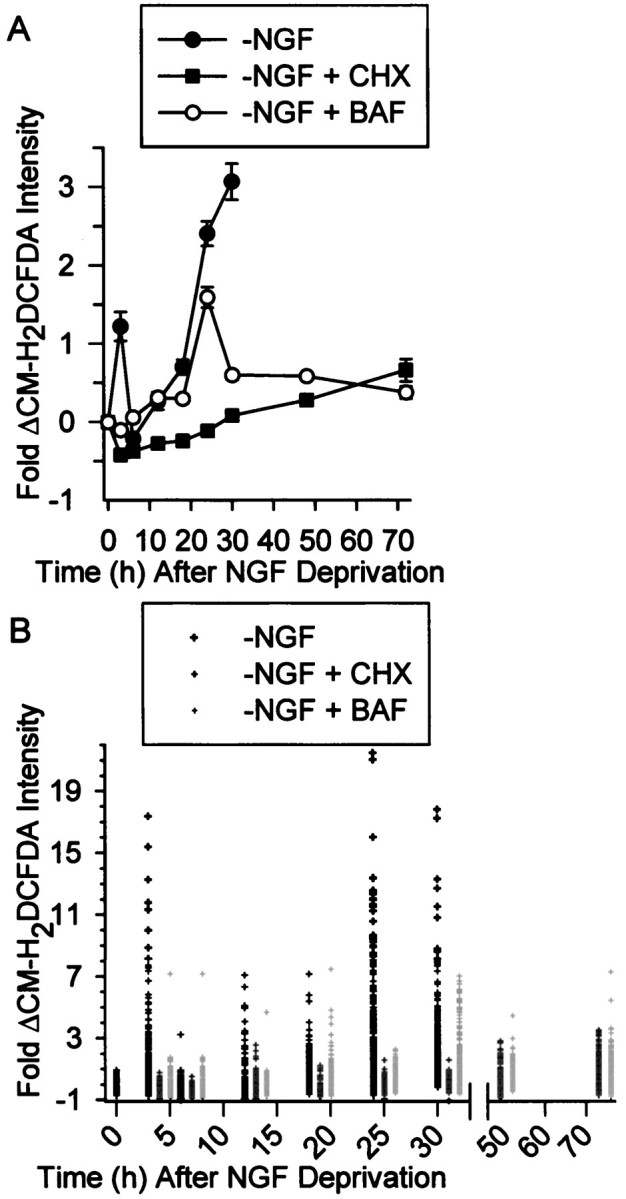
Increased Ros levels in NGF-deprived rat sympathetic neurons in cell culture. A, Average ROS levels, as measured by fluorescence intensity of the redox-sensitive dye CM-H2 DCFDA, showed a transient early increase after NGF deprivation followed by a later sustained increase. The protein synthesis inhibitor CHX (1 μg/ml), blocked both the early and delayed ROS bursts, whereas the pan-caspase inhibitor BAF (30 μm) completely blocked the early burst but only attenuated the later one. Data are shown as fold change from control values. B,Single-cell CM-H2 DCFDA values used for the averages inA. The data from the NGF-deprived cells is above the time point, and the CHX and BAF values for that time point are to the right of these values. There is no data shown for NGF-deprived cultures later than 30 hr after withdrawal because most cells had died by that time. The values shown are from cells that appeared intact when observed by phase-contrast microscopy. Except for the 24 hr time point for NGF deprivation, n = 93–290 neurons taken from three or four separate platings for the different conditions and time points. n = 383 neurons from six separate platings for the 24 hr NGF-withdrawal time point. Control CM-H2DCFDA intensity and intensity at T0 was that of cells maintained continuously in the presence of NGF. For each cell, CM-H2 DCFDA intensity was normalized (as a percentage) to the average CM-H2 DCFDA intensity of cells measured in sibling NGF-maintained cultures at the same time. Fold change from the average was then obtained for each cell by subtracting 100 from this and then dividing by 100 ([%CM-H2 DCFDA intensity − 100%]/100%). For example, the increase at 3 hr after NGF deprivation was ∼222% of control. This technique was used in all subsequent figures. Error bars for fold change are SEM in this and subsequent figures.
Table 1.
Effects of treatments on intracellular pH
| Treatments | BCECF, 490:440 ratios |
|---|---|
| +NGF | 0.85 ± 0.01- |
| 0.97 ± 0.01 | |
| −NGF | |
| 3 hr | 0.85 ± 0.01 |
| 6 hr | 0.90 ± 0.01 |
| 12 hr | 0.90 ± 0.02 |
| −NGF + Antimycin A | 0.77 ± 0.01* |
| −NGF + BAF | 0.93 ± 0.01 |
| −NGF + CHX | 0.95 ± 0.01 |
| −NGF (24 hr) + FCCP | 0.74 ± 0.01* |
| −NGF (24 hr) + L-NAC | 0.95 ± 0.01 |
| −NGF (24 hr) + Rotenone | 0.9 ± 0.01 |
Cultures maintained in NGF, deprived of NGF, or deprived of NGF for 24 hr and exposed to medium containing CHX (1 μg/ml), BAF (30 μm), or l-NAC (30 mm) were loaded with BCECF in the treatment medium. They were then placed in L-15 medium, containing the appropriate treatment, for 490:440 ratio determinations. Cultures treated with antimycin A (1 μm), FCCP (5 μm), or rotenone (10 μm) were deprived of NGF for 24 hr and exposed to the compounds only during BCECF loading and during recording. This acute treatment was done because of potent suppression of protein synthesis by these compounds with long-term treatment (Table 2). Lower 490:440 ratios indicate acidification. Asterisk denotes significantly different values (p < 0.01) from average control values measured in sibling NGF-maintained cells at the same time. All statistical comparisons were made by Mann–Whitney rank sum test. N= 37–59 cells from two or three separate platings except for the 3–24 hr time course in which N = 35–36 cells from two platings. The range of 490:440 ratios measured in NGF-maintained cells is shown at top.
It was recently shown that inhibition of protein synthesis with cycloheximide (CHX) blocks an increase in ROS that occurs during the glutamate-induced death of a hippocampal cell line (Tan et al., 1998). To determine whether a similar block occurred in NGF-deprived sympathetic neurons, cultures were exposed to 1 μg/ml CHX, a concentration that causes a rapid, sustained, and complete suppression of protein synthesis in these cells and that also blocks apoptosis (Martin et al., 1988; Franklin and Johnson, 1998). Figure 1,A and B, shows that CHX caused a profound inhibition of both the early and the late ROS bursts occurring after NGF deprivation. Average CM-H2DCFDA intensity in CHX-maintained cultures was at, or below, control levels for at least the first 30 hr after NGF withdrawal. However, by 48–72 hr after deprivation, CHX had become somewhat less effective at suppressing ROS. Average CM-H2DCFDA intensity was 0.28 ± 0.08-fold above control level by 48 hr and 0.66 ± 0.14 by 72 hr (p < 0.01 for both). No cells in the CHX-maintained cultures had CM-H2DCFDA intensities more than threefold above control average at any time point (Fig. 1B). The pH of cells deprived of NGF and exposed for 24 hr to CHX was the same as that of cells maintained in the presence of NGF (Table 1; p > 0.1), indicating that there was no pH-dependent quenching of CM-H2DCFDA fluorescence. The average CM-H2DCFDA intensity of NGF-deprived, CHX (1 μg/ml for 24 hr)-maintained neurons exposed to H2O2 (10 mm) for 30 min was not significantly different from that of NGF-maintained cells exposed to 10 mmH2O2 for the same period (7.4 ± 0.93- and 6.4 ± 1.4-fold increases, respectively;p > 0.5; n = 33–35 neurons). Therefore, the block of increased CM-H2DCFDA fluorescence intensity by CHX could not be explained by CHX-mediated alterations of dye loading or changes in the ability of the dye to fluoresce upon oxidation. The protein synthesis inhibitor anisomycin (100 ng/ml; Franklin and Johnson, 1998) and the transcription inhibitor actinomycin D (100 ng/ml; Franklin and Johnson, 1998) also decreased ROS to near baseline levels in cells deprived of NGF and exposed to these compounds for 24 hr. The average CM-H2DCFDA intensity in the anisomycin-exposed cells was 0.22 ± 0.13-fold of NGF control levels (p > 0.4 compared with control; n = 35 neurons) and was reduced to −0.6 ± 0.07-fold of control in the actinomycin D-exposed cells (p < 0.001 compared with control;n = 38 neurons). Therefore, inhibition of macromolecular synthesis had potent antioxidant effects on the ROS levels of NGF-deprived cells.
Inhibition of caspase activity also blocks increased ROS levels in glutamate-induced cell death (Tan et al., 1998). The pan-caspase inhibitor boc-aspartyl (OMe)-fluoromethylketone (BAF) has a potent anti-apoptotic effect on NGF-deprived sympathetic neurons (Deshmukh et al., 1996), indicating that this death is caspase-mediated. To determine whether caspases influenced ROS levels in these cells, we treated cultures from the time of withdrawal with 30 μmBAF, a concentration that blocks ∼80% of apoptotic death for 3 d after deprivation (Deshmukh et al., 1996). Surprisingly, BAF treatment completely blocked the early ROS burst (p > 0.1 compared with control) and greatly reduced, but did not prevent the late burst (Fig.1A,B). Average CM-H2DCFDA intensity peaked at 1.59 ± 0.13-fold above those in control cells 24 hr after deprivation. It then declined to a steady-state level that averaged 0.59 ± 0.05-fold above control levels by 48 hr and 0.38 ± 0.08-fold above control by 72 hr after the withdrawal (p < 0.01 compared with control in both cases). Although not as effective as CHX at preventing high CM-H2DCFDA intensities, BAF, nevertheless, limited CM-H2DCFDA values in NGF-deprived cells to no more than approximately eightfold above average control level. The pH of BAF-saved cells was the same as control cells (Table 1), and they responded with a similar increase of CM-H2DCFDA intensity when exposed to H2O2 (10 mm) for 30 min (6.4 ± 1.4- and 6.3 ± 0.6-fold for control and BAF, respectively; p > 0.3;n = 35–38 neurons). Therefore, the suppression in CM-H2DCFDA intensity by BAF cannot be explained by nonspecific effects of the caspase inhibitor on cellular pH, dye loading, or on the ability of the dye to fluoresce when oxidized. The effect of the pan-caspase inhibitors, z-VAD-fmk (100 μm), and VAD-CHO (100 μm) on ROS levels was similar to that of BAF. Twenty-four hours after NGF withdrawal, average CM-H2DCFDA intensity in z-VAD-fmk-maintained cells was 0.98 ± 0.24-fold above that of control (p < 0.001). This value was well below that of NGF-deprived cells at 24 hr without the inhibitor (Fig.1A). The average CM-H2DCFDA intensity at 3 hr after NGF deprivation in VAD-CHO-treated cells was 0.28 ± 0.09-fold of the average intensity in control cells, not significantly different from that of NGF-maintained cells (p > 0.2). Twenty-four hr after NGF withdrawal, average CM-H2DCFDA intensity in VAD-CHO-maintained cells was 1.98 ± 0.31-fold above that of control cells, well below that of cells deprived without presence of the inhibitor (p < 0.001). These data suggest that the early transient ROS burst after NGF deprivation was entirely dependent on caspase activity, whereas the later burst was only partially caspase-dependent. Inhibiting protein synthesis with CHX potently suppressed all components of the ROS bursts.
ROS derived from the mitochondrial electron transport chain
The primary source of ROS production in most cells is the electron transport chain of mitochondria (Halliwell and Gutteridge, 1999). Electrons can “leak” from this chain to cause a univalent reduction of elemental oxygen to the free radical, superoxide (O2−). To determine whether the ROS bursts were of mitochondrial origin, we treated cells with carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), a protonophore that uncouples oxidative phosphorylation and decreases O2− formation (Turrens, 1997; Tan et al., 1998). Acute application of FCCP (5 μm) to cultures deprived of NGF for 3–24 hr potently inhibited both the early and late ROS bursts (Fig.2A). ROS levels in BAF-maintained cells peaked 24 hr after NGF deprivation and subsequently declined but stayed above control levels (Fig.1A,B; p < 0.01). Acute application of FCCP (5 μm), blocked the residual ROS production remaining in BAF-saved neurons 48 hr after NGF removal (Fig.2A), suggesting that even at this time mitochondria were the source of ROS.
Fig. 2.
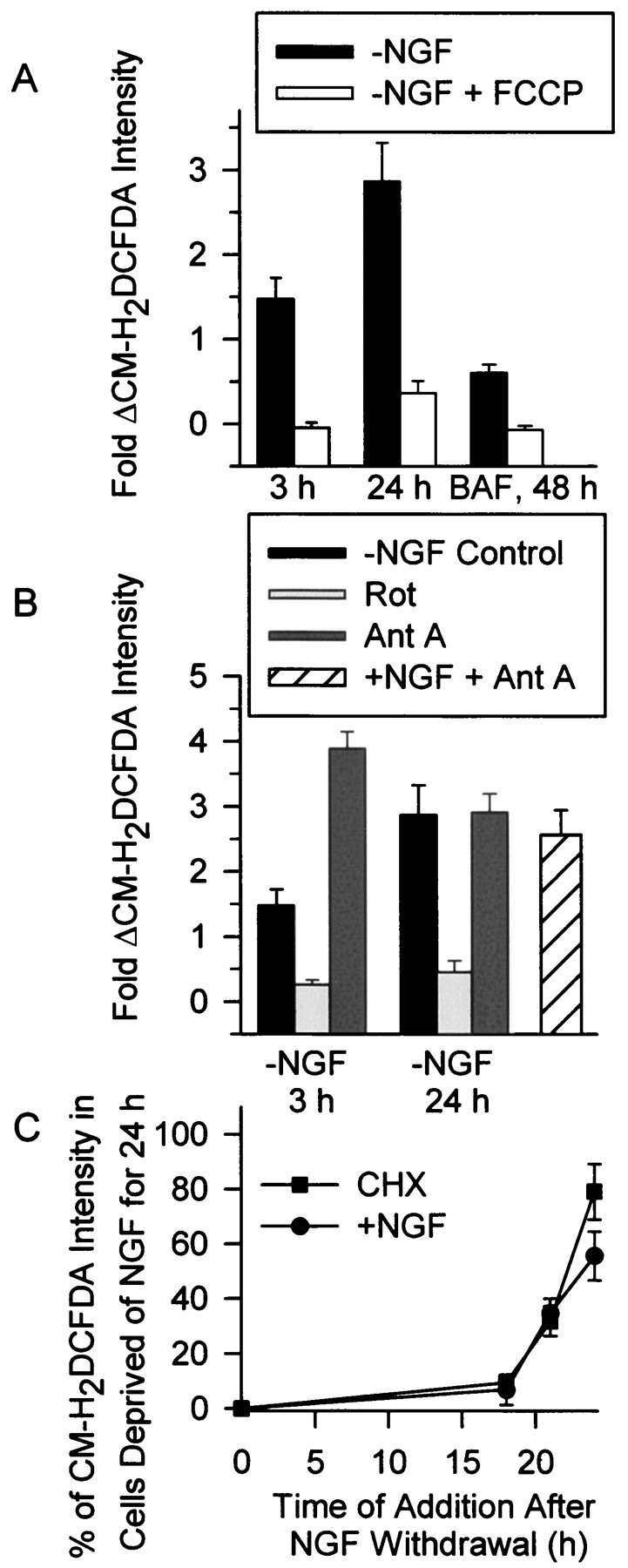
Increased ROS after NGF deprivation derived from the mitochondrial electron transport chain. A, Block of all components of the ROS burst by the protonophore FCCP (5 μm) suggested that the ROS derived from the mitochondrial electron transport chain. Cells deprived of NGF for 3, 24, or 48 hr (with 30 μm BAF) were exposed to FCCP during the time of CM-H2 DCFDA loading. FCCP was also included in the recording medium. Data were normalized as in Figure 1.n = 59–162 neurons from three to five separate platings. B, Acute effects of rotenone (Rot; 10 μm) and antimycin A (Ant A; 1 μm) on the ROS bursts at 3 and 24 hr after NGF deprivation. Also shown is the effect of Ant A on ROS in NGF-maintained neurons. Compounds were added to the culture medium during CM-H2 DCFDA loading and were also included in the recording medium. n = 59–145 neurons from three platings. C, Time course of suppression of the late ROS burst by CHX (1 μg/ml) and NGF (+NGF indicates 50 ng/ml NGF here and throughout the manuscript). Either CHX or NGF were added to the culture medium at the indicated times after NGF deprivation. The CM-H2 DCFDA intensity was then measured at 24 hr after the withdrawal. For the 24 hr time point, CHX or NGF were added during the CM-H2 DCFDA loading. Data are normalized to average CM-H2 DCFDA intensity of cells deprived of NGF for 24 hr. n = 87–163 neurons from three platings.
As a control, we exposed NGF-deprived (24 hr) neurons loaded with CM-H2DCFDA and acutely treated with FCCP (5 μm was included in CM-H2DCFDA loading medium and in recording medium) to 10 mmH2O2 for 30 min. This treatment increased CM-H2DCFDA intensity to that of H2O2 (10 mm)-treated cells deprived of NGF for the same period and not exposed to FCCP (5.9 ± 0.5-fold increase for FCCP;p > 0.5). There was a significant, acidification of FCCP-treated cells (Table 1). It was conceivable that this acidification quenched CM-H2DCFDA fluorescence (Reynolds and Hastings, 1995) and that H2O2 blocked the quenching by causing the cells to become less acidic. To test this possibility, we measured pH in cells deprived of NGF for 24 hr and acutely exposed to FCCP (5 μm was included in BCECF loading medium and in recording medium) and H2O2 (10 mm for 30 min). The BCECF 490:440 ratios in these neurons were not significantly different from those of similar FCCP-treated cells not exposed to H2O2 (Table 1;p > 0.4; n = 24 neurons). Therefore, the H2O2 treatment did not alter cellular pH. These data indicate that the level of acidification caused by FCCP did not quench the dye and that suppression of CM-H2DCFDA intensity by FCCP was not caused by nonspecific effects of altered pH. Therefore, attenuation of CM-H2DCFDA intensity was probably caused by suppression of mitochondrial ROS production. Because no other treatment reported herein altered cellular pH (except for a small acidification caused by antimycin A; Table 1), pH-dependent changes of CM-H2DCFDA intensity cannot explain any of the data in this manuscript.
To further explore the role of mitochondria in causing increased ROS levels after NGF deprivation, we used specific inhibitors of electron transport through mitochondrial respiratory complexes. Acute application of the complex 1 (NADH coenzyme Q reductase) inhibitor, rotenone (10 μm), almost completely blocked both the early and late ROS bursts (Fig. 2B). Rotenone did not affect the ability of H2O2(10 mm for 25 min) to increase CM-H2DCFDA intensity to that measured in cells deprived of NGF for 24 hr and treated for the same time with 10 mmH2O2 (6.6 ± 0.4-fold increase for rotenone; p > 0.5; n = 23 neurons). Acute application of antimycin A (1 μm), a respiratory complex 3 (coenzyme Q-cytochrome c reductase) inhibitor, enhanced the early ROS burst in NGF-deprived cells and also greatly increased ROS in NGF-maintained cells (Fig. 2B). It did not influence the late ROS burst (p > 0.5).
In the course of doing controls for the experiments described in this report we found that many unrelated compounds suppressed protein synthesis in sympathetic neurons and also inhibited apoptotic death (Table 2 lists some of the compounds that we tested; Martin et al., 1988; Deckwerth and Johnson, 1993; Franklin and Johnson, 1998). Because CHX blocked the ROS bursts, it was possible that the effects of FCCP and rotenone on the bursts resulted from such a nonspecific effect. Indeed, we found that FCCP (5 μm) and rotenone were potent inhibitors of protein synthesis in these cells (Table 2). However, Figure 2C shows that the effects of FCCP and rotenone on protein synthesis could not explain their effects on the ROS bursts because they suppressed ROS far more quickly than CHX (1 μg/ml). Thus, the acute effects of FCCP and rotenone on ROS were not caused by protein synthesis inhibition. These data suggest that most, if not all, of both the early and late ROS bursts were produced by “leakage” from the mitochondrial electron transport chain. Additionally, because block of electron flow through complex 1 decreased ROS, whereas block of flow through complex 3 either increased ROS levels (3 hr after deprivation) or had no effect (24 hr after deprivation), they suggest that the electrons entered the chain at complex 1 and leaked to molecular O2 to form O2− between complexes 1 and 3 (at coenzyme Q; Dugan et al., 1997; Halliwell and Gutteridge 1999). The lack of effect of antimycin on the late burst may indicate that ROS production was at a maximum and could not be further increased.
Table 2.
Effects of treatments on protein synthesis and survival
| % Synthesis | Surv | |
|---|---|---|
| Mitochondrial agents | ||
| Antimycin A (1 μm) | 33 ± 9 (24 hr) | ++ |
| Bongkreckic acid (30 μm) | ND | NE |
| FCCP (5 μm) | 52 ± 10 (0.3 hr) | +++ |
| 14 ± 2 (24 hr) | ||
| Cyclosporin A (30 μm) | 40 ± 1 (24 hr) | + |
| Rotenone (10 μm) | 21 ± 2 (24 hr) | ++ |
| Antioxidants | ||
| l-NAC (30 mm) | 103 ± 6 (24 hr) | +++ |
| Desferrioxamine mesylate (1 mm) | 31 ± 4 (24 hr) | + |
| GSH ethyl ester (10 mm) | 87 ± 5 (24 hr) | +++ |
| NG-Nitro-l-arginine methyl ester hydrochloride (300 μm) | ND | NE |
| NG-Nitro-l-arginine (300 μm) | ND | NE |
| α-Phenyl-tert-butylnitrone (10 mm) | 24 ± 10 (16 hr) | +++ |
| U74500A (30 μm) | 21 ± 4 (24 hr) | +++ |
| Other | ||
| N-Acetyl-l-alanine (30 mm) | ND | NE |
| BAF (30 μm) | 97 ± 5 (24 hr) | +++ |
For those compounds that promoted survival, the concentrations shown were the optimal ones. For those that had no significant effect on survival, the concentrations shown were the highest tested. Cells in survival assays were rescued with NGF at 48 or 72 hr after deprivation, and survival was determined by visual inspection several days later. The number in parentheses under “% Synthesis” is the number of hours of exposure. All data are averages from 11-20 cultures from two to four separate platings. +, Delayed death for ∼1 d; ++, delayed death for ∼2 d; +++, delayed death for at least 3 d; NE, no effect; ND, not done. Compounds not discussed in text include: mitochondrial permeability transition inhibitors bongkreckic acid (Henderson and Lardy, 1970) and cyclosporin A (Szabó and Zoratti, 1991); iron chelators desferrioxamine mesylate (Halliwell and Gutteridge, 1999) and U74500A (Braughler et al., 1987); nitric oxide synthase inhibitorsNG-Nitro-l-arginine methyl ester hydrochloride (Crossin, 1991) andNG-Nitro-l-arginine (Dwyer et al., 1991); and the antioxidant spin-trap α-Phenyl-tert-butylnitrone (Halliwell and Gutteridge, 1999).
Increased GSH concentration was, at least partially, responsible for the apparent transience of the early ROS burst
It seemed likely to us that the early and late ROS bursts were associated. We hypothesized that the early burst appeared transient not because of a decrease in production of ROS by mitochondria but, rather, because it was masked by an increase in antioxidant defenses by cells (Lin et al., 1995). Consistent with this hypothesis, Deckwerth and Johnson (1994) reported that GSH concentration increases in these cells after NGF deprivation. Because CM-H2DCFDA detects H2O2, (but not O2−), which is removed by an enzymatic reaction involving GSH (Royall and Ischiropoulis, 1993;Halliwell and Gutteridge, 1999), an increase in GSH concentration might account for the apparent transience of the ROS burst. That is, H2O2 production might remain elevated but total cellular H2O2 concentration would decrease because of increased GSH levels. To test this hypothesis, we deprived cells of NGF and, at various times after the deprivation, stained them with the GSH-sensitive dye MCB (Fernandez-Checa and Kaplowitz, 1990; Ratan et al., 1994). Figure3A shows that average MCB intensity increased 0.45 ± 0.15-fold above that of NGF-maintained cells by 6 hr after NGF deprivation (p < 0.05). The average intensity reached a peak of 0.58 ± 0.10 above control average by 12 hr after NGF removal (p < 0.05 compared with control). It then declined somewhat but remained elevated with respect to average control intensity for the entire time course of death. Increased MCB intensity after NGF deprivation was completely blocked in cultures treated withl-buthionine-[S,R]-sulfoximine (BSO; 200 μm), a potent, irreversible inhibitor of λ-glutamylcysteine synthetase, a key enzyme in the GSH biosynthetic pathway (Griffith and Meister, 1979). A population-based assay for reduced and oxidized glutathione (GSH plus GSSG) showed a similar increase in whole cultures (Fig.3B). These findings were consistent with the hypothesis that continued ROS production (O2−) was masked by a GSH-mediated reduction of H2O2 concentration. To further test this hypothesis, we measured the effect of BSO (200 μm) on CM-H2DCFDA intensity after NGF withdrawal. Figure 3C shows that this treatment prevented the return of ROS levels to baseline. However, average CM-H2DCFDA intensity in these cells was not as high as in cells not treated with BSO (Fig.1A), indicating some suppression of ROS levels by this compound. These data suggest that the early ROS burst appeared transient not because of an actual decrease in ROS production but, at least in part, because elevated GSH concentration decreased H2O2 accumulation.
Fig. 3.
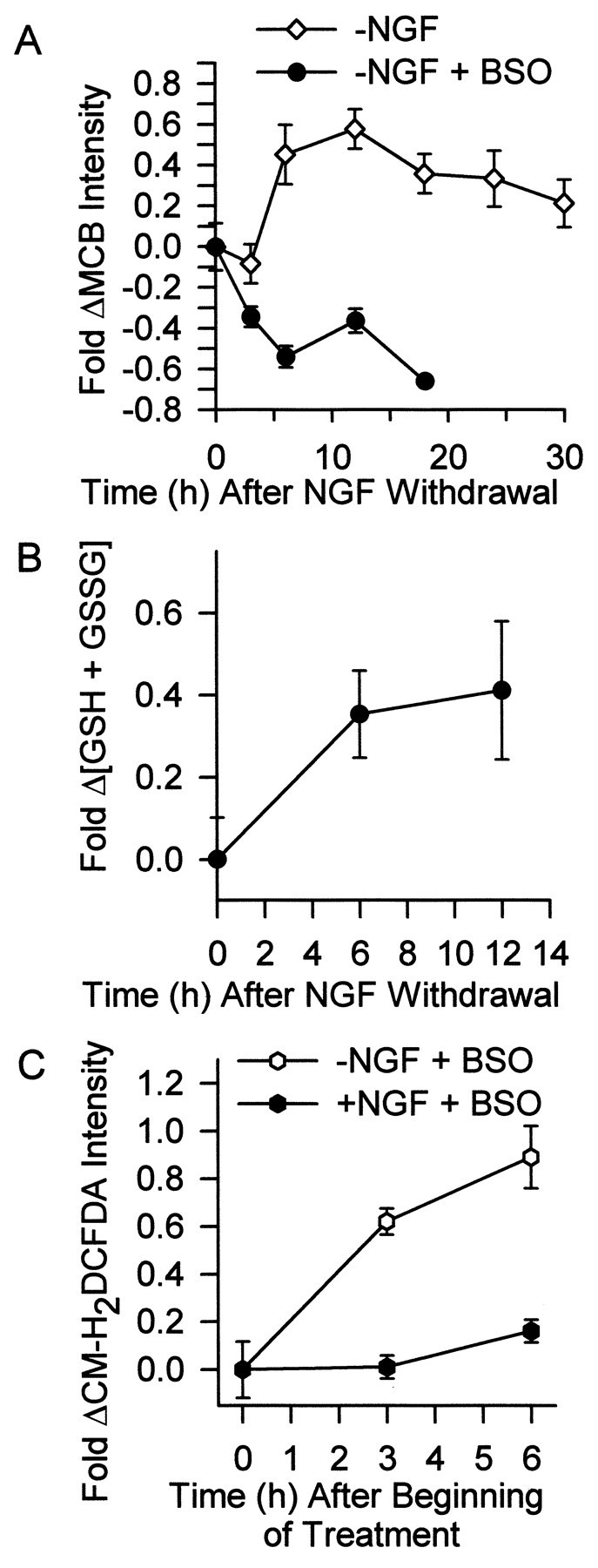
Increased GSH concentration at least partially caused the transience of the early ROS burst. A,Glutathione levels increased within several hours after NGF deprivation. The inhibitor of GSH production, BSO (200 μm), blocked this increase. Neurons were deprived of NGF for the indicated times. During the last 30 min of the incubation, cultures were exposed to the GSH-sensitive dye MCB (5 μm). Neurons were then viewed with an inverted microscope, and images were captured by a CCD camera. Intensity of MCB staining in single cells was determined with MetaMorph software and is shown as fold change from that of control cells maintained in the presence of NGF. The BSO-treated neurons appeared phase-bright and healthy for the first 12 hr after NGF deprivation. However, by 18 hr, many had died. Data are normalized to MCB intensity of NGF-maintained neurons stained and measured at the same time as the experimental cells. n = 67–137 neurons from three or four separate platings. B, The concentration of GSH plus GSSG in whole cultures increased after NGF deprivation, independently confirming the single-cell results in A. The GSH plus GSSG concentration is shown as fold increase above that measured in NGF-maintained cells sampled at the initial time point.n = 10–14 cultures from three separate platings.C, Inhibition of the GSH increase by BSO (200 μm) at least partially blocked the transience of the ROS burst after NGF deprivation. Data are shown as fold change of CM-H2 DCFDA intensity in NGF-replete or -deprived neurons exposed to BSO for the indicated times after withdrawal.n = 99–140 neurons from three or four separate platings.
The late ROS burst occurred at about the same time as cytochrome c release
Exit of cytochrome c from mitochondria into the cytoplasm is central to the apoptotic death of NGF-deprived sympathetic neurons (Deshmukh and Johnson, 1998; Neame et al., 1998; Martinou et al., 1999). To ascertain the temporal relationship between the ROS bursts and cytochrome c release, we deprived cells of NGF and used immunocytochemistry to determine the time course of cytochrome c redistribution. Figure4A shows the three criteria that were used for scoring the status of cytochrome c in these experiments: (1) neurons showing intense punctate staining when viewed by confocal microscopy were scored as having retained at least some cytochrome c in their mitochondria, (2) cells showing no punctate staining when viewed by confocal microscopy were scored as having completely lost cytochrome c from their mitochondria and, (3) cells that exhibited homogeneous cytoplasmic staining when viewed by fluorescence microscopy were scored as having released at least some cytochrome c from mitochondria. The latter neurons tended to stain much more faintly than cells that retained cytochrome c in mitochondria, consistent with reports that cytochrome c rapidly degrades in these cells once it is released into the cytoplasm (Deshmukh and Johnson, 1998; Neame et al., 1998). Surprisingly, confocal micrographs generally revealed only two mitochondrial staining patterns after NGF deprivation; intense, punctate staining or no staining at all. Intermediate staining patterns were seldom observed. Using these criteria, we determined the time course of appearance of cytochrome c in the cytoplasm and of complete loss of cytochrome c from the mitochondria after NGF withdrawal (Fig. 4B). The time course of the appearance of cytoplasmic staining for cytochrome c was only slightly earlier than that of complete loss from the mitochondria, suggesting that rapid release from the mitochondrial pool was followed by rapid degradation in the cytoplasm. It was not possible to determine cytochrome c status in cells later than 30 hr after NGF deprivation because of extensive cell death. To circumvent complications from cell loss, we maintained cells (alive) with BAF (30 μm) and determined the time course of appearance of cytoplasmic cytochrome c staining and of complete loss of cytochrome c from the mitochondria. By 48 hr after NGF deprivation, 94.0 ± 2.7% of BAF-saved cells had no detectable cytochrome c. During the first 30 hr after NGF withdrawal there were no apparent differences in the rate of release in the presence or absence of the caspase inhibitor. Therefore, caspase activity was, apparently, not required for cytochrome c release. The time courses of the late ROS burst and release of cytochrome c into the cytoplasm were nearly identical (Fig. 5). These data indicate that the period of maximal rate of cytochrome c release coincided with the peak of the late ROS burst.
Fig. 4.
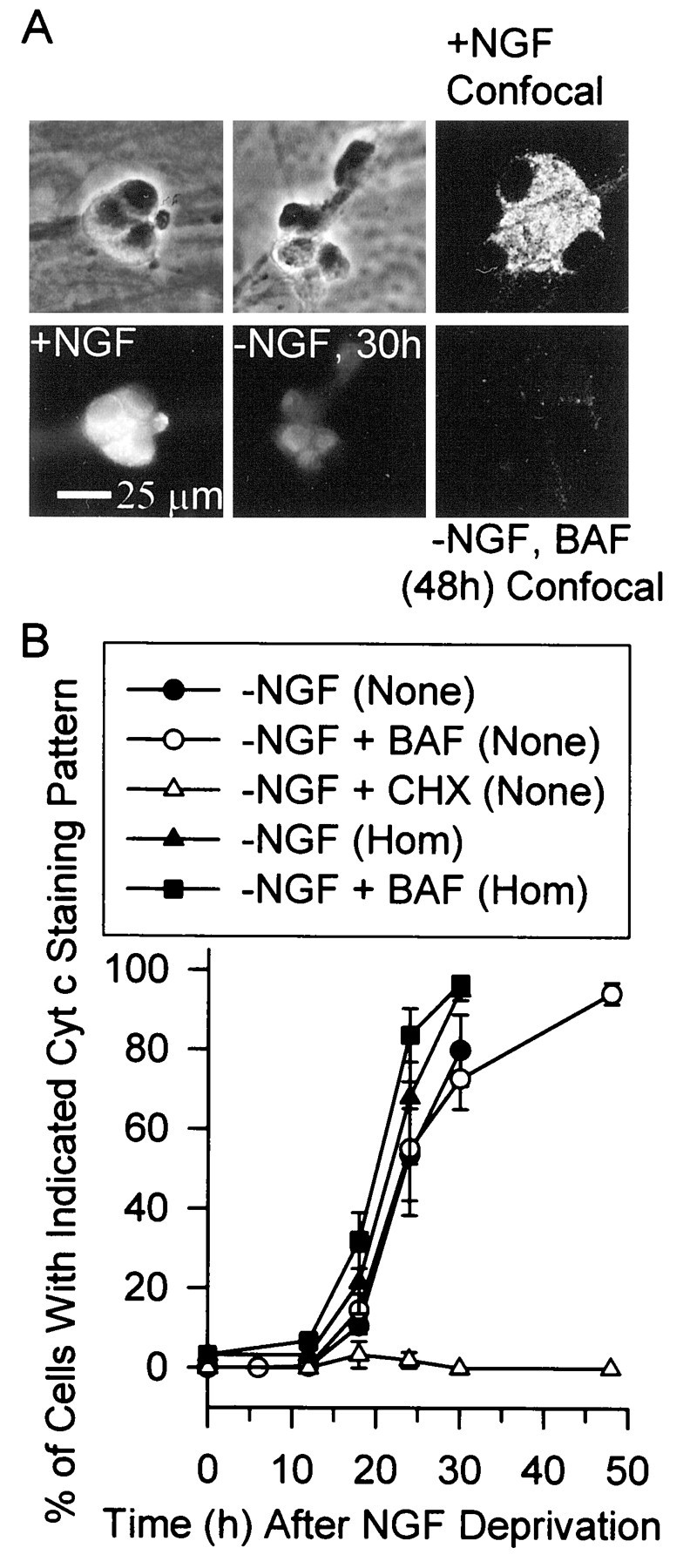
Time course of cytochrome c release from mitochondria after NGF deprivation. A, Micrographs demonstrating the criteria used to score cytochrome c localization. Thebottom two panels on the left are fluorescent micrographs of clusters of neurons immunostained for cytochrome c. The cells had been maintained in culture medium containing or lacking NGF as indicated. The micrographs above them are the same cells viewed with phase-contrast microscopy. Note the intense punctate staining in the control (+NGF) cells and the faint homogeneous staining in neurons deprived of NGF for 30 hr. The +NGF cells are representative of neurons that were scored as having not released cytochrome c. The cells deprived of NGF for 30 hr are representative of ones scored as having released cytochrome c into the cytoplasm. The two panels on the right show confocal micrographs of neurons in separate cultures immunostained for cytochrome c. The cultures were maintained continuously in the presence of NGF or in medium containing BAF (30 μm) and no NGF for 48 hr. The top cluster of cells is representative of neurons that were scored as having cytochrome c-replete mitochondria, whereas the bottomcluster of cells is representative of those that were scored as having completely lost cytochrome c from mitochondria. B, Time courses of the appearance of homogeneous staining for cytochrome c in the cytoplasm and complete loss of cytochrome c staining in the mitochondria. Scoring for homogeneous cytoplasmic staining for cytochrome c was done, as in A, with fluorescence microscopy. Neurons were scored as having punctate (no cytochrome c release) or homogeneous (Hom; cytochrome c in the cytoplasm) staining by a naive observer. Scoring for complete loss of cytochrome c (None) from mitochondria was done, also as inA, by confocal microscopic imaging of immunostained cells. All cytochrome c loss was blocked by CHX (1 μg/ml). BAF concentration was 30 μm. n = 292–345 neurons from three separate platings for the different time points for homogeneous staining. n = 169–287 neurons from three or four platings for complete loss of cytochrome c staining except for the 48 hr CHX treatment where n = 99 from two platings.
Fig. 5.

Comparison of the time courses of changes in ROS (CM-H2 DCFDA) and cytochrome c staining (Cyt c) in NGF-deprived neurons receiving the indicated treatments. The data are from Figures 1A and 4B. To allow easier comparison of time courses, all data are normalized to the maximal changes observed. Symbols and error bars are omitted to improve visual clarity.
Effects of CHX and l-NAC on the ROS burst, cytochrome c release, and death were closely correlated
Complete suppression of protein synthesis in NGF-deprived sympathetic neurons in culture with 1 μg/ml CHX completely blocks release of cytochrome c from mitochondria. This block appears to be the sole means by which protein synthesis inhibition suppresses apoptosis in these cells because microinjecting cytochrome c into CHX-saved cells causes rapid death (Deshmukh and Johnson, 1998; Neame et al., 1998; Fig. 4B). To determine how closely the abilities of CHX to inhibit protein synthesis, the ROS burst, cytochrome c release, and death were associated, we determined the effects of different concentrations of CHX on these parameters. To circumvent any complications from dying cells in the cytochrome c experiments, we maintained neurons in a viable state with BAF (30 μm). Cytochrome c release or retention after 2 d was determined by immunostaining. Other BAF-maintained cultures received the same concentrations of CHX, and BAF-insensitive ROS production was determined during the peak of the late ROS burst, 24 hr after deprivation. There was a close relationship between the concentration-dependent inhibition of the BAF-insensitive ROS burst by CHX, block of cytochrome c release in the BAF-saved cells by CHX, and block of apoptotic death by CHX in NGF-deprived cells without BAF (Fig.6A). Significant inhibition of both death and cytochrome c release occurred when protein synthesis was suppressed to less than ∼20% of the control rate (300 ng/ml CHX). These values corresponded to >60% suppression of the BAF-insensitive ROS burst by CHX.
Fig. 6.
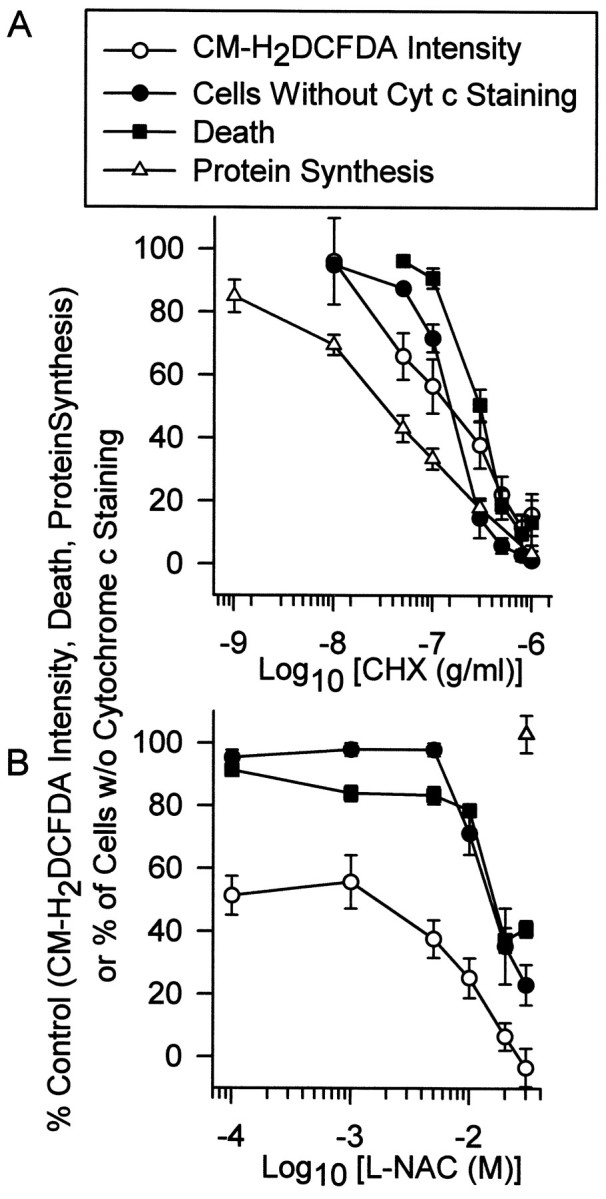
Concentration-dependent block of the late ROS burst by CHX or l-NAC was similar to the concentration-dependent block of cytochrome c release and death by these compounds. A, Block of increased ROS (CM-H2 DCFDA intensity), cytochrome c release, death, and protein synthesis by the indicated CHX concentrations. The CM-H2 DCFDA intensity is that of neurons deprived of NGF and maintained in BAF (30 μm)- and CHX-containing medium for 24 hr. The data are normalized to average CM-H2 DCFDA intensity of BAF-saved cells not exposed to CHX. n= 71–141 neurons from three or four separate platings for each concentration. Cytochrome c data are from cells maintained in BAF (30 μm) and the indicated CHX concentrations for 48 hr. This time point was chosen because we found no difference in the percentage of release at later time points. Therefore, cells that were going to release cytochrome c had done so by 48 hr after NGF deprivation. Cytochrome c status was determined by confocal microscopy as described in Figure 4. Data are shown as percentage of cells with no cytochrome c staining. n = 26–115 neurons from three or four separate platings for each concentration. Death was determined by depriving cells of NGF for 3 d (a time when most NGF-deprived cells have died; Deckwerth and Johnson, 1993) and maintaining them in medium containing the indicated CHX concentrations. Cultures were then returned to NGF-containing medium, and survival was assayed 5–7 d later, as described in Materials and Methods. Death is normalized to the percentage occurring in NGF-deprived cells not exposed to CHX for 48 hr. n = 6–9 cultures from three separate platings for each concentration. Protein synthesis data are from NGF-maintained cells exposed for 72 hr to the indicated CHX concentrations. Data are normalized to that of NGF-maintained cells not exposed to CHX. n = 26–32 cultures from three or four separate platings for each concentration. The dose–response curve for suppression of protein synthesis by exposure to these concentrations of CHX for 72 hr is nearly identical to that of cultures exposed to the same concentrations of CHX for 4 hr and 24 hr (Franklin and Johnson, 1998). B, Block of increased ROS (CM-H2 DCFDA intensity), cytochrome c release, and death by the indicated l-NAC concentrations. n = 82–154 neurons from three or four separate platings for each concentration. Cytochrome c data are from cells maintained in BAF (30 μm) and the indicated l-NAC concentrations for 48 hr. Cytochrome c status, CM-H2 DCFDA intensity, and death were determined and normalized as described in A.n = 117–176 neurons from three separate platings for each concentration of l-NAC in cytochrome c experiments and 6–15 cultures from three or four separate platings for each concentration in cell death experiments. Protein synthesis data are from cultures exposed to l-NAC (30 mm) for 24 hr. n = 20 cultures.
Cytochrome c shuttles electrons from respiratory complex 3 to respiratory complex 4 (cytochrome oxidase) in the mitochondrial electron transport chain. Loss of cytochrome c from mitochondria blocks flow of electrons between these two complexes and, therefore, can enhance ROS production in a manner similar to that of antimycin A treatment (Fig. 2B;Cai and Jones, 1998). Therefore, it seemed possible that loss of cytochrome c from mitochondria could have caused the BAF-insensitive component of the late ROS burst. The increase in ROS could, thus, have been an epiphenomenon largely irrelevant to apoptotic death. Alternatively, the burst could have been an essential component of apoptosis, even if caused by cytochrome c loss. To determine whether increased ROS production might contribute to apoptosis, we tested the effects of a number of antioxidant compounds on the survival of NGF-deprived cells. All of them, except for two nitric oxide synthase inhibitors, potently blocked apoptosis after NGF withdrawal. However, most of these compounds also potently inhibited protein synthesis (Table 2). An exception was the membrane-permeant analog of cysteine,N-acetyl-l-cysteine (l-NAC; Ferrari et al., 1995).l-NAC did not cause suppression of protein synthesis so it could not have blocked apoptosis of NGF-deprived cells via that mechanism. Therefore, we determined the effects of different concentrations of l-NAC on death of NGF-deprived cells, the ROS burst, and on cytochrome c release in BAF-maintained cells. All three parameters were inhibited byl-NAC (Fig. 6B). Exposure of NGF-deprived, l-NAC-maintained (30 mm for 24 hr) neurons to H2O2 (30 mm for 20 min) caused an average increase of CM-H2DCFDA intensity that was not significantly different from that caused by treating NGF-maintained cells not exposed to l-NAC the same way (p> 0.5; n = 35–59 neurons). Therefore,l-NAC did not alter dye loading or the ability of the dye to respond to oxidation. As with CHX, no effect on cytochrome c release or death was observed unless BAF-insensitive ROS production was suppressed by >60%.N-acetyl-l-alanine (30 mm) had no effect on survival of NGF-deprived cells, indicating that the cysteine thiol was necessary for survival promotion by l-NAC (data not shown). These data and the CHX findings suggest that, whatever the cause of the late ROS burst, it may have been involved in inducing cytochrome c release and death.
Both CHX and l-NAC blocked the ROS burst by increasing cellular GSH concentration
Ratan et al. (1994) reported that inhibition of protein synthesis by CHX blocks oxidative stress-induced death of cortical neurons in cell culture by shunting cysteine from incorporation into protein to GSH synthesis. To determine whether a similar effect was responsible for the block of the ROS burst by CHX in NGF-deprived sympathetic neurons, we determined relative GSH concentrations in CHX-treated neurons by MCB staining. Neurons were deprived of NGF and maintained for 24 hr in medium containing different concentrations of CHX and BAF (30 μm; to prevent death at lower CHX concentrations). Figure 7A shows that the effect of these CHX concentrations on the ROS burst was functionally related to the effect of each concentration on MCB staining. Higher MCB intensities correlated with lower CM-H2DCFDA intensities. This finding suggests that CHX decreased ROS by increasing cellular GSH. A similar relationship between ROS levels and MCB staining was observed in cultures deprived of NGF and supported for 24 hr in medium containing BAF and different concentrations ofl-NAC. Thus, l-NAC also appeared to decrease ROS by increasing GSH. Inhibition of GSH synthesis with BSO (200 μm) completely blocked the ability of CHX and l-NAC to increase MCB staining (Fig. 7B), clearly indicating that both CHX andl-NAC blocked the ROS burst by increasing cellular GSH concentration. Increased glutathione as a function of increased CHX or l-NAC concentrations was independently confirmed by measuring GSH plus GSSG in whole cultures with the glutathione reductase assay (Fig. 7C). Long-term exposure of NGF-deprived cultures to l-NAC (30 mm) was as effective as CHX (1 μg/ml) in causing sustained suppression of ROS (data not shown).
Fig. 7.
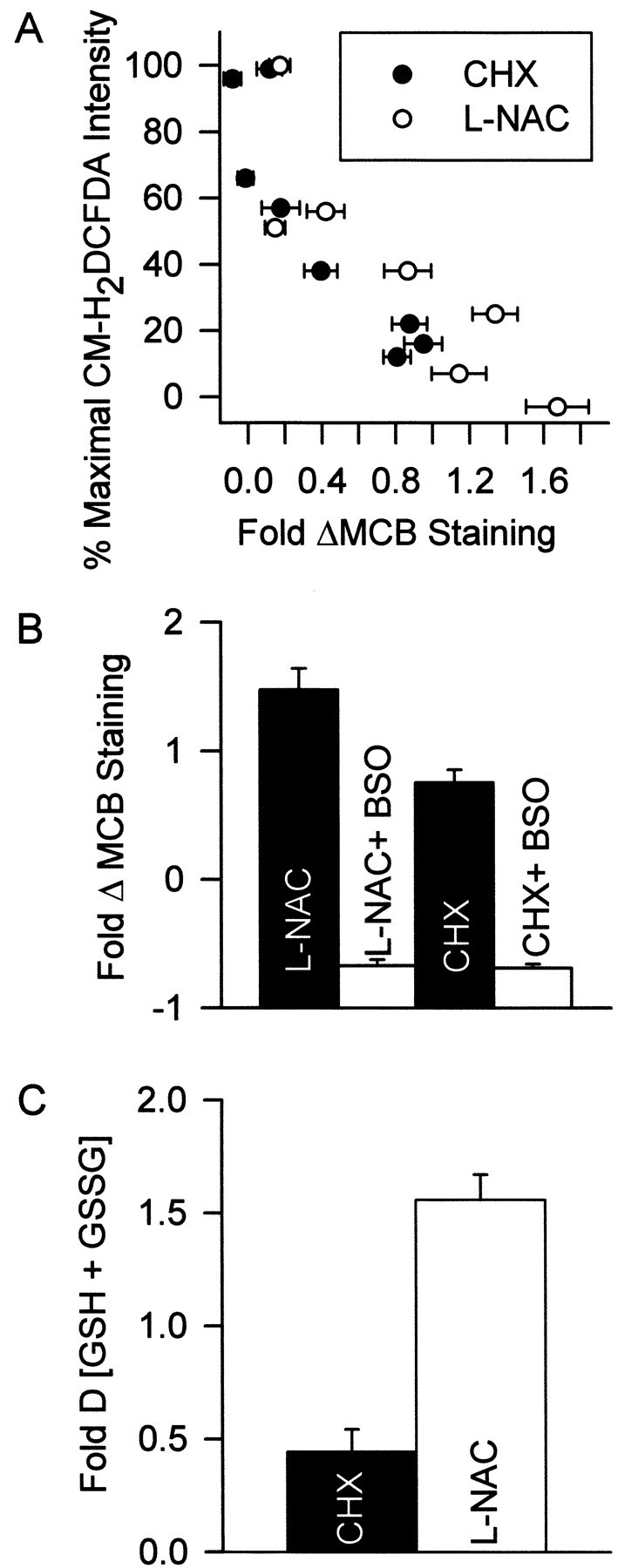
The ability of CHX and l-NAC to block the ROS burst derived from their ability to regulate cellular GSH levels. A, The abilities of different concentrations of CHX and l-NAC to block the ROS burst (CM-H2DCFDA intensity) was closely correlated with the ability of these concentrations to increase MCB staining. Cultures were deprived of NGF for 24 hr in medium containing different CHX or l-NAC concentrations and BAF (30 μm). The CM-H2DCFDA data are from Figure 6, A andB, and are matched to MCB staining resulting from treatment with the same CHX or l-NAC concentrations.n = 88–173 neurons from three separate platings.B, Increased MCB staining was caused by increased GSH concentration. The inhibitor of GSH production, BSO (200 μm), prevented CHX (1 μg/ml) and l-NAC (30 mm) from increasing MCB intensity. Cultures were treated with CHX (18 hr) or l-NAC (24 hr) before MCB staining.n = 77–123 from three separate platings.C, Effects of CHX (1 μg/ml) and l-NAC (30 mm) on GSH plus GSSG concentration in whole cultures. Cells were exposed to the compounds in the absence of NGF and BAF for 12 hr. This early time point was chosen to avoid complications from loss of cells caused by the apoptosis that would occur in the absence of BAF (Deckwerth and Johnson, 1993). Data are normalized to GSH plus GSSG concentration in cultures deprived of NGF without the compounds for the same period. n = 9–11 cultures from three separate platings for each treatment.
Increased GSH concentration blocked both apoptotic and oxidative death
In an attempt to ascertain whether upregulation of GSH concentration by CHX and l-NAC was related to block of apoptosis by these compounds, we treated cultures of NGF-deprived neurons maintained by CHX (1 μg/ml) or l-NAC (30 mm) with BSO (200 μm). This treatment almost completely prevented the ability of both compounds to block death, suggesting the importance of GSH (Fig.8A). However, it remained possible that BSO reduced GSH to levels inconsistent with survival under any circumstance or that it had toxic effects unrelated to prevention of GSH synthesis. Indeed, most cells in NGF-maintained cultures also died after 2 d of treatment with BSO (200 μm; Fig. 8A). To further explore the role of GSH in survival, we maintained NGF-deprived cultures in medium containing GSH ethyl ester (10 mm), a membrane-permeant form of GSH. This treatment potently inhibited apoptosis and also blocked cytochrome c release (Figs. 8B, 9C). Likel-NAC, GSH ethyl ester had little effect on protein synthesis (Table 2). Because BSO inhibits the synthesis of GSH rather than its activity, treating NGF-deprived neurons saved from death by GSH ethyl ester with BSO should not affect the ability of the GSH to promote survival. Figure 8B shows that such BSO (200 μm) treatment did not significantly alter the GSH ethyl ester block of apoptosis (p> 0.1 compared with GSH ethyl ester alone). Therefore, block of survival by BSO was mediated by decreased GSH concentration rather than by nonspecific toxic effects.
Fig. 8.
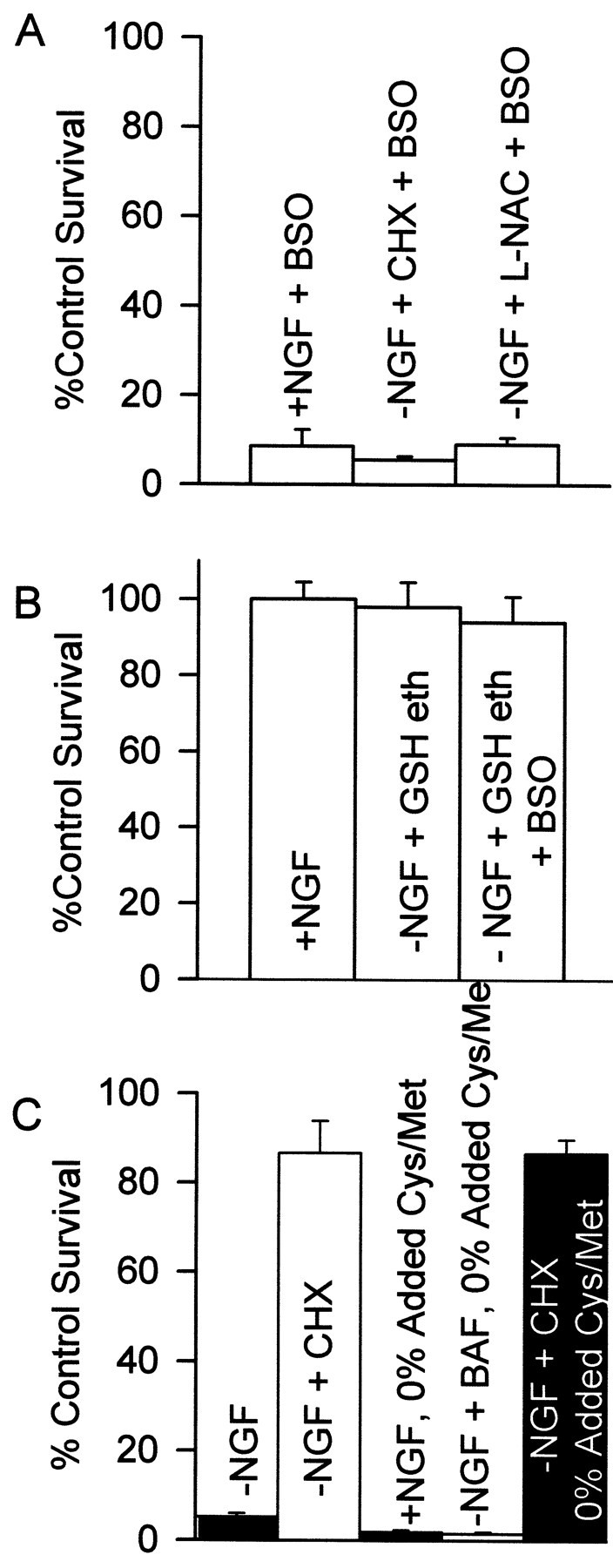
Increased GSH concentration blocked apoptosis.A, BSO (200 μm) inhibited the abilities of NGF to support survival and of CHX and l-NAC to prevent apoptosis of NGF-deprived cells. n = 9–13 cultures from three separate platings. B, A membrane-permeant form of GSH, GSH ethyl ester (10 mm), prevented apoptosis after NGF deprivation. Treatment of GSH ethyl ester-saved cultures with BSO (200 μm) had little effect on survival, indicating no nonspecific toxic effects of BSO. This concentration of GSH ethyl ester was the optimal survival-promoting concentration tested.n = 9–12 cultures from three platings.C, CHX (1 μg/ml) blocked oxidative death caused by cystine–methionine depletion. Cultures were incubated in medium containing no cystine or methionine except for that contained in 10% added fetal bovine serum. Consistent with an oxidative, rather than an apoptotic death, neurons treated in this manner had a grainy appearance and did not atrophy before death (data not shown). Two bars on theleft show data from cells maintained in normal medium. BAF concentration was 30 μm. n = 8–11 cultures from three platings. Cells in A–Creceived the indicated treatments for 48 hr before NGF rescue.
Fig. 9.
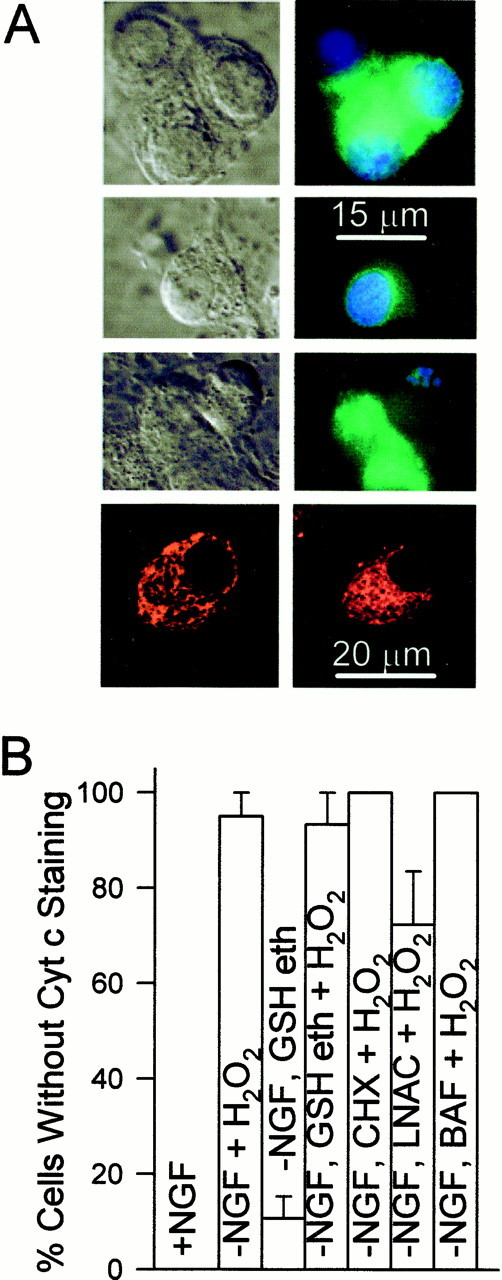
Treatment of cultures with H2O2 caused rapid cytochrome c redistribution.A,First row, Paired Nomarski and fluorescent micrographs of NGF-maintained neurons stained for cytochrome c. The nucleus was stained with bisbenzimide (10 μg/ml in TBS buffer for 30 min; Hoechst 33258). Second row,Paired Nomarski and fluorescent micrographs of NGF-maintained cells showing cytochrome c and bisbenzimide staining 1 hr after treatment with H2O2 (10 mm). Note the faint, homogenous staining for cytochrome c and lack of fragmented apoptotic nuclei. Third row, Paired Nomarski and fluorescent micrographs of NGF-deprived (24 hr), l-NAC (30 mm)-maintained cells showing cytochrome c and bisbenzimide staining 1 hr after treatment with H2O2 (10 mm). Note the faint, homogenous staining for cytochrome c and the fragmented apoptotic nuclei in one cell. The other two cells had not released cytochrome c. Fourth row, Confocal micrographs showing Mitotracker Red CMXRos staining in NGF-maintained cells (left) and NGF-maintained cells exposed for 1 hr to H2O2 (10 mm;right). The Mitotracker Red CMXRos (20 ng/ml) was added to the culture medium for 20 min at the end of H2O2 treatment. B, Loss of cytochrome c after 1 hr of treatment with H2O2(10 mm). Except for the control (+NGF), cultures were deprived of NGF and maintained in medium containing CHX (1 μg/ml), l-NAC (30 mm), GSH ethyl ester (10 mm), or BAF (30 μm) for 24 hr before H2O2treatment. Cells were scored as having retained or lost cytochrome c from mitochondria by confocal or fluorescence microscopy as detailed in Figure 4. n = 64–186 neurons from three platings.
To determine whether suppression of protein synthesis by CHX could protect sympathetic neurons from oxidative as well as apoptotic death, we induced oxidative death by incubating cultures in medium containing no added cystine or methionine except for that contained in fetal bovine serum. Such treatment deprives cells of required reducing equivalents for maintaining an antioxidant state (Tan et al., 1998). Neurons maintained in NGF or deprived of NGF and exposed to BAF (30 μm) died within 24 hr after the beginning of this treatment (Fig. 8C). Cycloheximide (1 μg/ml) almost completely prevented this death. Treatment with BSO (200 μm) blocked the inhibition of oxidative death by CHX, indicating a requirement for GSH (data not shown). Taken together, these data suggest that increasing cellular GSH levels, by whatever means, potently suppressed both the apoptotic and oxidative death of these cells. l-NAC (20 mm) increased GSH to about the same level as the most potent saving concentrations of CHX (Fig. 7A) and saved ∼60% of neurons from death (Fig. 6B). Therefore, at least one means by which CHX blocks death of these neurons is via increased cellular GSH concentration.
A pro-oxidant state caused rapid cytochrome c release
The effect of increased GSH concentration on cytochrome c redistribution and death of NGF-deprived neurons along with the close correlation between the time courses of the late ROS burst and cytochrome c release suggested that a pro-oxidant state might be conducive to cytochrome c release in these cells. To more directly determine whether this was the case, we treated cultures maintained with NGF, or deprived of NGF for 24 hr and maintained in medium containing CHX (1 μg/ml), l-NAC (30 mm), GSH ethyl ester (10 mm), or BAF (30 μm) with H2O2 (10 mm). Immunocytochemistry demonstrated that, in all these culture conditions, treatment with H2O2 for 1 hr caused extensive loss of cytochrome c from mitochondria (Fig.9A,B). The mitochondria in cells treated for 1 hr with H2O2 retained a membrane potential (Fig. 9A) as revealed by staining of mitochondria with the membrane potential-dependent dye, Mitotracker Red CMXRos (Neame et al., 1998). One hundred percent of NGF-maintained neurons (n = 96) and NGF-maintained neurons treated with H2O2 (n = 97) exhibited intense, punctate Mitotracker Red CMXRos staining. Thus, there was no general disruption of mitochondria caused by the H2O2 treatment. NGF-deprived cells maintained by l-NAC for 24 hr and then exposed to 10 mmH2O2 had an average CM-H2DCFDA intensity of 30 ± 8% of cells deprived of NGF for the same period. This finding suggests that ROS levels experienced by NGF-deprived cells are sufficient to cause cytochrome c release.
Many NGF-deprived neurons maintained in medium containing CHX,l-NAC, or GSH ethyl ester exhibited fragmented nuclei characteristic of apoptotic death after only 1 hr of H2O2 treatment (Fig.9A; 19, 53, and 44% had apoptotic nuclei respectively;n = 95–165 neurons), suggesting that cytochrome c redistribution induced apoptosis. Consistent with a report showing that cytoplasmic cytochrome c cannot induce apoptosis in these cells when NGF is present (Deshmukh and Johnson, 1998), none of the NGF-maintained cells treated with H2O2 had apoptotic nuclei (n = 117 cells). No cells maintained in BAF (30 μm)-containing medium without NGF (n = 82) had apoptotic nuclei after H2O2 treatment. These data suggest that the cytochrome c released into the cytoplasm of NGF-deprived cells by H2O2treatment activated caspases. They also suggest that it is the oxidative state of the cell rather than GSH concentration, per se, that regulates cytochrome c release during the apoptotic death of these neurons. Moreover, they demonstrate that a pro-oxidant state caused cytochrome c release when protein synthesis was completely suppressed. Therefore, under these conditions, no newly produced proteins were necessary for cytochrome c redistribution.
The late, but not early, ROS burst appeared to regulate cytochrome c release
Greenlund et al. (1995) hypothesized that the early ROS burst occurring in sympathetic neurons after NGF deprivation initiates a sequence of events leading to gene induction. The protein products of this induction then cause apoptotic death. Should this be true, block of ROS production by l-NAC or CHX should block death only if cells are exposed to these compounds during the time of the early ROS burst (3 hr after NGF deprivation). Figure10A shows the time courses of the abilities of l-NAC, CHX, and NGF to block death after NGF withdrawal. Both l-NAC and CHX inhibited death almost as effectively when added to the culture medium 6 hr after NGF deprivation as when added at the time of withdrawal. The time course of rescue from death after NGF deprivation for both l-NAC and CHX were nearly identical, suggesting similar mechanisms of action. The time at which 50% of neurons could be rescued from death by addition of these compounds to the culture medium occurred at 19–20 hr after withdrawal. Fifty percent of neurons deprived of NGF could be rescued by addition of NGF to the culture medium 24 hr after deprivation. Therefore, antioxidant treatment after the end of the early ROS burst was sufficient for block of most apoptotic death.
Fig. 10.
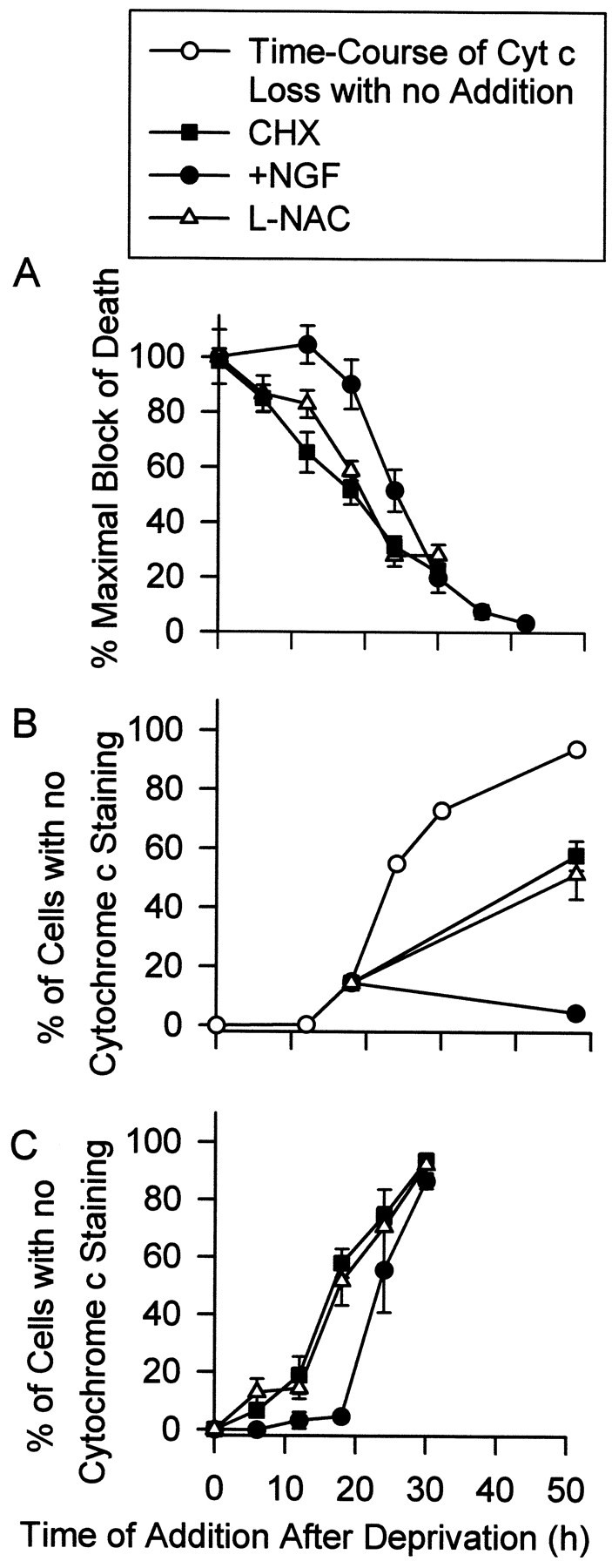
The late, but not early ROS burst appeared to regulate cytochrome c release. A, The time course of rescue from death induced by NGF deprivation was nearly identical for CHX (1 μg/ml) and l-NAC (30 mm). Either CHX,l-NAC, or NGF were added back to NGF-deprived cultures at the indicated times. Seventy-two hours after the initial withdrawal all media were replaced with NGF-containing medium, and the neurons were allowed to recover for at least 5 d before survival was determined. Data are normalized to the maximal block of death by each agent when it was added at the time of deprivation.n = 9–11 cultures from three separate platings for CHX and l-NAC. The rescue time course for NGF was similar to that reported by Deckwerth and Johnson (1993) for these cells and was, therefore, repeated only once. n = 3–16 cultures for each time point. B, Addition of NGF, but not CHX (1 μg/ml) or l-NAC (30 mm), to NGF-deprived cells immediately blocked cytochrome c release. Cultures were deprived of NGF and maintained in BAF (30 μm)-containing medium for 18 hr. At this time, medium containing BAF and either CHX, l-NAC, or NGF was added to the cultures. Forty-eight hours after the initial withdrawal, cultures were immunostained, and percentage of cells exhibiting complete loss of cytochrome c was determined as in Figure 4. The time course of complete loss of cytochrome c in BAF-saved, NGF-deprived neurons (open circles) was taken from Figure 4B.C, Time courses of the abilities of l-NAC (30 mm) and CHX (1 μg/ml) to block cytochrome c release were nearly identical. Data were obtained as in B except for the varying durations of deprivation before treatment with the agents. n = 101–159 neurons from three separate platings for B and C. Legend attop applies to A–C.
Deshmukh and Johnson (1998) reported that acute addition of NGF to NGF-deprived, BAF-maintained cultures of mouse sympathetic neurons caused an immediate block of additional cytochrome c loss in those cells. We repeated this experiment in rat sympathetic neurons with similar results. When NGF was added to NGF-deprived, BAF-maintained cultures 18 hr after deprivation, further cytochrome c loss from mitochondria was completely prevented (Fig. 10B). However, similar treatment at 18 hr with CHX orl-NAC did not immediately block release. Whereas only 20% of cells had lost cytochrome c staining by 18 hr after deprivation, ∼60% of cells exposed to CHX orl-NAC at this time had lost cytochrome c staining by 48 hr after withdrawal. Figure 10C shows that the time course of the loss of the abilities of both CHX andl-NAC to block cytochrome c release after NGF withdrawal were nearly identical. Both compounds blocked loss of cytochrome c in ∼50% of NGF-deprived neurons when added to the culture medium 18 hr after withdrawal, whereas readdition of NGF blocked loss from ∼50% of neurons 24 hr after withdrawal. Thus, the abilities of CHX, l-NAC, and NGF to block cytochrome c loss from NGF-deprived cells correlated closely with their abilities to block death. The inability of CHX to cause immediate block of cytochrome c release may have been related to the time required for it to upregulate GSH and block the ROS burst (Fig.2B). Nerve growth factor blocked cytochrome c release immediately, whereas its antioxidant effects took as long to develop as those of CHX. Thus, NGF must have blocked through mechanisms other than, or in addition to, antioxidant effects.
The data presented to this point suggested that the late ROS burst was involved in inducing cytochrome c redistribution in these cells. The potent, GSH-dependent, survival-promoting effects of protein synthesis suppression on apoptotic and oxidative death along with the antioxidant block of cytochrome c release in apoptosis suggested that much of the anti-apoptotic effect of CHX was mediated via antioxidant effects rather than by inhibition of production of pro-apoptotic proteins (Martin et al., 1988). Consistent with this hypothesis, the ability of CHX to inhibit ROS production after NGF deprivation diminished after several days of exposure to CHX (Fig. 1A,B), and neurons began to die (Fig.11A). On the third day after deprivation in the presence of CHX, 87 ± 7% of neurons could be rescued by NGF. By the seventh day only 23 ± 5% could be rescued. Few CHX-treated neurons exhibited nuclei characteristic of apoptosis 3 d after NGF deprivation, but by 5 d after withdrawal, 47 ± 7% of cells had fragmented or missing nuclei, suggesting that the death was apoptotic (Fig.11B). Because the concentration of CHX (1 μg/ml) used in these experiments causes complete, long-term block of protein synthesis in sympathetic neurons (Franklin and Johnson, 1998), apoptosis occurred in these cells with no new protein production. These data suggest that protein synthesis suppression by CHX blocked apoptosis caused by NGF deprivation, at least in part, via the antioxidant effects of GSH upregulation.
Fig. 11.

Apoptosis appeared to occur even in the presence of CHX. The ability of CHX (1 μg/ml) to block apoptosis appeared to decrease with time. Cultures for survival experiments were deprived of NGF and maintained for 3 or 7 d in the presence of CHX. Medium was then replaced with one containing NGF, and survival was assayed by blinded counting several days later. n = 8–13 cultures from three platings. For determination of the status of the nucleus, neurons maintained for 3 or 7 d in CHX-containing medium were fixed and stained with bisbenzimide (10 μg/ml TBS buffer for 30 min). Cells were scored as having fragmented, apoptotic nuclei or as having no visible nuclei (ghost cells) as described by Edwards and Tolkovsky (1994). n = 398–469 neurons from three platings.
DISCUSSION
Evidence suggests a role for redox state in the apoptotic death of many, but not all, cell types (Hockenbery et al., 1993; Kane et al., 1993; Jacobson and Raff, 1995; Beal et al., 1997; Polyak et al., 1997; Tan et al., 1998). We conducted a study of the role of ROS in the apoptotic death of NGF-deprived sympathetic neurons in cell culture. Increased ROS production occurred within 3 hr of NGF withdrawal (Fig.12). The pan-caspase inhibitor BAF completely blocked this increase. Between 3 and 12 hr after NGF deprivation, ROS production appeared to decline to basal, or lower, levels. This decrease was caused, at least in part, by decreased H2O2 levels secondary to increased GSH concentration rather than to an actual reduction of ROS production. A further increase of ROS began ∼12 hr after NGF. This second burst had both BAF-sensitive and -insensitive components, suggesting caspase-dependent and -independent ROS production. Because GSH levels increased within a few hours after NGF deprivation and remained elevated for the entire time course of apoptosis, an additional increase of ROS production must have caused the late burst rather than a decrease in cellular antioxidant capabilities. Uncoupling of oxidative phosphorylation with the protonophore FCCP blocked all components of the ROS bursts, suggesting that the ROS derived from the mitochondrial electron transport chain. The inhibitor of respiratory complex 1, rotenone, also blocked all components of the ROS bursts, whereas the complex 3 inhibitor, antimycin A, augmented ROS. These data suggest the ROS derived from electrons that leaked to O2 to form O2− between respiratory complexes one and three.
Fig. 12.
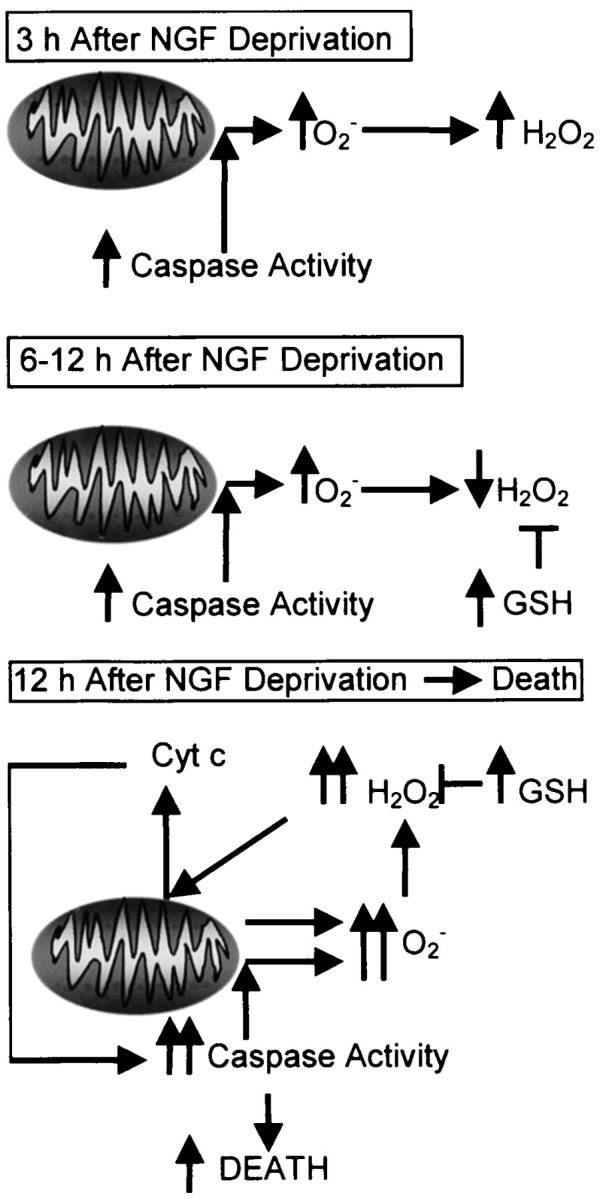
Summary of mitochondrial events suggested by data in the previous figures. Within 3 hr of NGF deprivation a caspase-dependent burst of mitochondrial O2− production occurred. Dismutation converted the O2− to H2O2. By 6 hr after deprivation GSH concentrations increased, masking continued increased production of O2− and H2O2. Later than 12 hr after NGF withdrawal a further increase of ROS production occurred. This burst had both caspase-dependent and independent components. The late increase of ROS production appeared to promote cytochrome c release.
Sources of increased ROS production
The late ROS burst was concurrent with release of cytochrome c. Because much of this burst was BAF-sensitive, it probably lay downstream of cytosolic caspase activity induced by the discharged cytochrome c. How caspases may have caused increased mitochondrial ROS production is unknown. It is possible that loss of outer mitochondrial membrane barrier function concurrent with cytochrome c release allowed activated caspases to enter the mitochondrial intermembrane space and affect components of the respiratory complexes. Alternatively, caspases may have activated one or more cytosolic factors that increased mitochondrial ROS production (Tan et al., 1998). The early ROS burst was, evidently, completely dependent on caspase activity but occurred long before any apparent cytochrome c redistribution. Recent evidence shows that many caspases are found in mitochondria as well as the cytosol (Mancini et al., 1998; Krajewski et al., 1999; Samali et al., 1999;Susin et al., 1999). A cytochrome c-independent, Bcl-2-regulated caspase-3 localized to mitochondrial membranes was recently described, demonstrating that alternate pathways for activation of caspases in subcellular organelles do exist (Krebs et al., 1999). Such a caspase may have caused the early increase of mitochondrial ROS production in NGF-deprived sympathetic neurons reported here.
A possible cause of the late, BAF-insensitive ROS was a shift of mitochondria from four-electron reduction of O2to one-electron reduction subsequent to cytochrome c release (Cai and Jones, 1998). Such a shift could cause elevated O2− production secondary to increased leakage of electrons from the electron transport chain. However, the peak of the late burst coincided with the appearance of cytochrome c in the cytoplasm but preceded total depletion from mitochondria. After complete loss, BAF-insensitive ROS production greatly declined rather than increased. This finding suggests that leakage of electrons from mitochondria depleted of cytochrome c did not make a major contribution to the BAF-insensitive ROS burst.
The role of ROS and GSH in cytochrome c release
Treatment of neurons with H2O2 caused rapid redistribution of cytochrome c from mitochondria into the cytoplasm. This finding demonstrates that a pro-oxidant state can cause cytochrome c release in these cells and suggests that increased ROS in NGF-deprived neurons may have induced release. The membrane-permeant analog of cysteine, l-NAC, blocked ROS, death, and cytochrome c release at similar concentrations, supporting this hypothesis. The block of ROS by l-NAC was mediated by increased GSH concentration. Further supporting a role for cellular redox state in regulating cytochrome c redistribution, a membrane-permeant form of GSH potently blocked both cytochrome c release and apoptosis.
Inhibition of protein synthesis by CHX blocked increased ROS, cytochrome c release, and death after NGF deprivation. An increase in cellular GSH concentration caused by CHX was responsible for suppressing ROS. Like l-NAC, similarity in the concentration-dependent effects of CHX on ROS, cytochrome c release, and death suggested that CHX blocked apoptosis, at least partially, via an antioxidant effect on cytochrome c release. The time course for block of death and cytochrome c redistribution by CHX andl-NAC were nearly identical. Acute addition of NGF immediately blocked cytochrome c release in cells deprived of NGF. However release continued for a while after the addition of CHX orl-NAC to NGF-deprived neurons. The time course for loss of the abilities of CHX or l-NAC to block cytochrome c release began several hours before the peak of the late ROS burst (Figs. 5,10C). Addition of CHX to NGF-deprived neurons did not immediately block the burst. Rather, several hours were required for full block (Fig. 2B). Lack of the ability of CHX to immediately block cytochrome c release may have resulted, at least in part, from the slow effect of CHX on ROS. The NGF block of cytochrome c release must have occurred via another mechanism as NGF treatment took as long as did CHX to block the late ROS burst in NGF-deprived neurons, yet immediately blocked cytochrome c release.
Greenlund et al. (1995) proposed that the early burst of ROS production occurring after NGF withdrawal from sympathetic neurons contributes to death by causing induction of pro-apoptotic genes (Martin et al., 1988;Estus et al., 1994). Consistent with a role for the early ROS burst in death, microinjection of superoxide dismutase inhibits death only if done during the period of the early burst (Greenlund et al., 1995). However, we found that CHX and l-NAC blocked most apoptotic death when added long after this time. Additionally, although BAF completely blocked the early ROS burst, it did not affect the rate of cytochrome c release. Because blocking protein synthesis prevents apoptosis in these cells solely by block of cytochrome c redistribution (Deshmukh and Johnson, 1998), this finding seems to indicate that the early ROS burst was irrelevant for apoptosis, controverting the findings of the previous report. Moreover, O2− production was not transient, making it unclear why injection of superoxide dismutase early after NGF deprivation should promote survival whereas later injection did not.
The ability of CHX to act as an antioxidant decreased with time, and neurons maintained for several days in the presence of CHX died with apoptotic characteristics. Because the concentration of CHX used in these experiments causes complete, long-term block of protein synthesis in these cells (Franklin and Johnson, 1998), production of new protein could not have been required for the death. These data further suggest that suppression of protein synthesis by CHX is anti-apoptotic, at least in part, because of antioxidant effects. However, CHX was a significantly more potent anti-apoptotic agent than wasl-NAC, although l-NAC (30 mm) increased GSH concentration much more than CHX (Figs.6A,B, 7A,C). These data suggest that CHX had death-suppressant effects in addition to those resulting from increased GSH concentration. Therefore, our data do not completely preclude a role for de novo protein synthesis in the death of these cells. However, the data do seem to indicate that the early ROS burst was not involved in inducing production of pro-apoptotic proteins.
How redox state may regulate cytochrome c release is unknown. Both inhibition of protein synthesis and deletion of the pro-apoptotic protein BAX block apoptosis of NGF-deprived mouse sympathetic neurons solely by preventing cytochrome c release (Deckwerth et al., 1996;Deshmukh and Johnson, 1998). Inhibition of protein synthesis hinders association of BAX with mitochondria in these cells (Putcha et al., 1999). Therefore, one possible explanation for the effects of redox state on cytochrome c release was that it regulated association of BAX with the mitochondria. Such association could, ostensibly, lie downstream from redox-sensitive signaling pathways (Yan and Greene, 1998; Tournier et al. 2000). Alternatively, BAX could have been responsible for causing the late ROS burst. Events downstream from the ensuing pro-oxidant state, possibly oxidation of critical thiols in mitochondrial proteins (Marchetti et al., 1997), might then have caused release.
In conclusion, we present evidence suggesting that increased ROS levels after withdrawal of NGF from sympathetic neurons promoted release of cytochrome c from mitochondria in these cells. Cytoplasmic cytochrome c, in turn, activated caspases that further increased mitochondrial ROS production. We also present data suggesting that much of the effect of protein synthesis inhibition on apoptosis of NGF-deprived sympathetic neurons may be mediated via an antioxidant block of cytochrome c release. Many diseases are associated with increased free radical production (Halliwell and Gutteridge, 1999). Should the findings reported here be applicable to other cell types, it is possible that free radicals contribute to pathological states, at least in part, by causing release of cytochrome c from mitochondria.
Footnotes
This work was supported by a grant to the University of Wisconsin Medical School under the Howard Hughes Medical Institute Research Resources Program for Medical Schools and by National Institutes of Health Grant NS37110. We thank Dr. Peter Lipton for critical review of this manuscript and Drs. Eugene M. Johnson Jr., Thomas Deckwerth, Mohanish Deshmukh, and Mr. Girish Putcha for helpful conversations. We also thank Adam Pelzek for technical support.
Correspondence should be addressed to James L. Franklin, Department of Neurological Surgery, University of Wisconsin Medical School, 4640 MSC, 1300 University Avenue, Madison, WI 53706. E-mail:jlfrankl@facstaff.wisc.edu.
REFERENCES
- 1.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF, Howell N, Bodis-Wollner I. Mitochondria and free radicals in neurodegenerative diseases. Wiley; New York: 1997. [Google Scholar]
- 3.Braughler JM, Pregenzer JF, Chase RL, Duncan LA, Jacobsen EJ, McCall JM. Novel 21-amino steroids as potent inhibitors of iron-dependent lipid peroxidation. J Biol Chem. 1987;262:10438–10440. [PubMed] [Google Scholar]
- 4.Cai J, Jones DP. Superoxide in apoptosis: mitochondrial generation triggered by cytochrome c loss. J Biol Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 5.Crossin KL. Nitric oxide (NO); a versatile second messenger in brain. Trends Biochem Sci. 1991;16:81–82. doi: 10.1016/0968-0004(91)90036-u. [DOI] [PubMed] [Google Scholar]
- 6.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deckwerth TL, Johnson EM., Jr Changes in glutathione metabolism suggest a critical role for the redox equilibrium in programmed neuronal death after trophic factor deprivation. Soc Neurosci Abstr. 1994;20:682. [Google Scholar]
- 8.Deckwerth TL, Elliot JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 9.Deshmukh M, Johnson EM., Jr Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- 10.Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM., Jr Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE-family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dugan LL, Creedon DJ, Johnson EM, Jr, Holtzman DM. Rapid suppression of free radical formation by nerve growth factor involves the mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA. 1997;94:4086–4091. doi: 10.1073/pnas.94.8.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dwyer MA, Bredt DS, Snyder SH. Nitric oxide synthase: irreversible inhibition by l-NG-Nitroarginine in brain in vitro and in vivo. Biochem Biophys Res Commun. 1991;176:1136–1141. doi: 10.1016/0006-291x(91)90403-t. [DOI] [PubMed] [Google Scholar]
- 13.Easton RM, Deckwerth TL, Parsadanian AS, Johnson EM., Jr Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: a phenotype indistinguishable from BAX deletion. J Neurosci. 1997;17:9656–9666. doi: 10.1523/JNEUROSCI.17-24-09656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edwards SN, Tolkovsky AM. Characterization of apoptosis in cultured rat sympathetic neurons after nerve growth factor withdrawal. J Cell Biol. 1994;124:537–546. doi: 10.1083/jcb.124.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: Identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez-Checa JC, Kaplowitz N. The use of monochlorobimane to determine the hepatic GSH levels and synthesis. Anal Biochem. 1990;190:212–219. doi: 10.1016/0003-2697(90)90183-a. [DOI] [PubMed] [Google Scholar]
- 17.Ferrari G, Yan CYI, Greene LA. N-acetylcysteine (d- and l-stereoisomers) prevents apoptotic death of neuronal cells. J Neurosci. 1995;15:2857–2866. doi: 10.1523/JNEUROSCI.15-04-02857.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franklin JL, Johnson EM., Jr Control of neuronal size homeostasis by trophic factor-mediated coupling of protein degradation to protein synthesis. J Cell Biol. 1998;142:1313–1324. doi: 10.1083/jcb.142.5.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franklin JL, Sanz-Rodriguez C, Juhasz A, Deckwerth TL, Johnson EM., Jr Chronic depolarization prevents programmed death of sympathetic neurons in vitro but does not support growth: requirement for Ca2+ influx but not Trk activation. J Neurosci. 1995;15:643–664. doi: 10.1523/JNEUROSCI.15-01-00643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenlund LJS, Deckwerth TL, Johnson EM., Jr Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 21.Griffith OW, Meister A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J Biol Chem. 1979;254:7558–7560. [PubMed] [Google Scholar]
- 22.Halliwell B, Gutteridge JMC. Free radicals in biology and medicine third edition. Oxford UP; Oxford: 1999. [Google Scholar]
- 23.Henderson JF, Lardy HA. Bongkrekic acid an inhibitor of the adenine nucleotide translocase of mitochondria. J Biol Chem. 1970;245:1319–1326. [PubMed] [Google Scholar]
- 24.Hockenbery DM, Oltvai ZN, Yin X-M, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 25.Jacobson MD, Raff MC. Programmed cell death and Bcl-2 protection in very low oxygen. Nature. 1995;374:814–816. doi: 10.1038/374814a0. [DOI] [PubMed] [Google Scholar]
- 26.Johnson MI, Argiro V. Techniques in the tissue culture of rat sympathetic neurons. Methods Enzymol. 1983;103:334–347. doi: 10.1016/s0076-6879(83)03022-0. [DOI] [PubMed] [Google Scholar]
- 27.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Örd T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 28.Krajewski S, Krajewska M, Ellerby LA, Welsh K, Xie Z, Deveraux QL, Salvesen GS, Bredesen DE, Rosenthal RE, Fiskum G, Reed JC. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:5752–5757. doi: 10.1073/pnas.96.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krebs JF, Armstrong RC, Srinivasan A, Aja T, Wong AM, Aboy A, Sayers R, Pham B, Vu T, Hoang K, Karanewsky DS, Leist C, Schmitz A, Wu JC, Tomaselli KJ, Fritz LC. Activation of membrane-associated procaspase-3 is regulated by Bcl-2. J Cell Biol. 1999;144:915–926. doi: 10.1083/jcb.144.5.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 31.Lin KI, Lee SH, Narayanan R, Baraban JM, Hardwick JM, Ratan RR. Thiol agents and Bcl-2 identify an alphavirus-induced apoptotic pathway that requires activation of the transcription factor NF-kappa B. J Cell Biol. 1995;131:1149–1161. doi: 10.1083/jcb.131.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mancini M, Nicholson DW, Roy S, Thornberry NA, Peterson EP, Casciola-Rosen LA, Rosen A. The caspase-3 precursor has a cytosolic and mitochondrial distribution: implications for apoptotic signaling. J Cell Biol. 1998;140:1485–1495. doi: 10.1083/jcb.140.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marchetti P, Decaudin D, Macho A, Zamzami N, Hirsch T, Susin SA, Kroemer G. Redox regulation of apoptosis: impact of thiol oxidation status on mitochondrial function. Eur J Immunol. 1997;27:289–296. doi: 10.1002/eji.1830270142. [DOI] [PubMed] [Google Scholar]
- 34.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM., Jr Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinou I, Desagher S, Eskes R, Antonsson B, André E, Fakan S, Martinou JC. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J Cell Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neame SJ, Rubin LL, Philpott KL. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nedergaard M, Desai S, Pulsinelli W. Dicarboxy-dichlorofluorescein: a new fluorescent probe for measuring acidic intracellular pH. Anal Biochem. 1990;187:109–114. doi: 10.1016/0003-2697(90)90425-9. [DOI] [PubMed] [Google Scholar]
- 38.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 39.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 40.Putcha GV, Deshmukh M, Johnson EM., Jr BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, Bcl-2, and caspases. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reed JC. Cytochrome c: can't live with it-can't live without it. Cell. 1997;91:559–562. doi: 10.1016/s0092-8674(00)80442-0. [DOI] [PubMed] [Google Scholar]
- 43.Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Royall JA, Ischiropoulis H. Evaluation of 2′, 7′-dichlorofluorescin and dihydrorhodamine123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch Biochem Biophys. 1993;302:348–355. doi: 10.1006/abbi.1993.1222. [DOI] [PubMed] [Google Scholar]
- 45.Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S. Presence of pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of Jurkat cells. EMBO J. 1999;18:2040–2048. doi: 10.1093/emboj/18.8.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Brenner C, Larochette N, Prévost M-C, Alzari PM, Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J Exp Med. 1999;189:381–393. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szabó I, Zoratti M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J Biol Chem. 1991;266:3376–3379. [PubMed] [Google Scholar]
- 48.Tan S, Sagara Y, Liu Y, Maher P, Schubert D. The regulation of reactive oxygen species production during programmed cell death. J Cell Biol. 1998;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tietze F. Enzymatic method for quantitative determination of nanogram amounts of total and oxidized glutathione. Anal Biochem. 1969;15:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 50.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–877. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 51.Turrens JF. Superoxide production by the mitochondrial respiratory chain. Bioscience Rep. 1997;17:3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- 52.Yan CYI, Greene LA. Prevention of PC12 cell death by N-acetylcysteine requires activation of the ras pathway. J Neurosci. 1998;18:4042–4049. doi: 10.1523/JNEUROSCI.18-11-04042.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]