

Abstract
Peripheral nerve injury can lead to a persistent neuropathic pain state in which innocuous tactile stimulation elicits pain behavior (tactile allodynia). Spinal administration of the anticonvulsant gabapentin suppresses allodynia by an unknown mechanism. In vitro studies indicate that gabapentin binds to the α2δ-1 (hereafter referred to as α2δ) subunit of voltage-gated calcium channels. We hypothesized that nerve injury may result in altered α2δ subunit expression in spinal cord and dorsal root ganglia (DRGs) and that this change may play a role in neuropathic pain processing. Using a rat neuropathic pain model in which gabapentin-sensitive tactile allodynia develops after tight ligation of the left fifth and sixth lumbar spinal nerves, we found a >17-fold, time-dependent increase in α2δ subunit expression in DRGs ipsilateral to the nerve injury. Marked α2δ subunit upregulation was also evident in rats with unilateral sciatic nerve crush, but not dorsal rhizotomy, indicating a peripheral origin of the expression regulation. The increased α2δ subunit expression preceded the allodynia onset and diminished in rats recovering from tactile allodynia. RNase protection experiments indicated that the DRG α2δ regulation was at the mRNA level. In contrast, calcium channel α1B and β3 subunit expression was not co-upregulated with the α2δ subunit after nerve injury. These data suggest that DRG α2δ regulation may play an unique role in neuroplasticity after peripheral nerve injury that may contribute to allodynia development.
Keywords: α2δ calcium channel subunit, peripheral nerve injury, rhizotomy, allodynia, dorsal root ganglia, spinal cord, sensory neurons
Peripheral nerve injury may lead to neuropathic syndromes characterized by both spontaneous and evoked painful sensations. Allodynia, or an exaggerated response to otherwise innocuous tactile stimuli, is considered both a hallmark and the most troublesome of these syndromes (Price et al., 1989; Wahren and Torebjork, 1992; Koltzenburg et al., 1994). The molecular mechanisms of neuropathic pain states are not clear. It has been hypothesized that disordered sensory processing arises from long-term changes in the function of sensory afferents and the organization of sensory input into the dorsal horn (Coderre et al., 1993; Woolf and Doubell, 1994).
Pharmacological evidence suggests that spinal N-type voltage-gated calcium channels (N-VGCCs) play a role in neuropathic pain transduction. Intrathecal delivery of N-type (ω-conopeptides), but not L- or P-type, VGCC antagonists suppresses allodynia in neuropathic rat models (Chaplan et al., 1994b; Calcutt and Chaplan, 1997). Autoradiographic studies showed the highest density of conopeptide-binding sites in the spinal dorsal horn (lamina I and II) where primary afferents terminate (Kerr et al., 1988; Takemura et al., 1989). N-VGCCs are also found in dorsal root ganglion (DRG) neurons where they are differentially modulated after sciatic nerve damage (Abdulla and Smith, 1997).
High-threshold neuronal VGCCs contain three subunits (Witcher et al., 1993): the α1 channel-forming subunit, the intracellular β subunit, and the α2δ subunit that consists of two disulfide-linked polypeptides (α2 and δ) encoded by the same gene (Ellis et al., 1988; De Jongh et al., 1990). Recent preclinical studies suggest a role for the α2δ subunit in neuropathic pain processing. Gabapentin, an anticonvulsant drug found to be effective against clinical neuropathic pain states (Mellick et al., 1995; Rosner et al., 1996; Mellick and Mellick, 1997; Rosenberg et al., 1997;Backonja et al., 1998; Rowbotham et al., 1998), also exerts a spinal action in preclinical neuropathic pain models (Hwang and Yaksh, 1997;Abdi et al., 1998) and yet lacks general analgesic properties (Field et al., 1997). Gabapentin binds with high affinity to the α2δ subunit in vitro (Gee et al., 1996). In addition, compounds modeled after gabapentin show relative antineuropathic pain potencies that correlate with their binding affinities and stereospecificity at the α2δ site (Suman-Chauhan et al., 1993; Dissanayake et al., 1997; Hwang and Yaksh, 1997).
We have hypothesized that spinal or peripheral VGCC α2δ subunit expression may be altered after nerve injury. This alteration may modulate the properties of functional VGCCs and account for the efficacy of N-VGCC antagonists and gabapentin against neuropathic pain, but not acute nociception. To test these hypotheses, we examined DRG and spinal cord α2δ subunit expression in a rat model of neuropathic pain resulting from tight spinal nerve ligation. Our studies indicate that peripheral nerve injury results in a marked DRG α2δ subunit upregulation that precedes the onset and correlates with the duration of tactile allodynia. These findings suggest that DRG α2δ subunit expression may be important in peripheral nerve injury-induced neuroplasticity that may contribute to neuropathic pain processing.
MATERIALS AND METHODS
Materials. [32P]UTP (specific activity, 800 Ci/mmol) was from NEN Research Products (Wilmington, DE). Tris-acetate and bis-Tris gels (NuPAGE) and buffers were from Invitrogen (Carlsbad, CA). The monoclonal antibody against the human neuronal α2 subunit (produced by Dr. W. Smith at Lilly Research Center, Earl Wood Manor, Windlesham, Surrey, UK) and the polyclonal antibody against the human β3 subunit were provided by SIBIA Neurosciences, Inc. (La Jolla, CA). The monoclonal antibody has been shown to interact positively with the α2subunit in rat tissues (Luo, 2000b). The polyclonal antibody against the rat brain α1 subunit (α1B) was from Exalpha Biologicals, Inc. (Boston, MA). Positive controls for the calcium channel subunit antibodies were derived from membrane extracts of human embryonic kidney cells (HEK293) overexpressing the human α2bδ, α1B, or β3 cDNAs and were provided by SIBIA Neurosciences, Inc. The monoclonal antibodies against rat neuronal nitric oxide synthase (nNOS) or endothelial nitric oxide synthase (eNOS) and their positive controls were from Sigma (St. Louis, MO) or Transduction Laboratories (Lexington, KY), respectively. Horseradish peroxidase-labeled secondary antibodies, against either mouse IgG or rabbit IgG, were from Pierce (Rockford, IL) and Santa Cruz Biotechnology (Santa Cruz, CA), respectively. The substrate solutions for horseradish peroxidase and enhancer solutions were from Pierce. RNases were from Ambion (Austin, TX), and RNA polymerases, restriction enzymes, and TRIzol reagent were from Life Technologies (Gaithersburg, MD). Gabapentin was from Parke-Davis Pharmaceuticals (Ann Arbor, MI). Other chemicals were from Sigma.
Animals. Rats (male Harlan Sprague Dawley; 100–150 gm; Harlan Sprague Dawley, Indianapolis, IN) were housed in separate cages on a 12/12 hr light/dark cycle and fed food and water ad libitum. All animal care and experiments were performed according to protocols approved by the Institutional Animal Care Committee of the University of California, San Diego.
Neuropathic lesions and drug administration. Spinal nerve injury was induced by the procedure described by Kim and Chung (1992). Briefly, the left L5/L6 lumbar spinal nerves were exposed in halothane/oxygen-anesthetized rats and tightly ligated with 6.0 silk suture at a point distal to their DRGs and proximal to their conjunction to form the sciatic nerve. Sham operations were performed in the same way except that spinal nerves were not ligated.
For unilateral dorsal rhizotomy, a midline incision was made after routine skin preparation in the backs of halothane-anesthetized rats. The dorsal aspect of L1 and L2 was cleared, and a left hemilaminectomy was performed. After the dura was opened, the L5 and L6 dorsal roots were cut proximally, followed by a second distal cut with removal of the intervening segment. A piece of fascia was mobilized and placed on top of the exposed spinal cord. The skin was closed, and the animal was allowed to recover for 7 d. After tissue removal for analysis, the identity of the cut roots was confirmed using anatomical landmarks.
For unilateral sciatic nerve crush, rats were anesthetized with halothane, and the skin was routinely shaved and cleaned. The sciatic nerve was exposed on both sides, and only the left nerve was crushed with a pair of forceps for two 15 sec periods. The skin was sutured, and the animal was returned to its home cage after complete recovery from anesthesia. Seven days later, rats were deeply anesthetized and transcardially perfused with iced saline immediately before tissue collection.
Gabapentin was dissolved in sterile saline and infused over the course of 1 week into allodynic rats, starting 1 week after the nerve ligation, through intrathecal catheters (Yaksh and Rudy, 1976) linked to ALZET osmotic pumps implanted following the procedure provided by the manufacturer (ALZA Corporation, Palo Alto, CA).
Behavioral testing. Tactile allodynia was tested as described previously (Chaplan et al., 1994a). Rats were placed in a clear plastic cage with a wire mesh bottom and allowed to acclimatize for 15 min. The 50% paw withdrawal threshold (PWT) was determined with von Frey filaments (Stoelting, Wood Dale, IL) using a modification of the up–down method of Dixon (1980). A series of filaments, starting with one that had a buckling weight of 2.0 gm, was applied in consecutive sequence to the plantar surface of the left (nerve-ligated) hindpaw with a pressure causing the filament to buckle. Paw lifting indicated a positive response and prompted the use of the next weaker filament. Absence of a paw withdrawal response after 5 sec prompted the use of the next filament of increasing weight. This paradigm continued until four more measurements had been made after the initial change of the behavioral response or until five consecutive negative (assigned a score of 15 gm) or four consecutive positive (assigned a score of 0.25 gm) responses had occurred. The resulting scores were used to calculate the 50% response threshold by using the formula: 50% gm threshold = 10(Xf+κ∂)/10,000, where Xf = the value (in log units) of the final von Frey filament used, κ = the value [from Chaplan et al. (1994a), their table] of the pattern of positive/negative responses, and ∂ = the mean difference (in log units) between stimuli. Behavioral tests were performed immediately before and daily after surgery and drug administrations.
RNA extraction and RNase protection assays. Total RNA was extracted from pulverized frozen rat tissues with TRIzol reagent and stored at −20°C. α2δ mRNA species were quantified by RNase protection assays as described (Luo et al., 1994,1999). A 413 bp α2δ cDNA fragment (nucleotides 1905–2317; GenBank accession number M86621), including the coding region of the unique seven amino acid insertion of the rat brain α2δ (α2bδ) splice variant, was generated by the reverse transcription (RT)-PCR method using rat brain RNA. PCR products were cloned into pBluescript SKII+ vectors that were digested withEcoRV and T-tailed with a single thymidine residue using ddTTP and terminal transferase. T/A cloning was performed by exploiting the single 3′ adenosine overhang generated in the PCR products by Taq polymerase, using a high concentration of T4 ligase and overnight incubation at 15°C. The ligation product was transfected into DH5α-competent cells and plated onto ampicillin–nafcillin Luria-Bertani medium agarose plates pretreated with isopropylthio-β-d-galactoside and 5-bromo-4-chloro-3-indolyl-β-d-galactoside. Colorless colonies were selected and amplified, and isolated plasmids were sequenced. A clone with the correct sequence was amplified, and plasmid DNA was linearized with HindIII. After in vitro transcription with [32P]UTP, a 500 bp labeled antisense cRNA probe was used for RNase protection. A full-length protected probe of 413 nucleotides indicated the presence of the α2bδ mRNA, and two shorter protected probe fragments (258 and 133 bp) indicated probable splice variants such as that found in skeletal muscle. To normalize for sample loading, an antisense probe of rat cyclophilin (a gift from Dr. J. N. Wood, University College, London, UK) was included in each RNase protection assay. A tRNA lane was included in each RNase protection assay to confirm the complete digestion of the free probes. Molecular masses of the protected probes were estimated by electrophoresis on polyacrylamide gels, and protected bands were exposed to BioMax films (Eastman Kodak, Rochester, NY) and quantified by densitometry within the linear range of the film sensitivity curve.
Western blot. To measure protein expression, tissues were extracted in 50 mm Tris buffer, pH 8.0, containing 0.5% Triton X-100, 150 mm NaCl, 1 mm EDTA, and protease inhibitors. The cell extracts were subjected to denaturing NuPAGE Tris-acetate gel electrophoresis under reducing conditions (0.05m DTT) and then electrophoretically transferred to nitrocellulose membranes (Schleicher & Schuell, Keene, NH). After blocking nonspecific binding sites with 5% low-fat milk in PBS containing 0.1% Tween 20 (PBS-T), the monoclonal or polyclonal antibodies were used to probe the membranes in PBS-T for 1 hr at room temperature or overnight at 4°C. After washing, the antibody–protein complexes were probed for 1 hr at room temperature with appropriate secondary antibodies labeled with horseradish peroxidase and detected with chemiluminescent reagents. Membrane extracts from HEK293 cells overexpressing the human α2bδ, α1B, or β3 cDNAs were used as positive controls for the calcium channel subunit antibodies. Because the δ peptide separates from the α2subunit under reducing conditions (Jay et al., 1991), the bands detected by the primary antibody against the α2δ protein reflect the α2 subunit only. Purified rat brain nNOS protein was used as a positive control for the nNOS antibody. The positive control for the eNOS monoclonal antibody was provided with the antibody.
In some experiments (see Figs. 2, 6), the nitrocellulose membranes were stripped with Re-Blot Western blot recycling kit (Chemicon, Temecula, CA) and reblotted with different antibodies.
Fig. 2.

Calcium channel protein levels in L5/L6 DRG and spinal cord of nerve-ligated rats. Total proteins were extracted from ipsilateral or contralateral DRGs (two each) or lumbar dorsal spinal cord quadrants of control or allodynic rats 1 week after the spinal nerve ligation. The protein levels were identified using indicated monoclonal antibodies (for α2 and eNOS) or polyclonal antibodies (for β3 and α1B).A, Representative Western blots each from at least four independent experiments showing protein levels in DRG and lumbar dorsal spinal cord of control and allodynic rats. Membrane extracts of stable HEK293 cells expressing transfected human recombinant α2bδ, α1B, and β3cDNAs were used as respective positive (+) controls. Extracts of endothelial cells were used as positive control for eNOS.Lanes are labeled C, for the contralateral side, and Ip, for the ipsilateral (nerve-ligated) side. B, Nerve ligation-induced increase of the α2 subunit in ipsilateral DRG and spinal cord compared with respective contralateral (contral.) samples. Data are presented as the means ± SEM from at least 11 (sham) to 16 (ligated) independent experiments (*p< 0.05 by Student's t test and Mann–Whitney test; #p < 0.05 by Student's t test but not by Mann–Whitney test). S.C., Spinal cord.
Fig. 6.
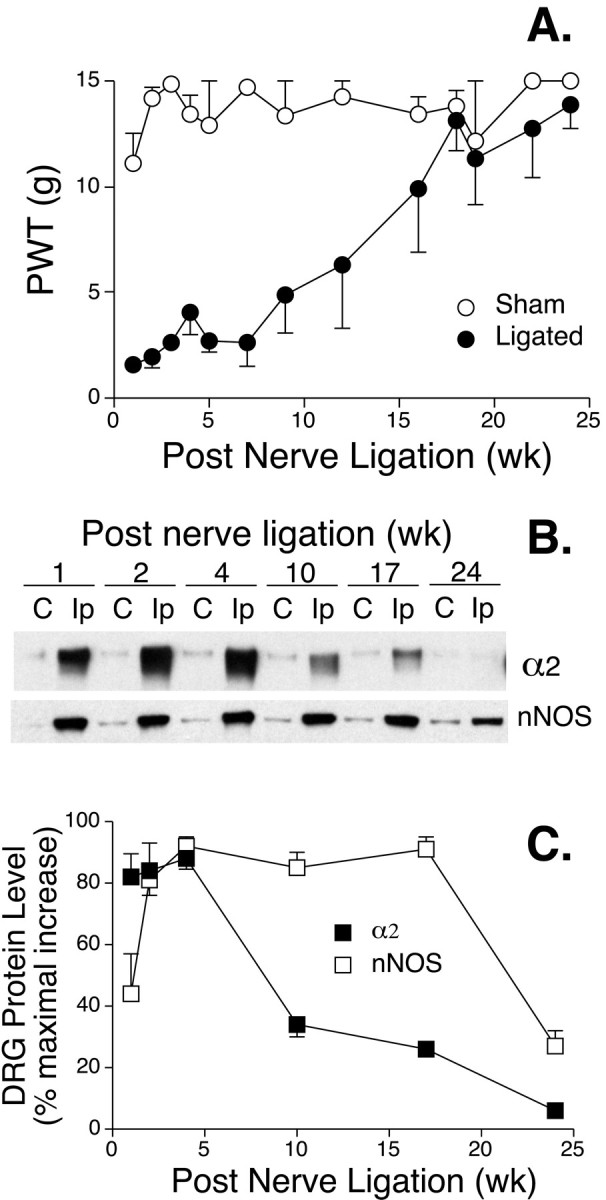
Upregulated DRG α2 subunit diminishes in nerve-ligated rats recovering from tactile allodynia. Nerve ligation injury was induced as described in Harlan Sprague Dawley rats, and the PWT to mechanical stimulation was tested up to 6 months after the surgery. Total protein was extracted from two pooled L5/L6 DRGs from each side of the rat at the designated time points, and DRG α2 subunit levels were examined by Western blot analyses.A, Recovery of nerve-ligated rats from tactile allodynia. Data presented are the means ± SEM from 4 to 16 rats in each group. B, Representative Western blots showing expression levels of α2 and nNOS in L5/L6 DRG of nerve-ligated rats at time points after the nerve injury. In some experiments, these blots were stripped and then reblotted with eNOS antibodies as shown in Figure 2, and eNOS expression was not increased in DRGs ipsilateral to the nerve ligation (data not shown).Lanes are labeled C, for the contralateral side, and Ip, for the ligation side.C, Summarized Western blot data shown inB. Data are presented as the percentage of maximal increase of α2 and nNOS expression in DRGs ipsilateral to the nerve ligation in each experiment and are expressed as the means ± SEM of six independent determinations. wk, Week.
Statistical analyses. Unpaired Student's ttests and the Mann–Whitney test were performed; significance was indicated by a two-tailed p value <0.05.
RESULTS
Peripheral but not central axon injury induces marked upregulation of the DRG α2δ calcium channel subunits that precedes the onset of allodynia
As shown in Figure 1, nerve-ligated rats developed tactile allodynia 4 d after the surgery, as indicated by the reduction of paw withdrawal thresholds to mechanical stimulation of injured paws. Tactile allodynia was not observed in sham-operated rats or in paws contralateral to the nerve ligation (data not shown). Intrathecal gabapentin infusion for 1 week reversed the allodynic state in a dose-dependent manner, consistent with its therapeutic effects after systemic administration in patients with neuropathic pain (Mellick et al., 1995; Rosner et al., 1996) and its spinal action in the same rat neuropathic pain model (Hwang and Yaksh, 1997). Thus, it seems that long-term intrathecal gabapentin infusion does not result in tolerance development. Side effects of gabapentin, such as motor function deficit and sedation, were not observed after chronic gabapentin infusion at the dose and duration applied.
Fig. 1.

Development of tactile allodynia after L5/L6 spinal nerve ligation and the effects of intrathecal gabapentin infusion on established allodynia. Left L5/L6 spinal nerve tight ligation was performed in Harlan Sprague Dawley rats, and the PWT to mechanical stimulation was tested daily as indicated. Allodynic rats (1 week after nerve ligation) were treated with saline or gabapentin for 1 week through an intrathecal (i.t.) pump. Data are presented as the means ± SEM from at least 5 treated to 20 nontreated rats in each group.
The level of α2δ protein in ipsilateral L5/L6 DRGs increased >17-fold 1 week after nerve ligation in nerve-injured, but not sham-operated, rats as indicated by the marked increase of α2 immunoreactivity in Western blots (Fig.2). The α2subunit in contralateral DRG showed a higher apparent molecular mass than did the spinal α2 subunit in denaturing gels (Fig. 2A) because of the fact that DRG expresses α2δ isoforms with distinctive glycosylation that are different from the α2δ subunit expressed in spinal cord, brain, and skeletal muscle (Luo, 2000b). The nerve injury-induced increase of DRG α2 subunit exists as two bands, a major band showing a migration rate the same as that of the α2 subunit in the contralateral DRG and a minor band with a migration rate similar to that of the spinal cord α2 subunit (Fig. 2A). These data are consistent with the findings from deglycosylation studies indicating that at least two α2δ species are present in DRG (Luo, 2000b) and indicate that peripheral nerve injury may alter the expression of different DRG α2δ subunits.
The spinal cord α2 subunit level was also increased significantly (55 ± 20%; n = 16;p < 0.05 by Student's t test and Mann–Whitney test), although to a much lesser extent than in DRG, after the nerve injury (Fig. 2). The increase in spinal cord α2 subunit is mainly in the upper band with a migration rate similar to that of the major DRG α2 species (Fig. 2A), suggesting a selective regulation of the spinal cord α2 subunit after peripheral nerve injury. On the basis of the low sequence identity, we do not anticipate that the α2 monoclonal antibody will cross-react with the recently identified α2δ-2 and α2δ-3 subunits (Klugbauer et al., 1999).
The β3 subunit in L5/L6 DRG migrated as a doublet in Western blots, and the upper band remained constant whereas the lower band was abolished on the ligated side. Both bands should represent the β3 subunit because the anti-β3 antibody does not cross-react with β1, β2, and β4 subunits (data not shown), and cell extracts from HEK293 cells overexpressing the transfected human β3 cDNA also contain two β3 bands (Fig. 2A). Interestingly, the expression level of the channel-forming α1B subunit in DRG is much higher than that in the spinal cord, and nerve ligation-induced injury did not cause a significant increase in α1B expression in affected DRGs (136 ± 22% of contralateral levels;n = 5; p = 0.1528 and 0.3095 by Student's t test and Mann–Whitney test, respectively). Data from quantitative RT-PCR analyses confirmed that nerve ligation did not cause upregulation of DRG α1B mRNA (data not shown). Finally, eNOS expression levels were similar in the same samples (Fig. 2A). These data indicate that the increased DRG α2 expression in nerve-ligated rats is specific.
To confirm the origin of signals responsible for the altered DRG α2 subunit expression, we examined the α2 subunit levels in DRGs of rats with unilateral dorsal rhizotomy or sciatic nerve crush 1 week after the nerve injuries. Rhizotomy-induced central axonal injury only caused a marginal (but significant) increase of the DRG α2 subunit (Fig.3). However, sciatic nerve crush caused a pronounced α2 upregulation in DRGs ipsilateral to the crush (Fig. 3), confirming that the DRG α2δ subunit expression is regulated by peripheral nerve injury signals. The smaller increase of the α2 subunit in DRGs of sciatic nerve-crushed rats (in the same period of 1 week) compared with that in spinal nerve-ligated rats may be because the former may cause axonal injury of fewer DRG neurons (because of distribution of noninjured axons to the DRG) than the latter and a longer time is required for the injury signals from the sciatic nerve to reach the DRG.
Fig. 3.

DRG α2 expression levels in rats with 1 week dorsal rhizotomy or sciatic nerve crush. Pooled DRGs were collected from both sides of rats with unilateral rhizotomy (Rhizot.; L5/L6) or sciatic nerve crush (L4–L6). Total proteins were extracted and subjected to Western blot analyses as described. The insetsabove thebar graph are representative Western blots.Lanes are labeled C, for the contralateral side, and Ip, for the ipsilateral (nerve-injured) side. The bar graph shows the percentage changes of the α2 subunit levels in ipsilateral DRGs (Ipsilat.) compared with those in contralateral DRGs (Contral.) that were assigned the value of 100%. Data presented are the means ± SEM averaged from four (rhizotomy) to six (crush) independent determinations (*p < 0.05 by Student's t test and Mann–Whitney test).
The increase of the α2 subunit in the DRG of nerve-injured rats was time dependent, being detectable 1 d after the nerve ligation and attaining a >17-fold increase 1 week after the ligation (Fig. 4). Thus, a substantial increase in DRG α2δ protein precedes the onset of allodynia at day four (see Fig. 1).
Fig. 4.

Time-dependent increases in the α2subunit from L5/L6 DRGs ipsilateral to spinal nerve ligation. Ipsilateral or contralateral pooled L5/L6 DRGs were collected from nerve-ligated rats at the designated times, and total proteins were extracted and subjected to Western blot analysis as described.A, A representative Western blot. Thelower nonspecific bands indicate an equivalent protein loading within each pair of samples. Lanes are labeledC, for the contralateral side, and Ip, for the ipsilateral (nerve-ligated) side. B, Percentage increase of the α2 subunit in ipsilateral DRGs compared with that in contralateral (contral.) DRGs that was assigned the value of 100%. Data presented are the means ± SEM averaged from 4 to 16 independent determinations.
The DRG α2δ calcium channel subunit is upregulated at the mRNA level after nerve injury
To examine whether the DRG α2δ subunit is transcriptionally regulated after nerve injury, we measured DRG α2δ mRNA levels after spinal nerve ligation. As indicated in Figure 5, α2δ mRNA increased in a time-dependent manner in L5/L6 DRGs ipsilateral to the nerve injury. The increase was evident at 1 d, but not at 8 hr, after the nerve ligation and reached a sixfold to sevenfold increase at 7 d, correlating well with the increase in α2δ protein in both the time course and magnitude. In contrast, such an increase was not detected in ipsilateral L4 DRG from injured rats or L5/L6 DRGs from sham-operated rats.
Fig. 5.

Time-dependent increases of DRG α2δ mRNA in nerve-ligated rats. Total RNA was extracted from two pooled L5/L6 DRGs or one L4 DRG at the designated time after nerve ligation, and α2δ mRNA levels were examined by RNase protection assays. A, Representative autoradiography showing the α2δ and cyclophilin (Cyclo.) probes protected by their corresponding mRNAs. α2δ bands had longer exposure times than did cyclophilin bands because of the low abundance of the α2δ mRNA. Each pair of samples was taken from the same rat on the contralateral side (lane labeledC) or surgery side (lane labeledIp). B, Summarized time-dependent increase of α2δ mRNA after nerve ligation. The percentage increase of α2δ mRNA was defined by comparing α2δ band densities in the injury side with those in the contralateral (contral.) side after using the ratio of the α2δ band to the cyclophilin band to correct for differences in RNA loading. Data presented are the means ± SEM averaged from four independent experiments.wk, Week.
Upregulated DRG α2δ subunit is diminished in nerve-ligated rats recovering from tactile allodynia
To study the possible linkage between nerve injury-induced upregulation of DRG α2δ subunit and allodynia development/maintenance, we examined DRG α2expression in nerve-ligated rats recovering from tactile allodynia. As indicated in Figure 6A, the allodynic state of nerve-ligated rats lasted for several weeks after the nerve injury and then gradually diminished to the control level. Interestingly, similar temporal changes also occurred in upregulated, ipsilateral DRG α2 expression. As indicated in Figure 6B and summarized in Figure6C, the nerve injury-induced upregulation of ipsilateral DRG α2 subunit peaked ∼2–4 weeks after the nerve ligation and then gradually diminished in rats recovering from allodynia. In contrast, nerve injury-induced elevation of neuronal nitric oxide synthase in ipsilateral DRGs, a phenomenon that we have shown not to be linked directly to allodynia (Luo et al., 1999), remained high in these rats at all of the time points except the last one (Fig. 6B,C), which may be caused by long-term ligation-induced cell death (Coggeshall et al., 1997; Lekan et al., 1997). Thus, there is an excellent temporal correlation between DRG α2δ subunit upregulation and the onset (Figs.1, 2, 4) and duration (Fig. 6) of allodynia, suggesting a role of the DRG α2δ subunit in the development/maintenance of the neuropathic pain state.
DISCUSSION
This study provides the first evidence to indicate that nerve injury results in a marked increase of the DRG α2δ subunit that precedes the onset of tactile allodynia (Figs. 1, 2, 4). DRG α2δ mRNA upregulation parallels that of α2δ proteins (Fig. 5), indicating that nerve injury-induced α2δ subunit expression is regulated at the transcript level. In contrast to nerve injury-induced expression of DRG neuronal nitric oxide synthase, which remains elevated in nerve-ligated rats recovering from tactile allodynia (Fig. 6) (Luo et al., 1999), the upregulated DRG α2δ subunit diminishes in these rats, showing an excellent temporal correlation between DRG α2δ subunit upregulation and allodynia (Fig.6). Marked DRG α2δ subunit upregulation is also evident in rats with sciatic nerve crush but not dorsal rhizotomy (Fig. 3), confirming that the DRG α2δ subunit expression is regulated by peripheral factors. Together, our studies suggest that expression of the DRG α2δ subunit or its modulation of functional VGCCs may be an important component in neuronal plasticity contributing to allodynia after nerve injury. This conclusion is supported by the finding that intrathecal gabapentin, which binds to the α2δ subunit with high affinity in vitro (Gee et al., 1996), can suppress allodynia in a dose-dependent manner in this animal model (Fig.1) (Hwang and Yaksh, 1997) but has no effect on pain behaviors in nonallodynic rats (Field et al., 1997).
The physiological or pathological role of the α2δ subunit upregulation remains elusive. The α2δ subunit is a highly glycosylated VGCC structural subunit expressed in skeletal muscle (Curtis and Catterall, 1984; Ellis et al., 1988), brain (Ellis et al., 1988; Ahlijanian et al., 1990; Witcher et al., 1993), and heart (Cooper et al., 1987; Ellis et al., 1988; Tokumaru et al., 1992). It is believed to be a single transmembrane subunit in which a vast majority of the protein except the transmembrane domain and five C-terminal amino acid residues is extracellular (Brickley et al., 1995; Gurnett et al., 1996; Wiser et al., 1996), suggesting that this subunit does not likely interact with intracellular molecules such as protein kinases and G-proteins. Rather, its extracellular localization and extensive glycosylation suggest the importance of this subunit in functions involving the extracellular domain of VGCCs. This is confirmed by the finding that glycosylation in the extracellular domain is critical for calcium channel functions (Gurnett et al., 1996). In addition, in vitro studies have shown that the carboxyl δ peptide contains the transmembrane domain that plays a role in anchoring the α2δ subunit and in stabilizing subunit interactions (Jay et al., 1991;Gurnett et al., 1996). Thus, it is possible that the DRG α2δ subunit serves as a rate-limiting factor in VGCC assembly, and its enhanced expression after nerve injury may result in increased expression of functional VGCCs on the cell surface of DRG neurons without corresponding increases in other subunits, which in turn alters the excitability of sensory neurons. This hypothesis is supported by in vitro findings that coexpression of the α2δ subunit with other VGCC subunits results in stimulation of current amplitude (Mikami et al., 1989; Mori et al., 1991; Hullin et al., 1992; Williams et al., 1992; Brust et al., 1993;Gurnett et al., 1996) and increased expression of ω-conotoxin binding at the cell surface (Brust et al., 1993).
Alternatively, the DRG α2δ subunit may have distinct functional roles in addition to governing VGCC expression, and such roles may be important in neuroplasticity after nerve injury. This hypothesis is supported by the following observations. First, the channel-forming α1B subunit and the β3 subunit are not co-upregulated with the α2δ subunit after nerve injury (Fig. 2), implying distinct functions of the α2δ subunit in addition to being a VGCC structural subunit. Second, an important aspect of the α2δ subunit is that its tissue-specific expression is governed by mRNAs encoded by a family of genes (Klugbauer et al., 1999) as well as by alternatively spliced transcripts from a given gene (Kim et al., 1992; Brust et al., 1993; Gilad et al., 1995; Angelotti and Hofmann, 1996). Thus, the primary nucleotide sequences of different α2δ subunits may dictate their functional and tissue-specific expression, which in turn may underlie diverse functions of VGCCs. Third, the α2δ subunit exists as two distinctive species (with different migration rates on denaturing gels) in spinal cord and DRG, which are differentially regulated after nerve injury (Fig.2A), indicating the heterogeneity of the α2δ subunit in different tissues. Deglycosylation studies have confirmed that the α2δ subunit in DRG is indeed different from that in spinal cord, brain, and skeletal muscle, at least at the post-translational modification level (Luo, 2000b). Fourth, the differential expression of distinct α2δ subunits in spinal cord and DRG (Fig. 2A) and the presence of neuronal α2δ mRNA in both tissues (data not shown) suggest that the spinal cord α2δ subunit is not synthesized in DRG and then retrogradely transported to the spinal cord. Rather, the α2δ subunit is likely synthesized in a tissue-specific manner and may play different functional roles in these tissues. If this is the case, the antihyperalgesic actions of intrathecal gabapentin (Fig. 1) could result from drug interactions with spinal cord and/or DRG α2δ subunits because DRG is accessible by intrathecally delivered agents as demonstrated by other studies (Porreca et al., 1999; Lai et al., 2000). In fact, the relative contribution of spinal cord and DRG α2δ subunits to neuropathic pain and the site of gabapentin's action in vivo are important issues and remain to be studied.
The marked upregulation of DRG α2δ subunit after sciatic nerve crush, but not rhizotomy, suggests that the similar nerve ligation-induced α2δ subunit expression is regulated by retrograde peripheral factors, such as positive regulatory factors generated at the ligation site or/and inhibitory factors blocked by the ligation. Experiments are being undertaken to examine the mechanism of the α2δ subunit regulation.
The higher expression level of the α1B subunit in DRG compared with that in the spinal cord samples indicates that the α1B subunit expression is also tissue specific. The nature of the β3 subunit doublet in Western blots is not clear at the present time but may indicate heterogeneity in post-translational modification of the β3subunit. Because phosphorylation plays a role in β3 subunit modification (Isom et al., 1994), different phosphorylation patterns may contribute to the differences in gel mobility of these bands. A similar doublet with a slightly different migration rate also exists in membranes of HEK293 cells expressing the human recombinant β3 subunit, indicating that both bands are likely to be components of the β3 subunit. The complete disappearance of the lower β3 band after nerve injury (Fig.2A) is interesting and indicates differential responses of these β3 species to the nerve ligation.
Because there are four major classes of β subunits (β1, β2, β3, and β4) that contain several splice variants (Birnbaumer et al., 1994), we do not know at the present time whether the β3 subunit is associated with the α2δ subunit in DRG, nor can we exclude the possibility that other β subunits are coregulated with the α2δ subunit. Several lines of evidence, however, have indicated that the β3 subunit is a major component of N-type calcium channels. Binding studies have shown that a single β subunit binds to the α1 subunit in a 1:1 stoichiometry (De Waard et al., 1995), and the β3subunit selectively binds to the interaction domain of the α1B subunit with high affinity (De Waard et al., 1995; Scott et al., 1996). Immunoprecipitation studies have shown that the β3 subunit is a major component of purified rabbit brain N-type calcium channels labeled with ω-conotoxin GVIA (Witcher et al., 1993; Scott et al., 1996). The differential regulation of the α2δ, α1B, and β3 subunits after nerve injury supports our hypotheses that either the elevated α2δ subunit alone is sufficient to cause upregulation of functional calcium channels or the elevated α2δ subunit has unknown functions that may contribute to neuropathic pain.
The heterogeneity of the β subunits has been hypothesized to contribute to the diversity of N-type calcium channel properties in different neurons (Nowycky et al., 1985; Jones and Marks, 1989; Plummer et al., 1989). In conjunction with the heterogeneity of the α1 (Birnbaumer et al., 1994) and the α2δ (Luo, 2000b) subunits, it is likely that functional and tissue-specific expression of the N-type calcium channels may depend on channel subunit composition and make unique contributions to nerve injury-induced neuropathic pain. Because neuropathic pain is likely a multimechanism disorder (Luo, 2000a), upregulation of the DRG α2δ subunit may not be the only factor contributing to neuropathic pain development. Upregulation of other functional proteins in DRG, such as specific types of sodium channels, has also been reported in nerve-injured rats (Porreca et al., 1999; Boucher et al., 2000; Lai et al., 2000). However, our findings of tissue-specific and differential regulation of the DRG α2δ subunit after nerve injury may provide important clues indicating unexpected roles of the VGCC subunit in nerve injury-induced neuropathic pain processing.
Footnotes
This work was supported in part by National Institutes of Health Grants F32HL09848, R3DE13270A, NS40135 (Z.D.L.), NS01769 (S.R.C.), and NS35630 (L.S.S.) as well as by institutional grants from the Howard Hughes Medical Institute (Z.D.L. and S.R.C.), University of California, San Diego. We thank Anna Grauers for her technical assistance in some experiments.
Correspondence should be addressed to Dr. Z. David Luo, Department of Anesthesiology-0818, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0818. E-mail: zluo@ucsd.edu.
REFERENCES
- 1.Abdi S, Lee DH, Chung JM. The anti-allodynic effects of amitriptyline, gabapentin, and lidocaine in a rat model of neuropathic pain. Anesth Analg. 1998;87:1360–1366. [PubMed] [Google Scholar]
- 2.Abdulla FA, Smith PA. Ectopic α2-adrenoceptors couple to N-type Ca2+ channels in axotomized rat sensory neurons. J Neurosci. 1997;17:1633–1641. doi: 10.1523/JNEUROSCI.17-05-01633.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahlijanian MK, Westenbroek RE, Catterall WA. Subunit structure and localization of dihydropyridine-sensitive calcium channels in mammalian brain, spinal cord, and retina. Neuron. 1990;4:819–832. doi: 10.1016/0896-6273(90)90135-3. [DOI] [PubMed] [Google Scholar]
- 4.Angelotti T, Hofmann F. Tissue-specific expression of splice variants of the mouse voltage-gated calcium channel alpha2/delta subunit. FEBS Lett. 1996;397:331–337. doi: 10.1016/s0014-5793(96)01205-7. [DOI] [PubMed] [Google Scholar]
- 5.Backonja M, Beydoun A, Edwards KR, Schwartz SL, Fonseca V, Hes M, LaMoreaux L, Garofalo E. Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus: a randomized controlled trial [see comments]. JAMA. 1998;280:1831–1836. doi: 10.1001/jama.280.21.1831. [DOI] [PubMed] [Google Scholar]
- 6.Birnbaumer L, Campbell KP, Catterall WA, Harpold MM, Hofmann F, Horne WA, Mori Y, Schwartz A, Snutch TP, Tanabe T. The naming of voltage-gated calcium channels. Neuron. 1994;13:505–506. doi: 10.1016/0896-6273(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 7.Boucher TJ, Okuse K, Bennett DL, Munson JB, Wood JN, McMahon SB. Potent analgesic effects of GDNF in neuropathic pain states. Science. 2000;290:124–127. doi: 10.1126/science.290.5489.124. [DOI] [PubMed] [Google Scholar]
- 8.Brickley K, Campbell V, Berrow N, Leach R, Norman RI, Wray D, Dolphin AC, Baldwin SA. Use of site-directed antibodies to probe the topography of the alpha 2 subunit of voltage-gated Ca2+ channels. FEBS Lett. 1995;364:129–133. doi: 10.1016/0014-5793(95)00371-f. [DOI] [PubMed] [Google Scholar]
- 9.Brust PF, Simerson S, McCue AF, Deal CR, Schoonmaker S, Williams ME, Veliçelebi G, Johnson EC, Harpold MM, Ellis SB. Human neuronal voltage-dependent calcium channels: studies on subunit structure and role in channel assembly. Neuropharmacology. 1993;32:1089–1102. doi: 10.1016/0028-3908(93)90004-m. [DOI] [PubMed] [Google Scholar]
- 10.Calcutt NA, Chaplan SR. Spinal pharmacology of tactile allodynia in diabetic rats. Br J Pharmacol. 1997;122:1478–1482. doi: 10.1038/sj.bjp.0701538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994a;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 12.Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther. 1994b;269:1117–1123. [PubMed] [Google Scholar]
- 13.Coderre TJ, Katz J, Vaccarino AL, Melzack R. Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain. 1993;52:259–285. doi: 10.1016/0304-3959(93)90161-H. [DOI] [PubMed] [Google Scholar]
- 14.Coggeshall RE, Lekan HA, Doubell TP, Allchorne A, Woolf CJ. Central changes in primary afferent fibers following peripheral nerve lesions. Neuroscience. 1997;77:1115–1122. doi: 10.1016/s0306-4522(96)00528-3. [DOI] [PubMed] [Google Scholar]
- 15. Cooper CL, Vandaele S, Barhanin J, Fosset M, Lazdunski M, Hosey MM. Purification and characterization of the dihydropyridine-sensitive voltage-dependent calcium channel from cardiac tissue. J Biol Chem 262 1987. 509 512[Erratum(1987)262:3927]. [PubMed] [Google Scholar]
- 16.Curtis BM, Catterall WA. Purification of the calcium antagonist receptor of the voltage-sensitive calcium channel from skeletal muscle transverse tubules. Biochemistry. 1984;23:2113–2118. doi: 10.1021/bi00305a001. [DOI] [PubMed] [Google Scholar]
- 17.De Jongh KS, Warner C, Catterall WA. Subunits of purified calcium channels. Alpha 2 and delta are encoded by the same gene. J Biol Chem. 1990;265:14738–14741. [PubMed] [Google Scholar]
- 18.De Waard M, Witcher DR, Pragnell M, Liu H, Campbell KP. Properties of the alpha 1-beta anchoring site in voltage-dependent Ca2+ channels. J Biol Chem. 1995;270:12056–12064. doi: 10.1074/jbc.270.20.12056. [DOI] [PubMed] [Google Scholar]
- 19.Dissanayake VU, Gee NS, Brown JP, Woodruff GN. Spermine modulation of specific [3H]-gabapentin binding to the detergent-solubilized porcine cerebral cortex alpha 2 delta calcium channel subunit. Br J Pharmacol. 1997;120:833–840. doi: 10.1038/sj.bjp.0700988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- 21.Ellis SB, Williams ME, Ways NR, Brenner R, Sharp AH, Leung AT, Campbell KP, McKenna E, Koch WJ, Hui A, Schwartz A, Harpold MM. Sequence and expression of mRNAs encoding the alpha 1 and alpha 2 subunits of a DHP-sensitive calcium channel. Science. 1988;241:1661–1664. doi: 10.1126/science.2458626. [DOI] [PubMed] [Google Scholar]
- 22.Field MJ, Oles RJ, Lewis AS, McCleary S, Hughes J, Singh L. Gabapentin (neurontin) and S-(+)-3-isobutylgaba represent a novel class of selective antihyperalgesic agents. Br J Pharmacol. 1997;121:1513–1522. doi: 10.1038/sj.bjp.0701320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN. The novel anticonvulsant drug, gabapentin (neurontin), binds to the alpha2delta subunit of a calcium channel. J Biol Chem. 1996;271:5768–5776. doi: 10.1074/jbc.271.10.5768. [DOI] [PubMed] [Google Scholar]
- 24.Gilad B, Shenkar N, Halevi S, Trus M, Atlas D. Identification of the alternative spliced form of the alpha 2/delta subunit of voltage sensitive Ca2+ channels expressed in PC12 cells. Neurosci Lett. 1995;193:157–160. doi: 10.1016/0304-3940(95)11689-t. [DOI] [PubMed] [Google Scholar]
- 25.Gurnett CA, De Waard M, Campbell KP. Dual function of the voltage-dependent Ca2+ channel alpha 2 delta subunit in current stimulation and subunit interaction. Neuron. 1996;16:431–440. doi: 10.1016/s0896-6273(00)80061-6. [DOI] [PubMed] [Google Scholar]
- 26.Hullin R, Singer-Lahat D, Freichel M, Biel M, Dascal N, Hofmann F, Flockerzi V. Calcium channel beta subunit heterogeneity: functional expression of cloned cDNA from heart, aorta and brain. EMBO J. 1992;11:885–890. doi: 10.1002/j.1460-2075.1992.tb05126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang JH, Yaksh TL. Effect of subarachnoid gabapentin on tactile-evoked allodynia in a surgically induced neuropathic pain model in the rat. Reg Anesth. 1997;22:249–256. doi: 10.1016/s1098-7339(06)80010-6. [DOI] [PubMed] [Google Scholar]
- 28.Isom LL, De Jongh KS, Catterall WA. Auxiliary subunits of voltage-gated ion channels. Neuron. 1994;12:1183–1194. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- 29.Jay SD, Sharp AH, Kahl SD, Vedvick TS, Harpold MM, Campbell KP. Structural characterization of the dihydropyridine-sensitive calcium channel alpha 2-subunit and the associated delta peptides. J Biol Chem. 1991;266:3287–3293. [PubMed] [Google Scholar]
- 30.Jones SW, Marks TN. Calcium currents in bullfrog sympathetic neurons. II. Inactivation. J Gen Physiol. 1989;94:169–182. doi: 10.1085/jgp.94.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kerr LM, Filloux F, Olivera BM, Jackson H, Wamsley JK. Autoradiographic localization of calcium channels with [125I]omega-conotoxin in rat brain. Eur J Pharmacol. 1988;146:181–183. doi: 10.1016/0014-2999(88)90501-8. [DOI] [PubMed] [Google Scholar]
- 32. Kim HL, Kim H, Lee P, King RG, Chin H. Rat brain expresses an alternatively spliced form of the dihydropyridine-sensitive L-type calcium channel alpha 2 subunit. Proc Natl Acad Sci USA 89 1992. 3251 3255[Erratum(1992)89:5699]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 34.Klugbauer N, Lacinova L, Marais E, Hobom M, Hofmann F. Molecular diversity of the calcium channel α2δ subunit. J Neurosci. 1999;19:684–691. doi: 10.1523/JNEUROSCI.19-02-00684.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koltzenburg M, Torebjörk HE, Wahren LK. Nociceptor modulated central sensitization causes mechanical hyperalgesia in acute chemogenic and chronic neuropathic pain. Brain. 1994;117:579–591. doi: 10.1093/brain/117.3.579. [DOI] [PubMed] [Google Scholar]
- 36.Lai J, Hunter JC, Ossipov MH, Porreca F. Blockade of neuropathic pain by antisense targeting of tetrodotoxin-resistant sodium channels in sensory neurons. Methods Enzymol. 2000;314:201–213. doi: 10.1016/s0076-6879(99)14104-1. [DOI] [PubMed] [Google Scholar]
- 37.Lekan HA, Chung K, Yoon YW, Chung JM, Coggeshall RE. Loss of dorsal root ganglion cells concomitant with dorsal root axon sprouting following segmental nerve lesions. Neuroscience. 1997;81:527–534. doi: 10.1016/s0306-4522(97)00173-5. [DOI] [PubMed] [Google Scholar]
- 38.Luo Z, Fuentes ME, Taylor P. Regulation of acetylcholinesterase mRNA stability by calcium during differentiation from myoblasts to myotubes. J Biol Chem. 1994;269:27216–27223. [PubMed] [Google Scholar]
- 39.Luo ZD. Molecular dissection of pain mediators. Pain Rev. 2000a;7:37–64. [Google Scholar]
- 40.Luo ZD. Rat dorsal root ganglia express distinctive forms of the α2 calcium channel subunit. NeuroReport. 2000b;11:3449–3452. doi: 10.1097/00001756-200011090-00010. [DOI] [PubMed] [Google Scholar]
- 41.Luo ZD, Chaplan SR, Scott BP, Cizkova D, Calcutt NA, Yaksh TL. Neuronal nitric oxide synthase mRNA upregulation in rat sensory neurons after spinal nerve ligation: lack of a role in allodynia development. J Neurosci. 1999;19:9201–9208. doi: 10.1523/JNEUROSCI.19-21-09201.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mellick GA, Mellick LB. Reflex sympathetic dystrophy treated with gabapentin. Arch Phys Med Rehabil. 1997;78:98–105. doi: 10.1016/s0003-9993(97)90020-4. [DOI] [PubMed] [Google Scholar]
- 43.Mellick GA, Mellicy LB, Mellick LB. Gabapentin in the management of reflex sympathetic dystrophy [letter]. J Pain Symptom Manage. 1995;10:265–266. doi: 10.1016/0885-3924(95)00001-F. [DOI] [PubMed] [Google Scholar]
- 44.Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, Narumiya S, Numa S. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature. 1989;340:230–233. doi: 10.1038/340230a0. [DOI] [PubMed] [Google Scholar]
- 45.Mori Y, Friedrich T, Kim MS, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T, Mikoshiba K, Imoto K, Tanabe T, Numa S. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- 46.Nowycky MC, Fox AP, Tsien RW. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature. 1985;316:440–443. doi: 10.1038/316440a0. [DOI] [PubMed] [Google Scholar]
- 47.Plummer MR, Logothetis DE, Hess P. Elementary properties and pharmacological sensitivities of calcium channels in mammalian peripheral neurons. Neuron. 1989;2:1453–1463. doi: 10.1016/0896-6273(89)90191-8. [DOI] [PubMed] [Google Scholar]
- 48.Porreca F, Lai J, Bian D, Wegert S, Ossipov MH, Eglen RM, Kassotakis L, Novakovic S, Rabert DK, Sangameswaran L, Hunter JC. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci USA. 1999;96:7640–7644. doi: 10.1073/pnas.96.14.7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Price DD, Bennett GJ, Rafii A. Psychophysical observations on patients with neuropathic pain relieved by a sympathetic block. Pain. 1989;36:273–288. doi: 10.1016/0304-3959(89)90086-9. [DOI] [PubMed] [Google Scholar]
- 50.Rosenberg JM, Harrell C, Ristic H, Werner RA, de Rosayro AM. The effect of gabapentin on neuropathic pain. Clin J Pain. 1997;13:251–255. doi: 10.1097/00002508-199709000-00011. [DOI] [PubMed] [Google Scholar]
- 51.Rosner H, Rubin L, Kestenbaum A. Gabapentin adjunctive therapy in neuropathic pain states. Clin J Pain. 1996;12:56–58. doi: 10.1097/00002508-199603000-00010. [DOI] [PubMed] [Google Scholar]
- 52.Rowbotham M, Harden N, Stacey B, Bernstein P, Magnus-Miller L. Gabapentin for the treatment of postherpetic neuralgia: a randomized controlled trial. JAMA. 1998;280:1837–1842. doi: 10.1001/jama.280.21.1837. [DOI] [PubMed] [Google Scholar]
- 53.Scott VE, De Waard M, Liu H, Gurnett CA, Venzke DP, Lennon VA, Campbell KP. Beta subunit heterogeneity in N-type Ca2+ channels. J Biol Chem. 1996;271:3207–3212. doi: 10.1074/jbc.271.6.3207. [DOI] [PubMed] [Google Scholar]
- 54.Suman-Chauhan N, Webdale L, Hill DR, Woodruff GN. Characterisation of [3H]gabapentin binding to a novel site in rat brain: homogenate binding studies. Eur J Pharmacol. 1993;244:293–301. doi: 10.1016/0922-4106(93)90155-3. [DOI] [PubMed] [Google Scholar]
- 55.Takemura M, Kiyama H, Fukui H, Tohyama M, Wada H. Distribution of the omega-conotoxin receptor in rat brain. An autoradiographic mapping. Neuroscience. 1989;32:405–416. doi: 10.1016/0306-4522(89)90089-4. [DOI] [PubMed] [Google Scholar]
- 56.Tokumaru H, Anzai K, Abe T, Kirino Y. Purification of the cardiac 1,4-dihydropyridine receptor using immunoaffinity chromatography with a monoclonal antibody against the alpha 2 delta subunit of the skeletal muscle dihydropyridine receptor. Eur J Pharmacol. 1992;227:363–370. doi: 10.1016/0922-4106(92)90152-l. [DOI] [PubMed] [Google Scholar]
- 57.Wahren LK, Torebjork E. Quantitative sensory tests in patients with neuralgia 11 to 25 years after injury. Pain. 1992;48:237–244. doi: 10.1016/0304-3959(92)90063-H. [DOI] [PubMed] [Google Scholar]
- 58.Williams ME, Feldman DH, McCue AF, Brenner R, Velicelebi G, Ellis SB, Harpold MM. Structure and functional expression of alpha 1, alpha 2, and beta subunits of a novel human neuronal calcium channel subtype. Neuron. 1992;8:71–84. doi: 10.1016/0896-6273(92)90109-q. [DOI] [PubMed] [Google Scholar]
- 59.Wiser O, Trus M, Tobi D, Halevi S, Giladi E, Atlas D. The alpha 2/delta subunit of voltage sensitive Ca2+ channels is a single transmembrane extracellular protein which is involved in regulated secretion. FEBS Lett. 1996;379:15–20. doi: 10.1016/0014-5793(95)01475-6. [DOI] [PubMed] [Google Scholar]
- 60.Witcher DR, De Waard M, Sakamoto J, Franzini-Armstrong C, Pragnell M, Kahl SD, Campbell KP. Subunit identification and reconstitution of the N-type Ca2+ channel complex purified from brain. Science. 1993;261:486–489. doi: 10.1126/science.8392754. [DOI] [PubMed] [Google Scholar]
- 61.Woolf CJ, Doubell TP. The pathophysiology of chronic pain—increased sensitivity to low threshold A beta-fibre inputs. Curr Opin Neurobiol. 1994;4:525–534. doi: 10.1016/0959-4388(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 62.Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]