

Abstract
Widely expressed in the brain, the α4β2 nicotinic acetylcholine receptor (nAChR) is proposed to play a major role in the mechanisms that lead to and maintain nicotine addiction. Using the patch-clamp technique and pharmacological protocols, we examined the consequences of long-term exposure to 0.1–10 μm nicotine in K-177 cells expressing the major human brain α4β2 receptor. The acetylcholine dose–response curves are biphasic and revealed both a high- and a low-affinity component with apparent EC50values of 1.6 and 62 μm. Ratios of receptors in the high- and low-affinity components are 25 and 75%, respectively. Chronic exposure to nicotine or nicotinic antagonists [dihydro-β-erytroidine (DHβE) or methyllycaconitine (MLA)] increases the fraction of high-affinity receptors up to 70%. Upregulated acetylcholine-evoked currents increase by twofold or more and are less sensitive to desensitization. Functional upregulation is independent of protein synthesis as shown by the lack of effect of 20 μmcycloheximide. Single-channel currents recorded with 100 nmacetylcholine show predominantly high conductances (38.8 and 43.4 pS), whereas additional smaller conductances (16.7 and 23.5 pS) were observed with 30 μm acetylcholine. In addition, long-term exposure to dihydro-β-erytroidine increases up to three times the frequency of channel openings. These data indicate, in contrast to previous studies, that human α4β2 nAChRs are functionally upregulated by chronic nicotine exposure.
Keywords: acetylcholine, nicotinic receptor, α4β2, nicotine, upregulation, nicotine addiction
Nicotine is a tobacco compound that binds specifically to neuronal nicotinic acetylcholine receptors (nAChRs) of the brain (for review, see Dani and Heinemann, 1996;Changeux et al., 1998)). Tobacco addiction results from the repetitive intake of nicotine present in cigarette smoke and its rapid diffusion to the CNS (Gamberino and Gold, 1999; Leshner and Koob, 1999). Postmortem autoradiographies of smokers' brains have revealed a higher density of [H3]–nicotine binding compared with matched controls (Perry et al., 1999). Moreover, numerous studies have shown that long-term exposures to nicotine (or other nAChRs ligands) produce an increase of the total amount of brain labeling by [3H]–nicotine (Marks et al., 1985, 1992; Lapchak et al., 1989; Flores et al., 1992, 1997; Koylu et al., 1997; Perry et al., 1999). In rodents, it was proposed that chronic nicotine injection led to the conversion of a fraction of low-affinity nAChRs into high-affinity receptors (Romanelli et al., 1988). However, a consensus derived from initial observations is that long-term exposure to nicotine causes an increase in the number of binding sites at the cell surface (Wonnacott, 1990; Peng et al., 1994). Known as “upregulation,” this mechanism is opposite to “downregulation,” which was proposed for seven transmembrane receptors such as opiate receptors (Creese and Sibley, 1981).
The reinforcing effects of nicotine implicate β2-containing nAChRs (Picciotto et al., 1995, 1998) probably by modulating the release of dopamine in the mesolimbic system (Pidoplichko et al., 1997). In the striatum, the modulation of dopamine release depends on α4β2 nAChRs (Sharples et al., 2000). Dopamine release from brain striatal synaptosomes or from striatal slices could be potentiated (Rowell and Wonnacott, 1990; Yu and Wecker, 1994) or inhibited (Marks et al., 1993) by nicotine treatment.
Upregulation can be induced in vitro by exposing oocytes or cell lines expressing α4β2 nAChRs to chronic concentrations of nicotine (Peng et al., 1994; Hsu et al., 1996; Gopalakrishnan et al., 1997; Whiteaker et al., 1998). However, despite multiple investigations, what is still unclear is whether upregulation results in a functional increase or decrease and the relevance of these mechanisms in nicotine addiction.
In vitro electrophysiological measurements have demonstrated that prolonged ACh or nicotine applications (in a time scale of minutes) produced a progressive decline of the current carried by nAChRs (Katz and Thesleff, 1957; Peng et al., 1994; Lester and Dani, 1995; Fenster et al., 1997; Pidoplichko et al., 1997; Corringer et al., 1998). Called “desensitization,” this decline corresponds to a progressive closure of the receptors that are continuously exposed to nicotinic agonists. On one hand, it has been shown with the oocyte system that upregulation of α4β2 nAChRs occurs after receptor desensitization (Peng et al., 1994; Fenster et al., 1999a,b). On the other hand, Gopalakrishnan et al. (1996, 1997) have suggested that human α4β2 nAChRs expressed in human embryonic kidney (HEK) 293 cells could be functional after chronic exposure to nicotine or nicotinic ligands.
MATERIALS AND METHODS
K-177 is a stable cell line (HEK-293) expressing the human α4 and β2 nAChR subunits that was kindly provided by Abbott Laboratories (Chicago, IL). Constructions of cDNAs, transfection procedures, selection, and culture have been described previously (Gopalakrishnan et al., 1996; Buisson et al., 1998). Whole-cell currents recorded with an Axopatch 200B amplifier were filtered at 1 kHz and sampled at 5 kHz by a PCI card (National Instrument) and stored on the hard disk of a Macintosh computer. Compared with our previous studies (Buisson et al., 1996; Buisson and Bertrand, 1998), the saline solutions were modified as indicated to increase the current stability. Cells were recorded at room temperature in the following extracellular medium (in mm): 130 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, pH 7.4 with NaOH. Borosilicate electrodes (3–8 MΩ) were filled with (in mm): 130 K-gluconate, 5 NaCl, 2 MgCl2, 10 HEPES, 5 EGTA, pH 7.4 with KOH. Under these conditions, the single-channel activity of human muscle nAChRs recorded in outside-out patches pulled from TE-671 cells could last up to 40 min when elicited with a low ACh concentration. To minimize the capacitance in single-channel recordings, electrodes were coated with Sylgard (Dow Corning). Single-channel currents were sampled at 10 kHz. The reversal potential of α4β2 nAChRs was determined at −1 mV (n = 5).
Unless indicated, after removal from the incubator (± chronic nAChR ligand), cells were washed thoroughly twice with recording medium and placed on the stage of a inverted Zeiss microscope. On average, <5 min was necessary before the whole-cell recording configuration was established. To avoid modification of the cell conditions, a single cell was recorded per Petri dish, and cells were recorded alternately between control and chronic-treated dishes. To evoke short responses, agonists were delivered using a modified liquid filament made of a piezo-driven glass theta tube (final diameter of ∼150 μm, pulled from 1.5 mm diameter theta borosilicate tubing). One channel was connected to a 16-tube barrel and the other one to an 8-tube barrel. Barrels were produced by gluing 200 μm polyethylene tubing in the opening of a 1 ml plastic syringe. In each channel, gravity-driven solutions flowed at a rate of 120 μl/min per channel. Dose–response curves including nine concentration points could be measured in <3 min.
No differences in the fraction of responsive cells could be detected among experimental conditions. More than 95% of the cells responded to Ach, and every cell presenting a measurable current was taken into account. Cells were held at −100 mV throughout the experiment. All drugs were prepared daily from stock solutions.
Neuronal α4β2 nAChR dose–response curves could be described by the sum of two empirical Hill equations comparable to that used byCovernton and Connolly (2000):
| Equation 1 |
Imax is the maximal current amplitude, and x is the agonist concentration. EC50H, nH1, and a1 are the half-effective concentration, the Hill coefficient, and the percentage of receptors in the high-affinity state, whereas EC50L and nH2 are the half-effective concentration and the Hill coefficient in the low-affinity state. In some cases, a single Hill equation y =Imax*{1/(1 + (EC50/x)nH)} was used for comparison of the fit with Equation 1.Imax, EC50, and nH have the same signification.
The time course of desensitization to ACh was analyzed with a mono-exponential in the form:
| Equation 2 |
where y = current (in picoamperes),A (in picoamperes), τ (time constant, in milliseconds), and B (current at equilibrium, in picoamperes), andt = time (in milliseconds).
Frequency of openings was computed as the ratio between integrals of the Gaussian functions that fit opening events and the integral of the Gaussians that fit the zero-current baseline (electrode + setup noise).
Data are expressed as mean ± SEM with n as the number of independent measurements.
RESULTS
Human α4β2 nAChRs display biphasic dose–response profiles
Analysis of the peak current amplitude as a function of the ACh concentration revealed that dose–response curves could not be fitted properly by a single Hill equation (Fig.1A, dashed line). A better fit of the mean values was obtained, however, using two Hill equations corresponding to a low- and a high-affinity component (Fig. 1A, continuous curves, Table 1). Best fits yielded high-affinity coefficients of EC50H = 1.6 ± 0.07 μm and nH1 = 0.95 ± 0.02, whereas the low-affinity values were EC50L = 62 ± 1.4 μm and nH2 = 1.5 ± 0.03 (n = 95; 12 recording sessions). The maximal evoked current was 2571 ± 177 pA, and the fractions of high- and low-affinity components were of 25 and 75 ± 0.1%, respectively (n = 95). Two possibilities could account for the biphasic nature of the dose–response profile. First, α4β2 nAChRs can be divided into two different populations of receptors that are not structurally related, each one with distinct properties. Second, the receptors exist in two interconvertible states with different affinities. Unfortunately, electrophysiological measurements provide no further insight into discriminating between these two alternatives. Nonetheless, for clarity, currents can be divided into high- and low-affinity components.
Fig. 1.
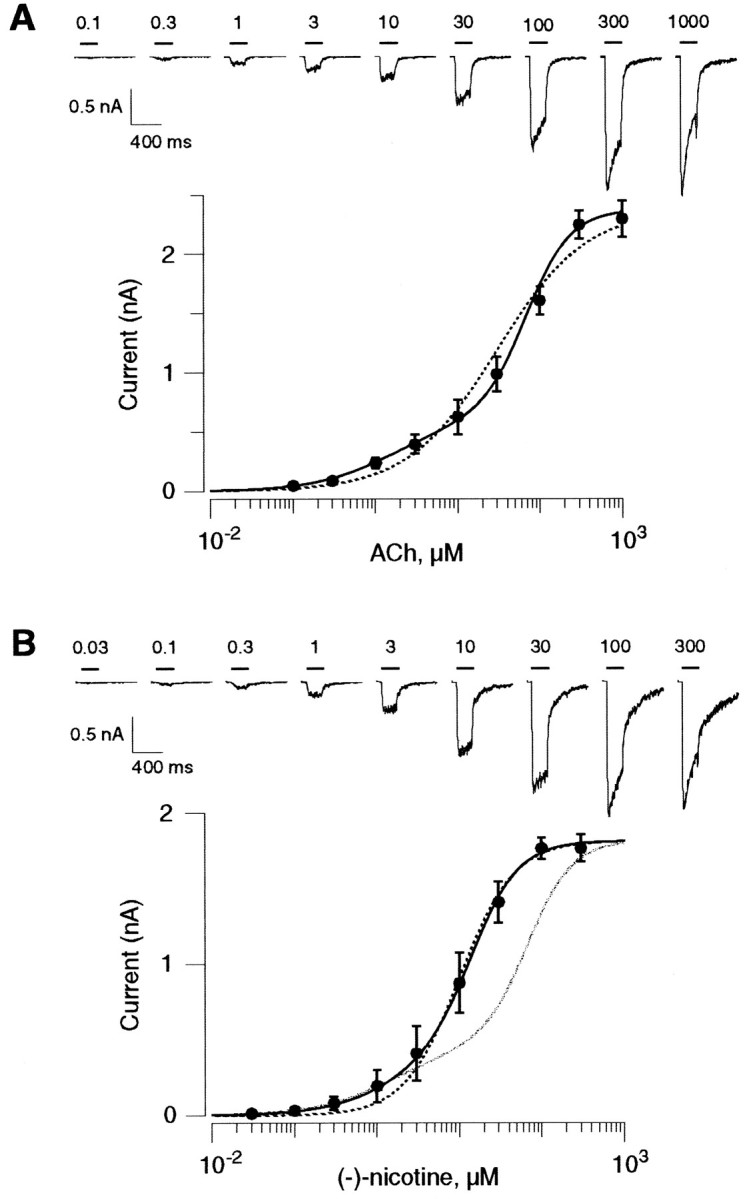
Two-component dose–response curves of human α4β2 nAChRs. A, Increasing ACh concentrations (200 msec pulses) were delivered every 15 sec. Typical traces of ACh-evoked currents are presented at the top. Horizontal bars indicate the ACh pulses with the concentration values (in micromolar). The fast desensitization and the current rebound observed at the end of the application pulse (1000 μm ACh) might indicate an open-channel block by the high ACh concentration. Mean current amplitudes were plotted as a function of the ACh concentration on a semilogarithmic scale (n = 12). Thedashed line corresponds to the best fit that can be drawn using a single Hill equation (EC50 = 30 μm and nH = 0.8). A better fit is obtained with the sum of two Hill equations (continuous line) yielding high-affinity coefficients of EC50H = 1.60 μm and nH1 = 0.92, whereas the low-affinity values are EC50L = 68 μm and nH2 = 1.60. The fraction of high- and low-affinity states are 25 and 75%, respectively. B, The same protocol as inA was repeated for the determination of the α4β2 nAChR sensitivity toward nicotine. Currents were then elicited with increasing concentrations of nicotine (top).Horizontal bars indicate the nicotine pulses with the concentration values (in micromolar). Mean current amplitudes were plotted as a function of the nicotine concentration on a semilogarithmic scale (n = 11). The dashed line corresponds to the best fit that can be drawn using a single Hill equation (EC50 = 10 μm and nH = 1.30). A better fit is obtained with the sum of two Hill equations (thick line) yielding high-affinity coefficients of EC50H = 2.4 μm and nH1 = 0.91, whereas the low-affinity values are EC50L = 14.5 μm and nH2 = 1.53. The fractions of high- and low-affinity states are 25 and 75%, respectively. For comparison, the ACh dose–response profile is scaled up to the maximal nicotine-evoked current (gray line). Note that the low-affinity component is much more sensitive to nicotine than to ACh.
Table 1.
Fits of dose–response data with empirical Hill equations
| Chronic exposure to | High-affinity state | Low-affinity state | Imax pA | ||||
|---|---|---|---|---|---|---|---|
| (a1) | EC50H (μm) | nH1 | (1 − a1) | EC50L (μm) | nH2 | ||
| Control (59) | 25 ± 1 | 1.5 ± 0.1 | 0.93 ± 0.01 | 75 | 61 ± 0.8 | 1.5 ± 0.04 | 2202 ± 273 |
| 8–10 hr 100 nm (−)-nicotine (8) | 44 ± 5 | 1.4 ± 0.1 | 0.94 ± 0.03 | 56 | 61 | 1.5 | 3305 ± 448 |
| 19–23 hr 1 μm (−)-nicotine (5) | 45 ± 5 | 2.1 ± 0.4 | 0.90 ± 0.03 | 55 | 61 | 1.5 | 4798 ± 606 |
| 19–23 hr 10 μm (−)-nicotine (7) | 37 ± 6 | 1.4 ± 0.1 | 0.84 ± 0.03 | 63 | 61 | 1.5 | 3477 ± 341 |
| 8–10 hr 10 nm DHβE (7) | 50 ± 6 | 1.1 ± 0.2 | 0.84 ± 0.03 | 50 | 61 | 1.5 | 4868 ± 878 |
| 8–24 hr 10 μm DHβE (16) | 70 ± 5 | 0.67 ± 0.2 | 0.91 ± 0.06 | 30 | 61 | 1.5 | 6026 ± 539 |
| 8–10 hr 10 μm MLA (12) | 57 ± 3 | 1.6 ± 0.2 | 0.97 ± 0.02 | 43 | 61 | 1.5 | 5326 ± 735 |
| 10–24 hr 20 μm cyclo control (10) | 36 ± 3 | 1.5 ± 0.2 | 0.92 ± 0.04 | 64 | 61 | 1.5 | 3563 ± 645 |
| 10–24 hr 20 μm cyclo + DHβE 10 μm (11) | 54 ± 3 | 0.9 ± 0.1 | 1 ± 0.05 | 46 | 61 | 1.5 | 6012 ± 515 |
Control cells, recorded alternately with cells chronically exposed to drugs, were pooled together because they presented few variations between experiments. Data points of the dose–response values measured for cells exposed to drugs were fitted as follows: the low-apparent affinity was fixed at 61 μm with a Hill coefficient of 1.5 (corresponding to the mean values of control cells), and the other parameters were adjusted to fit of the data.
To assess whether high- and low-affinity components display differences in their pharmacological profile, we then evoked currents with increasing concentrations of nicotine (Fig. 1B). Nicotine appeared to be as potent as ACh in eliciting responses. Maximal current amplitudes were not significantly different between ACh and nicotine. The biphasic nature of the dose–response profile was less marked for nicotine (Fig. 1B, gray line). However, a single Hill equation hardly fits data points for the low nicotine concentrations (Fig. 1B, thin line). The calculated EC50 values (and nH) for the high- and low-affinity states were 2.4 ± 0.5 μm (0.91 ± 0.03) and 14.5 ± 0.8 μm (1.53 ± 0.07), with fraction of 25% and 75%, respectively. The difference of profile observed between ACh and nicotine revealed that nicotine is less potent than ACh in discriminating between high- and low-affinity components. Moreover, this observation suggested that the two states could present different pharmacological profiles.
Effects of chronic exposure to nicotine
Recent studies performed with animal models indicate that a chronic nicotine exposure causes a sensitization to an acute nicotine pulse (Benwell et al., 1995; Balfour et al., 2000; Grottick et al., 2000). To determine whether the major brain nAChR remains functional when exposed for hours to low nicotine concentrations, we cultured cells expressing human α4β2 nAChRs for at least 8 hr in the presence of 100 nm nicotine. This nicotine concentration is comparable to that found in smokers' blood (Henningfield et al., 1993), and the incubation time is long enough to reach the steady-state upregulation process (Gopalakrishnan et al., 1997; Vallejo et al., 1999) (see below). In these experiments, the culture medium was not replaced before the Petri dish was mounted on the microscope stage, and the ACh dose–response curve was established in a perfusion medium that contained 100 nm nicotine (except during the 200 msec ACh pulses). Surprisingly, significant responses were measured even in the continuous presence of 100 nm nicotine (Fig.2A, top traces). After these recordings, the cell was superfused with a nicotine-free solution. No deviation of the current baseline was observed during nicotine removal (data not show), indicating that, if present, the fraction of channels remaining open after 10 hr in nicotine was below detection limits. When the cell was again challenged with the identical dose–response protocol 2 min later, a large increase in responses was observed (Fig. 2A,bottom traces). Higher EC50 values (compared with values determined in control conditions) were determined for the ACh dose–response curve measured in the presence of 100 nm nicotine (Fig. 2B). When the ACh dose–response protocols were performed several minutes after nicotine removal (i.e., for a time ≥5 min), no significant differences in the EC50 values could be observed between control and nicotine-exposed cells (see below and Table 1). Thus, differences in the EC50 values observed in the presence of nicotine might result from cumulative mechanisms involving both competitions at the binding sites and receptor desensitization.
Fig. 2.

Effects on human α4β2 nAChRs of a long-term exposure to nicotine. A, Chronic nicotine exposure failed to suppress ACh-evoked currents. Cells were incubated overnight in a culture medium containing 100 nm nicotine. In this particular experiment, the culture medium was not washed out before the recording. The cell was still superfused with the saline solution containing 100 nm nicotine until the establishment of the whole-cell configuration. First, ACh-evoked currents could be recorded even in the continuous presence of 100 nm nicotine between ACh pulses (top traces). Second, immediately after nicotine removal from the perfusion medium, the same protocol evoked currents of larger amplitudes (bottom traces). Thehorizontal bars indicate the duration of the ACh applications with the concentration values. B, The ACh dose–response relationship was determined in a series of cells (n = 7) either under a 100 nmnicotine-containing solution (squares) or immediately (2 min) after nicotine removal (circles). In the presence of 100 nm nicotine, high-affinity coefficients are EC50H = 3.48 μm and nH1 = 0.98, whereas the low-affinity values are EC50L = 127 μm and nH2 = 1.44; the fractions of high- and low-affinity states are 21 and 79%, respectively. After nicotine removal, high-affinity coefficients were EC50H = 2.3 μm and nH1 = 0.82, whereas the low-affinity values were EC50L = 91.4 μm and nH2 = 1.37; the fractions of high- and low-affinity states were 33 and 67%, respectively. C, The ACh dose–response profile was determined either for control cells or for cells exposed to 0.1 or 1 μm nicotine. ACh-evoked currents recorded in a typical control cell (top traces) are presented in comparison to currents elicited in another cell incubated for 10 hr in 100 nm nicotine with an extensive wash before recording (bottom traces). Horizontal bars indicate the ACh applications. D, ACh dose–response curves measured in control (○, n = 11) and after chronic nicotine incubation (100 nm, ▪, n = 8; 1 μm, ▿, n = 5; parameters for the fit are given in Table 1).
Although nicotine exposure effects could already be observed after 2–3 hr of incubation, maximal steady-state effects were only observed after 8 hr (data not shown) (Gopalakrishnan et al., 1997; Vallejo et al., 1999). Comparison of the ACh-evoked currents in control and in nicotine-treated cells indicated that long-term exposure to 100 nm nicotine increased the amplitude of the currents. The potentiation is stronger at the lowest ACh concentrations (Fig.2C) and reached at least twofold at saturation of ACh. As illustrated in Figure 2D, nicotine caused a dose–dependent upregulation of human α4β2 nAChRs. A 1 μm concentration of nicotine is slightly more potent for inducing upregulation. The maximal effect should be reached between 1 and 10 μm (Table 1). ACh dose–response relationships presented in Figure 2Drevealed that long-term exposure to nicotine did not change the apparent affinities of the receptors for ACh but increased the percentage of receptors in the high-affinity state up to 45% (Table1).
Moreover, ACh-evoked currents recorded in upregulated cells always displayed longer relaxation tails and slower desensitization kinetics (compare the current profiles in Fig. 2C). The slower time course of the current tails suggests that ACh could dissociate more slowly from the binding sites of upregulated receptors or that nAChRs closed more slowly. Average ACh-evoked currents presented in Figure3A illustrate the slowdown of the current desensitization observed at upregulated nAChRs. Quantification of the decay time of the mean ACh-evoked responses recorded under control (n = 5) and after a chronic exposure to 100 nm nicotine (n = 5) confirmed the reduction in desensitization (Fig. 3B). It is necessary to underline that with a high concentration of ACh (>30 μm) the high-affinity receptors are maximally activated. Thus, modifications observed at saturating ACh concentrations correspond to changes of both receptor populations (high- and low-affinity components).
Fig. 3.

Long-term exposure to nicotine reduces desensitization. A, Mean ACh-evoked currents recorded in control (left traces) and after nicotine treatment (right traces) have been averaged (n= 5 in each condition). Dashed lines through the data points were computed using a mono-exponential (see Materials and Methods). B, Parameter values determined for the fit of current decays (Eq. 2) in control condition and after 8–10 hr exposure to 100 nm nicotine (nic).
Moreover, a chronic nicotine exposure could also affect the desensitization rate of the low-affinity receptors. Thus, peak current measurements of responses evoked at upregulated receptors might have underestimated the fraction of the low-affinity receptor current. However, the liquid filament technique used in this study is one of the fastest drug application systems available, and it allowed us to record α7-evoked currents that are known for their fast activation and desensitization (Buisson et al., 1998). However, underestimation of the current mediated by low-affinity receptors cannot be ruled out.
Dihydro-β-erytroidine and methyllycaconitine induce upregulation
A pharmacological study has indicated that chronic exposure to nicotinic antagonists led to the upregulation of human α4β2 nAChRs (Gopalakrishnan et al., 1997). Among the different compounds tested, dihydro-β-erytroidine (DHβE) presented the higher potency. DHβE is a competitive antagonist of the human α4β2 nAChR (B. Buisson and D. Bertrand, unpublished observation) that inhibits the receptor with an apparent IC50 of 80 nm (Buisson et al., 1996). Micromolar concentrations of this compound could not elicit detectable currents in K-177 cells (data not shown). However, long-term exposure to DHβE promoted a large increase of the ACh-evoked current (Fig. 4A). As observed previously with nicotine, DHβE exposure induced a slowdown of the current desensitization and of its relaxation tails (compare traces in Fig. 4A). These effects were already observed at 10 nm, revealing the remarkable potency of DHβE. The upregulation of α4β2 nAChRs was increased by higher concentrations of DHβE: at 1 μm (data not shown) and at 10 μm, the ACh dose–response curves presented a marked biphasic profile with a maximal ACh-evoked current that increased up to threefold (Fig. 4B, Table 1). Fit of the ACh dose–response curves revealed further that DHβE promoted a significant increase of the fraction of nAChRs in the high-affinity state (up to 70%) (Fig. 4B, Table 1). For technical limitations, concentrations of DHβE higher than 10 μm could not be tested.
Fig. 4.

Human α4β2 nAChR upregulation is induced by dihydro-β-erytroidine (DHβE).A, Typical currents evoked by increasing ACh concentrations are presented for a control cell (top traces) and for a cell exposed to DHβE (bottom traces). Horizontal bars indicate the ACh application with the concentration values (in micromolar). Note the large increase of the amplitude of the currents and the decrease of desensitization after chronic exposure to DHβE. Long-lasting tails of the current observed with DHβE-incubated cells suggest that ACh dissociates more slowly from the receptors than the competitive antagonist. B, Dose–response relationships were determined in control (○, n = 7) or after chronic exposure to 10 nm DHβE (▵, n = 7) or 10 μm DHβE (▪, n = 7).Lines through the data points are the best fits obtained with the sum of two Hill equations (see Table 1 for the values).
To determine whether upregulation was not restricted to a single antagonist, the effect of a long-term exposure to MLA, another inhibitor of neuronal nAChRs, was investigated. A 10 μmconcentration of MLA caused upregulation of α4β2 nAChRs (Table 1) with ACh-evoked responses presenting characteristics of currents recorded after a long-term exposure to nicotine or DHβE. Thus, an agonist (nicotine) and antagonists (DHβE and MLA) could each induce a functional upregulation of human α4β2 nAChRs. These observations indicate that receptor opening is not necessary for the induction of α4β2 upregulation of nAChRs.
Because DHβE appeared to be the most potent compound for inducing upregulation, it was used further to characterize this phenomenon.
Functional upregulation is independent of de novo protein synthesis
Upregulation of human α4β2 nAChRs is characterized by (1) a change of ratio between receptors in the high- and low-affinity states, (2) a decrease of the desensitization rate with a slowdown of the current relaxation, and (3) an increase of the maximal ACh-evoked current. Two main hypotheses can be considered to explain these three upregulation effects.
First, upregulation could result from an increase in the number of nAChRs at the cell surface. Different mechanisms have been proposed such as de novo synthesis of new proteins (Wonnacott, 1990), incorporation of an internal pool of preexisting receptors, or a decrease of the turnover of receptors (Peng et al., 1994). It has been suggested recently that in contrast to cell surface nAChRs, intracellular receptors of the oocyte present a higher agonist affinity (Fenster et al., 1999b). Then, incorporation of intracellular “high-affinity receptors” into the cell membrane could explain the increase in the fraction of high-affinity receptors that we have observed.
Second, it has been suggested that in the plasma membrane, nAChRs could exist in two different states with different affinities (Bhat et al., 1994; Shafaee et al., 1999; Vallejo et al., 1999). In this context, upregulation may be viewed as a change in the ratio of receptors between different states (Table 1).
Because incubation of the cells with the protein synthesis inhibitor cycloheximide (20 μm) had no effect on the DHβE-induced upregulation (Fig. 5A, Table1), it seems unlikely that de novo synthesis of receptor proteins may account for the effects caused by DHβE exposure. As presented in Figure 5, B and C, the upregulation process could fully reverse within a few hours after DHβE removal. Both current amplitudes and current profiles returned back to control conditions within 6–9 hr after DHβE removal (see typical traces presented in Fig. 5C). Moreover, the fraction of nAChRs in the high-affinity state decreased back to the control 25% (n = 7).
Fig. 5.

Human α4β2 nAChR upregulation is independent of protein synthesis and reverses within a few hours. A, Addition in the culture medium of the protein synthesis inhibitor cycloheximide (20 μm) had no effect on the maximal ACh-evoked current (1 mm, 200 msec) recorded in control (n = 10) or after chronic exposure to DHβE (10 μm, n = 11) (Table 1).B, DHβE upregulation was reversible within a few hours. Current amplitudes evoked by saturating ACh concentration (1 mm, 200 msec) were measured in a series of cells in control (n = 22) after overnight exposure to DHβE (n = 22) or after overnight exposure to DHβE (15 hr) with an additional 6–9 hr recovery period after DHβE removal (n = 7) (see Table 1 for the values).C, ACh-evoked currents return back to control amplitude and desensitization profiles after DHβE removal. Representative ACh-evoked currents recorded in control (top traces), after DHβE exposure (middle traces), and after recovery (bottom traces) are illustrated.Horizontal bars correspond to the ACh applications with concentration values (identical for traces in a vertical column).
Distinct single-channel properties at low and high ACh concentrations
The initial investigation of α4β2 nAChR single channels revealed one key feature of these ligand-gated channels: their propensity to run down within a few minutes when recorded in outside-out patches from the oocyte membrane (Ballivet et al., 1988;Cooper et al., 1991). These properties have been reported thereafter in different cell systems (Pereira et al., 1994; Buisson et al., 1996). To overcome this difficulty, single-channel activities have been recorded using the cell-attached configuration (Papke et al., 1989; Charnet et al., 1992; Ragozzino et al., 1997), an alternative protocol in which nAChRs are continuously exposed to ACh within the pipette. Because of this, nAChRs may enter into desensitized states.
Outside-out patches pulled from K-177 cells were exposed to 0.1 and 30 μm concentrations of ACh that can activate either the high- or the high- and low-affinity components. At a low ACh concentration, single-channel activities could be recorded for up to 6 min (allowing 36 sweeps of ACh applications in the best conditions). However, when the ACh concentration was increased to 30 μm, single-channel activities always disappeared within 2 min. The low setup noise allowed us to investigate further the single-channel properties of human α4β2 nAChRs at a higher resolution. Statistical analysis of cumulative all-point amplitude histograms revealed the existence of multiple conductance levels.
In agreement with our initial characterization of the human α4β2 nAChR (Buisson et al., 1996), openings of large amplitudes (Fig.6A) were always observed when the patch was exposed to a low ACh concentration (100 nm;n = 30), which may activate exclusively nAChRs in the high-affinity state. However, when the same patch was exposed to a higher ACh concentration (30 μm) (Fig.6A), lower conductances were observed more clearly. This observation has been made regarding the seven patches that could be successively recorded under 0.1 and 30 μmACh. Best fit of the all-point amplitude histograms (data not shown) could be performed using five elementary current amplitudes yielding to conductances of 16.7 ± 0.5, 23.5 ± 0.4, 31.6 ± 0.4, 38.8 ± 0.5, and 43.4 ± 0.5 pS (n = 7). In correlation with the statistical analysis, different elementary current amplitudes were repetitively observed (Fig. 6A). Such conductance levels have been reported previously in other studies performed with α4β2 nAChRs of different species (Ballivet et al., 1988; Papke et al., 1989; Charnet et al., 1992; Pereira et al., 1994;Ramirez-Latorre et al., 1996; Ragozzino et al., 1997), including human (Buisson et al., 1996; Kuryatov et al., 1997; Nelson et al., 1999). To illustrate the change in the opening amplitudes observed when the ACh concentration was raised from 100 nm to 30 μm, conductance events were normalized for each ACh concentration (Fig. 6B). These data suggest that low-conductance openings occurred more frequently at 30 μm ACh. Thus, we propose that the high conductances (38.8 and 43.4 pS) could correspond to the opening of the high-affinity receptors, whereas the low conductances (16.7, 23.5, and 31.6 pS) could reveal the opening of low-affinity receptors.
Fig. 6.

Single-channel currents of human α4β2 display multiple conductance levels at low and high ACh concentrations.A, Portion of 800 msec recordings performed with a single patch have been enlarged to illustrate the different current amplitudes that could be observed. The thick horizontal bars above top traces indicate the applications of ACh. The thin dashed lines correspond to conductance levels of 0, 40, and 80 pS from top tobottom. B, Cumulative all-point amplitude histograms computed from several traces obtained in the same patch (the bin was set at 0.1 pA) were normalized to the total number of events in each recording condition (20 sweeps recorded in 100 nm ACh and 14 recorded in 30 μm ACh). The barscorresponding to the setup noise have been truncated to present the current amplitudes at a higher resolution. Note the increase of the low conductance event number when openings are elicited by a 30 μm ACh concentration. This observation has been repeated in all patches recorded in both ACh concentrations (n = 7).
Upregulation increases the frequency of opening at a low ACh concentration
To get a further insight into the upregulation mechanism, we investigated single-channel properties of upregulated nAChRs. Because of the rundown, detailed analysis could not be performed, and we were restricted mainly to conductance measurements. Upregulation could result either from the isomerization of a fraction of low-affinity, low-conductance receptors into high-affinity, high-conductance receptors or the incorporation into the cell membrane of intracellular high-affinity (Fenster et al., 1999b), high-conductance nAChRs. In both cases, the frequency of opening measured at a low ACh concentration should increase. We have recorded single-channel activities in outside-out patches pulled from cells incubated for at least 18 hr in 10 μm DHβE and compared single-channel characteristics with patches pulled from matched control cells.
The single-channel activities elicited by 100 nm ACh was always higher in the membrane patches pulled from cells exposed to 10 μm DHβE than control cells (Fig.7A). Average traces computed from multiple sweeps further revealed that the higher single-channel activities corresponded to an overall increase of the computed macroscopic current amplitude (Fig. 7B). It is of value to stress that single-channel conductances were indistinguishable between upregulated and control patches (n = 8; data not shown) and that the rundown at 100 nm ACh remained unchanged after DHβE exposure. However, at 100 nm Ach, the open probability showed a threefold increase: from 6.1 ± 1.1% in control to 18.3 ± 2.3% in DHβE-treated cells (n = 5) (Fig. 7C). In addition, openings of longer duration were observed more frequently in patches pulled from DHβE-exposed cells (Fig. 7A). Altogether, these observations suggest that upregulation corresponds to an increase of the single-channel activities evoked by a low concentration of ACh.
Fig. 7.

Chronic incubation with DHβE increases the frequency of opening at 100 nm ACh. A, Typical single-channel currents recorded in an outside-out patch pulled from a control cell (left) or pulled from a cell incubated for 22 hr in 10 μm DHβE (right). The horizontal bars indicate pulses of ACh. Each trace is the first record of multiple sweeps (16 for the control cell and 17 for the 10 μm DHβE-treated cell). B, Average single-channel currents confirm the increase observed for whole-cell currents after DHβE-induced upregulation. Unitary currents including those presented inA were average for a control patch (left; mean of 10 sweeps) or for a patch pulled from a cell exposed for 22 hr to 10 μm DHβE (right; mean of 10 sweeps). C, Cumulative all-point amplitude histograms corresponding to multiple sweeps including and after the ones presented in A (16 sweeps in control and 17 sweeps for 10 μm DHβE). Surface areas of the Gaussians for channel openings and setup noise have values of 99 and 1703 (in control) and 288 and 1344 (in 10 μm DHβE). The open time probabilities are of 5.8% (control) and of 21.4% (10 μm DHβE) for these two representative patches.
The use of Table 1 for the computation of the current carried by the high- and low-affinity components in control or after DHβE treatment (10 μm, 22 hr) reveals a large increase (7.6 ×) of the current carried by the high-affinity component, whereas the fraction carried by the low-affinity component remains approximately constant (1.09×). The augmentation of the current carried by the high-affinity component is attributable either to an increase in the receptor number or to the mean open time, or both. Outside-out patch recordings yielded a threefold increase of the open probability for DHβE-treated cells versus control (Fig. 7C). Thus, to account for the 7.6-fold increase of the current carried by the high-affinity component, the number of active receptors in this state must have increased by at least a factor of 2. A change in the fraction of the high-affinity component can therefore result from either the conversion of low-affinity state receptors or the insertion of new receptors in the membrane.
DISCUSSION
High- and low-affinity α4β2 nAChRs
Since the initial pharmacological characterizations of brain nAChRs (Clarke et al., 1985; Deutch et al., 1987; Swanson et al., 1987), a consensus has emerged regarding the fact that the α4β2 subtype represents the predominant form in the CNS (Whiting et al., 1987; Schoepfer et al., 1988; Flores et al., 1992) and constitutes the high-affinity binding site for nicotine (Clarke et al., 1985;Picciotto et al., 1995; Marubio et al., 1999). However, it was suggested previously that brain nicotine binding sites are heterogeneous and constitute at least two populations of nAChRs that could isomerize between each other (Romanelli et al., 1988; Bhat et al., 1994). The natural alkaloid (±)-epibatidine, initially purified from the skin of the Ecuadoran frog Epipedobatestricolor (Badio and Daly, 1994), was rapidly identified as a high-affinity ligand for brain nAChRs (Houghtling et al., 1995). Binding of labeled (±)-epibatidine to oocytes or transfected cells expressing the rat α4 and β2 subunits has revealed low- and high-affinity sites with biochemical properties identical to low- and high-affinity (±)-epibatidine binding sites identified in rat brain membranes (Shafaee et al., 1999). Moreover, high- and low-affinity (±)-epibatidine binding sites have been identified in the human cortex (Marutle et al., 1999) where α4-containing nAChRs have been detected (Wevers et al., 1994, 1999) and where type II currents were recorded (Alkondon et al., 2000). These currents might correspond to the activation of native α4β2 nAChRs (Alkondon and Albuquerque, 1993;Albuquerque et al., 1995). Although it was suggested that the α5 subunit may contribute to functional α4α5β2 nAChRs (Ramirez-Latorre et al., 1996; Kuryatov et al., 1997), such receptors account for only a very small fraction of the total α4β2-containing nAChRs in the adult chick or rat brain (Conroy and Berg, 1998; Shafaee et al., 1999). In conclusion, binding measurements suggest that α4β2 nAChRs of the adult brain or α4β2 nAChRs reconstituted in heterologous systems constitute at least a two-component population of receptors.
A functional study performed with brain synaptosomes from wild-type and β2 knock-out mice has revealed two main β2-containing populations of brain nAChRs with distinct affinities for ACh (Marks et al., 1999). The α4β2 nAChRs of chick, rat, and human have been characterized extensively using different systems of expression. Published data suggest that ACh apparent affinities (EC50values) are distributed either in low- or in high-micromolar values (Bertrand et al., 1990; Buisson et al., 1996; Gopalakrishnan et al., 1996; Chavez-Noriega et al., 1997; Kuryatov et al., 1997; Zwart and Vijverberg, 1998; Cardoso et al., 1999; Nelson et al., 1999; Sabey et al., 1999; Covernton and Connolly, 2000). In agreement with previous findings (Covernton and Connolly, 2000), our data indicate that α4β2 nAChRs may exhibit two apparent affinities for ACh. The EC50 values that we have determined by using the sum of two Hill equations correspond to the high and low EC50 values reported in other studies of the human α4β2 nAChR.
Although questioned recently (Nelson et al., 1999), it seems that the occurrence of a low or a high ACh apparent affinity is independent of the host cell type. Indeed, the biphasic profile of the ACh dose–response curve is not restricted to the cell line expression system because two-component dose–response curves could be observed repetitively with oocytes expressing the human α4β2 nAChR (S. Bertrand and D. Bertrand, unpublished observations). In addition, this phenomenon is not species specific and was also observed for the rat α4β2 nAChR expressed in tsA-201 cells (Buisson et al., 2000) or in the oocyte (Zwart and Vijverberg, 1998; Covernton and Connolly, 2000). We emphasize that the observation of a monophasic dose–response curve is likely restricted by experimental parameters (such as the use of a perfusion system that is too slow or the performance of measurements with too few concentration points) rather than by the use of a specific expression system. Moreover, depending on the affinity for the two nAChR populations and the number of concentration points, an agonist could display dose–response curves that look shallower or steeper.
Consistent with the macroscopic current data, single-channel data also suggest that α4β2 nAChRs constitute a heterogeneous population of receptors. Consequently, we have chosen to apply the conclusions of the macroscopic current analysis to the microscopic current analysis. Thus far, our investigations have shown that at a low ACh concentration (100 nm), the 38–43 pS conductances were the main conductances observed. In contrast, at a higher ACh concentration (30 μm), lower conductance levels (16–23 pS) were observed more frequently. Two hypotheses could explain these observations. First, the different conductance levels might correspond to the activation of different pools of nAChRs (low- vs high-affinity receptors). Second, the same population of receptors could undergo different transitions depending on the ACh concentration. Moreover, the possible influence of subunit phosphorylation cannot be ruled out. As an example, the calcium/calmodulin kinase II is able to enhance the conductance of glutamate AMPA receptors (Derkach et al., 1999). Additional experiments will be needed to further characterize the relationships between single-channel and whole-cell currents. Strategies that could prevent or slow down the rundown (Liu and Berg, 1999) would be very helpful in this task.
Human α4β2 nAChR functional upregulation
Investigations performed with α4β2 nAChRs expressed in oocytes have shown that long-term exposure to nicotine induced a progressive loss of receptor function (Peng et al., 1994; Fenster et al., 1999b). By contrast, our data indicate that after a chronic exposure to 100 nm nicotine, human α4β2 nAChRs expressed in HEK-293 cells can be activated even in the continuous presence of nicotine. Moreover, after nicotine removal, α4β2 nAChRs are “hyperfunctional,” with an overall higher apparent affinity for ACh and currents of higher amplitudes with less desensitization. Thus, long-term exposure effects to nicotine may depend on the host cell system investigated. First is the type of cell-expressing system: Xenopus oocytes versus mammalian cell lines. Second is the temperature for protein expression: 18°C for oocytes versus 37°C for cell lines. Third is the absence (oocytes) or presence (cell lines) of serum in the culture medium. Fourth is the endogenous activity of intracellular factors such as kinases or phosphatases that could be different in oocytes and in cell lines. Indeed, protein kinase A and protein kinase C can interfere with the upregulation mechanism (Gopalakrishnan et al., 1997; Fenster et al., 1999a,b). Extensive investigations of the effect(s) of each of these factors could provide further insights into the mechanisms that modulate the upregulation of α4β2 nAChRs.
Because competitive antagonists induce upregulation of α4β2 nAChRs, a transmembrane signal has to trigger the incorporation of presynthesized receptors into the membrane, if we suppose that upregulation corresponds to an increase of the receptor number within the cell membrane (Wonnacott, 1990). Different mechanisms could be proposed for the transduction mechanism. The receptors could interact with cytoskeletal elements (Liu and Berg, 1999; Shoop et al., 2000) or could be coupled with metabotropic receptors such as those described recently for GABAA receptors (Liu et al., 2000).
Recent studies indicate that after a long-term exposure to nicotine, α4 and β2 mRNA levels do not change in the mouse brain (Marks et al., 1992) and that α4β2 proteins remain constant in the membrane of cell lines (Whiteaker et al., 1998; Vallejo et al., 1999). Our data, together with other studies (Zwart and Vijverberg, 1998; Marutle et al., 1999; Shafaee et al., 1999; Vallejo et al., 1999; Covernton and Connolly, 2000), indicate that surface α4β2 nAChRs are distributed in at least two populations. In the light of previous observations (Romanelli et al., 1988; Bhat et al., 1994; Vallejo et al., 1999), we propose that long-term exposure to nicotine could induce a fraction of low-affinity nAChRs to isomerize into high-affinity nAChRs by slow conformational transitions (time scale in hours). Such a hypothesis has the advantage of providing a coherent framework to explain the full reversibility of the upregulation process.
The eventuality that intracellular high-affinity receptors could be incorporated into the cell membrane cannot be ruled out with the present study. Reversibility of upregulation (Fig. 5) supposes, however, that newly incorporated nAChRs must be removed preferentially or shut down selectively within a few hours after the removal of the upregulating compound.
The small but consistent increase in the ACh-evoked current observed in cells treated with the protein inhibitor cycloheximide alone (Table 1) indicates a possible influence of this drug on the receptor number or equilibrium. In agreement with this observation, a small increase in the fraction of the high-affinity component was also observed. Although presently unexplained, this observation suggests a possible role of the protein synthesis in the regulation of the receptor at the cell surface.
Detailed examination of the amount of current carried by the high-affinity component between control and DHβE-treated cells revealed a 7.6-fold increase, whereas an increase of only threefold of the open probability was observed in corresponding outside-out patches. This would suggest that the number of highly activatable receptors must have at least doubled. That the amount of current carried by the low-affinity component remains approximately constant makes it tempting to conclude that new receptors must have been inserted into the membrane. Because of technical limitations (channel rundown), this important point cannot be resolved at present by electrophysiology alone. Therefore, further measurements are necessary before a final conclusion can be reached.
Our observations raise a fundamental question concerning the properties of native α4β2 nAChRs. It is conceivable that in the brain, a chronic low concentration of ACh could displace the equilibrium between low- and high-affinity α4β2 nAChRs. This mechanism could provide a molecular basis for the so-called “volume transmission” (Agnati et al., 1995; Bertrand and Changeux, 1995); that is, a neurotransmitter may diffuse in the extracellular space and act on receptors present outside the synaptic process. Moreover, we reveal that in vitro upregulation induces ACh-evoked currents of higher amplitudes that desensitize more slowly. Consistent with this observation, α4β2 nAChRs present in presynaptic or postsynaptic membranes could promote enhanced synaptic transmissions after their upregulation by low concentrations of ACh or nicotine. Consequently, α4β2-linked pathologies, such as autosomal dominant nocturnal frontal lobe epilepsy, might be investigated within this new paradigm (Kuryatov et al., 1997; Bertrand et al., 1998; Bertrand, 1999). Finally, pharmacological strategies targeted to α4β2 nAChRs (Lloyd and Williams, 2000) might take into account the unexpected properties of this receptor subtype.
Footnotes
This work was supported by the Swiss National Science Foundation and the “Office Fédéral de l'Education et des Sciences” (D. B.). We thank S. Bertrand, I. Favre, L. Curtis, C. Blanchet, C. Yamate-Poitry, and J. Sullivan for their help and discussions in the preparation of this manuscript. K-177 cells were kindly provided by J. Sullivan (Abbott Laboratories, Chicago, IL).
Correspondence should be addressed to Daniel Bertrand, Department of Physiology, Medical Faculty, 1 rue Michel Servet, 1211 Geneva 4, Switzerland. E-mail: bertrand@cmu.unige.ch.
Dr. Buisson's present address: Département de Screening, Trophos, Parc Scientifique de Luminy, case 931, 13288 Marseille cedex 9, France.
REFERENCES
- 1.Albuquerque EX, Pereira EF, Castro NG, Alkondon M, Reinhardt S, Schroder H, Maelicke A. Nicotinic receptor function in the mammalian central nervous system. Ann NY Acad Sci. 1995;757:48–72. doi: 10.1111/j.1749-6632.1995.tb17464.x. [DOI] [PubMed] [Google Scholar]
- 2.Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J Pharmacol Exp Ther. 1993;265:1455–1473. [PubMed] [Google Scholar]
- 3.Alkondon M, Pereira EF, Eisenberg HM, Albuquerque EX. Nicotinic receptor activation in human cerebral cortical interneurons: a mechanism for inhibition and disinhibition of neuronal networks. J Neurosci. 2000;20:66–75. doi: 10.1523/JNEUROSCI.20-01-00066.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badio B, Daly JW. Epibatidine, a potent analgetic and nicotinic agonist. Mol Pharmacol. 1994;45:563–569. [PubMed] [Google Scholar]
- 5.Balfour DJ, Wright AE, Benwell ME, Birrell CE. The putative role of extra-synaptic mesolimbic dopamine in the neurobiology of nicotine dependence. Behav Brain Res. 2000;113:73–83. doi: 10.1016/s0166-4328(00)00202-3. [DOI] [PubMed] [Google Scholar]
- 6.Ballivet M, Nef P, Couturier S, Rungger D, Bader CR, Bertrand D, Cooper E. Electrophysiology of a chick neuronal nicotinic acetylcholine receptor expressed in Xenopus oocytes after cDNA injection. Neuron. 1988;1:847–852. doi: 10.1016/0896-6273(88)90132-8. [DOI] [PubMed] [Google Scholar]
- 7.Benwell ME, Balfour DJ, Birrell CE. Desensitization of the nicotine-induced mesolimbic dopamine responses during constant infusion with nicotine. Br J Pharmacol. 1995;114:454–460. doi: 10.1111/j.1476-5381.1995.tb13248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertrand D, Ballivet M, Rungger D. Activation and blocking of neuronal nicotinic acetylcholine receptor reconstituted in Xenopus oocytes. Proc Natl Acad Sci USA. 1990;87:1993–1997. doi: 10.1073/pnas.87.5.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhat RV, Marks MJ, Collins AC. Effects of chronic nicotine infusion on kinetics of high-affinity nicotine binding. J Neurochem. 1994;62:574–581. doi: 10.1046/j.1471-4159.1994.62020574.x. [DOI] [PubMed] [Google Scholar]
- 10.Buisson B, Bertrand D. Open-channel blockers at the human α4β2 neuronal nicotinic acetylcholine receptor. Mol Pharmacol. 1998;53:555–563. doi: 10.1124/mol.53.3.555. [DOI] [PubMed] [Google Scholar]
- 11.Buisson B, Gopalakrishnan M, Arneric SP, Sullivan JP, Bertrand D. Human α4β2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: a patch-clamp study. J Neurosci. 1996;16:7880–7891. doi: 10.1523/JNEUROSCI.16-24-07880.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buisson B, Gopalakrishnan M, Bertrand D. Stable expression of human neuronal nicotinic receptors. In: Arneric SP, Brioni JD, editors. Neuronal nicotinic receptors: pharmacology and therapeutic opportunities. Wiley; New York: 1998. pp. 99–124. [Google Scholar]
- 13.Buisson B, Vallejo YF, Green WN, Bertrand D. The unusual nature of epibatidine responses at the α4β2 nicotinic acetylcholine receptor. Neuropharmacology. 2000;39:2561–2569. doi: 10.1016/s0028-3908(00)00158-1. [DOI] [PubMed] [Google Scholar]
- 14.Cardoso RA, Brozowski SJ, Chavez-Noriega LE, Harpold M, Valenzuela CF, Harris RA. Effects of ethanol on recombinant human neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1999;289:774–780. [PubMed] [Google Scholar]
- 15.Changeux JP, Bertrand D, Corringer PJ, Dehaene S, Edelstein S, Lena C, Le Novere N, Marubio L, Picciotto M, Zoli M. Brain nicotinic receptors: structure and regulation, role in learning and reinforcement. Brain Res Brain Res Rev. 1998;26:198–216. doi: 10.1016/s0165-0173(97)00040-4. [DOI] [PubMed] [Google Scholar]
- 16.Charnet P, Labarca C, Cohen BN, Davidson N, Lester HA, Pilar G. Pharmacological and kinetic properties of alpha 4 beta 2 neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Physiol (Lond) 1992;450:375–394. doi: 10.1113/jphysiol.1992.sp019132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chavez-Noriega LE, Crona JH, Washburn MS, Urrutia A, Elliott KJ, Johnson EC. Pharmacological characterization of recombinant human neuronal nicotinic acetylcholine receptors h α 2 β 2, h α 2 β 4, h α 3 β 2, h α 3 β 4, h α 4 β 2, h α 4 β 4 and h α 7 expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1997;280:346–356. [PubMed] [Google Scholar]
- 18.Clarke PB, Schwartz RD, Paul SM, Pert CB, Pert A. Nicotinic binding in rat brain: autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]-α-bungarotoxin. J Neurosci. 1985;5:1307–1315. doi: 10.1523/JNEUROSCI.05-05-01307.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conroy WG, Berg DK. Nicotinic receptor subtypes in the developing chick brain: appearance of a species containing the α4, β2, and α5 gene products. Mol Pharmacol. 1998;53:392–401. doi: 10.1124/mol.53.3.392. [DOI] [PubMed] [Google Scholar]
- 20.Cooper E, Couturier S, Ballivet M. Pentameric structure and subunit stoichiometry of a neuronal nicotinic acetylcholine receptor. Nature. 1991;350:235–238. doi: 10.1038/350235a0. [DOI] [PubMed] [Google Scholar]
- 21.Corringer PJ, Bertrand S, Bohler S, Edelstein SJ, Changeux JP, Bertrand D. Critical elements determining diversity in agonist binding and desensitization of neuronal nicotinic acetylcholine receptors. J Neurosci. 1998;18:648–657. doi: 10.1523/JNEUROSCI.18-02-00648.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Covernton PJ, Connolly JG. Multiple components in the agonist concentration-response relationships of neuronal nicotinic acetylcholine receptors. J Neurosci Methods. 2000;96:63–70. doi: 10.1016/s0165-0270(99)00185-5. [DOI] [PubMed] [Google Scholar]
- 23.Creese I, Sibley DR. Receptor adaptations to centrally acting drugs. Annu Rev Pharmacol Toxicol. 1981;21:357–391. doi: 10.1146/annurev.pa.21.040181.002041. [DOI] [PubMed] [Google Scholar]
- 24.Dani JA, Heinemann S. Molecular and cellular aspects of nicotine abuse. Neuron. 1996;16:905–908. doi: 10.1016/s0896-6273(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 25.Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci USA. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deutch AY, Holliday J, Roth RH, Chun LL, Hawrot E. Immunohistochemical localization of a neuronal nicotinic acetylcholine receptor in mammalian brain. Proc Natl Acad Sci USA. 1987;84:8697–8701. doi: 10.1073/pnas.84.23.8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA. Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci. 1997;17:5747–5759. doi: 10.1523/JNEUROSCI.17-15-05747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fenster CP, Beckman ML, Parker JC, Sheffield EB, Whitworth TL, Quick MW, Lester RA. Regulation of α4β2 nicotinic receptor desensitization by calcium and protein kinase C. Mol Pharmacol. 1999a;55:432–443. [PubMed] [Google Scholar]
- 29.Fenster CP, Whitworth TL, Sheffield EB, Quick MW, Lester RA. Upregulation of surface α4β2 nicotinic receptors is initiated by receptor desensitization after chronic exposure to nicotine. J Neurosci. 1999b;19:4804–4814. doi: 10.1523/JNEUROSCI.19-12-04804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol. 1992;41:31–37. [PubMed] [Google Scholar]
- 31.Flores CM, Davila-Garcia MI, Ulrich YM, Kellar KJ. Differential regulation of neuronal nicotinic receptor binding sites following chronic nicotine administration. J Neurochem. 1997;69:2216–2219. doi: 10.1046/j.1471-4159.1997.69052216.x. [DOI] [PubMed] [Google Scholar]
- 32.Gamberino WC, Gold MS. Neurobiology of tobacco smoking and other addictive disorders. Psychiatr Clin North Am. 1999;22:301–312. doi: 10.1016/s0193-953x(05)70078-2. [DOI] [PubMed] [Google Scholar]
- 33.Gopalakrishnan M, Monteggia LM, Anderson DJ, Molinari EJ, Piattoni-Kaplan M, Donnelly-Roberts D, Arneric SP, Sullivan JP. Stable expression, pharmacologic properties and regulation of the human neuronal nicotinic acetylcholine alpha 4 beta 2 receptor. J Pharmacol Exp Ther. 1996;276:289–297. [PubMed] [Google Scholar]
- 34.Gopalakrishnan M, Molinari EJ, Sullivan JP. Regulation of human α4β2 neuronal nicotinic acetylcholine receptors by cholinergic channel ligands and second messenger pathways. Mol Pharmacol. 1997;52:524–534. [PubMed] [Google Scholar]
- 35.Grottick AJ, Wyler R, Higgins GA. The α4β2 agonist SIB 1765F, but not the α7 agonist AR-R 17779, cross-sensitises to the psychostimulant effects of nicotine. Psychopharmacology (Berl) 2000;150:233–236. doi: 10.1007/s002130000444. [DOI] [PubMed] [Google Scholar]
- 36.Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED. Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend. 1993;33:23–29. doi: 10.1016/0376-8716(93)90030-t. [DOI] [PubMed] [Google Scholar]
- 37.Houghtling RA, Davila-Garcia MI, Kellar KJ. Characterization of (+/−)(−)[3H]epibatidine binding to nicotinic cholinergic receptors in rat and human brain. Mol Pharmacol. 1995;48:280–287. [PubMed] [Google Scholar]
- 38.Hsu YN, Amin J, Weiss DS, Wecker L. Sustained nicotine exposure differentially affects alpha 3 beta 2 and alpha 4 beta 2 neuronal nicotinic receptors expressed in Xenopus oocytes. J Neurochem. 1996;66:667–675. doi: 10.1046/j.1471-4159.1996.66020667.x. [DOI] [PubMed] [Google Scholar]
- 39.Katz B, Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol (Lond) 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koylu E, Demirgoren S, London ED, Pogun S. Sex difference in up-regulation of nicotinic acetylcholine receptors in rat brain. Life Sci. 1997;61:185–190. doi: 10.1016/s0024-3205(97)00665-6. [DOI] [PubMed] [Google Scholar]
- 41.Kuryatov A, Gerzanich V, Nelson M, Olale F, Lindstrom J. Mutation causing autosomal dominant nocturnal frontal lobe epilepsy alters Ca2+ permeability, conductance, and gating of human α4β2 nicotinic acetylcholine receptors. J Neurosci. 1997;17:9035–9047. doi: 10.1523/JNEUROSCI.17-23-09035.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lapchak PA, Araujo DM, Quirion R, Collier B. Effect of chronic nicotine treatment on nicotinic autoreceptor function and N-[3H]methylcarbamylcholine binding sites in the rat brain. J Neurochem. 1989;52:483–491. doi: 10.1111/j.1471-4159.1989.tb09146.x. [DOI] [PubMed] [Google Scholar]
- 43.Leshner AI, Koob GF. Drugs of abuse and the brain. Proc Assoc Am Physicians. 1999;111:99–108. doi: 10.1046/j.1525-1381.1999.09218.x. [DOI] [PubMed] [Google Scholar]
- 44.Lester RA, Dani JA. Acetylcholine receptor desensitization induced by nicotine in rat medial habenula neurons. J Neurophysiol. 1995;74:195–206. doi: 10.1152/jn.1995.74.1.195. [DOI] [PubMed] [Google Scholar]
- 45.Liu Q-S, Berg D. Actin filaments and the opposing actions of CaM kinase II and calcineurin in regulating α7-containing nicotinic receptors on chick ciliary ganglion neurons. J Neurosci. 1999;19:10280–10288. doi: 10.1523/JNEUROSCI.19-23-10280.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marks MJ, Stitzel JA, Collins AC. Time course study of the effects of chronic nicotine infusion on drug response and brain receptors. J Pharmacol Exp Ther. 1985;235:619–628. [PubMed] [Google Scholar]
- 47.Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci. 1992;12:2765–2784. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marks MJ, Grady SR, Collins AC. Downregulation of nicotinic receptor function after chronic nicotine infusion. J Pharmacol Exp Ther. 1993;266:1268–1276. [PubMed] [Google Scholar]
- 49.Marks MJ, Whiteaker P, Calcaterra J, Stitzel JA, Bullock AE, Grady SR, Picciotto MR, Changeux JP, Collins AC. Two pharmacologically distinct components of nicotinic receptor-mediated rubidium efflux in mouse brain require the β2 subunit. J Pharmacol Exp Ther. 1999;289:1090–1103. [PubMed] [Google Scholar]
- 50.Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Lena C, Le Novere N, de Kerchove d'Exaerde A, Huchet M, Damaj MI, Changeux JP. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398:805–810. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]
- 51.Marutle A, Warpman U, Bogdanovic N, Lannfelt L, Nordberg A. Neuronal nicotinic receptor deficits in Alzheimer patients with the Swedish amyloid precursor protein 670/671 mutation. J Neurochem. 1999;72:1161–1169. doi: 10.1046/j.1471-4159.2000.0721161.x. [DOI] [PubMed] [Google Scholar]
- 52.Nelson ME, Kuryatov A, Lindstrom JM. Host cell effects on functional properties of human α4β2 nicotinic ACHRS. Soc Neurosci Abstr. 1999;25:1722. [Google Scholar]
- 53.Papke RL, Boulter J, Patrick J, Heinemann S. Single-channel currents of rat neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes. Neuron. 1989;3:589–596. doi: 10.1016/0896-6273(89)90269-9. [DOI] [PubMed] [Google Scholar]
- 54.Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J. Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Mol Pharmacol. 1994;46:523–530. [PubMed] [Google Scholar]
- 55.Pereira EF, Alkondon M, Reinhardt S, Maelicke A, Peng X, Lindstrom J, Whiting P, Albuquerque EX. Physostigmine and galanthamine: probes for a novel binding site on the alpha 4 beta 2 subtype of neuronal nicotinic acetylcholine receptors stably expressed in fibroblast cells. J Pharmacol Exp Ther. 1994;270:768–778. [PubMed] [Google Scholar]
- 56.Perry DC, Davila-Garcia MI, Stockmeier CA, Kellar KJ. Increased nicotinic receptors in brains from smokers: membrane binding and autoradiography studies. J Pharmacol Exp Ther. 1999;289:1545–1552. [PubMed] [Google Scholar]
- 57.Picciotto MR, Zoli M, Lena C, Bessis A, Lallemand Y, LeNovere N, Vincent P, Pich EM, Brulet P, Changeux JP. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–67. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- 58.Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, Fuxe K, Changeux JP. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- 59.Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–404. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- 60.Ragozzino D, Fucile S, Giovannelli A, Grassi F, Mileo AM, Ballivet M, Alema S, Eusebi F. Functional properties of neuronal nicotinic acetylcholine receptor channels expressed in transfected human cells. Eur J Neurosci. 1997;9:480–488. doi: 10.1111/j.1460-9568.1997.tb01625.x. [DOI] [PubMed] [Google Scholar]
- 61.Ramirez-Latorre J, Yu CR, Qu X, Perin F, Karlin A, Role L. Functional contributions of alpha5 subunit to neuronal acetylcholine receptor channels. Nature. 1996;380:347–351. doi: 10.1038/380347a0. [DOI] [PubMed] [Google Scholar]
- 62.Romanelli L, Ohman B, Adem A, Nordberg A. Subchronic treatment of rats with nicotine: interconversion of nicotinic receptor subtypes in brain. Eur J Pharmacol. 1988;148:289–291. doi: 10.1016/0014-2999(88)90577-8. [DOI] [PubMed] [Google Scholar]
- 63.Rowell PP, Wonnacott S. Evidence for functional activity of up-regulated nicotine binding sites in rat striatal synaptosomes. J Neurochem. 1990;55:2105–2110. doi: 10.1111/j.1471-4159.1990.tb05802.x. [DOI] [PubMed] [Google Scholar]
- 64.Sabey K, Paradiso K, Zhang J, Steinbach JH. Ligand binding and activation of rat nicotinic α4β2 receptors stably expressed in HEK293 cells. Mol Pharmacol. 1999;55:58–66. [PubMed] [Google Scholar]
- 65.Schoepfer R, Whiting P, Esch F, Blacher R, Shimasaki S, Lindstrom J. cDNA clones coding for the structural subunit of a chicken brain nicotinic acetylcholine receptor. Neuron. 1988;1:241–248. doi: 10.1016/0896-6273(88)90145-6. [DOI] [PubMed] [Google Scholar]
- 66.Shafaee N, Houng M, Truong A, Viseshakul N, Figl A, Sandhu S, Forsayeth JR, Dwoskin LP, Crooks PA, Cohen BN. Pharmacological similarities between native brain and heterologously expressed alpha4beta2 nicotinic receptors. Br J Pharmacol. 1999;128:1291–1299. doi: 10.1038/sj.bjp.0702900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharples CG, Kaiser S, Soliakov L, Marks MJ, Collins AC, Washburn M, Wright E, Spencer JA, Gallagher T, Whiteaker P, Wonnacott S. UB-165: a novel nicotinic agonist with subtype selectivity implicates the α4β2* subtype in the modulation of dopamine release from rat striatal synaptosomes. J Neurosci. 2000;20:2783–2791. doi: 10.1523/JNEUROSCI.20-08-02783.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Swanson LW, Simmons DM, Whiting PJ, Lindstrom J. Immunohistochemical localization of neuronal nicotinic receptors in the rodent central nervous system. J Neurosci. 1987;7:3334–3342. doi: 10.1523/JNEUROSCI.07-10-03334.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vallejo YF, Cheng H, Green WN. Nicotine-induced upregulation of α4β2 receptors is caused by converting low- to high-affinity binding sites. Soc Neurosci Abstr. 1999;25:1722. [Google Scholar]
- 70.Wevers A, Jeske A, Lobron C, Birtsch C, Heinemann S, Maelicke A, Schroder R, Schroder H. Cellular distribution of nicotinic acetylcholine receptor subunit mRNAs in the human cerebral cortex as revealed by non-isotopic in situ hybridization. Brain Res Mol Brain Res. 1994;25:122–128. doi: 10.1016/0169-328x(94)90286-0. [DOI] [PubMed] [Google Scholar]
- 71.Wevers A, Monteggia L, Nowacki S, Bloch W, Schutz U, Lindstrom J, Pereira EF, Eisenberg H, Giacobini E, de Vos RA, Steur EN, Maelicke A, Albuquerque EX, Schroder H. Expression of nicotinic acetylcholine receptor subunits in the cerebral cortex in Alzheimer's disease: histotopographical correlation with amyloid plaques and hyperphosphorylated-tau protein. Eur J Neurosci. 1999;11:2551–2565. doi: 10.1046/j.1460-9568.1999.00676.x. [DOI] [PubMed] [Google Scholar]
- 72.Whiteaker P, Sharples CG, Wonnacott S. Agonist-induced up-regulation of α4β2 nicotinic acetylcholine receptors in M10 cells: pharmacological and spatial definition. Mol Pharmacol. 1998;53:950–962. [PubMed] [Google Scholar]
- 73.Whiting P, Esch F, Shimasaki S, Lindstrom J. Neuronal nicotinic acetylcholine receptor beta-subunit is coded for by the cDNA clone alpha 4. FEBS Lett. 1987;219:459–463. doi: 10.1016/0014-5793(87)80272-7. [DOI] [PubMed] [Google Scholar]
- 74.Wonnacott S. The paradox of nicotinic acetylcholine receptor upregulation by nicotine. Trends Pharmacol Sci. 1990;11:216–219. doi: 10.1016/0165-6147(90)90242-z. [DOI] [PubMed] [Google Scholar]
- 75.Yu ZJ, Wecker L. Chronic nicotine administration differentially affects neurotransmitter release from rat striatal slices. J Neurochem. 1994;63:186–194. doi: 10.1046/j.1471-4159.1994.63010186.x. [DOI] [PubMed] [Google Scholar]
- 76.Zwart R, Vijverberg HP. Four pharmacologically distinct subtypes of α4β2 nicotinic acetylcholine receptor expressed in Xenopus laevis oocytes. Mol Pharmacol. 1998;54:1124–1131. [PubMed] [Google Scholar]