

Abstract
The principal role of estrogen is its control of the female ovulatory cycle via negative and positive feedback on gonadotropin secretion. However, a detailed, cohesive picture of how the steroid specifically regulates the excitability of hypothalamic neurons involved in the central control of gonadotropin secretion is still emerging. Here, we used an ovariectomized female guinea pig model to test the hypothesis that estrogen acts on GABAergic neurons in the preoptic area (POA) to elicit a biphasic profile of luteinizing hormone (LH) secretion. Intracellular electrophysiological recordings revealed that estradiol benzoate (EB; 25 μg, s.c.) decreased the hyperpolarizing response of GABAergic neurons to the GABABreceptor agonist baclofen 24 hr after treatment. This effect of GABAB receptor stimulation in unidentified POA neurons was still depressed 42 hr after EB administration. By the use of a ribonuclease protection assay, however, EB reduced glutamic acid decarboxylase mRNA expression 42 hr but not 24 hr after its administration. Thus, estrogen attenuated the autoinhibition of GABAergic POA neurons during the initial LH suppressive (i.e., negative feedback) phase and subsequently reduced GABAergic function during the LH surge (i.e., positive feedback). These studies demonstrate that the effects of estrogen on hypothalamic GABAergic neurons coincide with the inhibitory and stimulatory actions, respectively, of the steroid on LH secretion. Furthermore, the data provide novel insights into the mechanism by which estrogen regulates hypothalamic GABAergic neurons, which are critical for the biphasic modulation of LH release observed over the course of the female ovulatory cycle.
Keywords: luteinizing hormone, estrogen, GABA, preoptic area, electrophysiology, glutamic acid decarboxylase, in situhybridization
It is well recognized that estrogen controls the mammalian female reproductive cycle by both negative and positive feedback actions on gonadotropin [i.e., follicle-stimulating hormone and luteinizing hormone (LH)] secretion, the specific nature of which varies among mammalian species. In females that exhibit a luteal phase, estrogen suppresses gonadotropin release from the anterior pituitary throughout the vast majority of the reproductive cycle (Tsai and Yen, 1971; Yen and Tsai, 1971; Yamaji et al., 1972;Condon et al., 1988; Witkin et al., 1994) that, for guinea pigs and primates, lasts 14–18 d (considerably longer than that observed for other commonly used rodent species) and 1 month, respectively (Knobil, 1974; Terasawa and Wiegand, 1978; Witkin et al., 1991). During the mid-to-late follicular phase, however, plasma estrogen levels gradually rise, resulting in the preovulatory LH surge (Yen and Tsai, 1972;Knobil, 1974; Condon et al., 1988).
It is primarily accepted that for guinea pigs and primates, the dual estrogenic feedback on LH secretion occurs via alterations in the excitability of gonadotropin-releasing hormone (GnRH) neurosecretory cells located in the preoptic area (POA) and mediobasal hypothalamus (MBH) (Silverman et al., 1979; Goldsmith et al., 1990; Witkin et al., 1991; King et al., 1998). GnRH neurons do not express estrogen receptors (Watson et al., 1992; Herbison et al., 1995; Sullivan et al., 1995), which implies that afferent neural substrates are the primary targets of estrogen action. Two prominent, estrogen-sensitive inhibitory neurotransmitter systems that provide synaptic input to GnRH neurons are GABA and opioid peptides such as β-endorphin and dynorphin (Chronwall, 1985; Leranth et al., 1985; Morrell et al., 1985;Hammer et al., 1994; Herbison, 1997). The GABAergic neurons most likely are local intrinsic interneurons (Brown et al., 1994) and interact with opioid systems to inhibit LH release (Masotto et al., 1989; Brann et al., 1992; Donoso et al., 1992). On the other hand, there is considerable evidence that GABA directly inhibits GnRH and thereby LH secretion (Leranth et al., 1985; Lagrange et al., 1995). Despite extensive research, however, we still lack a clear picture of precisely how estrogen alters hypothalamic GABAergic neurotransmission to control GnRH and thereby LH secretion.
Because of the noted similarities in the reproductive cycle of guinea pigs and primates, the former makes an ideal animal model for the study of mammalian female reproductive physiology. Therefore, we used the ovariectomized female guinea pig to test the hypothesis that estrogen biphasically regulates LH secretion in part via temporally synchronized effects on hypothalamic GABAergic neurons. To this end, we examined estrogenic modulation of the GABABreceptor-mediated activation of an inhibitory K+ conductance and the expression of the biosynthetic enzyme for GABA, glutamic acid decarboxylase (GAD), in the POA. The effects of the GABAB receptor agonist baclofen (Bowery, 1989) and the antagonist CGP 52,432 (Lanza et al., 1993) were examined in intracellular recordings made from GABAergic POA neurons subsequently confirmed by combined histofluorescence andin situ hybridization for GAD65. GAD expression in POA neurons was determined with in situhybridization and ribonuclease protection assay. The results reveal that estrogen attenuates GABAergic autoinhibition and POA GAD expression in a temporal pattern that coincides with the steroid's negative and positive feedback on gonadotropin secretion, respectively. Moreover, these data indicate that POA GABAergic neurons are a key link in the estrogenic regulation of the ovulatory cycle.
MATERIALS AND METHODS
Animals and treatments. Female Topeka guinea pigs (470–660 gm) were obtained from our institutional breeding facility and maintained under constant temperature (72.4 ± 0.1°F) and light (on between 06:30 and 20:30 hr). Animals were housed individually, with food and water provided ad libitum. They were ovariectomized under ketamine and xylazine anesthesia (33 and 6 mg/kg, respectively, s.c.) 5–17 d before experimentation and given either estradiol benzoate (EB; 25 μg, s.c.) or its sesame oil vehicle (0.1 ml, s.c.) 24 or 42 hr before experimentation. Serum estrogen concentrations were determined by radioimmunoassay from trunk blood collected on the day of experimentation. This treatment regimen produced physiological levels of 17β-estradiol (vehicle, undetectable; 24 hr, 351.1 ± 31.3 pg/ml; 42 hr, 66.5 ± 11.2 pg/ml; n = 11–24) within the range of values observed over the course of the female menstrual cycle (Yen and Tsai, 1972;Knobil, 1974). All animal procedures described in this study are in accordance with institutional guidelines based on National Institutes of Health standards.
Drugs. All drugs were purchased from Sigma (St. Louis, MO) unless otherwise specified. EB was dissolved in sesame oil to a concentration of 250 μg/ml. Tetrodotoxin (TTX) was dissolved in Milli-Q H2O and further diluted with 0.1% acetic acid (final concentration, 1 mm; pH 4–5). (±)-Baclofen was dissolved in 0.1N HCl to a concentration of 40 mm. [3-[[(3,4-dichlorophenyl)methyl]amino]propyl](diethoxymethyl) phosphinic acid (CGP 52,432; provided by A. Sedlacek, CIBA-GEIGY AG, Basel, Switzerland) was dissolved in Milli-Q H2O to a concentration of 1 mm. Aliquots of the stock solutions were stored as appropriate until needed.
Serum LH measurement. Serial blood samples were collected from indwelling catheters implanted via the jugular vein into the right atrium as described previously (Condon et al., 1988). Catheter implantation was performed on animals anesthetized with the ketamine and xylazine mixture 2 weeks after ovariectomy. The sampling period ranged from 2 hr before to 48 hr after an intravenous injection of 17β-estradiol (E2). Serum LH levels were determined in duplicate with a heterologous radioimmunoassay using an anti-ovine LH antisera (GDN-15; kindly provided by Dr. Gordon Niswender) as reported previously (Condon et al., 1988).
Tissue preparation. For electrophysiological studies, on the day of experimentation the animal was decapitated, its brain was removed from the skull, and the hypothalamus was dissected. Four coronal slices (350–450 μm) through the POA were cut using a vibratome. The slices were transferred to a multiwell auxiliary chamber containing oxygenated (95% O2 and 5% CO2) artificial CSF (aCSF; see below) and kept there until electrophysiological recording.
For in situ hybridization, the brain was sliced into coronal blocks using a brain slicer (EM Corporation, Chestnut Hill, MA). The POA block (3 mm) and MBH blocks (2 mm) were fixed in 4% paraformaldehyde for 6 hr, soaked in 20% sucrose solution, frozen, and sectioned at 15 μm. For the ribonuclease protection assay the POA and MBH were dissected from their respective blocks using a dissecting microscope. The tissue was quickly frozen, and total RNA was extracted using Trizol Reagent (Life Technologies, Gaithersburg, MD).
Electrophysiology. Intracellular recordings in current clamp were performed as described previously (Kelly et al., 1992). Briefly, slices were maintained in a chamber perfused with warmed (35°C), oxygenated aCSF containing the following constituents, in mm: NaCl, 124; KCl, 5; NaH2PO4, 2.6; dextrose, 10; HEPES, 10; MgSO4, 2; and CaCl2, 2. In some experiments, slice preparation and subsequent slice incubation before being transferred to the recording chamber used aCSF containing 1 mmCaCl2. Artificial CSF and all drug solutions were perfused via a peristaltic pump at a rate of 1.5 ml/min. Drug solutions were prepared in 20 ml syringes by diluting the appropriate stock solution with aCSF, and the flow was controlled via a three-way stopcock. Microelectrodes (100–225 MΩ) were assembled from borosilicate glass pipettes and filled with a 3% biocytin solution in 1.75 m KCl and 0.025 m Tris, pH 7.4.
After successful impalement, slices were perfused with 2 μm TTX (6 min) to block spontaneous firing and supplemented with 1 μm TTX in all subsequent drug solutions. Agonist dose–response relationships were generated by applying a dose of baclofen until a new steady-state membrane potential (Vm) had been obtained (4–7 min). After drug discontinuation, the Vm eventually returned to its predrug resting level, and an incrementally higher dose of baclofen was administered, until finally a maximum steady-state hyperpolarization (ΔVmax) was reached. Estimates of the baclofen EC50 and ΔVmax were obtained from single neurons via the logistic equation:
fitted by computer (SigmaPlot; Jandel Scientific, San Rafael, CA) from the data points. The pharmacodynamics sometimes were reevaluated after the drug washout in the presence of CGP 52,432 (1 μm). Estimates of the Ki for CGP 52,432 were derived from the logistic equation:
fitted by computer from the data points.
Prebaclofen current–voltage (I/V) relationships were established by giving hyperpolarizing and depolarizing current pulses (0.2 Hz; 1 sec) and monitoring the voltage deflections. After the maximal response to baclofen (10–100 μm) reached steady state, the Vm was then returned to its original resting state by injecting positive current, and a secondI/V was established. Cell conductance was analyzed by linear regression as the slope of theI/V plots between −60 and −80 mV and between −100 and −130 mV. The baclofen-induced conductance change (Δg) was determined by subtracting the predrug from the postdrug I/V slopes.
Guinea pig GAD67 clone. A fragment of the guinea pig GAD67 gene was cloned using reverse transcription (RT)-PCR. Oligonucleotide primers 100% homologous to human GAD65 were designed (5′-primer, base pairs 671–692; 3′-primer, base pairs 1007–1028 of the human sequence). These primers were used to clone both GAD65 [as reported previously (Wagner et al., 1999)] and GAD67. Primer synthesis by Life Technologies included at the 5′-end of both primers a 12 base extension of deoxy-UMP residues used with the PCR cloning kit CloneAmp pAMP10 System (Life Technologies). The GAD67 fragment was amplified from 100 ng of total RNA extracted from the guinea pig POA using RT-PCR (GeneAmp kit; Perkin-Elmer, Foster City, CA). The human GAD 3′-primer was used for the cDNA first-strand synthesis. Reverse transcription was performed for 15 min at 42°C. PCR was conducted for 35 cycles of denaturation (92°C; 1 min), annealing (55°C; 2 min), and extension (72°C; 3 min), with a 3 min final extension. The 358 bp PCR product was subcloned into the pAMP10 vector using the CloneAmp (Life Technologies) system, and sequencing confirmed the product to be GAD67 cDNA.
In situ hybridization. In situ hybridization was performed using the GAD65 and GAD67 riboprobes. Slides were post-fixed in fresh 4% paraformaldehyde (40 min), rinsed with Sorensen's phosphate buffer, and treated with Proteinase-K (1.0 μg/ml; 2 min; 37°C). All sections were then treated (3 min) with 0.1 mtriethanolamine, followed by 0.25% acetic anhydride in 0.1m triethanolamine (10 min). Thereafter, the sections were rinsed in 2× SSC and hybridized (56–58°C; ≥18 hr) as described previously (Fang and Rønnekleiv, 1999). Sections were rinsed in 2× SSC (30 min) on a shaker, reacted with RNase (20 μg/ml; 30 min; 37°C), and sequentially rinsed in 1×, 0.5×, and 0.25× SSC (∼55°C). Slides were finally washed (30 min; 65°C) in 0.1× SSC containing 1.0 mm dithiothreitol. The sections were dehydrated in increasing concentrations of ethanol and together with autoradiographic 14C-microscales (Amersham, Arlington Heights, IL) were exposed to hyperfilm-βmax x-ray film (NEN, Boston, MA) for 5–6 d at 4°C. Slides were then dipped in Kodak NTB-2 emulsion and exposed for up to 16 d at 4°C. Sections were evaluated and photographed under dark-field illumination using a Zeiss microscope configured with a dark-light attachment (Foster, Inc.).
Ribonuclease protection assay. The antisense GAD65 and GAD67 riboprobes were labeled by in vitro transcription with [32P]rUTP and were purified using the Fullengther Preparative Gel Apparatus (Biokey American Instrument, Aloha, OR). The GAD probes were incubated with 3 μg of total RNA or 125–4000 fg of sense standard RNA overnight at 45°C. Hybridization was terminated by ribonuclease digestion; the protected fragments were loaded onto an acrylamide gel and exposed to film for visualization. Quantification was performed using a phosphorimager (Bio-Rad, Hercules, CA). Each GAD band was normalized with its corresponding cyclophilin band.
Cell phenotype identification. After electrophysiological recording, slices were fixed in 4% paraformaldehyde in 0.03m Sorensen's phosphate buffer (90–180 min; 4°C) and then soaked overnight in buffer containing 20% sucrose. All solutions were prepared with diethylpolycarbonate-treated Milli-Q H2O and molecular-grade reagents. Frozen slices were sectioned at 20 μm on a cryostat (Leitz Model 1720 Digital Cryostat), mounted on Superfrost-plus slides, and then washed (5 min) with 0.1 m phosphate buffer. Streptavidin-Texas Red (Jackson ImmunoResearch, West Grove, PA), diluted with seaweed gelatin solution (Rönnekleiv et al., 1991) in the presence of RNAsin (60 U/ml) and sodium heparin (1.25 mg/ml), was then applied (2 hr). The reaction was terminated by washing with 0.1 m phosphate buffer. Biocytin-filled GABAergic neurons were identified by combined histofluorescence and in situ hybridization as described previously (Wagner et al., 1999).
Statistical analyses. Comparison between two groups was performed using either the Student's two-tailed t test, the paired t test, or the Mann–Whitney U test. Comparisons between two or more groups were performed using a multifactorial ANOVA followed by the least significant difference (LSD) test. Differences were considered statistically significant if the probability of error was <5%.
RESULTS
Animal model
The dual feedback actions of estrogen on the reproductive axis of the female guinea pig are illustrated in Figure1. Systemic E2 administration to ovariectomized animals results in a rapid inhibition of pulsatile LH secretion (negative feedback). This suppression lasts nearly 40 hr and is immediately followed by a surge of LH release over and above that observed before E2 administration (positive feedback).
Fig. 1.

Estrogen produces both negative and positive feedback on gonadotropin secretion. Representative composite hormone profile based on serial blood samples taken from ovariectomized female guinea pigs. Filled circles represent plasma LH concentrations determined by radioimmunoassay at various time points before and after E2 (25 μg) administration.
Estrogen decreases the GABAB response in POA neurons during negative feedback
The potential for GABAB receptor involvement in this initial, negative feedback phase was evaluated via intracellular electrophysiological recording from POA neurons. As shown in Figure 2a, the GABAB receptor agonist baclofen elicited a dose-dependent membrane hyperpolarization of POA neurons. Evaluation of the I/V plot in Figure 2b reveals that the baclofen response reversed polarity very near the Nernst equilibrium potential for K+. The baclofen-induced hyperpolarization was antagonized by CGP 52,432 (1 μm) that was overcome by increasing concentrations of the agonist (Fig. 2c). This antagonism by CGP 52,432 produced a rightward shift in the agonist dose–response curve with an estimated Ki of 64 ± 7 nm (Fig. 2d).
Fig. 2.

Stimulation of GABAB receptors inhibits POA neurons by activating a K+ conductance.a, Successively increasing doses of baclofen (1, 3, 10, and 30 μm) hyperpolarized this POA neuron (resting Vm = −55 mV) at 8.5, 11, 16, and 18 mV, respectively. b, An I/Vplot derived from a POA neuron just before (control; open circles) and near the end of the application of a maximal concentration of baclofen (100 μm; filled circles) is shown. The reversal potential for the baclofen response was −94 mV, and a Δg of 0.67 nS between −60 and −80 mV and a Δg of 1.63 nS between −100 and −130 mV were also observed. c, Dose–response relationship from the cell shown in a is then generated in the presence of CGP 52,432 (1 μm). Successively increasing doses of baclofen (10, 30, 100, and 300 μm) elicited hyperpolarizations of 2.5, 4.5, 11, and 12.5 mV, respectively. d, Composite baclofen dose–response curves in the absence (open circles) and presence (filled circles) of CGP 52,432 (1 μm) are shown. Cells were perfused with successively higher concentrations of baclofen (1, 3, 10, 30, 100, and 300 μm; 4–7 min/dose; n = 2–10).Symbols represent means, and vertical lines are 2 SEMs of the baclofen-induced hyperpolarization normalized to the ΔVmax. Before CGP 52,432, the mean baclofen EC50 value was 2.3 ± 0.5 μm, whereas in the presence of CGP 52,432, the EC50 was shifted to 33.0 ± 10.0 μm. The estimatedKi for CGP 52,432 was 64.0 nm. BAC, Baclofen.
Compared with the dose–response relationships generated in POA neurons from vehicle-treated animals (Fig.3a), those obtained from animals treated with EB 24 hr before were markedly attenuated (Fig.3b). Although EB did not affect baclofen potency, it decreased the hyperpolarization magnitude at all doses tested (Fig.3c). This finding was corroborated by a parallel diminution in the baclofen-induced Δg measured between −60 and −80 mV and between −100 and −130 mV (Fig. 3d). These effects of EB on the GABAB receptor-mediated response still persisted 42 hr after administration (ΔVmax = 8.2 ± 1.3 mV; n= 4; p < 0.05).
Fig. 3.

Estrogen attenuates the efficacy of GABAB receptor-mediated neurotransmission in the POA 24 hr after its administration. a, Successively increasing doses of baclofen (1, 3, and 10 μm) hyperpolarized this POA neuron from a vehicle-treated animal (resting Vm = −45 mV) 10, 22.5, and 25.5 mV, respectively. The upward deflection represents the return of low-threshold spikes and/or action potentials (truncated) seen during the later stages of drug washout. b, Successively increasing doses of baclofen (3, 10, 30, and 100 μm) hyperpolarized this POA neuron (resting Vm = −50 mV) from an EB-treated (25 μg; 24 hr) animal by 1.5, 2.5, 3, and 4 mV, respectively. c, Composite dose–response curves from recordings of POA neurons obtained from vehicle- and EB-treated animals are shown. Cells were perfused with successively higher concentrations of baclofen (1, 3, 10, and 30 μm; 4–7 min/dose; n = 2–10). Symbolsrepresent means, and vertical lines are 1 SEM of the hyperpolarizations elicited by a given concentration of baclofen. The ΔVmax obtained via logistic fit for POA neurons from vehicle-treated animals was 13.5 mV, whereas that obtained for POA neurons from EB-treated animals was 7.5 mV. *, Hyperpolarizations obtained with 10 and 30 μm baclofen that are significantly different (multifactorial ANOVA and LSD;p < 0.05) from those obtained with 1 or 3 μm baclofen are shown. #, Hyperpolarizations of POA neurons obtained from EB-treated animals are significantly lower (multifactorial ANOVA and LSD; p < 0.05) than those obtained from vehicle-treated animals at all doses tested.d, Composite bar graph illustrates the baclofen-induced Δg in POA neurons from vehicle- and EB-treated animals (n = 5–8). Columns represent means, and vertical lines are 1 SEM of the baclofen-induced Δg estimated by linear regression between −60 and −80 mV and between −100 and −130 mV. *, Values of Δg obtained in POA neurons from EB-treated animals that are significantly different (multifactorial ANOVA and LSD;p < 0.05) from those obtained from vehicle-treated controls are shown.
GABAergic neuronal distribution in the guinea pig hypothalamus
GABAergic neurons express estrogen receptors, and GABA is arguably the predominant neurotransmitter in the hypothalamus (Decavel and van den Pol, 1990; Herbison, 1997). GAD is the rate-limiting enzyme for the production of GABA and is expressed in two forms, GAD65 and GAD67, derived from two genes (Martin and Rimvall, 1993). To study the distribution of GABAergic neurons in the guinea pig hypothalamus and to determine whether GABA synthesis is regulated by estrogen, we prepared specific PCR clones for GAD65 (Wagner et al., 1999) and GAD67. The guinea pig GAD65mRNA sequence was 90 and 89% identical to corresponding sequences of human and rat GAD65 and 75 and 74% identical to human and rat GAD67, respectively. The guinea pig GAD67 mRNA sequence was 94 and 90% identical to corresponding sequences of human and rat GAD67, respectively, and 72% identical to human GAD65. As shown in Figure 4, the GAD65 riboprobe robustly labeled neurons throughout the rostrocaudal extent of the POA and MBH. In the POA, the hybridization signal was particularly abundant in the anteroventral periventricular nucleus of the hypothalamus (AVPV) and the medial preoptic nucleus (MPN). In the MBH, high levels of GAD65 expression were observed in the dorsomedial hypothalamic nucleus (DMH), the arcuate nucleus (Arc), and the ventral premammillary nucleus (PMv). By the use of in situhybridization analysis, GAD67 exhibited a similar mRNA distribution pattern (data not shown).
Fig. 4.
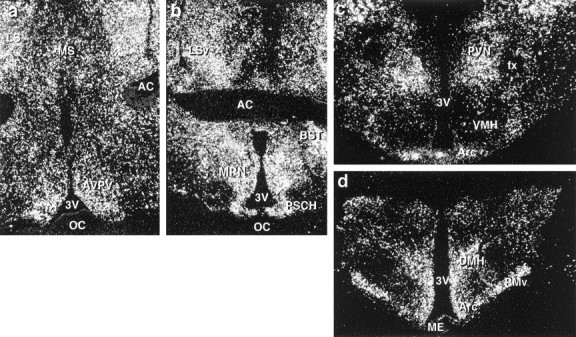
Distribution of GAD65 in the guinea pig hypothalamus. a, b, Dark-field photomicrographs that illustrate the distribution of GAD65 in the rostral (a) and caudal (b) POA.c, d, Dark-field photomicrographs of coronal sections through the MBH from rostral to caudal illustrating the distribution of GAD65 mRNA. AC, Anterior commissure;BST, bed nucleus of the stria terminalis;fx, fornix; LS, lateral septum;LSv, lateral septum (ventral part); ME, median eminence; MS, medial septum; OC, optic chiasm; PSCH, suprachiasmatic preoptic nucleus;PVN, paraventricular nucleus; VMH, ventromedial nucleus of the hypothalamus; 3V, third ventricle.
Expression of GAD65 in electrophysiologically and immunocytochemically identified POA neurons
The aforementioned observations render the GABAergic neuronal phenotype a likely target for the modulatory effect of EB on the GABAB receptor-mediated hyperpolarization. We therefore used GAD65 as a marker for post hoc identification after electrophysiological recording using combined histofluorescence and in situ hybridization. Examples of GAD-positive POA neurons from animals treated with either vehicle (Fig. 5a,b) or EB (Fig. 5c,d) 24 hr before are shown in Figure 5. The ΔVmax for baclofen observed in the GABAergic cell from the EB-treated animal (5.8 mV) was only 64% of that observed in the GABAergic cell from the vehicle-treated animal (9.0 mV). The majority (62%) of POA neurons from EB-treated animals were GAD positive, and the reduction in the ΔVmax in these cells (6.9 ± 0.9 mV) was comparable with that observed in the population as a whole (see Fig. 3c).
Fig. 5.
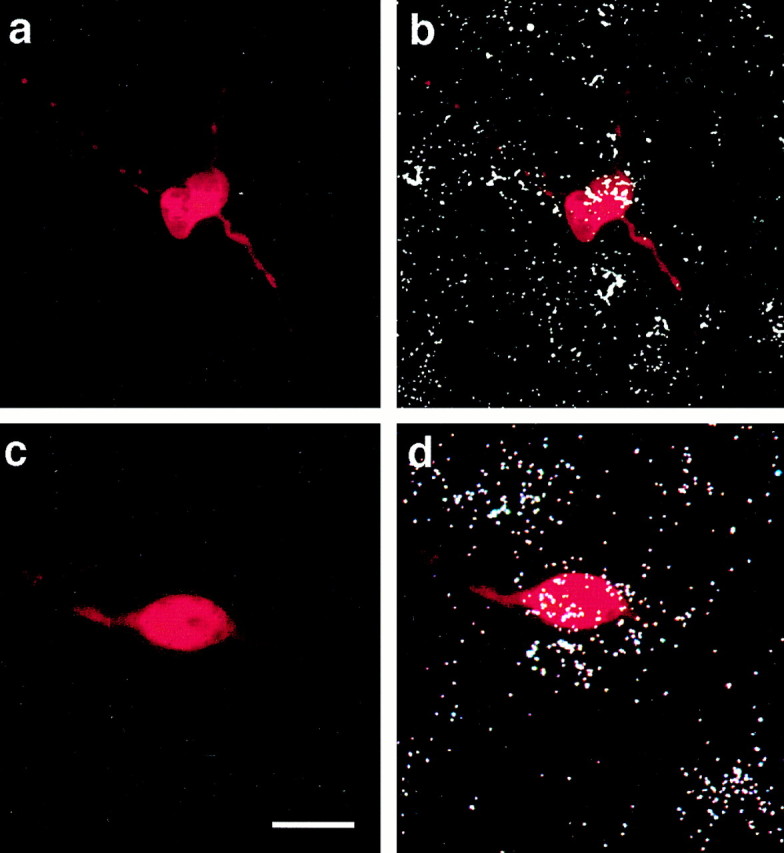
GABAergic POA neurons are identified using combined histofluorescence and in situ hybridization for GAD65 after electrophysiological recording.a, Photomicrograph of the biocytin-streptavidin-Texas Red fluorescent labeling of a recorded cell from a vehicle-treated animal. b, An overlay of the fluorescent labeling ina and the hybridization signal that clearly illustrates the double labeling for GAD65. c, Photomicrograph of the biocytin-streptavidin-Texas Red fluorescent labeling of a recorded POA neuron from an EB-treated (25 μg; 24 hr) animal. d, An overlay of the fluorescent labeling inc and the hybridization signal illustrating the double labeling for GAD65. Scale bar, ∼15 μm (for all photomicrographs).
Estrogen decreases GAD65 and GAD67 mRNA levels at the time of positive feedback
We used a sensitive ribonuclease protection assay (RPA) to quantify the levels of GAD65 and GAD67 mRNA in the guinea pig hypothalamus. Figure6a illustrates the levels of GAD65 and GAD67 mRNA found in hypothalamic RNA extracts from vehicle-treated animals. Both GAD65 and GAD67 mRNAs were found in highest quantities in the POA. However, GAD67 mRNA was significantly more abundant throughout the hypothalamus in comparison with GAD65 (Fig. 6a).
Fig. 6.
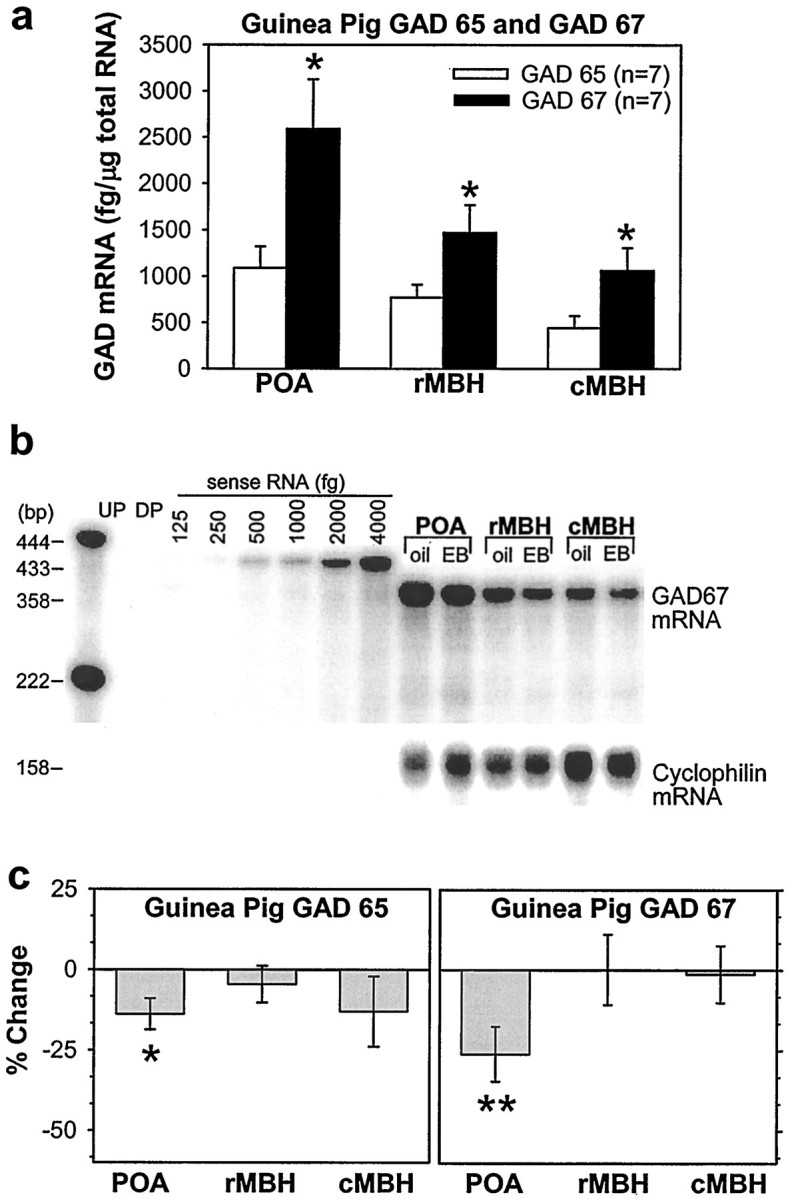
Estrogen produces a subsequent decrease in the POA expression of GAD67 and GAD65 42 hr after its administration. a, Distribution and quantitative analysis using a sensitive ribonuclease protection assay of GAD65 and GAD67 mRNA in the POA and MBH obtained from female guinea pigs. *, Denotes the levels of GAD67 that are significantly greater (Student'st test; p < 0.05) than those obtained for GAD65. b, A representative ribonuclease protection assay of total RNA (3 μg/lane) from vehicle- and EB-treated (25 μg; 42 hr) animals illustrating the levels of GAD67 mRNA detected in the POA and the rostral (r) and caudal (c) MBH. Sense RNA (125–4000 fg) was used to construct a standard curve.c, Quantitative analysis of GAD65(left) and GAD67 (right) mRNA in hypothalamic brain tissue obtained from vehicle- and EB-treated animals. Bands were normalized to their corresponding cyclophilin band and quantified from their respective sense mRNA standard curves.Columns represent means, and vertical lines are 2 SEMs of the EB-induced percent change in the GAD65 and GAD67 levels with respect to those observed in vehicle-treated animals. *, Denotes a significant change (paired t test; p < 0.05) in the level of GAD65 caused by EB relative to that observed in the POA of vehicle-treated controls. **, Denotes a significant change (paired t test; p < 0.01) in the level of GAD67 caused by EB relative to that observed in the POA of vehicle-treated controls. DP, Digested probe;UP, undigested probe.
Finally, we evaluated the effect of EB on the expression of both GAD65 and GAD67 in the hypothalamus using the sensitive RPA for mRNA quantification. In the POA, EB was without effect on the expression of either isoform at 24 hr (data not shown) but decreased the expression of both GAD65 and GAD67 mRNA 42 hr after its administration (Fig. 6b,c). By contrast, both GAD65 and GAD67 mRNA expression in the MBH were unaltered over the same time period. Taken together, these data demonstrate that EB attenuates the efficacy with which GABAergic ligands activate GABAB receptors to inhibit GABAergic POA neurons and increases the inhibitory tone provided by these neurons during negative feedback. Subsequently the expression of the biosynthetic enzyme for GABA decreases, which would decrease inhibitory tone in this region during positive feedback.
DISCUSSION
The results of the present study reveal that estrogen decreases the GABAB receptor-mediated autoinhibition of GABAergic POA neurons. In addition, estrogen induces an apparent decrease in the subsequent ability of these neurons to synthesize GABA. These conclusions are based on the observations that (1) estrogen reduces the hyperpolarization and the underlying K+ conductance observed in POA neurons, the majority of which are GAD positive, in response to a given dose of GABAB receptor agonist and (2) the steroid decreases mRNA levels for both GAD65 and GAD67 in the POA as determined by ribonuclease protection assay.
Somatodendritic GABAB autoreceptors couple to a K+ conductance in GABAergic POA neurons
The coupling of postsynaptic GABAB receptors to a K+ conductance in POA neurons is consistent with what we have shown previously in hypothalamic Arc neurons (Kelly et al., 1992; Wagner et al., 1999) and with that observed in many other brain regions (for review, see Misgeld et al., 1996). Moreover, the resultant hyperpolarization observed in the present study is antagonized by CGP 52,432 with an estimatedKi (64 nm) comparable with that of similar reports from other areas (Lanza et al., 1993; Bon and Galvan, 1996). These receptors have long been thought to serve as autoreceptors, inhibiting GABA release via presynaptic inhibition of Ca2+ channels (Bowery, 1989;Misgeld et al., 1996). However, activation of postsynaptic GABAB receptors in the presence of TTX, which inhibits synaptic transmission, results in a robust hyperpolarization of GABAergic Arc neurons (Wagner et al., 1999). Qualitatively, we observed an identical response under the same conditions from GABAergic POA neurons in the present study. Thus, the excitability of hypothalamic GABAergic neurons is regulated by the activation of both somatodendritic GABAB receptors and those on presynaptic nerve terminal membranes. The source of endogenous neurotransmitter activating the somatodendritic GABAB receptor may arise from a presynaptic bouton making contact with the GABAergic POA neuron in a GABA–GABA synapse (Decavel and van den Pol, 1992; Commons et al., 1999). Alternatively, it could come from the GABAergic POA neurons themselves, contained within recurrent collaterals making synaptic contact with their own perikarya as has been shown in the Arc (Yagi and Sawaki, 1975; Sawaki and Yagi, 1976; van den Pol and Cassidy, 1982).
Estrogen uncouples postsynaptic GABAB receptors from their effector K+ channels in POA neurons, manifest by a reduction in agonist efficacy
In view of its fundamental role in controlling the reproductive axis, it should not be surprising that estrogen exerts a multitude of effects on hypothalamic GABAergic neurons. These neurons express estrogen receptors and concentrate radiolabeled estradiol (Herbison, 1997). In addition, the steroid elicits ultrastructural changes in GABAergic neurons and alters synaptic morphology between GABAergic nerve terminals and their respective postsynaptic contacts, including GnRH neurons (Leranth et al., 1991; Párducz et al., 1993;Naftolin et al., 1996). Furthermore, in Arc neurons estrogen uncouples postsynaptic GABAB and μ-opioid receptors from their effector K+ channels, resulting in a reduction in agonist potency (Kelly et al., 1992). This receptor/effector uncoupling is caused by the estrogen receptor-mediated activation of an intracellular protein kinase A (PKA) pathway (Lagrange et al., 1997). Because the response to both GABAB and μ-opioid receptor agonists is negatively modulated by estrogen to the same extent, we believe that PKA phosphorylates critical protein(s) common to both of these signaling pathways (for review, see Kelly and Wagner, 1999). In the present study postsynaptic GABAB receptors on POA GABAergic neurons were uncoupled from their effector system not by a reduction in potency but by an attenuation in the efficacy of GABAB receptor-mediated neurotransmission. This would indicate a downregulation of GABABreceptors. Future studies will determine whether activation of kinase pathways is involved in mediating estrogen's negative modulation of the GABAB receptor-mediated response in these neurons.
GABAB receptor/effector uncoupling caused by estrogen in GABAergic POA neurons reduces autoinhibition, which increases the inhibitory tone onto GnRH neurons during negative feedback
The steroid-induced, dampened responsiveness of GABAergic POA neurons to GABAB receptor activation likely serves to reduce the extent of autoinhibition, thereby increasing neurotransmitter release from these neurons (Fig.7). Indeed, synaptic levels of GABA in the POA of ovariectomized rats, measured by microdialysis or push–pull perfusion, are elevated as early as 1 hr after systemic injection of E2 (Herbison et al., 1991; Jarry et al., 1995). Moreover, this is analogous to the steroid regulation of hypothalamic GABAergic neuronal activity observed in the male rat, in which antiandrogen treatment and castration both decrease POA GABA turnover, the latter of which is prevented by testosterone replacement (Grattan and Selmanoff, 1994; Grattan et al., 1996a). This androgen effect may be caused by the aromatization of testosterone to estrogen in the POA (Roselli et al., 1987).
Fig. 7.

Schematic representation illustrating the biphasic central feedback actions of estrogen on the mammalian female reproductive axis. a, During negative feedback, estrogen uncouples postsynaptic GABAB receptors from their effector K+ channels in GABAergic POA neurons. This leads to a decreased autoinhibition and an increased inhibitory tone onto GnRH neurons, thereby reducing GnRH release and ultimately LH secretion.b, During positive feedback, estrogen decreases POA GAD expression, thereby diminishing intraneuronal transmitter levels and the release of GABA. This reduces inhibitory GABAergic tone onto GnRH neurons, which facilitates excitatory (e.g., glutamatergic, noradrenergic) inputs in generating the preovulatory LH surge.
The results of the present study, in conjunction with available anatomical (Leranth et al., 1985; Naftolin et al., 1996) and functional (Masotto et al., 1989; Herbison et al., 1991; Brann et al., 1992;Donoso et al., 1992; Jarry et al., 1995) evidence, suggest that GABA plays a critical role in the steroid-induced negative feedback of gonadotropin secretion. The increase in the synaptic concentrations of GABA in the POA produced by estrogen occurs concomitantly with a decrease in plasma levels of LH (Herbison et al., 1991; Jarry et al., 1995). The estrogen-induced suppression of LH secretion observed in the present study persists well beyond 24 hr after administration (Fig. 1), which suggests that the reduced autoinhibition of POA GABAergic neurons and hence the increased GABA release are sustained over a considerable period of time. In support of this idea, we have observed that POA neurons from ovariectomized animals treated with EB 42 hr before also exhibit an attenuated GABABresponse magnitude.
Estrogen subsequently decreases GAD expression in the POA, and the resultant decrease in GABA levels helps promote the preovulatory LH during positive feedback
Estrogen also produced a latent decrease in the expression of both GAD65 and GAD67 in the POA 42 hr after its administration. This is consistent with the reduction in mRNA levels for both isoforms reported during the proestrus phase of the estrous cycle in the diagonal band of Broca of female rats, which was associated with a decrease in GABA turnover (Grattan et al., 1996b). In addition, GABA release in the POA of ovariectomized rats decreases beginning several hours before the onset, and continues through the ascending phase, of the estrogen-induced LH surge (Jarry et al., 1992).
Although GABAergic neurons express estrogen receptors, there are a number of reports showing that estrogen affects neither the enzymatic activity of GAD nor its transcription in rat POA (for review, seeHerbison, 1997). However, our results suggest that the steroid decreases the transcription of both isoforms in the female guinea pig. On the other hand, an increase in GABAergic neuronal activity such as that which might occur via depolarization or, in the case of the present study, a relief from autoinhibition increases the synthesis of GABA that is known to competitively inhibit GAD (Martin and Rimvall, 1993). Thus, a prolonged increase in neuronal activity attributed to an estrogen-induced decrement in GABABreceptor-mediated autoinhibition of GABAergic POA neurons may account, in part, for the decrease in GAD expression via end-product inhibition. Such a reduction in the capacity of GABAergic POA neurons to produce the inhibitory neurotransmitter would likely facilitate the generation of the estrogen-induced LH surge (Fig. 7).
GABAergic neurons are a primary target of estrogen's actions in the CNS
The present results serve to extend our appreciation of the vital importance of estrogen in regulating GABAergic neuronal function in the brain. For example, in the basal ganglia, estrogen rapidly inhibits Ca2+ currents in medium spiny GABAergic neurons (Mermelstein et al., 1996). Estrogen also exerts profound effects on sensorimotor activity and mating behavior that may be related to the uncoupling of the autoreceptor in these neurons (Becker et al., 1987; Xiao and Becker, 1997). Moreover, in hippocampal neurons estrogen reduces GABAergic synaptic currents, a reduction that is associated with increased dendritic spine formation on postsynaptic pyramidal cell membranes (Murphy et al., 1998). Hence, via its actions on GABAergic neurons not only can estrogen profoundly influence reproduction and the basal ganglia's regulation of fine motor control, but estrogen may also impact learning and memory (Fink et al., 1996;Murphy et al., 1998) as well as exert neuroprotective effects (Yang et al., 2000).
In conclusion, we have shown presently that estrogen negatively modulates the GABAB receptor-mediated autoinhibition of GABAergic POA neurons in the female guinea pig. This finding provides new insight into the mechanism(s) of steroid-induced negative feedback of the mammalian reproductive cycle. Moreover, the subsequent decrease in the capability of these neurons to synthesize GABA likely serves to promote steroid-induced positive feedback and the associated LH surge (Fig. 7). These effects on GABAergic neurons may help to explain many actions of this pleiotropic hormone in the CNS.
Footnotes
The experiments described in this study were supported by Public Health Service Grants NS35944, NS38809, and DA00192 (Research Scientist Development Award to M.J.K.). We thank Jason T. Deignan and Barry Naylor for their technical assistance and Dr. David Grandy for his critical evaluation of this manuscript.
Correspondence should be addressed to Dr. Edward J. Wagner, Department of Physiology and Pharmacology, L334, Oregon Health Sciences University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97201. E-mail: wagnere@ohsu.edu.
REFERENCES
- 1.Becker JB, Snyder PJ, Miller MM, Westgate SA, Jenuwine MJ. The influence of the estrous cycle and intrastriatal estradiol on sensorimotor performance in the female rat. Pharmacol Biochem Behav. 1987;27:53–59. doi: 10.1016/0091-3057(87)90476-x. [DOI] [PubMed] [Google Scholar]
- 2.Bon C, Galvan M. Electrophysiological actions of GABAB agonists and antagonists in rat dorso-lateral septal neurones in vitro. Br J Pharmacol. 1996;118:961–967. doi: 10.1111/j.1476-5381.1996.tb15493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bowery N. GABA(B) receptors and their significance in mammalian pharmacology. Trends Pharmacol Sci. 1989;10:401–407. doi: 10.1016/0165-6147(89)90188-0. [DOI] [PubMed] [Google Scholar]
- 4.Brann DW, Zamorano PL, Putnam-Roberts CD, Mahesh VB. Gamma-aminobutyric acid-opioid interactions in the regulation of gonadotropin secretion in the immature female rat. Neuroendocrinology. 1992;56:445–452. doi: 10.1159/000126281. [DOI] [PubMed] [Google Scholar]
- 5.Brown D, Herbison AE, Robinson JE, Marrs RW, Leng G. Modelling the luteinizing hormone-releasing hormone pulse generator. Neuroscience. 1994;63:869–879. doi: 10.1016/0306-4522(94)90531-2. [DOI] [PubMed] [Google Scholar]
- 6.Chronwall BM. Anatomy and physiology of the neuroendocrine arcuate nucleus. Peptides. 1985;6:1–11. doi: 10.1016/0196-9781(85)90128-7. [DOI] [PubMed] [Google Scholar]
- 7.Commons KG, Kow L-M, Milner TA, Pfaff DW. In the ventromedial nucleus of the rat hypothalamus, GABA-immunolabeled neurons are abundant and are innervated by both enkephalin- and GABA-immunolabeled axon terminals. Brain Res. 1999;816:58–67. doi: 10.1016/s0006-8993(98)01084-1. [DOI] [PubMed] [Google Scholar]
- 8.Condon TP, Dykshoorn-Bosch MA, Kelly MJ. Episodic luteinizing-hormone release in the ovariectomized female guinea pig: rapid inhibition by estrogen. Biol Reprod. 1988;38:121–126. doi: 10.1095/biolreprod38.1.121. [DOI] [PubMed] [Google Scholar]
- 9.Decavel C, van den Pol AN. GABA: a dominant neurotransmitter in the hypothalamus. J Comp Neurol. 1990;302:1019–1037. doi: 10.1002/cne.903020423. [DOI] [PubMed] [Google Scholar]
- 10.Decavel C, van den Pol AN. Converging GABA- and glutamate-immunoreactive axons make synaptic contact with identified hypothalamic neurosecretory neurons. J Comp Neurol. 1992;316:104–116. doi: 10.1002/cne.903160109. [DOI] [PubMed] [Google Scholar]
- 11.Donoso AO, López FJ, Negro-Vilar A. Cross-talk between excitatory and inhibitory amino acids in the regulation of luteinizing hormone-releasing hormone secretion. Endocrinology. 1992;131:1559–1561. doi: 10.1210/endo.131.3.1354606. [DOI] [PubMed] [Google Scholar]
- 12.Fang Y, Rønnekleiv OK. Cocaine upregulates the dopamine transporter in fetal rhesus monkey brain. J Neurosci. 1999;19:8966–8978. doi: 10.1523/JNEUROSCI.19-20-08966.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fink G, Sumner BEH, Rosie R, Grace O, Quinn JP. Estrogen control of central neurotransmission: effect on mood, mental state, and memory. Cell Mol Neurobiol. 1996;16:325–344. doi: 10.1007/BF02088099. [DOI] [PubMed] [Google Scholar]
- 14.Goldsmith PC, Thind KK, Song T, Kim EJ, Boggan JE. Location of the neuroendocrine gonadotropin-releasing hormone neurons in the monkey hypothalamus by retrograde tracing and immunostaining. J Neuroendocrinol. 1990;2:157–168. doi: 10.1111/j.1365-2826.1990.tb00846.x. [DOI] [PubMed] [Google Scholar]
- 15.Grattan DR, Selmanoff M. Castration-induced decrease in the activity of medial preoptic and tuberoinfundibular GABAergic neurons is prevented by testosterone. Neuroendocrinology. 1994;60:141–149. doi: 10.1159/000126744. [DOI] [PubMed] [Google Scholar]
- 16.Grattan DR, Rocca MS, Sagrillo CA, McCarthy MM, Selmanoff M. Antiandrogen microimplants into the rostral medial preoptic area decrease γ-aminobutyric acidergic neuronal activity and increase luteinizing hormone secretion in the intact male rat. Endocrinology. 1996a;137:4167–4173. doi: 10.1210/endo.137.10.8828473. [DOI] [PubMed] [Google Scholar]
- 17.Grattan DR, Rocca MS, Strauss KI, Sagrillo CA, Selmanoff M, McCarthy MM. GABAergic neuronal activity and mRNA levels for both forms of glutamic acid decarboxylase (GAD65 and GAD67) are reduced in the diagonal band of Broca during the afternoon of proestrus. Brain Res. 1996b;733:46–55. doi: 10.1016/0006-8993(96)00532-x. [DOI] [PubMed] [Google Scholar]
- 18.Hammer RP, Zhou L, Cheung S. Gonadal steroid hormones and hypothalamic opioid circuitry. Horm Behav. 1994;28:431–437. doi: 10.1006/hbeh.1994.1040. [DOI] [PubMed] [Google Scholar]
- 19.Herbison AE. Estrogen regulation of GABA transmission in rat preoptic area. Brain Res Bull. 1997;44:321–326. doi: 10.1016/s0361-9230(97)00210-4. [DOI] [PubMed] [Google Scholar]
- 20.Herbison AE, Heavens RP, Dye S, Dyer RG. Acute action of oestrogen on medial preoptic gamma-aminobutyric acid neurons: correlation with oestrogen negative feedback on luteinizing hormone secretion. J Neuroendocrinol. 1991;3:101–106. doi: 10.1111/j.1365-2826.1991.tb00246.x. [DOI] [PubMed] [Google Scholar]
- 21.Herbison AE, Horvath TL, Naftolin F, Leranth C. Distribution of estrogen receptor-immunoreactive cells in monkey hypothalamus: relationship to neurones containing luteinizing hormone-releasing hormone and tyrosine hydroxylase. Neuroendocrinology. 1995;61:1–10. doi: 10.1159/000126810. [DOI] [PubMed] [Google Scholar]
- 22.Jarry H, Hirsch B, Leonhardt S, Wuttke W. Amino acid neurotransmitter release in the preoptic area of rats during the positive feedback actions of estradiol on LH release. Neuroendocrinology. 1992;56:133–140. doi: 10.1159/000126220. [DOI] [PubMed] [Google Scholar]
- 23.Jarry H, Leonhardt S, Wuttke W. The inhibitory effect of β-endorphin on LH release in ovariectomized rats does not involve the preoptic GABAergic system. Exp Clin Endocrinol Diabetes. 1995;103:317–323. doi: 10.1055/s-0029-1211372. [DOI] [PubMed] [Google Scholar]
- 24.Kelly MJ, Wagner EJ. Estrogen modulation of G-protein-coupled receptors. Trends Endocrinol Metab. 1999;10:369–374. doi: 10.1016/s1043-2760(99)00190-3. [DOI] [PubMed] [Google Scholar]
- 25.Kelly MJ, Loose MD, Ronnekleiv OK. Estrogen suppresses μ-opioid- and GABAB-mediated hyperpolarization of hypothalamic arcuate neurons. J Neurosci. 1992;12:2745–2750. doi: 10.1523/JNEUROSCI.12-07-02745.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.King JC, Liu E, Ronsheim PM, Slonimski M, Rubin BS. Expression of Fos within luteinizing hormone-releasing hormone neurons, in relation to the steroid-induced luteinizing hormone surge in guinea pigs. Biol Reprod. 1998;58:316–322. doi: 10.1095/biolreprod58.2.316. [DOI] [PubMed] [Google Scholar]
- 27.Knobil E. On the control of gonadotropin secretion in the rhesus monkey. Recent Prog Horm Res. 1974;30:1–36. doi: 10.1016/b978-0-12-571130-2.50005-5. [DOI] [PubMed] [Google Scholar]
- 28.Lagrange AH, Rønnekleiv OK, Kelly MJ. Estradiol-17β and μ-opioid peptides rapidly hyperpolarize GnRH neurons: a cellular mechanism of negative feedback. Endocrinology. 1995;136:2341–2344. doi: 10.1210/endo.136.5.7720682. [DOI] [PubMed] [Google Scholar]
- 29.Lagrange AH, Ronnekleiv OK, Kelly MJ. Modulation of G protein-coupled receptors by an estrogen receptor that activates protein kinase A. Mol Pharmacol. 1997;51:605–612. doi: 10.1124/mol.51.4.605. [DOI] [PubMed] [Google Scholar]
- 30.Lanza M, Fassio A, Gemignani A, Bonanno G, Raiteri M. CGP 52432: a novel potent and selective GABA(B) autoreceptor antagonist in rat cerebral cortex. Eur J Pharmacol. 1993;237:191–195. doi: 10.1016/0014-2999(93)90268-m. [DOI] [PubMed] [Google Scholar]
- 31.Leranth C, MacLusky NJ, Sakamoto H, Shanabrough M, Naftolin F. Glutamic acid decarboxylase-containing axons synapse on LHRH neurons in the rat medial preoptic area. Neuroendocrinology. 1985;40:536–539. doi: 10.1159/000124127. [DOI] [PubMed] [Google Scholar]
- 32.Leranth C, Shanabrough M, Naftolin F. Estrogen induces ultrastructural changes in progesterone receptor-containing GABA neurons of the primate hypothalamus. Neuroendocrinology. 1991;54:571–579. doi: 10.1159/000125962. [DOI] [PubMed] [Google Scholar]
- 33.Martin DL, Rimvall K. Regulation of gamma-aminobutyric acid synthesis in the brain. J Neurochem. 1993;60:395–407. doi: 10.1111/j.1471-4159.1993.tb03165.x. [DOI] [PubMed] [Google Scholar]
- 34.Masotto C, Wisniewski G, Negro-Vilar A. Different gamma-aminobutyric acid receptor subtypes are involved in the regulation of opiate-dependent and independent luteinizing hormone-releasing hormone secretion. Endocrinology. 1989;125:548–553. doi: 10.1210/endo-125-1-548. [DOI] [PubMed] [Google Scholar]
- 35.Mermelstein PG, Becker JB, Surmeier DJ. Estradiol reduces calcium currents in rat neostriatal neurons via a membrane receptor. J Neurosci. 1996;16:595–604. doi: 10.1523/JNEUROSCI.16-02-00595.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Misgeld U, Bijak M, Jarolimek W. A physiological role for GABAB receptors and the effects of baclofen in the mammalian central nervous system. Prog Neurobiol. 1996;46:423–462. doi: 10.1016/0301-0082(95)00012-k. [DOI] [PubMed] [Google Scholar]
- 37.Morrell JI, McGinty JF, Pfaff DW. A subset of β-endorphin- or dynorphin-containing neurons in the medial basal hypothalamus accumulates estradiol. Neuroendocrinology. 1985;41:417–426. doi: 10.1159/000124212. [DOI] [PubMed] [Google Scholar]
- 38.Murphy DD, Cole NB, Greenberger V, Segal M. Estradiol increases dendritic spine density by reducing GABA neurotransmission in hippocampal neurons. J Neurosci. 1998;18:2550–2559. doi: 10.1523/JNEUROSCI.18-07-02550.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naftolin F, Leranth C, Horvath TL, Garcia-Segura LM. Potential neuronal mechanisms of estrogen actions in synaptogenesis and synaptic plasticity. Cell Mol Neurobiol. 1996;16:213–223. doi: 10.1007/BF02088177. [DOI] [PubMed] [Google Scholar]
- 40.Párducz A, Perez J, Garcia-Segura LM. Estradiol induces plasticity of GABAergic synapses in the hypothalamus. Neuroscience. 1993;53:395–401. doi: 10.1016/0306-4522(93)90203-r. [DOI] [PubMed] [Google Scholar]
- 41.Rönnekleiv OK, Bosch MA, Naylor BR, Kelly MJ. Progonadotropin-releasing hormone synthesis and processing: measurements of mRNA and peptides. Methods Neurosci. 1991;5:85–108. [Google Scholar]
- 42.Roselli CE, Horton LE, Resko JA. Time-course and steroid specificity of aromatase induction in rat hypothalamus-preoptic area. Biol Reprod. 1987;37:628–633. doi: 10.1095/biolreprod37.3.628. [DOI] [PubMed] [Google Scholar]
- 43.Sawaki Y, Yagi K. Inhibition and facilitation of antidromically identified tubero-infundibular neurones following stimulation of the median eminence in the rat. J Physiol (Lond) 1976;260:447–460. doi: 10.1113/jphysiol.1976.sp011524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silverman AJ, Krey LC, Zimmerman EA. A comparative study of the luteinizing hormone releasing hormone (LHRH) neuronal networks in mammals. Biol Reprod. 1979;20:98–110. doi: 10.1093/biolreprod/20.1.98. [DOI] [PubMed] [Google Scholar]
- 45.Sullivan KA, Witkin JW, Ferin M, Silverman A-J. Gonadotropin-releasing hormone neurons in the rhesus macaque are not immunoreactive for the estrogen receptor. Brain Res. 1995;685:198–200. doi: 10.1016/0006-8993(95)00352-q. [DOI] [PubMed] [Google Scholar]
- 46.Terasawa E, Wiegand SJ. Effects of hypothalamic deafferentation on ovulation and estrous cyclicity in the female guinea pig. Neuroendocrinology. 1978;26:229–248. doi: 10.1159/000122830. [DOI] [PubMed] [Google Scholar]
- 47.Tsai CC, Yen SSC. Acute effects of intravenous infusion of 17β-estradiol on gonadotropin release in pre- and post-menopausal women. J Clin Endocrinol Metab. 1971;32:766–771. doi: 10.1210/jcem-32-6-766. [DOI] [PubMed] [Google Scholar]
- 48.van den Pol AN, Cassidy JR. The hypothalamic arcuate nucleus of rat: a quantitative Golgi analysis. J Comp Neurol. 1982;204:65–98. doi: 10.1002/cne.902040108. [DOI] [PubMed] [Google Scholar]
- 49.Wagner EJ, Bosch MA, Kelly MJ, Rønnekleiv OK. A powerful GABAB receptor-mediated inhibition of GABAergic neurons in the arcuate nucleus. NeuroReport. 1999;10:2681–2687. doi: 10.1097/00001756-199908200-00045. [DOI] [PubMed] [Google Scholar]
- 50.Watson RE, Langub MC, Landis JW. Further evidence that most luteinizing hormone-releasing hormone neurons are not directly estrogen-responsive: simultaneous localization of luteinizing hormone-releasing hormone and estrogen receptor immunoreactivity in the guinea-pig brain. J Neuroendocrinol. 1992;4:311–317. doi: 10.1111/j.1365-2826.1992.tb00173.x. [DOI] [PubMed] [Google Scholar]
- 51.Witkin JW, Ferin M, Popilskis SJ, Silverman A-J. Effects of gonadal steroids on the ultrastructure of GnRH neurons in the rhesus monkey: synaptic input and glial apposition. Endocrinology. 1991;129:1083–1092. doi: 10.1210/endo-129-2-1083. [DOI] [PubMed] [Google Scholar]
- 52.Witkin JW, Xiao E, Popilskis S, Ferin M, Silverman A-J. FOS expression in the gonadotropin-releasing hormone (GnRH) neuron does not increase during the ovarian steroid-induced GnRH surge in the rhesus monkey. Endocrinology. 1994;135:956–961. doi: 10.1210/endo.135.3.8070392. [DOI] [PubMed] [Google Scholar]
- 53.Xiao L, Becker JB. Hormonal activation of the striatum and the nucleus accumbens modulates paced mating behavior in the female rat. Horm Behav. 1997;32:114–124. doi: 10.1006/hbeh.1997.1412. [DOI] [PubMed] [Google Scholar]
- 54.Yagi K, Sawaki Y. Recurrent inhibition and facilitation: demonstration in the tubero-infundibular system and effects of strychnine and picrotoxin. Brain Res. 1975;84:155–159. doi: 10.1016/0006-8993(75)90810-0. [DOI] [PubMed] [Google Scholar]
- 55.Yamaji T, Dierschke DJ, Bhattacharya AN, Knobil E. The negative feedback control by estradiol and progesterone of LH secretion in the ovariectomized rhesus monkey. Endocrinology. 1972;90:771–777. doi: 10.1210/endo-90-3-771. [DOI] [PubMed] [Google Scholar]
- 56.Yang S-H, Shi J, Day AL, Simpkins JW. Estradiol exerts neuroprotective effects when administered after ischemic insult. Stroke. 2000;31:745–750. doi: 10.1161/01.str.31.3.745. [DOI] [PubMed] [Google Scholar]
- 57.Yen SSC, Tsai CC. The effect of ovariectomy on gonadotropin release. J Clin Invest. 1971;50:1149–1153. doi: 10.1172/JCI106587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yen SSC, Tsai CC. Acute gonadotropin release induced by exogenous estradiol during the mid-follicular phase of the menstrual cycle. J Clin Endocrinol Metab. 1972;34:298–305. doi: 10.1210/jcem-34-2-298. [DOI] [PubMed] [Google Scholar]