

Abstract
Retigabine [D-23129;N-(2-amino-4-(4-fluorobenzylamino)-phenyl) carbamic acid ethyl ester] is a novel anticonvulsant compound that is now in clinical phase II development. It has previously been shown to enhance currents generated by KCNQ2/3 K+ channels when expressed in Chinese hamster ovary (CHO) cells (Main et al., 2000;Wickenden et al., 2000). In the present study, we have compared the actions of retigabine on KCNQ2/3 currents with those on currents generated by other members of the KCNQ family (homomeric KCNQ1, KCNQ2, KCNQ3, and KCNQ4 channels) expressed in CHO cells and on the native M current in rat sympathetic neurons [thought to be generated by KCNQ2/3 channels (Wang et al., 1998)]. Retigabine produced a hyperpolarizing shift of the activation curves for KCNQ2/3, KCNQ2, KCNQ3, and KCNQ4 currents with differential potencies in the following order: KCNQ3 > KCNQ2/3 > KCNQ2 > KCNQ4, as measured either by the maximum hyperpolarizing shift in the activation curves or by the EC50 values. In contrast, retigabine did not enhance cardiac KCNQ1 currents. Retigabine also produced a hyperpolarizing shift in the activation curve for native M channels in rat sympathetic neurons. The retigabine-induced current was inhibited by muscarinic receptor stimulation, with similar agonist potency but 25% reduced maximum effect. In unclamped neurons, retigabine produced a hyperpolarization and reduced the number of action potentials produced by depolarizing current injections, without change in action potential configuration.
Keywords: potassium channels, KCNQ channels, M channels, sympathetic neurons, retigabine, anti-convulsant
M-type potassium (K+) currents (IK(M)) are a species of subthreshold voltage-gated K+ current that serve to stabilize the membrane potential and control neuronal excitability. They were first described in sympathetic neurons but have subsequently been reported in various other neurons, including hippocampal and cortical pyramidal cells (for review, see Brown, 1988; Marrion, 1997).
Recent evidence suggests that the native M channels in rat sympathetic neurons are composed of a heteromeric assembly of KCNQ2 and KCNQ3 K+ channel subunits (Wang et al., 1998;Hadley et al., 2000; Selyanko et al., 2000b; Shapiro et al., 2000). These are K+ channel gene products that are widely distributed in the nervous system, mutations of which give rise to a form of congenital epilepsy termed benign familial neonatal convulsions (Biervert et al., 1998; Charlier et al., 1998; Singh et al., 1998; for review, see Rogawski, 2000; Jentsch, 2000). This implies that M channels may assist in controlling seizure discharges and that drugs that enhance M channel activity might be effective anti-epileptic agents. In accordance with this, the anti-convulsant drug retigabine [D-23129;N-(2-amino-4-(4-fluorobenzylamino)-phenyl) carbamic acid ethyl ester] (Rostock et al., 1996; Tober et al., 1996) has been reported to open K+ channels in NG108–15 neuroblastoma-glioma hybrid cells (Rundfeldt, 1997) and to enhance currents through expressed heteromeric KCNQ2/3 channels, in part by shifting their voltage sensitivity to more hyperpolarized membrane potentials (Main et al., 2000; Rundfeldt and Netzer, 2000;Wickenden et al., 2000).
Two questions arise, however, concerning the effect of retigabine. First, KCNQ2 and KCNQ3 are members of a larger family of homologous K+ channels comprising, in addition, a subunit (KCNQ1) of the cardiac delayed rectifier current (Barhanin et al., 1996; Sanguinetti et al., 1996), a subunit (KCNQ4) present in the auditory system (Kubisch et al., 1999; Kharkovets et al., 2000), and another recently identified subunit (KCNQ5) widely distributed in the CNS and PNS (Lerche et al., 2000; Schroeder et al., 2000). All of these subunits, when expressed as homomultimers, generate “M-like” currents as defined kinetically and pharmacologically (Hadley et al., 2000; Selyanko et al., 2000b; Schroeder et al., 2000). It is not yet known whether they are all equally sensitive to retigabine. Second, it has yet to be established whether retigabine affects native neuronal M currents in the same way that it affects expressed KCNQ2/3 channels. Accordingly, in the present experiments, we have compared the action of retigabine on heteromeric KCNQ2/3 currents expressed in mammalian CHO cells with its action on expressed homomeric KCNQ1, KCNQ2, KCNQ3, and KCNQ4 currents, on the one hand, and on native M currents in rat sympathetic neurons on the other.
While this work was in progress, Wickenden et al. (2001) reported that retigabine also enhances currents through concatameric KCNQ3/5 channels in CHO cells.
MATERIALS AND METHODS
Chinese hamster ovary cell culture and transfection. Chinese Hamster Ovary (CHO) cells stably transfected with cDNA encoding human M1 muscarinic receptors (Mullaney et al., 1993) were cultured in α-MEM supplemented with 10% fetal calf serum, 1% l-glutamine, and 1% penicillin/streptomycin. Cells were split when confluent and plated onto 35 mm dishes. Cells were left at 37°C and 5% CO2 for 1 d before transfection using LipofectAmine Plus (Life Technologies, Gaithersburg, MD) according to the manufacturer's recommendations. Plasmids containing KCNQ and CD8 cDNAs, and driven by the cytomegalovirus promoter, were cotransfected in a ratio of 10:1. For expression of heteromultimers, equal amounts of KCNQ2 and KCNQ3 cDNAs were used. Successfully transfected cells were identified by adding CD8-binding Dynabeads (Dynal, Great Neck, NY) before electrophysiological recording.
Ganglion cell culture. Primary cultures of neurons were prepared from superior cervical ganglia (SCG) from 17-d-old Sprague Dawley rats, using a standard enzymatic dissociation procedure, as fully described elsewhere (Delmas et al., 1998b). Rats were killed by CO2 inhalation and decapitated, according to the U.K. Animals (Scientific Procedures) Act 1986. Ganglia were dissected from the carotid bifurcation, and the surrounding sheath was removed. Each ganglion was cut into several pieces with iridectomy scissors and incubated at 37°C in collagenase (500 U/ml and bovine serum albumin 6 mg/ml) and then in trypsin (1 mg/ml with 6 mg/ml BSA). After trituration the cells were centrifuged, and the pellet was resuspended in culture medium (L-15 medium supplemented with 10% fetal bovine serum, 24 mm NaHCO3, 38 mm glucose, 50 U/ml penicillin and streptomycin, 25 ng/ml nerve growth factor). After the dissociated cells were plated onto laminin-coated 35 mm plastic Petri dishes, the cells were kept in a 37°C incubator with 5% CO2.
Electrophysiological recording. Currents in CHO cells and SCG neurons were recorded using the amphotericin-B perforated-patch technique (Rae et al., 1991). Patch electrodes (2–3 MΩ) were filled by dipping the tip for 40 sec into the appropriate filtered internal solution, and the pipette was then backfilled with the internal solution containing 0.1–0.2 mg/ml amphotericin-B. After permeabilization, access resistances were generally <15 MΩ. The recording electrodes had resistance of 2–3 MΩ when filled with internal solution.
Recordings from CHO cells were made at room temperature 1–2 d after transfection. No appreciable current was noticed in untransfected CHO cells. The extracellular solution consisted of (in mm): NaCl 144, KCl 2.5, CaCl2 2, MgCl2 0.5, HEPES 5, and glucose 10; pH 7.4 with Tris base. To record tail currents at −120 mV (for the purpose of construction of activation curves), external KCl was raised to 25 mm; the increase in osmolarity was compensated by reducing the concentration of NaCl to 121.5 mm. The internal (pipette) solution contained (in mm): K+ acetate 80, KCl 30, HEPES 40, MgCl2 3, EGTA 3, CaCl2 1; pH 7.4 with KOH.
K+ and Ca2+current recordings in SCG neurons were as described (Delmas et al., 2000). Briefly, SCG neurons were perfused with an external solution consisting of (in mm): NaCl 120, KCl 3, HEPES 5, NaHCO3 23, glucose 11, MgCl2 1.2, CaCl2 2.5, tetrodotoxin 0.0005, pH 7.4. Internal solution for M-current recording consisted of (in mm): K+ acetate 90, KCl 40, HEPES 20, MgCl2 3 (adjusted to pH 7.3–7.4 with KOH, and 300 mOsm/l with K+ acetate); for Ca2+ current recording the solution consisted of (in mm): CsCl 30, cesium acetate 110, HEPES 10, MgCl2 1 (pH 7.2–7.3 with CsOH, 300 mOsm/l). Experiments were performed at 30–32°C.
Voltage-clamp recordings. Data were acquired and analyzed using pClamp software (version 6.0.3). Currents in transfected CHO cell line were recorded using Axopatch 1D patch-clamp amplifier (Axon Instruments) and filtered at 1 kHz. The capacity transients were cancelled using the resistance capacitance circuit within the amplifier. Series resistance compensation was set to 60–80%.
SCG neurons were voltage clamped using an Axopatch 200A amplifier (Axon Instruments). Current traces were low-pass filtered at 2–5 kHz using a four-pole Bessel filter, and series resistance and membrane capacitance were partially compensated (>70%). Leak and capacitance currents were subtracted digitally using the P/6 subtraction procedure of Pclamp6. The capacity transients were cancelled using the resistance capacitance circuit within the amplifier. Series resistance compensation was also used and was set to 60–80%.
The data were analyzed using the Clampfit (pClamp6 and 7), ORIGIN 5, and Excel data handling and graphical presentation software packages. The methods of recording and analysis were similar to those used previously for studying KCNQ currents (Selyanko et al., 2000b). Results are presented as the mean ± SEM.
Activation curves were fitted by the Boltzmann equation:
| Equation 1 |
where I is the tail current recorded at −120 mV after a pre-step to membrane potential V (estimated from the amplitudes of exponentials backfitted to the beginning of the test step),I(50) is the current after a step to +50 mV,V1/2 is the membrane potential at which I is equal to one-half I(50), andk is the slope of the curve. The concentration–response of different KCNQ channels to retigabine was estimated by the shift in theV1/2 values caused by 0.1, 0.3, 1, 3, 10, 30, 100, and 300 μm. Dose–response curves were fitted to the following equation:
| Equation 2 |
where EC50 is the concentration corresponding to half-maximal activity, x is the agonist concentration, and p is the slope of the curve.
Inhibition of the KCNQ1 current was evaluated by the decrease in current amplitude after step depolarizations to +50 mV from −70 mV after addition of 3, 10, 30, 100, 300, and 1000 μm of retigabine. The inhibition curve was fitted with Equation 2, with IC50 concentration corresponding to half-maximal block.
Chemicals and drugs. cDNAs for human KCNQ2 and rat KCNQ3 were obtained from Dr. D. McKinnon (Department of Neurobiology and Behavior, State University of New York, Stony Brook, NY), for human KCNQ1 from Dr. M. T. Keating (Howard Hughes Medical Institute, University of Utah, Ogden, UT), and for KCNQ4 from Dr. T. J. Jentsch (Zentrum für Molekulare Neurobiologie, Hamburg, University of Hamburg, Hamburg, Germany). Retigabine was obtained from Glaxo SmithKline (Stevenage, UK). Oxotremorine methiodide (Oxo-M) was obtained from RBI (Natick, MA). All other drugs and chemicals were obtained from Sigma or BDH (Poole, UK).
RESULTS
Retigabine enhances KCNQ2–4 currents in CHO cells
Figure 1 illustrates the effect of 10 μm retigabine on currents carried by heteromeric KCNQ2/3 channels and homomeric KCNQ2, KCNQ3, and KCNQ4 channels expressed in CHO cells. In these experiments, an “M-current protocol” (Adams and Brown, 1982) was used to record the expressed currents; that is, the cell was predepolarized to −20 mV to activate the current, then hyperpolarized to −90 mV in steps of 10 mV to deactivate the current. Deactivation is registered by the slow tail currents, and reactivation is registered on stepping back to −20 mV by the slow outward current. The following points emerge from this comparison. (1) No appreciable current could be recorded from control (untransfected) cells, and retigabine had no significant effect on these cells (Fig. 1B). (2) As predicted from the previous experiments of Main et al. (2000), Rundfeldt and Netzer (2000), and Wickenden et al. (2000), retigabine increased the outward current recorded at −20 mV in cells transfected with KCNQ2 + KCNQ3 cDNAs. However, it also increased the −20 mV current in cells transfected solely with KCNQ2, KCNQ3, or KCNQ4 cDNAs to comparable (or greater) extents. (3) The additional current at −20 mV induced by retigabine in KCNQ2- and KCNQ2/3-transfected cells was removed by hyperpolarizing to −90 mV. However, an additional component of inward current was recorded at −90 mV in KCNQ3- and KCNQ4-transfected cells. This implies that a component of KCNQ3 or KCNQ4 current persisted in the presence of retigabine negative to resting potentials (−60 mV) (Selyanko et al., 2000b), where these channels would normally be fully deactivated. (4) As also reported previously for KCNQ2/3 currents (Main et al., 2000; Rundfeldt and Netzer, 2000; Wickenden et al., 2000) and KCNQ3/5 currents (Wickenden et al., 2001), retigabine slowed the deactivation of the currents during step-hyperpolarizations and accelerated the reactivation on repolarization. This effect was most pronounced with KCNQ3, for which the time dependence of current deactivation was lost except at very negative potentials, and the effect was least pronounced for KCNQ4. Thus, these tests indicate that the previously reported enhancing effect of retigabine on KCNQ2/3 and KCNQ3/5 channels also extends to homomeric KCNQ2, KCNQ3, and KCNQ4 channels, albeit with quantitative differences (see further below).
Fig. 1.
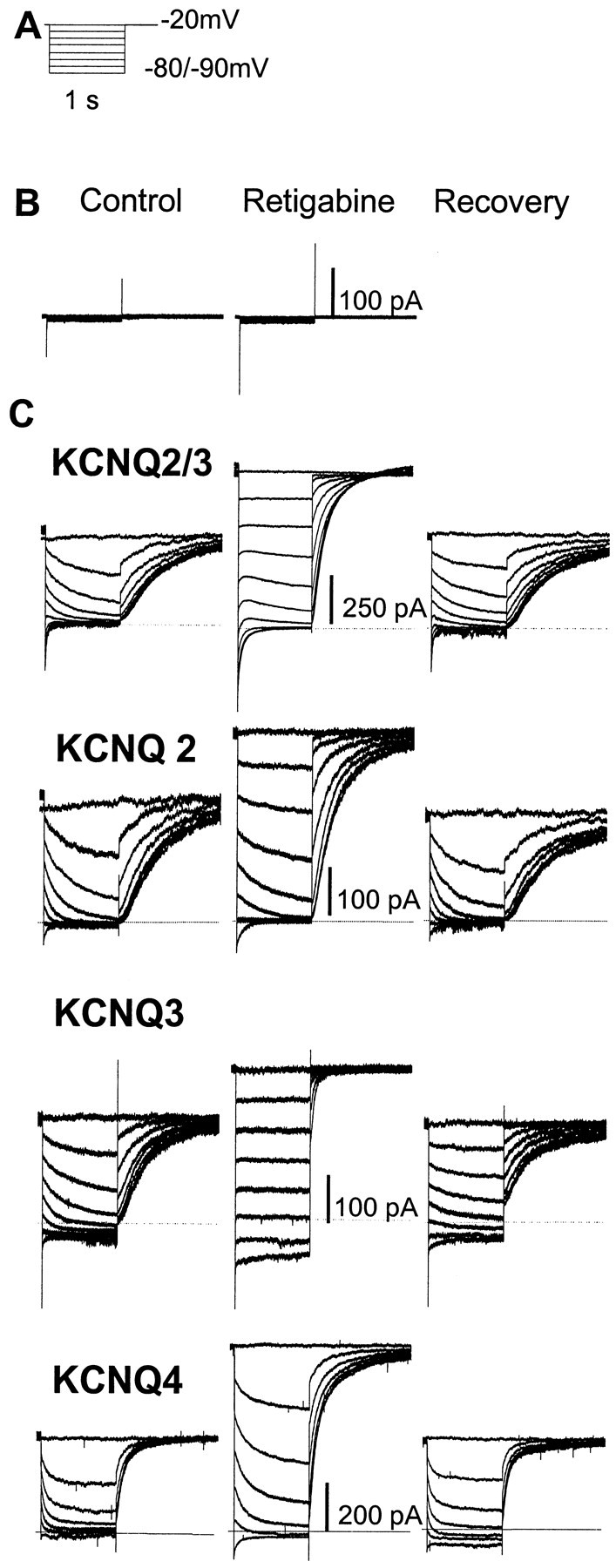
Enhancement of heteromeric (KCNQ2/3) and homomeric (KCNQ2–4) currents in CHO cells by retigabine. A, Voltage protocol: currents were activated by clamping the membrane at −20 mV and then deactivated by 1 sec hyperpolarizations to −90 mV in 10 mV increments. B, Retigabine (10 μm) did not activate any endogenous currents in untransfected CHO cells.C, KCNQ currents generated by the voltage protocol inA recorded in the absence (left), presence (center), and 10 min after washout (right) of retigabine. Dotted linesdenote zero current level.
Augmentation of KCNQ2–4 currents by retigabine was confirmed after current activation from −70 mV to +50 mV in 10 mV steps (Figs.2, 3). Several points are worth noting. First, retigabine clearly accelerated the onset of all currents, but most noticeably with KCNQ3. Second, current–voltage curves were shifted to the left (Fig. 3). For KCNQ2, KCNQ4, and KCNQ2/3, this corresponded to a ∼20–30 mV negative shift in threshold for current activation. For KCNQ3 the shift was much greater because the current was still active at the holding potential (−70 mV): in effect, retigabine converted the KCNQ3 current from a time- and voltage-dependent current with a threshold of −50 mV to a substantially time- and voltage-independent “leak” current from −90 mV upward. Third, retigabine clearly increased the maximal current generated by KCNQ3 and KCNQ4 channels (and to varying extents, by KCNQ2/3 channels) but not by KCNQ2 channels. Variation in the amount by which retigabine increased the maximum current amplitude through different KCNQ channels may reflect a variable component of secondary blocking action at positive potentials (see KCNQ1 below).
Fig. 2.
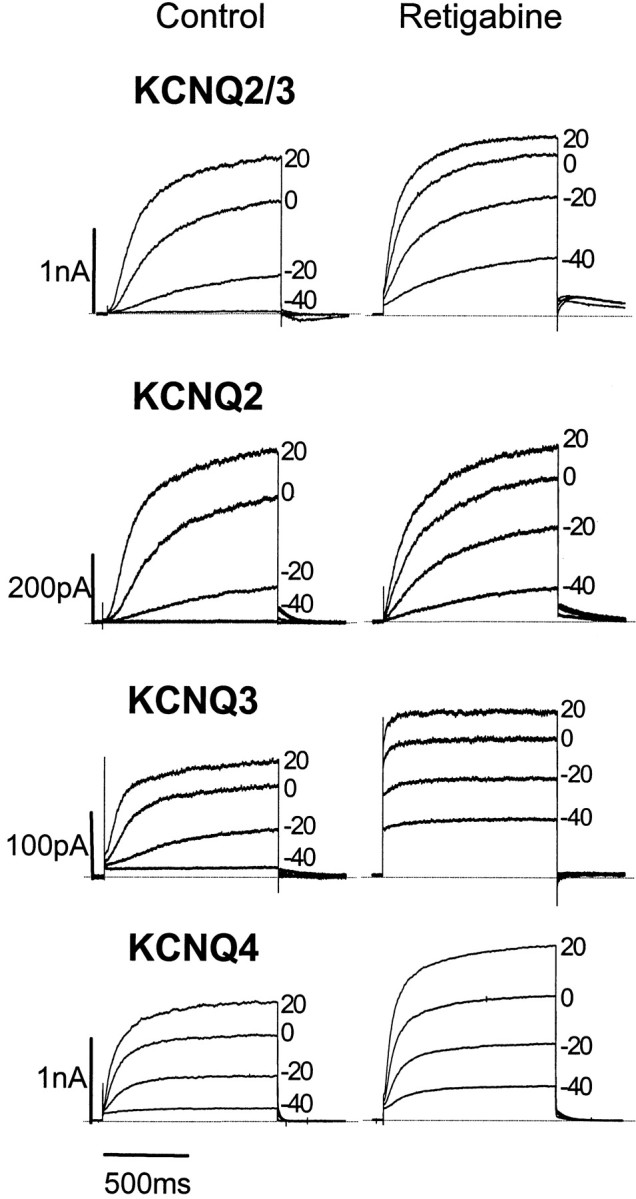
Activation of heteromeric KCNQ2/3 and homomeric KCNQ2–4 channels expressed in CHO cells in the absence and presence of retigabine. Families of KCNQ currents were recorded in the absence (left) or presence (right) of 10 μm retigabine. Currents were activated by 1 sec depolarizing pulses in 10 mV increments to +50 mV from a holding potential of −70 mV. Records show currents recorded at −40, −20, 0, and +20 mV. Note that KCNQ3 and KCNQ4, but not KCNQ2/3 and KCNQ2 currents, are increased at all voltages by retigabine.
Fig. 3.

Effects of retigabine on the KCNQ2–4 current–voltage relationship. Normalized current values were plotted against command potential before (○) and after (●) addition of 10 μm retigabine. To obtain normalized values, peak current amplitudes in response to depolarizing pulses from a holding voltage of −70 to +50 mV in 10 mV increments were normalized against the maximum current amplitude at +50 mV in control recordings. Retigabine shifts the threshold for channel activation and augments KCNQ current amplitude across a range of membrane potentials.
Retigabine shifts KCNQ activation curves
The current–voltage curves in Figure 3 suggest that one effect of retigabine is to produce a negative shift in the KCNQ current activation curve. This was further tested using the protocol shown in Figure 4, in which the current was first fully activated by stepping to +50 mV, then stepped for 1 sec to various potentials between +50 and −110 mV, followed by a final step to −120 mV (Fig. 4A). The relative amount of conductance activated at each potential at the end of the 1 sec step could then be determined directly from the residual tail-current at −120 mV. Boltzmann plots (Fig. 4B) (see Materials and Methods) confirmed that 10 μm retigabine produced a significant left-shift of the activation curves in the order KCNQ3 (−43 mV) > KCNQ2/3 (−30 mV) > KCNQ2 (−24 mV) >KCNQ4 (−14 mV). (Values in brackets give mean shift in half-activation potential V1/2.) In contrast, the slopes of the activation curves were not significantly changed by retigabine (Fig. 4, legend).
Fig. 4.
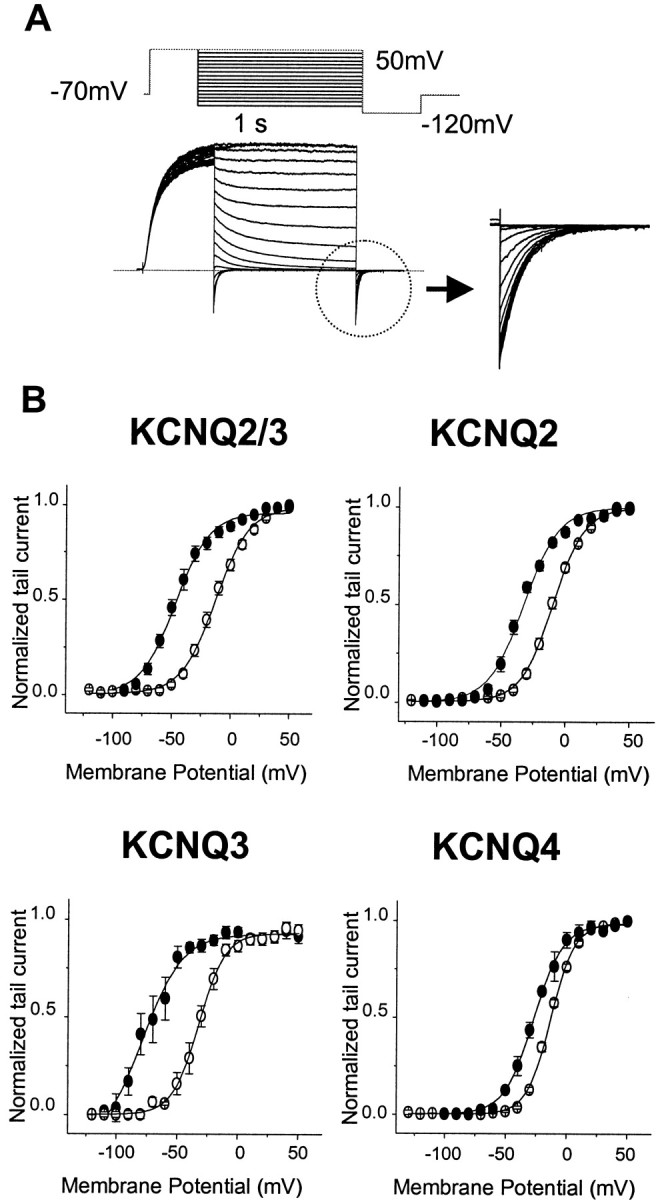
Effects of retigabine on the KCNQ2–4 activation curves. A, Currents were activated by depolarizing pulses to +50 mV from a holding potential of −70 mV and deactivated by 1 sec hyperpolarizing pulses to various potentials between +50 and −120 mV, followed by a step to −120 mV. Theinset shows the tail currents recorded at −120 mV, which were used to construct the activation curves. B, Activation curves in the absence (○) and presence (●) of retigabine were obtained from tail currents as described in Materials and Methods and fitted with the Boltzmann Equation 1. TheV1/2 values were as follows: KCNQ2/3, −17.3 ± 2.2 mV for control (n = 9) and −47.7 ± 2.7 mV for retigabine-treated cells (n = 9); KCNQ2, −11.5 ± 1.5 mV for control (n = 16) and −35.7 ± 2.1 mV for retigabine-treated cells (n = 6); KCNQ3, −28.7 ± 2.5 mV for control (n = 7) and −71.5 ± 3.1 mV for retigabine-treated cells (n = 7); and KCNQ4, −12.6 ± 0.7 mV for control (n = 4) and −26.2 ± 0.6 mV for retigabine-treated cells (n = 4). Slopes were as follows: KCNQ2/3, 12.9 ± 1.2 mV (control, n = 9) and 11.4 ± 1.3 mV (retigabine, n = 5); KCNQ2, 10.9 ± 0.6 mV (control, n = 16) and 11.2 ± 0.9 mV (retigabine, n = 6); KCNQ3, 11.4 ± 0.9 mV (control, n = 7) and 10.9 ± 0.9 mV (retigabine, n = 7); and KCNQ4, 10.1 ± 0.3 mV (control, n = 4) and 12.1 ± 1.6 mV (retigabine, n = 4). The data shown are mean ± SEM.
The differential effects shown in Figure 4 were obtained using a fixed (10 μm) concentration of retigabine. To measure the potency of retigabine, the shift in the activation curves produced by incremental concentrations of retigabine were determined, as illustrated in Figure 5. To amplify the inward tail currents at −120 mV (and hence improve the accuracy of the deduced activation curves), the external K+ concentration was raised to 25 mm for these experiments. This did not itself affect the action of retigabine because the shift of the KCNQ2/3 activation curve produced by 10 μm retigabine in 25 mm[K+] was indistinguishable from that observed in 2.5 mm [K+], which is illustrated in Figure 4. The magnitude of the shift in the activation curves was clearly dependent on the concentration of retigabine (Fig. 5B). The relation between the concentration of retigabine and the shift in the half-activation potentialV1/2 for the different KCNQ channels tested deduced from these experiments, and normalized to the maximum shift produced by 10–300 μm retigabine, is shown in Figure 6. Data points could be fitted with a Hill equation (see Materials and Methods), with slope ∼1 but EC50 values varying from 0.6 μm for KCNQ3 to 5.2 μmfor KCNQ4. The maximum shift of V1/2also varied with different KCNQ channels. The order of potency as determined from EC50 values (KCNQ3 > KCNQ2/3 > KCNQ2 > KCNQ4) accords with the apparent “efficacy” as measured by the maximum shift (Table1).
Fig. 5.
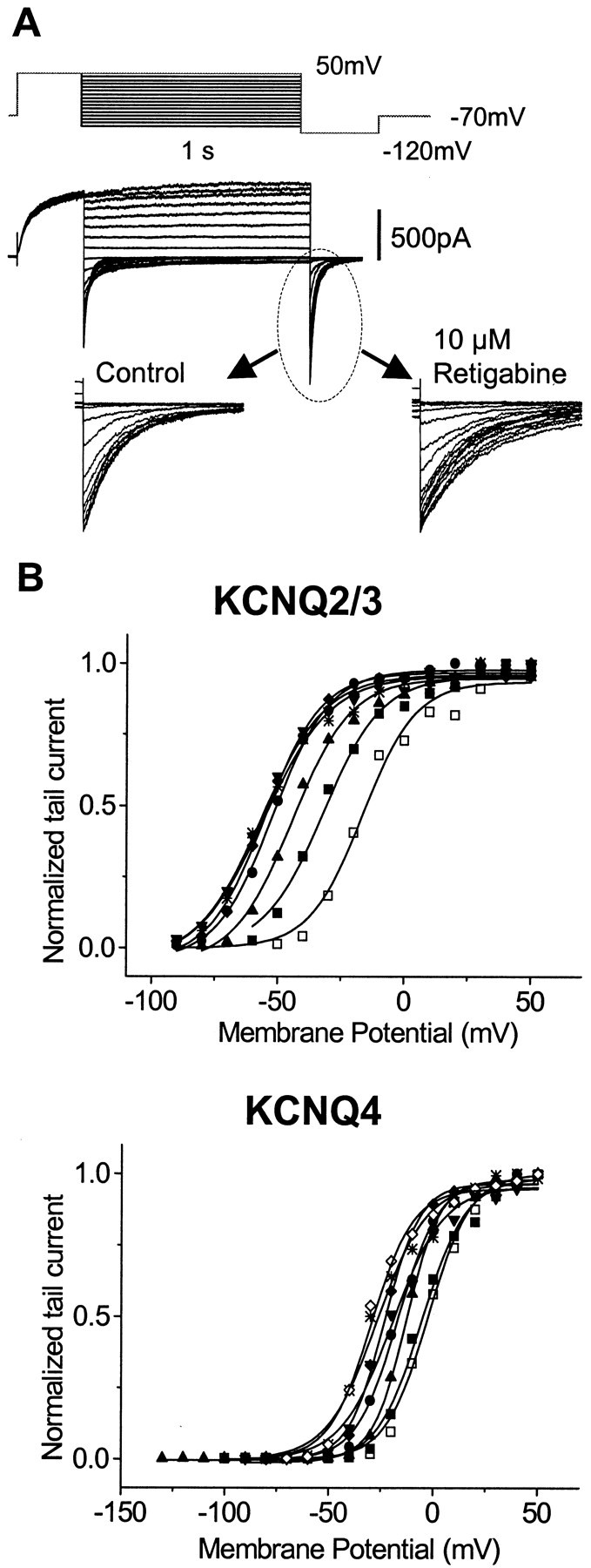
Effects of retigabine on KCNQ currents are concentration dependent. A, Experiments were performed in high extracellular K+ (25 mm) solution to enhance the tail currents. A family of currents for KCNQ2/3 and the voltage protocol under these conditions are shown. Application of retigabine led to a slowing in the rate of decline of the KCNQ tail current (inset). B, Representative activation curves for KCNQ2/3 and KCNQ4 generated in the absence (■) and presence of 1 μm (▪), 3 μm (▴), 10 μm (●), 30 μm (▾), 100 μm(♦), and 300 μm (✳) retigabine. For KCNQ4, 1 mm retigabine (⋄) was also applied.
Fig. 6.

Concentration–response curves for KCNQ2/3, KCNQ2, KCNQ3, and KCNQ4. The magnitude of the leftward shift in the half-activation potential (V1/2) was calculated with each concentration of retigabine and normalized against the maximum shift. The maximum shift in the activation curve was obtained with 10 μm retigabine for KCNQ2/3, KCNQ2, and KCNQ3 and with 300 μm for KCNQ4. Normalized values were plotted against retigabine concentration, and the data were fitted with Equation 2 with the following parameters: EC50 = 1.9 ± 0.2 μm and slope = 1.3 ± 0.2 for KCNQ2/3 (n = 5); EC50 = 2.5 ± 0.6 μmand slope = 1.1 ± 0.5 for KCNQ2 (n = 5); EC50 = 0.6 ± 0.3 μm and slope = 1.2 ± 0.4 for KCNQ3 (n = 3); and EC50 = 5.2 ± 0.9 μm and slope = 0.9 ± 0.2 for KCNQ4 (n = 3). Each point represents the mean ± SEM.
Table 1.
Potency of retigabine against KCNQ2/3, KCNQ1, KCNQ2, KCNQ3, and KCNQ4
| Maximum shift in V1/2 (mV) | Retigabine EC50 (μm) | |
|---|---|---|
| KCNQ2/3 | −30.4 | 1.9 ± 0.2 (n = 5) |
| KCNQ1 | −0.7 | 100.1 ± 6.5 (n = 4)1-a |
| KCNQ2 | −24.2 | 2.5 ± 0.6 (n = 5) |
| KCNQ3 | −42.8 | 0.6 ± 0.3 (n = 3) |
| KCNQ4 | −24.6 | 5.2 ± 0.9 (n = 3) |
Columns show the maximum leftward shift ofV1/2 (see Fig. 5) induced by retigabine and the corresponding EC50 value for the shift for KCNQ2–4 (see Fig. 6), calculated as the mean ± SEM of the shifts produced in the individual experiments (number of experiments in parentheses). Note that 100 μm retigabine was sufficient to produce a maximum shift for KCNQ2/3, KCNQ2, and KCNQ3; for KCNQ4, 300 μm retigabine was required.
KCNQ1, IC50 for inhibition (see Fig. 7).
Interaction of retigabine and linopirdine on KCNQ2/3 currents
The blocking action of the M-channel antagonist linopirdine (Lamas et al., 1997) on KCNQ2/3 currents was unaffected by the presence of retigabine. IC50 values were 7.3 ± 1.7 μm (n = 3) in the absence of retigabine and 11.1 ± 2.3 μm (n = 4) in the presence of 10 μm retigabine. These values were not significantly different (p = 0.27; t test). The IC50 value in the absence of retigabine is not dissimilar from that (4.0 ± 0.5 μm) reported previously by Wang et al. (1998)against KCNQ2/3 currents expressed in frog oocytes.
KCNQ1 currents are resistant to enhancement by retigabine
In contrast to its effects on KCNQ2–4 currents, retigabine (10 μm) did not enhance currents generated by homomeric KCNQ1 channels in CHO cells (Fig.7A) nor did it shift the KCNQ1 activation curve (Fig. 7C). Instead, at positive potentials, higher concentrations of retigabine (100 μm) reduced the KCNQ1 current amplitude in an apparently voltage-dependent manner, the fractional reduction increasing with increasing positivity (Fig. 7B,D). This effect was even more pronounced at 1 mm. The blocking action of retigabine on KCNQ1 current was quantitated by applying 3, 10, 30, 100, 300, and 1000 μm retigabine and measuring the reduction of current at +50 mV. Fractional inhibition of the current was plotted against retigabine concentration and points fitted with a Hill equation (see Materials and Methods), with a slope of 1.26 ± 0.16 and IC50 of 100.1 ± 6.5 μm (n = 4) (Fig. 7E) (note that only ∼65% of the KCNQ1 current was inhibited with 1 mm retigabine). Retigabine had similar effects (no activation shift and partial current block at positive potentials) on currents generated by coexpressed KCNQ1 + KCNE1 channels (n = 4). A variable expression of the voltage-dependent inhibitory action of retigabine revealed in KCNQ1 channels may account for the variable degree of current enhancement at positive potentials through different KCNQ channels illustrated in Figure 2.
Fig. 7.
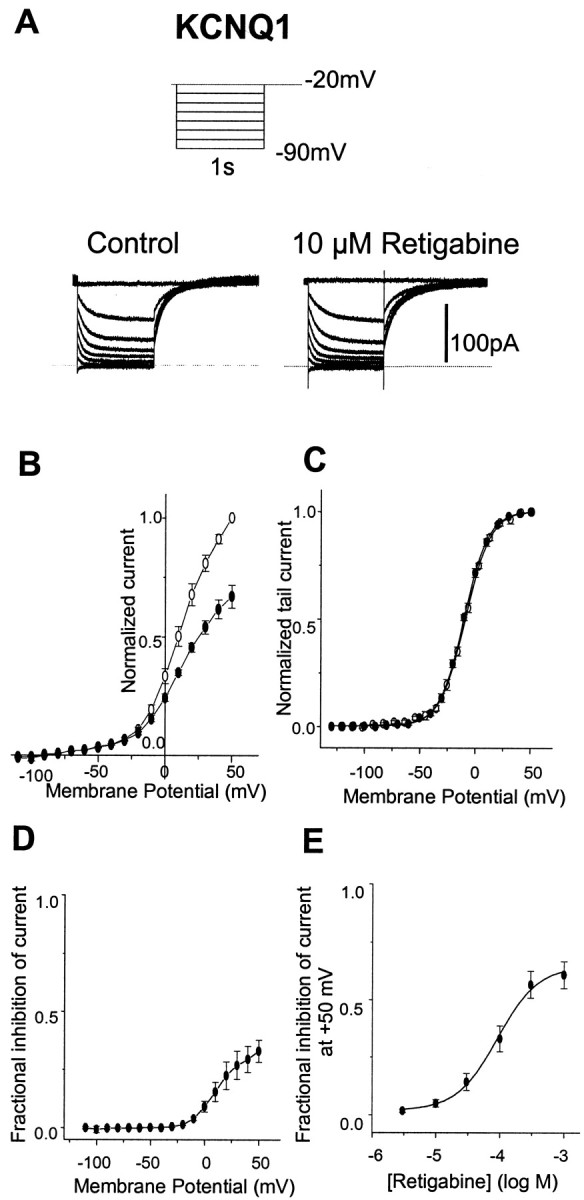
Retigabine does not enhance KCNQ1 current in CHO cells. A, KCNQ1 currents were recorded using the protocol shown in inset in the absence (left) and presence (right) of 10 μm retigabine. B, Normalized current–voltage curve for KCNQ1 in the absence (○) and presence (●) of 100 μm retigabine. Voltage protocol as in Figure2. Note the reduction of peak current in the presence of retigabine.C, Activation curves were constructed for KCNQ1 in the absence (○, n = 11) and presence (●,n = 5) of 100 μm retigabine as described in Figure 4. Lines are Boltzmann fits to the data, givingV1/2 = −12.5 ± 1.1 mV and slope of 12.4 ± 0.9 mV for control andV1/2 = −13.2 ± 1.2 mV and slope of 12.4 ± 0.9 mV in the presence of retigabine.V1/2 values are not significantly different (unpaired t test; p = 0.7).D, Voltage dependence of inhibition: fraction of current inhibited by 100 μm retigabine plotted against voltage (replotted from B). E, Concentration dependence of inhibition. Fractional inhibition at +50 mV (ordinate) was plotted against log M retigabine concentration (abscissa). Data were fitted with Equation2 (see Materials and Methods). Points show means ± SEM, withn = 4. Mean IC50 was 100.1 ± 6.5 μm, and the slope was 1.26 ± 0.16.
Retigabine enhances native M currents in sympathetic neurons and shifts the voltage dependence for their activation
The native M current in rat SCG neurons has been attributed to currents carried through heteromeric KCNQ2/3 channels (Wang et al., 1998). Hence, retigabine might be expected to affect these native M currents in a manner resembling its effect on KCNQ2/3 channels. In the present experiments we therefore recorded native M currents in dissociated rat SCG neurons using the perforated-patch configuration of the patch pipette (see Materials and Methods).
Figure 8 illustrates the effect of 10 μm retigabine on the outward currents evoked in a dissociated rat SCG neuron by 1 sec depolarizing steps from −80 mV. In the absence of retigabine, activation of M current is manifest by the appearance of a slow time-dependent component of outward current at command potentials of −50 mV and upward, subsequent to the transient (∼50–100 msec duration) “A-current” (Fig. 8A). In the presence of retigabine, two differences are seen: the time-dependent component appears at a more negative command potential (−70 mV), and the total outward current is increased at all command potentials. The extra current induced by retigabine is indicated by the subtracted currents in Figure 8B: this current is clearly “M-like” in appearance, with slow activation and deactivation, and saturates at −30 mV. Note that unlike the raw currents, the subtracted current is devoid of an initial transient A-current component, implying that the A-current was not enhanced by retigabine.
Fig. 8.

Retigabine enhances outward currents in SCG neurons. A, Families of currents recorded from a rat SCG neuron elicited by 1 sec voltage steps from a holding potential of −80 mV to test potentials from −70 to −10 mV (as indicated). Retigabine (RTG) was applied via bath perfusion at 10 μm. b, Retigabine-induced outward currents obtained by subtracting control currents from currents in the presence of retigabine. Note the saturation of the current at potentials positive to −30 mV.
The effect of retigabine on the current–voltage curves for the native M current was further examined by applying slow voltage ramps in the presence and absence of the M current blocker linopirdine (10 μm) (Fig.9A). In this way, the component of outward rectification in the current–voltage curves attributable to M current could be identified as that component of current blocked by linopirdine (Fig. 9B). In the presence of retigabine, this component of rectification was shifted ∼20 mV in the negative direction (mean, −21 ± 2 mV; n = 5). As a result, the threshold for the outwardly rectifying M current was shifted from between −50 and −60 mV to approximately −80 mV. However, a small component of linopirdine-sensitive outward current, which appeared insensitive to voltage, could also be detected at potentials negative to −80 mV in the presence of retigabine. This was confirmed in the form of a persistent inward current on raising external [K+] to 20 mm (Fig. 9C). As with expressed KCNQ2/3 and KCNQ2 channels (Fig. 3), retigabine did not increase the maximum amplitude of the linopirdine-sensitive component of current. Thus, the effects of retigabine on the linopirdine-sensitive (presumed M) component of current, a negative shift in the current–voltage curve without increased maximum, were in qualitative agreement with its effects on expressed KCNQ2/3 channels. (Although the concentration of linopirdine used in these experiments was unlikely to have completely blocked the M current, the fraction blocked was presumably the same in the absence and presence of retigabine, because retigabine did not affect the action of linopirdine on expressed KNCQ2/3 currents; see above.)
Fig. 9.
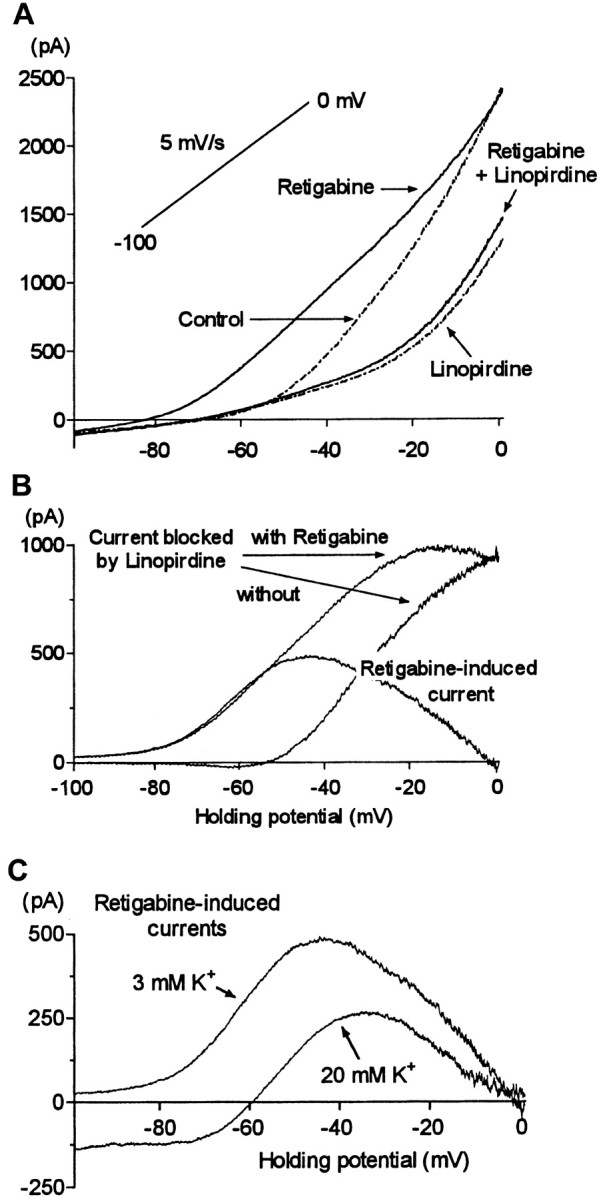
Retigabine shifts the voltage dependence of activation of the M current. A, Superimposed steady-state I–V relationships (determined using a voltage ramp from −100 to 0 mV at 5 mV/sec) from a single cell in control conditions and after addition of linopirdine (10 μm), retigabine (10 μm, after washout of linopirdine), and retigabine + linopirdine. B, Difference currents obtained by digitally subtracting theI–V relationships shown inA. Note the leftward shift in the activation of the M current (linopirdine-sensitive current) induced by retigabine and the bell shape of the retigabine induced current. C, Retigabine-induced current in the presence of 3 and 20 mmexternal K+. CalculatedEK values were −102 and −56 mV, respectively.
Enhancement of M current in SCG neurons was confirmed using the standard M-current protocol, in which M current is recorded in isolation from inactivating currents by predepolarizing to −20 mV and then deactivating the M current by step hyperpolarization to −50 mV (Fig. 10; compare Fig. 1). Incremental addition of increasing concentrations of retigabine produced a progressive increase in the steady outward current at −20 mV, with an EC50 of ∼0.76 μm (Fig.10B) (mean 0.74 ± 0.02 μm; n = 5). There was also an increase in the residual SCG M current at −50 mV, implying that the hyperpolarizing step no longer fully deactivated the M current (as expected from the negative shift of the activation curve). These experiments also revealed that retigabine slowed the deactivation of the M current at −50 mV, the time constant for the faster component of the relaxation being slowed from ∼30 to ∼80 msec (Fig.11).
Fig. 10.
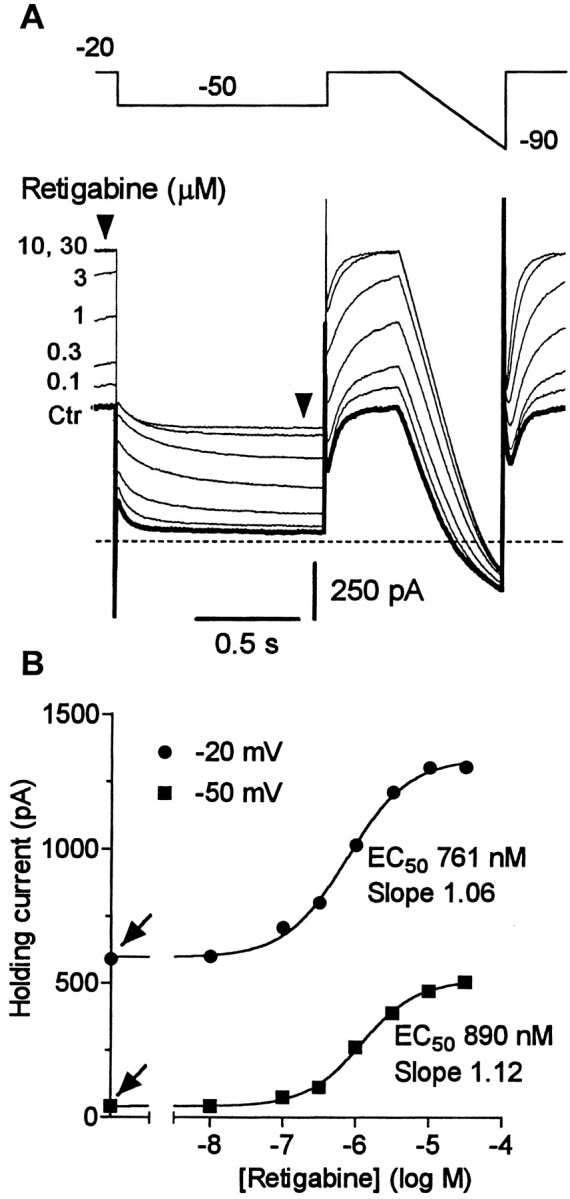
Modulation of the M current by retigabine is concentration dependent. A, Superimposed M-current traces before and after cumulative addition of retigabine (0.1–30 μm). The M current was recorded in response to 1 sec hyperpolarizing steps from a holding potential of −20 to −50 mV followed by a ramp to −90 mV. Note that the increase in holding current is associated with alteration of M-current deactivation relaxation. B, Holding current at −50 and −20 mV plotted against retigabine concentrations for the cell illustrated inA. Solid lines represent best fits to the Hill equation with holding currents in the absence of retigabine of 610 pA at −20 mV and 42 pA at −50 mV (as indicated byarrows). EC50 values and Hill coefficient (slope) are indicated. Mean values are given in Results.
Fig. 11.

Muscarinic inhibition of retigabine-enhanced M current. A, Currents were produced by holding at −20 mV and giving repeated steps to −80 mV in 10 mV increments. Steady-state currents were enhanced by retigabine (RTG) and inhibited by oxotremorine-M (Oxo-M). Theinset shows currents induced by the simultaneous application of retigabine and Oxo-M. B, Families of M-current deactivations obtained before (Control) and after addition of retigabine (10 μm) and retigabine (10 μm) + Oxo-M (10 μm).
The retigabine-enhanced M current is sensitive to inhibition by muscarinic receptor agonists
The native M current in rat SCG neurons is inhibited by stimulating endogenous M1 muscarinic receptors (Marrion et al., 1989; Bernheim et al., 1992). We therefore tested whether the retigabine-modified M current was also sensitive to muscarinic receptor stimulation. As shown in Figure 11, this appeared to be the case: retigabine (10 μm) augmented the outward current recorded at −20 mV, and subsequent addition of the muscarinic receptor agonist Oxo-M (10 μm) eliminated this extra current. The inhibitory effect of Oxo-M was quantitated by measuring the initial amplitude of the deactivation tail currents at −50 mV recorded in the presence of incrementing concentrations of Oxo-M in the absence and presence of retigabine. As indicated in Figure12A, the IC50 values for Oxo-M were similar in the presence and absence of retigabine. However, the maximum inhibition produced by Oxo-M (30 μm) was reduced by ∼25%. This was unlikely to have been caused by an effect (noncompetitive) of retigabine on the muscarinic receptors because retigabine did not reduce the effect of Oxo-M on the Ca2+ current recorded in pertussis toxin-treated cells (Fig. 12B), which is mediated by M1 receptor activation (Bernheim et al., 1992;Delmas et al., 1998b). Hence, although equally sensitive to Oxo-M in terms of IC50 values, retigabine-modified M currents appear somewhat less susceptible to muscarinic receptor modulation in terms of the amount of inhibition. One additional point emerging from these experiments was that the current-enhancing effect of retigabine was more rapid in onset than the current-inhibiting effect of Oxo-M when the two were applied simultaneously (Fig.11A, inset). This implies that retigabine has a more direct effect on the M channels than Oxo-M, the action of which involves a diffusible second messenger (Selyanko et al., 1992).
Fig. 12.
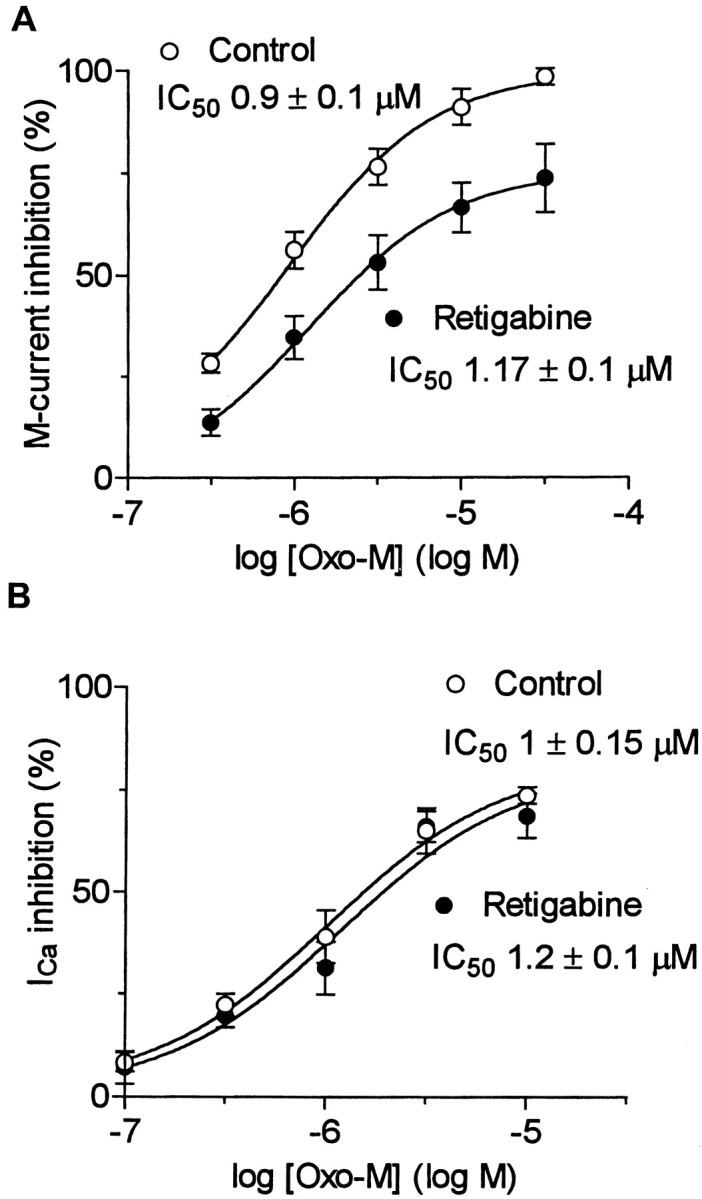
Retigabine-enhanced M currents are more resistant to modulation. A, Concentration dependence of M-current inhibition by Oxo-M in the presence (●) or absence (○) of retigabine (10 μm). B, Concentration dependence of Ca2+ current inhibition by Oxo-M in the presence (●) or absence (○) of retigabine (10 μm). Ca2+ currents were recorded using the perforated-patch method in cells pretreated with pertussis toxin (0.5 μg/μl) for 24 hr (Delmas et al., 1998a). Points were fitted with Equation 2 (see Materials and Methods).
Selectivity of retigabine action on rat SCG neurons
As pointed out in connection with Figure 8 above, retigabine (10 μm) had no significant effect on the transient “A-type” current recorded in rat SCG neurons (n = 12), which is probably mediated by Kv4.2 K+ channels (Malin and Nerbonne, 2000). Likewise, 10 μm retigabine did not affect the hyperpolarization-activated cation currentIh (n = 4) (Lamas, 1998) or the N-type Ca2+ current (n = 6). However, it did exert a small (20 ± 2%;n = 4) inhibitory effect on the Ca2+-activated K+ current driving the post-spike after-hyperpolarization.
Physiological consequences of retigabine action
Because the M current acts as a braking current on action potential discharges (Brown, 1988), retigabine might be expected to reduce spike activity. Figure 13illustrates a test for this on an SCG neuron. In Figure 13A, the neuron was challenged with 0.5 sec depolarizing or hyperpolarizing current injections from a resting membrane potential of approximately −60 mV. The depolarizing pulses typically evoked a phasic discharge of one or two spikes. The application of retigabine (3 μm) hyperpolarized the cells by −9 ± 4 mV (n = 7). When the membrane potential was brought back to −60 mV, the original depolarizing current injections no longer induced spikes (Fig. 13B). This occurs because the increase of M current conductance exerts a more effective brake on firing, as evidenced by the appearance of hyperpolarizing sags during depolarization (Fig. 13B). Retigabine also decreased the voltage response to hyperpolarizing current pulses, as expected from an increase in cell conductance, but had no effect on action potential size or shape (Fig. 13D).
Fig. 13.

Retigabine abolishes firing in SCG neurons. Voltage responses to depolarizing and hyperpolarizing current pulses recorded from an SCG neuron before (A) and during the application of retigabine (RTG; 3 μm) (B, C). Note in C that larger depolarizing current pulses are required to evoke an action potential in the presence of retigabine. D, Action potential evoked by a 1 msec/700 pA depolarizing pulse in the presence (thick line) and absence (thin line) of retigabine (3 μm).
DISCUSSION
The present experiments show that the previously described property of retigabine to enhance K+currents through heteromeric KCNQ2/3 channels (Main et al., 2000;Rundfeldt and Netzer, 2000; Wickenden et al., 2000) and KCNQ3/5 channels (Wickenden et al., 2001) also extends to currents through homomeric KCNQ2 and KCNQ3 channels, and through the homologous KCNQ4 channels. Available information suggests that the homomeric KCNQ5 channels (Lerche et al., 2000; Schroeder et al., 2000) are also sensitive to retigabine (B. Jensen, personal communication). In contrast, the cardiac KCNQ1 channels, expressed either homomerically or as heteromeric assemblies with their natural cardiac subunit KCNE1, appear to be insensitive to the current-enhancing action of retigabine. Hence, current enhancement by retigabine is confined to neurally expressed KCNQ channels.
In agreement with Wickenden et al. (2000, 2001), we find that the principal action of retigabine is to produce a shift in the KCNQ activation curve to more hyperpolarized potentials. The maximum shift varied with different KCNQ channels in the order KCNQ3 > KCNQ2/3 > KCNQ2 > KCNQ4; the potency of retigabine, as measured by the concentration required to produce a half-maximal shift, also followed the same order. As noted previously for KCNQ2/3 and KCNQ3/5 channels (Main et al., 2000; Wickenden et al., 2000, 2001), this shift is accompanied by a slowing of current deactivation and acceleration of current activation. To an extent, this would be anticipated from the initial descriptions of M-current kinetics (Adams and Brown, 1982) if the channels “sensed” a progressively more hyperpolarized potential. However, this interpretation may be too simple, because the change in kinetics does not quantitatively match that predicted from the original kinetic scheme for a given shift in steady-state activation. M-channel kinetics are clearly more complex than originally envisaged (Marrion, 1997; Selyanko and Brown, 1999), and the kinetics of KCNQ channels per se have not yet been analyzed in sufficient detail to suggest a more precise interpretation of the action of retigabine.
At strongly positive potentials, the action of retigabine on KCNQ channels appears to be complicated by a secondary inhibitory action. This is most clearly seen with KCNQ1 channels, in which there is no voltage shift and hence no current enhancement (Fig. 7), but it probably extends to KCNQ2 and KCNQ2/3 channels, thereby accounting for the limited increase in maximal current through these channels and hence providing some degree of “self-limitation” to the enhancement of currents through these channels. [The maximumPopen through KCNQ2 and KCNQ2/3 channels expressed in CHO cells is well below unity when recorded in the absence of retigabine (Selyanko et al., 2000a; our unpublished observations) and hence does not preclude an increase in maximum current through these channels in the presence of retigabine.] Because the IC50 for inhibition of KCNQ1 channels (∼100 μm) is between 20 and 100 times greater than the EC50 for enhancement of KCNQ2–4 currents, this seems unlikely to provide a major constraint against the potential therapeutic applications of retigabine.
A second important point established in the present experiments is that retigabine also enhances currents through the native M channels in rat sympathetic neurons. As with KCNQ channels, this is caused primarily by a hyperpolarizing shift in the voltage dependence of M-current activation. The calculated shift of −21 mV at 10 μmretigabine was less than that (−34 mV) for the shift of the KCNQ2/3 activation curve. However, the full activation curve of the native M current in SCG neurons could not be determined because of interference by other currents at positive potentials, so the shift was estimated from that component of voltage-dependent current blocked by 10 μm linopirdine. When measured from the enhancement of outward currents at −20 mV, retigabine had a similar potency on native M currents (EC50 0.74 μm) (Fig. 10) as that predicted (∼1 μm) from the shift of the KCNQ2/3 activation curves, and as that reported previously for the enhancement of KCNQ2/3 outward currents (0.34 μm) (Wickenden et al., 2000). Hence, these results provide additional support for the view (Wang et al., 1998) that the native ganglionic M current is carried by KCNQ channels.
Interestingly, the retigabine-enhanced current, although still sensitive to inhibition by muscarinic receptor stimulation, was inhibited less completely than the native current. Because the difference (∼25%) was much less than the proportionate increase in M current at the test potential (−20 mV) produced by retigabine, this cannot be attributed to a complete loss of response of retigabine-modified channels. Instead, it suggests that retigabine affects the channels in such a way as to diminish the extent to which individual M channels are inhibited by muscarinic receptor stimulation, possibly through some form of “allosteric” effect.
From a functional viewpoint, the shift in voltage dependence of the M current has the consequence that, in the presence of 10 μm retigabine, a substantial fraction (30–40%) of the M current became activated at the normal resting potential (approximately −60 mV under perforated-patch recording conditions) (Selyanko et al., 1992). Hence, retigabine induced a substantial outward current, leading to membrane hyperpolarization. In addition, the enhanced M current further dampened the ability of the neuron to generate action potentials during an imposed depolarization and abbreviated the discharge train. If replicated in central neurons, this would provide a satisfactory mechanistic explanation for the reported anti-epileptic action of retigabine.
Footnotes
This work was supported by grants from the UK Medical Research Council, the Wellcome Trust, and Glaxo SmithKline, UK. We thank Dr. Mala Shah for tissue culture, Drs. Derek Trezise and Martin Main (Glaxo SmithKline, Stevenage, UK) for retigabine and helpful discussions, Dr. D. McKinnon for hKCNQ2 and rKCNQ3 cDNAs, Dr. M. T. Keating for hKCNQ1, and Dr. T. J. Jentsch for KCNQ4. We are grateful to Dr. A. A. Selyanko for all his suggestions and discussions.
Correspondence should be addressed to Prof. D. A. Brown, Department of Pharmacology, University College London, Gower Street, London WC1E 6BT, UK. E-mail: d.a.brown@ucl.ac.uk.
REFERENCES
- 1.Adams PR, Brown DA. M-currents and other potassium currents in bullfrog sympathetic neurons. J Physiol (Lond) 1982;330:537–572. doi: 10.1113/jphysiol.1982.sp014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KvLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 3.Bernheim L, Mathie A, Hille B. Characterization of muscarinic receptor subtypes inhibiting Ca2+ current and M current in rat sympathetic neurons. Proc Natl Acad Sci USA. 1992;89:9544–9548. doi: 10.1073/pnas.89.20.9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch T, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- 5.Brown DA. M-currents. In: Narahashi T, editor. Ion channels, Vol 1. Plenum; New York: 1988. pp. 55–99. [DOI] [PubMed] [Google Scholar]
- 6.Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- 7.Delmas P, Abogadie FC, Dayrell M, Haley JE, Milligan G, Caulfield MP, Brown DA, Buckley NJ. G-proteins and G-protein subunits mediating cholinergic inhibition of N-type calcium currents in sympathetic neurons. Eur J Neurosci. 1998a;10:1654–1666. doi: 10.1046/j.1460-9568.1998.00170.x. [DOI] [PubMed] [Google Scholar]
- 8.Delmas P, Brown DA, Dayrell M, Abogadie FC, Caulfield MP, Buckley NJ. On the role of endogenous G-protein βγ subunits in N-type Ca2+ current inhibition by neurotransmitters in rat sympathetic neurons. J Physiol (Lond) 1998b;506:319–329. doi: 10.1111/j.1469-7793.1998.319bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delmas P, Abogadie FC, Buckley NJ, Brown DA. Calcium channel gating and modulation by transmitters depend on cellular compartmentalization. Nat Neurosci. 2000;3:670–678. doi: 10.1038/76621. [DOI] [PubMed] [Google Scholar]
- 10.Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, Brown DA. Differential tetraethylammonium sensitivity of KCNQ1–4 potassium channels. Br J Pharmacol. 2000;129:413–415. doi: 10.1038/sj.bjp.0703086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- 12.Kharkovets T, Hardelin J-P, Safieddine S, Schweizer M, El-Amraoui A, Petit C, Jentsch TJ. KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci USA. 2000;97:4333–4338. doi: 10.1073/pnas.97.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437–446. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- 14.Lamas JA. A hyperpolarization-activated cation current (Ih) contributes to resting potential in rat superior cervical sympathetic neurons. Pflügers Arch. 1998;436:429–435. doi: 10.1007/s004240050653. [DOI] [PubMed] [Google Scholar]
- 15.Lamas JA, Selyanko AA, Brown DA. Effects of a cognition-enhancer, Linopirdine (DuP 996), on M-type potassium currents (IK(M)) and some other voltage- and ligand-gated membrane currents in rat sympathetic neurons. Eur J Neurosci. 1997;9:605–616. doi: 10.1111/j.1460-9568.1997.tb01637.x. [DOI] [PubMed] [Google Scholar]
- 16.Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J Biol Chem. 2000;275:22395–22400. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]
- 17.Main MJ, Cryan JE, Dupere JRB, Cox B, Clare JJ, Burbidge SA. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol. 2000;58:253–262. doi: 10.1124/mol.58.2.253. [DOI] [PubMed] [Google Scholar]
- 18.Malin SA, Nerbonne JM. Elimination of the fast transient in superior cervical ganglion neurons with expression of Kv4.2(W362F): molecular dissection of IA. J Neurosci. 2000;20:5191–5199. doi: 10.1523/JNEUROSCI.20-14-05191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marrion NV. Control of M-current. Annu Rev Physiol. 1997;59:483–504. doi: 10.1146/annurev.physiol.59.1.483. [DOI] [PubMed] [Google Scholar]
- 20.Marrion NV, Smart TG, Marsh SJ, Brown DA. Muscarinic suppression of the M-current in the rat sympathetic ganglion is mediated by receptors of the M1-subtype. Br J Pharmacol. 1989;98:557–573. doi: 10.1111/j.1476-5381.1989.tb12630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mullaney I, Dodd MW, Buckley N, Milligan G. Agonist activation of transfected human M1 muscarinic acetylcholine receptors in CHO cells results in downregulation of both the receptor and the α subunit of the G-protein Gq. Biochem J. 1993;289:125–131. doi: 10.1042/bj2890125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- 23.Rogawski MA. KCNQ2/KCNQ3 K+ channels and the molecular pathogenesis of epilepsy: implications for therapy. Trends Neurosci. 2000;23:393–398. doi: 10.1016/s0166-2236(00)01629-5. [DOI] [PubMed] [Google Scholar]
- 24.Rostock A, Tober C, Rundfeldt C, Barsch R, Engel J, Polymeropoulos EE, Kutscher B, Loscher W, Homack D, White HS, Wolf HH. D-23129, a new anticonvulsant with a broad spectrum activity in animal models of epileptic seizures. Epilepsy Res. 1996;23:211–223. doi: 10.1016/0920-1211(95)00101-8. [DOI] [PubMed] [Google Scholar]
- 25.Rundfeldt C. The new anticonvulsant retigabine (D-23129) acts as an opener of K+ channels in neuronal cells. Eur J Pharmacol. 1997;336:243–249. doi: 10.1016/s0014-2999(97)01249-1. [DOI] [PubMed] [Google Scholar]
- 26.Rundfeldt C, Netzer R. The novel anticonvulsant retigabine activates M-currents in Chinese hamster ovary-cells transfected with human KCNQ2/3 subunits. Neurosci Lett. 2000;282:73–76. doi: 10.1016/s0304-3940(00)00866-1. [DOI] [PubMed] [Google Scholar]
- 27.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 28.Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem. 2000;31:24089–24096. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- 29.Selyanko AA, Brown DA. M-channel gating and simulation. Biophys J. 1999;77:701–713. doi: 10.1016/S0006-3495(99)76925-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selyanko AA, Stansfeld CE, Brown DA. Closure of potassium M-channels by muscarinic acetylcholine-receptor stimulants requires a diffusible messenger. Proc R Soc Lond B Biol Sci. 1992;250:119–125. doi: 10.1098/rspb.1992.0139. [DOI] [PubMed] [Google Scholar]
- 31.Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Delmas P, Buckley NJ, London B, Brown DA. Two types of K+ channel subunit, erg1 and KCNQ2/3, contribute to the M-like current in a mammalian neuronal cell. J Neurosci. 1999;19:7742–7756. doi: 10.1523/JNEUROSCI.19-18-07742.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Selyanko AA, Hadley JK, Brown DA. Single-channel analysis of KCNQ2/3 potassium currents expressed in mammalian (CHO) cells. Soc Neurosci Abstr. 2000a;30:1908. [Google Scholar]
- 33.Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Jentsch TJ, Brown DA. Inhibition of KCNQ1–4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J Physiol (Lond) 2000b;522:349–355. doi: 10.1111/j.1469-7793.2000.t01-2-00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J Neurosci. 2000;20:1710–1721. doi: 10.1523/JNEUROSCI.20-05-01710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, McHarg ML, Gagnon D, Rosales T, Peiffer A, Anderson VE, Leppert M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- 36.Tober C, Rostock A, Bartsch R. D-23129: a potent anticonvulsant in the amygdala kindling model of complex partial seizures. Eur J Pharmacol. 1996;303:163–169. doi: 10.1016/0014-2999(96)00073-8. [DOI] [PubMed] [Google Scholar]
- 37.Wang H-S, Pan Z, Shi W, Brown BS, Wymore R, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 38.Wickenden AD, Yu W, Zou A, Jegla T, Wagoner PK. Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/3 potassium channels. Mol Pharmacol. 2000;58:591–600. doi: 10.1124/mol.58.3.591. [DOI] [PubMed] [Google Scholar]
- 39.Wickenden AD, Zou A, Wagoner PK, Jegla T. Characterization of KCNQ3/5 potassium channels expressed in mammalian cells. Br J Pharmacol. 2001;132:381–384. doi: 10.1038/sj.bjp.0703861. [DOI] [PMC free article] [PubMed] [Google Scholar]