

Abstract
To understand the cellular and in vivo functions of specific K+ channels in glia, we have studied mice with a null mutation in the weakly inwardly rectifying K+ channel subunit Kir4.1. Kir4.1−/− mice display marked motor impairment, and the cellular basis is hypomyelination in the spinal cord, accompanied by severe spongiform vacuolation, axonal swellings, and degeneration. Immunostaining in the spinal cord of wild-type mice up to postnatal day 18 reveals that Kir4.1 is expressed in myelin-synthesizing oligodendrocytes, but probably not in neurons or glial fibrillary acidic protein-positive (GFAP-positive) astrocytes. Cultured oligodendrocytes from developing spinal cord of Kir4.1−/− mice lack most of the wild-type K+ conductance, have depolarized membrane potentials, and display immature morphology. By contrast, cultured neurons from spinal cord of Kir4.1−/− mice have normal physiological characteristics. We conclude that Kir4.1 forms the major K+ conductance of oligodendrocytes and is therefore crucial for myelination. The Kir4.1 knock-out mouse is one of the few CNS dysmyelinating or demyelinating phenotypes that does not involve a gene directly involved in the structure, synthesis, degradation, or immune response to myelin. Therefore, this mouse shows how an ion channel mutation could contribute to the polygenic demyelinating diseases.
Keywords: oligodendrocytes, myelination, inwardly rectifying potassium channels, knock-out mouse, glia, spinal cord
According to classical concepts, inwardly rectifying K+ channels (Kir channels) have two major functions. In excitable cells the Kir channels help to set the resting membrane potential (RMP) without substantially shunting Na+ currents during activity (Hille, 1992). In glial cells the Kir channels may siphon extracellular K+ that is released during activity toward sinks such as capillaries and the vitreous humor. The recent literature suggests that Kir channels also serve a third general function by setting the RMP in glial cells as well, which in turn would govern the transmembrane gradients of many transported molecules. Kir channels therefore would be required for many of the general functions performed by glia, such as cell proliferation, signaling, differentiation, and, in particular, myelination by oligodendrocytes (Arcangeli et al., 1993, 1995; Lepple-Wienhues et al., 1996; Hida et al., 1998). However, the evidence for such a crucial third class of roles has been mainly correlative: Kir channels are upregulated dramatically in postmitotic oligodendrocytes, producing a hyperpolarization (Knutson et al., 1997), and Kir channels are expressed throughout the oligodendrocyte lineage (Barres et al., 1990;Chvatal et al., 1995, 1997; Reimann and Ashcroft, 1999).
There are few specific pharmacological blockers of Kir channels. Therefore, specific proof for a crucial role of Kir channels in glial cells calls for genetic ablation. The Kir4.1 channel subunit appears to be expressed predominantly (Takumi et al., 1995), but perhaps not exclusively (Bredt et al., 1995; Li et al., 2001), in glial cells in the CNS. In the retina, studies imply an important role of Kir4.1 in buffering the increased extracellular K+resulting from electrical activity, primarily by providing a pathway for K+ siphoning (Kofuji et al., 2000). However, we still have only a primitive idea of other distinct roles of the Kir4.1 channel in other CNS regions. The Kir4.1 knock-out mouse (Kofuji et al., 2000) represents the first reported genetic ablation of a constitutively active glial Kir channel, and we show here that its effects include severe motor impairment probably caused by dysmyelination and axonal degeneration. These effects are not so dramatic as the lethal ablation of Kir2.1 (Zaritsky et al., 2000) but are more harmful than ablation of the G-protein-gated Kir 3.2 (Signorini et al., 1997) and Kir 3.4 (Wickman et al., 1998) channels.
MATERIALS AND METHODS
Targeting of the Kir4.1 subunit and PCR analysis. A standard gene-targeting approach was chosen to disrupt Kir4.1 gene expression as described previously (Kofuji et al., 2000). Primers for genotyping included the following: Kir4.1, forward 5′-GAT CTA TGG ACG ACC TTC ATT GAC ATG CAA TGG-3′ and reverse 5′-GGC TGC TCT CAT CTA CCA CAT GGT AGA AAG TCA GG-3′, and neomycin resistance gene, forward 5′-ATC GCC TTC TAT CGC CTT CTT GAC GAG TTC TTC-3′.
Primary cell cultures. Mixed cultures of spinal cord cells were established from postnatal day 0 (P0) to P12 mice. Dissociated spinal cord cells were plated onto 35 mm poly-d,l-lysine-coated glass dishes at 3 × 106 cells per dish in B27-supplemented Neurobasal medium containing 10% fetal calf serum. Cells were subject to either immunocytochemistry or electrophysiological recordings after 10–14 d in culture. For electrophysiology the extracellular solution was (in mm) 150 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 10 d-glucose, pH 7.4. To measure Kir4.1 currents, we changed the external solution to one containing 50 mm KCl in place of 50 mm NaCl. Single-cell recording was performed at 21–24°C. Patch pipettes were filled with a solution containing (in mm) 108 KH2PO4, 4.5 MgCl2, 9 HEPES, 9 EGTA, 4 Mg-ATP, 0.3 Na-GTP, and 14 creatine phosphate, pH 7.4. Pipette resistance ranged from 4 to 7 MΩ when filled with internal solution. Currents were measured with a patch-clamp amplifier (Axopatch 200A, Axon Instruments, Foster City, CA), filtered at 2 kHz, digitized at 10 kHz, recorded on a computer via a commercial software package (pClamp 8, Axon Instruments) and monitored simultaneously on both a storage oscilloscope and a chart recorder. Cells were voltage clamped at a holding potential of −80 mV.
Mature oligodendrocytes were identified on the basis of typical morphology and on electrophysiological properties: biphasic, slowly decaying currents (Chvatal et al., 1995, 1997, 1999) and lack of action potentials. Identity was confirmed by injection of Lucifer yellow with subsequent counterstaining with either myelin basic protein (MBP) or oligodendrocyte-specific protein antibody. Cells were classified as neurons according to their morphology and their electrophysiological characteristics: strong voltage-activated Na+ currents and action potentials elicited by depolarizing current pulses in the current-clamp mode. Series resistance was monitored throughout the experiments, and cells were discarded if the series resistance changed by >10%. The drugs were applied by a local perfusion system. Solution exchange occurred within 2 sec. Values of the resting membrane potentials were obtained at the start of the experiment and corrected for the pipette tip potential. The input resistance and membrane conductance were measured at a membrane potential of −80 mV, using a 10 mV step.
Immunohistochemistry and immunofluorescence. Animals were anesthetized with halothane and cardioperfused with PBS, followed by 4% paraformaldehyde (PFA) in PBS. Spinal cords were removed and post-fixed in 4% PFA overnight. Organs were embedded in paraffin and sectioned at 8–10 μm. For immunohistochemistry the paraffin sections were dewaxed in xylene, rehydrated in ethanol, and boiled for 2 min in an antigen-demasking solution (Vector Laboratories, Burlingame, CA). For cell culture staining the cells were fixed in 4% PFA for 20 min. After being blocked for 15 min in 0.2% Triton X-100 and 10% normal goat serum (NGS) in PBS, the samples were washed and incubated with the primary antibody in 1% NGS at 4°C overnight. Samples were incubated with a biotinylated secondary antibody, followed by an avidin-conjugated fluorochrome (Vector Laboratories) or a fluorescent-conjugated (Cy-3, Alexa 488) secondary antibody (Jackson ImmunoResearch, West Grove, PA) for 1 hr.
The anti-Kir4.1 polyclonal antibody was raised in rabbits and tested in HEK 293 and COS-7 cells transfected with the rat Kir4.1 subunit as described previously (Kofuji et al., 2000). The following antibodies also were used in this study: rat anti-glial fibrillary acidic protein (GFAP; Zymed, San Francisco, CA); mouse anti-oligodendrocyte-specific protein (Chemicon, Temecula, CA); rat anti-MBP (Chemicon); rabbit anti-myelin proteolipid protein (PLP; Chemicon); mouse anti-neurofilament, 70 kDa (Chemicon); mouse anti-2′3′-cyclic nucleotide 3′-phosphohydrolase (CNPase; SMI-91, Sternberger Monoclonals, Lutherville, MD). Confocal images of tissue sections and cultured cells were obtained on a Zeiss LSM 410 microscope (Oberkochen, Germany) equipped with argon (red), HeNe (green), and UV lasers.
For quantitative morphological studies, spinal cord cultures of PLP-positive cells growing in isolation on the culture dish were visualized on a Nikon epifluorescence microscope (20× objective) equipped with a CCD camera. Individual images were taken at random, digitized via a Snappy frame grabber, and analyzed by the NIH Image program to obtain a one-pixel boundary of the entire cell. Oligodendrocytes expressing a typical myelin sheath were clearly distinguishable from myelin sheath-negative oligodendrocytes. The cells were counted, and myelin sheath-positive cells were expressed as a percentage of the total oligodendrocyte count. The area of the cell soma disregarding branches was measured and calculated by NIH Image (Morley et al., 1997). “Branching” of PLP-positive cells was assessed by individually counting major branches directly extending from the oligodendrocyte cell body, and the average number of branches/cells was calculated. Analyses were verified by a second observer who was unaware of the genotype.
To assay apoptosis, we used terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) to visualize cells with fragmented DNA (In situ cell death detection kit, Fluorescein, Roche Molecular Biochemicals, Palo Alto, CA). The procedure was performed according to the manufacturer's instructions. Sections were pretreated with 0.1% Triton X-100 and subsequently were incubated for 1 hr at 37°C with 50 μl of the provided TUNEL mixture. Positive controls were treated additionally with 500 ng/ml DNase diluted in PBS with 1 mm MgCl, pH 7.4, for 10 min before the TUNEL mixture was added. Then the cells were washed three times in PBS and evaluated under a fluorescence microscope. Nissl counterstaining was performed with the NeuroTrace kit (Molecular Probes, Eugene, OR).
Electron microscopy. Spinal cords were dissected; the sections were post-fixed in 1% phosphate saline-buffered osmium tetroxide containing 0.8% KFeCN and dehydrated through an ethanol series. The tissue was cleared in propylene oxide and embedded in epoxy. Ultrathin sections were collected every 800 Å, stained with uranyl acetate and lead citrate, and examined with a Philips electron microscope at 80 kV.
Magnetic resonance imaging. Magnetic resonance imaging was performed on PFA-cardioperfused animals at 8°C with an 11.7 Tesla, vertical bore (89 mm) Bruker AMX500 microimaging system (Bruker Instruments, Billerica, MA). We used an Acustar shielded gradient set with 25 mm birdcage coil. Images were recorded by using a diffusion-weighted two-dimensional multislice echo protocol with a data matrix of 512 × 512 points, TE = 1500 msec, TR = 21, and 16 averages. Slice thickness was 0.2 mm in all cases, with an in-plane field of view 2 × 2 cm for the transverse slices and 3 × 2 cm for the sagittal slices. The diffusion-sensitizing gradient pulses were applied symmetrically about the refocusing RF pulse, directed along the long axis of the animal (perpendicular to the transverse slices), with an intensity of 46 G/cm, gradient width of 1 msec, and separation of 10.5 msec. Slices shown thus are moderately T2-weighted and heavily diffusion-weighted.
RESULTS
Motor impairment, tremor, and premature death in mice lacking Kir4.1
Mice heterozygous for the Kir4.1 deletion were viable and fertile and showed no readily observable pathological behavior. No striking differences were observed between the homozygous Kir4.1−/− mutants and control littermates at birth. Genotyping by Southern blot hybridization or by PCR established that homozygotes for the Kir4.1 deletion were born at the expected frequency from heterozygous matings. However, by P8-P10 the Kir4.1−/− mice stopped gaining body weight (Fig. 1A) and showed progressive weakness. By P10-P12 the Kir4.1−/− mice could be identified by their smaller size, decreased body weight, and dehydrated appearance. At this stage the Kir4.1−/− mice showed severe defects in controlling voluntary movements, posture, and balance. These defects increased in severity over subsequent days. This Kir4.1−/− phenotype resulted in frequent falls and rollovers, and the mice regained the upright position with difficulty (Fig. 1C). Additionally, Kir4.1−/− mice dragged their hind limbs, a phenotype consistent with paralysis. We next asked whether Kir4.1−/− mice had impaired ability to perform motor tasks. Kir4.1−/− mice were unable to climb a grid at an 80° angle; they simply fell off. WT littermates performed this task easily, scaling the grid within 1 min. As development proceeded, the Kir4.1−/− mice deteriorated further; by the third postnatal week a body tremor was observed.
Fig. 1.

Kir4.1−/− mice show a severe neurological phenotype and are less viable than wild-type mice. A, Postnatal body weight in WT, Kir4.1+/−, and Kir4.1−/− mice. Note that after P8 the Kir4.1−/− mice gain little weight.B, Survival rate. Kir4.1−/− mice are indistinguishable from WT littermates at birth. Kir4.1−/− mice, however, die by P24, even when the mice are hand-fed or are cared for by a foster mother.C, Kir4.1−/− mice show severe motor impairment resulting in frequent falls. The picture series illustrates a fall of a Kir4.1−/− mouse at P12.
The first deaths of Kir4.1−/− mice were at P8, and there was 100% mortality at P24 (Fig. 1B). Thus at a systems level, loss of the Kir4.1 channel causes motor impairment, loss of voluntary movement, tremor, and ultimately death.
Hypomyelination, vacuolation, and axonal swelling in spinal cord of Kir4.1−/− mice
What is the cellular basis of the observed whole animal phenotypes? The diameter of the spinal cord was similar in the Kir4.1−/− mice and control littermates. However, at P9 the Kir4.1−/− mice showed massive spongiform vacuolation throughout the whole spinal cord, predominantly in the inner areas of the white matter adjacent to the gray matter (Fig.2A,B). There were also defects, such as shrunken motoneuron cell bodies, in some gray matter areas (Fig. 2D). By P18 vacuolation had progressed in inner areas of the white matter, separating the white matter from the gray matter, but leaving the outermost white matter areas relatively well preserved (data not shown). Staining for MBP was reduced markedly in cross sections of the white matter of the spinal cord (Fig.2E,F). The reduction was most striking in the most affected areas adjacent to the gray matter, whereas MBP staining was relatively well preserved in the outermost areas of the white matter. A similar distribution was obtained when sections were stained for the axonal marker neurofilament (70 kDa), indicating a loss of axons (Fig. 2G,H).
Fig. 2.

Lack of Kir4.1 leads to spongiform vacuolation in the spinal cord. A–D, Hematoxylin–eosin staining of thoracic spinal cord sections from P9 mice. In Kir4.1−/− mice vacuoles of various sizes are present in both the white and the gray matter, and some motoneuron cell bodies appear shrunken (D). White matter areas adjacent to the gray matter are affected the most severely. C, D, Theinsets of A and B.E, F, Myelin basic protein (MBP) expression in the developing spinal cord. Confocal images of adjacent sections show immunohistochemical localization of MBP (green). In WT mice a dense MBP signal is detected predominantly in the white matter (left). In Kir4.1−/− mice MBP staining is reduced drastically in the vacuolated areas, whereas in more distal areas of the white matter the MBP immunoreactivity is better preserved.G, H, Immunohistochemical detection of neurofilament. In WT sections strong immunoreactivity for neurofilament (red) was present in the white matter for the labeling of axonal structures, and weak labeling was observed in the gray matter on large neuronal cell bodies representing motoneurons. In Kir4.1−/− sections the neurofilament immunoreactivity was reduced in the white matter, remaining relatively well preserved in neuronal cell bodies.lf, Lateral funiculus; vf, ventral funiculus; df, dorsal funiculus; vh, ventral horn of gray matter. Scale bars: A, B, 250 μm;C–H, 60 μm.
We used magnetic resonance imaging (MRI) to reveal the spatial extension of the lesions. High-field MRI is a sensitive technique to detect a variety of CNS pathologies. It is specifically well suited for white matter changes caused by demyelination, vacuolation, or inflammation, which can be detected in T2-weighted images as hyperintense areas. At P12, using sagittal tomographic sections of Kir4.1−/− animals, hyperintense lesions were detected throughout the whole spinal cord including brainstem areas (Fig.3A,B). Tomographic slices from upper brain from these animals showing parts of the cerebellum, midbrain, and cortical regions show that these areas are not affected in the Kir4.1−/− mice at this time point of development (Fig.3C,D). Transverse sections of the cervical spinal cord at P12 show respective lesions affecting predominantly white matter areas (Fig. 3E,F). At P18, signal intensity appears to be less pronounced in the white matter, and a hypointense signal similar to the WT sections is detected in the outermost areas of the spinal cord, indicating that to some extent myelination is preserved or remyelination occurs (Fig. 3G,H).
Fig. 3.
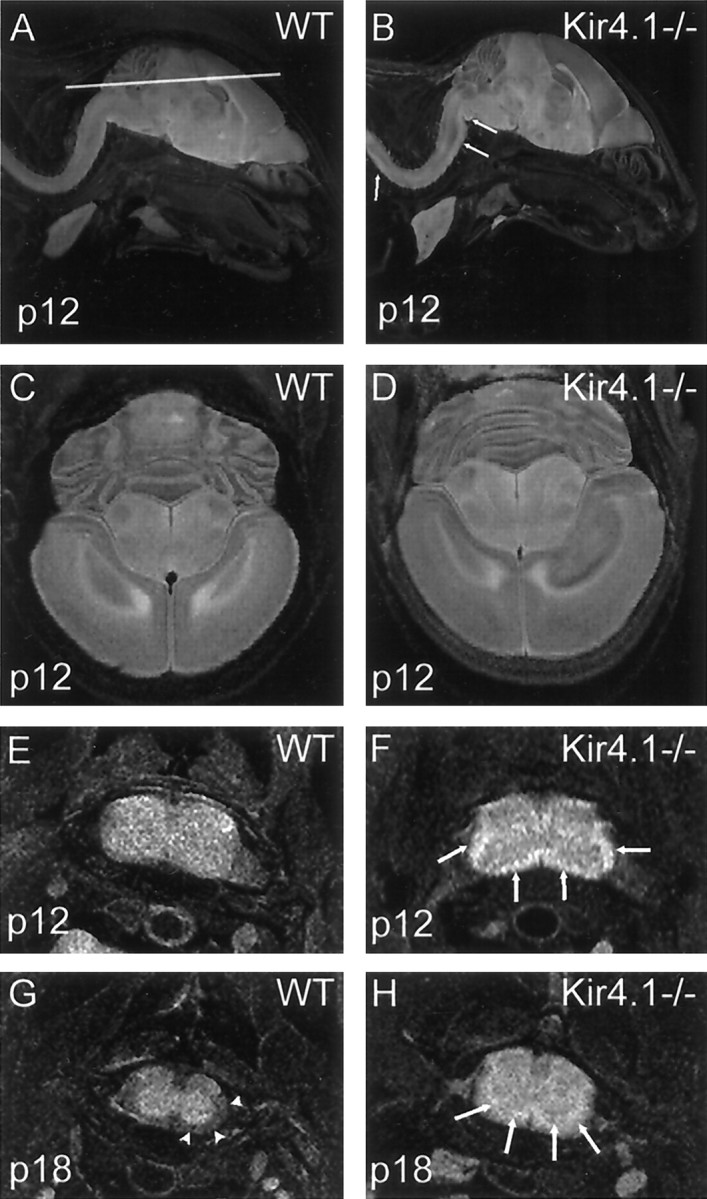
Anatomical MR images from fixed WT and Kir4.1−/− mice at P12 and P18. A, B, Sagittal tomographic slices from P12 animals. Vacuolation is observed throughout the spinal cord and in the brainstem. Lesions appear as regions of hyperintensity in the white matter (arrows in Kir4.1−/− sections). Note that the lesions extend to brainstem areas. C, D, Tomographic slices taken in the plane shown in A from these animals reveal parts of the cerebellum, midbrain, and cortical regions. Note that these areas are not affected in the Kir4.1−/− mice at this point of development. E, F, Transverse images from the same animals of the cervical spinal cord with the same imaging modalities. Arrows point at hyperintense lesions in Kir4.1−/− animals. Lesions include most of the white matter.G, H, At P18, hyperintense lesions (arrows) in Kir4.1−/− sections appear less pronounced than at P12. Additionally, a hypointense signal in the outermost white matter areas is detected. Note that in WT mice at P18 normal myelination has progressed, leading to a hypointense signal in WT spinal cord white matter (arrowheads).
At an ultrastructural level, numerous round and oval vacuoles of varying sizes were detected in the white matter of Kir4.1−/− mice (Fig. 4B), but not in control mice (Fig. 4A). We could not discern the internal structure of very large vacuoles, but smaller vacuoles contained membranous elements of unknown origin and aberrant myelin sheaths (Fig. 4B). In some cases thin myelin sheaths remained, but no axonal structures were discerned. In cases in which axons were preserved, compaction of myelin sheaths was incomplete, showing loosely attached layers of myelin (Fig. 4D). The major dense line of these myelin sheaths present in WT mice (Fig.4C) was indistinct in some areas in Kir4.1−/− mice (Fig.4D).
Fig. 4.
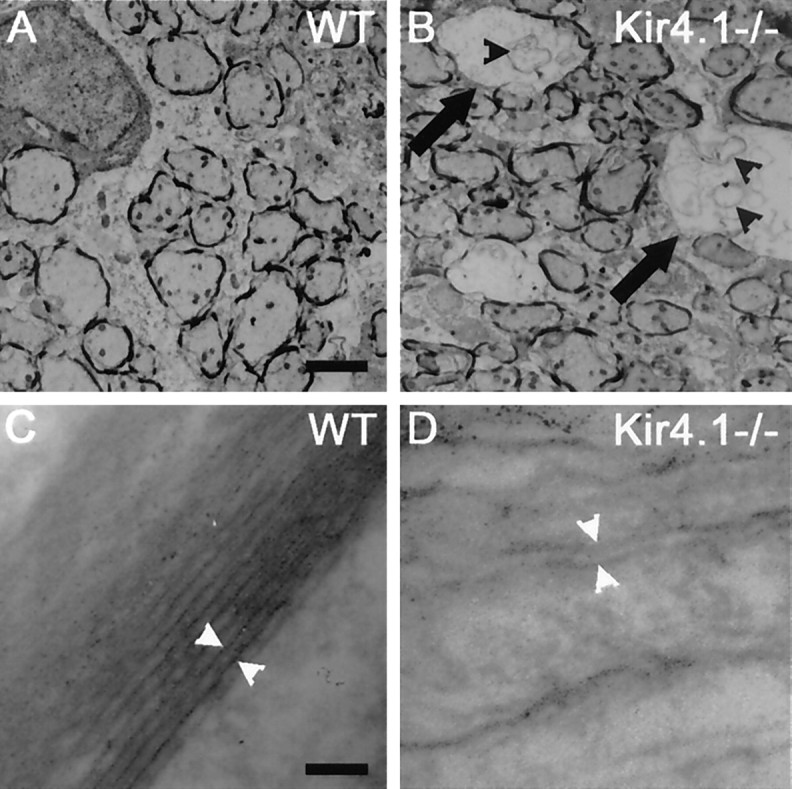
Electron micrographs in spinal cord sections from WT and Kir4.1−/− mice. Electron microscopy shows that dysmyelination is accompanied by axonal degeneration. All images are taken from the white matter of the thoracic spinal cord at P12. A, B, Various stages of dysmyelination in a Kir4.1−/− mouse are depicted inB. A less-affected area in the outermost region of the white matter is shown. Middle size vacuoles (arrows) are surrounded by thin myelin sheaths and contain aberrant myelin sheaths and membranous elements (black arrowheads).A, Control animal. C, Major dense lines of a WT spinal cord (white arrowheads).D, In Kir4.1−/− sections major dense lines were barely detected, and myelin sheaths were attached loosely, indicating a failure in myelin compaction. Scale bars: A, B, 1 μm;C, D, 0.01 μm.
The gray matter contained mainly small vacuoles. When examined by confocal microscopy, these were generally closely related to or contained nuclei, indicating cell death (data not shown). TUNEL staining in spinal cord from P9 Kir4.1−/− mice revealed specific labeling of small nuclei in the gray matter and in distal areas of the white matter, indicating apoptosis of glial cells (Fig.5A–C). However, only very few large motoneurons were TUNEL-positive (Fig. 5D–F). Throughout the observation period (P9, P12, P18) neuron numbers appeared to be similar to those in control littermates. Neither inflammatory cells nor extensive phagocytotic activity was detected in these sections.
Fig. 5.

Apoptosis in spinal cord from Kir4.1−/− mice.A–C, Double-labeling of a spinal cord section of a Kir4.1−/− mouse (at P9) with Nissl (red;A) and TUNEL (green;B). Nissl strongly labels cytoplasm of large motoneurons located in the gray matter, contrasting the nuclei staining of glial cells in gray as well as in white matter. Note in the overlay (C) that predominantly small nuclei (glial nuclei) are TUNEL-positive. Additionally, most TUNEL-positive cells are located in white matter areas. D–F, Higher power magnification (inset of A) of part of the ventral horn of the gray matter shows that most large motoneurons are TUNEL-negative (arrows; closed arrowheadpoints to two weakly positive motoneurons), whereas small nuclei are strongly TUNEL-positive (open arrowheads), indicating that glial cells predominantly are undergoing apoptosis.G–I, Comparable WT section of the gray matter of the ventral horn. Sections were pretreated with DNase to induce DNA breakage. Small glial nuclei as well as large nuclei of motoneurons are strongly TUNEL-labeled (H).I, Overlay (arrows point to double-labeled large motoneurons). Scale bars: A–C, 200 μm; D–I, 50 μm.
Thus at the cellular level the Kir4.1−/− mice have hypo/dysmyelination, extensive vacuolation, and apoptosis affecting glial cells. Failure of myelin compaction is observed, along with damage to axonal structures and axon degeneration. These data provide a plausible cellular framework for the phenotypes that have been described above: motor impairment, tremor, and premature death.
Kir4.1 expression in the postnatal mouse spinal cord
A lack of Kir4.1 has major effects on behavior and on spinal cord physiology (below), but where is Kir4.1 expressed in the spinal cord? At P9, Kir4.1 expression was detected predominantly in gray matter of the spinal cord and in certain nuclei of the brainstem, such as cochlear nucleus and spinal trigeminal nucleus. With immunostaining, we observed high levels of Kir4.1 expression in gray matter areas of the spinal cord, especially in the ventral horn surrounding large motoneurons (Fig.6A,B). Kir4.1-positive cells also were detected in the white matter (Fig.6D–F), with a less intense signal than in gray matter. Kir4.1-positive cells in white matter also were labeled with antibody to MBP, indicating that they are oligodendrocytes. At P18, Kir4.1 immunolabeling appeared to be weaker in general, but the pattern of more intense staining in gray matter continued (data not shown). Kir4.1 expression was not apparent by immunohistochemistry on sections of Kir4.1−/− mouse spinal cord or brainstem at P9 and P18, showing that the mutation successfully eliminated Kir4.1 protein expression (Fig. 6C). Cell cultures of spinal cord (P0-P12) WT mice were immunopositive for the Kir4.1 subunit on oligodendrocyte-specific protein-positive cells (Fig.6G–I) as well as on CNPase-positive cells (Fig.6J–L). These results confirm earlier findings that Kir4.1 is expressed at various stages of the oligodendrocyte lineage (Poopalasundaram et al., 2000). We tested whether the Kir4.1 subunit is expressed on spinal cord neurons or GFAP-positive astrocytes. No antibody staining was detected on neurofilament-positive neurons or GFAP-positive astrocytes (Fig. 6M–R).
Fig. 6.

Regional and cellular expression of Kir4.1 channel subunit in spinal cord sections and cultures as revealed by immunostaining. A–C, In cross sections of P9 mice, Kir4.1 (red) is expressed predominantly in the gray matter. No immunolabeling is detected in Kir4.1−/− sections, showing that the mutation successfully disrupted Kir4.1 gene expression.B, The inset of A.D–F, Immunostaining of longitudinal sections of WT mice spinal cord white matter. Kir4.1 labeling (red) was detected on cell bodies (D), but not on myelin sheaths stained for MBP (green;E). Overlay of confocal images is illustrated inF. G–I, Distribution of Kir4.1 in spinal cord mixed cell cultures. A strong Kir4.1 signal (red) is detected on cells (G) additionally labeled for oligodendrocyte-specific protein (green;H). The Kir4.1 antibody revealed predominantly membrane- and cell body-localized staining, whereas anti-oligodendrocyte-specific protein mainly stained cell cytoplasm.I, Overlay of confocal images. J–L, Kir4.1 is expressed in developing oligodendrocytes. Shown is labeling of an oligodendrocyte with Kir4.1 antibody (green; J) in spinal cord culture of neonatal mice. An identical cell is stained with CNPase antibody (red; K).L, Overlay of confocal images. M–O, Kir4.1 is not colocalized on spinal cord neurons in culture.M, Kir4.1 staining (green) on typical oligodendrocyte in close proximity (and partly overlapping) to NF-positive nerve processes (red;N). O, Overlay of confocal images.P–R, No immunolabeling with the Kir4.1 antibody was detected on GFAP-positive astrocytes. P, Bright-field images show two typical oligodendrocytes and one stellate astrocyte (arrow). Oligodendrocytes are stained heavily with the Kir4.1 antibody (green), revealing typical morphological characteristics like globular structures (Q). GFAP-positive astrocytes (red) are not stained with the Kir4.1 antibody (R). Scale bars: A, 250 μm; B, C, 30 μm; D–I, 20 μm;J–L, 40 μm; M–O, 50 μm;P–R, 60 μm.
Cultured oligodendrocytes from Kir4.1−/− mice are depolarized and morphologically immature
Our data indicate that Kir4.1 is a major K+ channel in oligodendrocytes, but what happens to the properties of these cells in the Kir4.1−/− mice? The electrophysiological properties of WT versus Kir4.1−/− oligodendrocytes were analyzed in spinal cord cell cultures (P8-P12). Cells were loaded with Lucifer yellow via the patch pipette and subsequently immunostained with an anti-MBP or oligodendrocyte-specific protein antibody. We measured current–voltage (I–V) relations for these cells at test potentials in 15 mV increments over the voltage range from −150 to +60 mV, in both Na+ and 50 mmK+ (Fig.7A–C). We isolated the currents caused by inwardly rectifying K+channels by subtracting records taken during Ba2+ (1 mm) blockade (Fig. 7B,C). The data, summarized in the Table1, show a robust inward rectifier K+ current (84.1 pA/pF ± 26.3 at −150 mV) in WT cells.
Fig. 7.

Electrophysiological properties of WT and Kir4.1−/− oligodendrocytes in cell cultures from the developing spinal cord. A, Voltage protocol. Membrane potential was held at −80 mV and jumped to test potentials between −150 and + 60 mV at 15 mV increments. B, Whole-cell voltage clamp of WT oligodendrocytes in Na+ recording solution, in 50 mm extracellular K+ solution, and in 50 mm K+ plus 1 mmBa2+. C, Whole-cell voltage clamp of Kir4.1−/− oligodendrocytes in Na+ recording solution, in 50 mm extracellular K+solution, and in 50 mm K+ plus 1 mm Ba2+. D, Representative I–V relations of a WT oligodendrocyte in Na+ recording solution, in 50 mmextracellular K+ solution, and in 50 mmK+ plus 1 mm Ba2+.E, Kir currents were isolated by subtracting traces in 50 mm extracellular K+ solution from the traces that were obtained in 50 mm K+plus 1 mm Ba2+. Note the weakly inwardly rectifying properties of Kir4.1 (mean ± SEM;n = 5).
Table 1.
Electrophysiological properties of oligodendrocytes
| WT (9) | +/− (8) | −/− (17) | |
|---|---|---|---|
| Current (−pA/pF) | 84.1 ± 26.3 | 26.3 ± 7.5* | 3.6 ± 1* |
| RMP (−mV) | 59.7 ± 7.6 | 40.1 ± 8.9 | 34.3 ± 3* |
| Membrane conductance (nS) | 25.5 ± 9.3 | 10.1 ± 3.3* | 4.4 ± 0.7* |
Cell culture recording on myelin-expressing oligodendrocytes. K+ currents were measured at −150 mV. Data are given as mean ± SEM. The number in parentheses indicates the number of patches in whole-cell voltage-clamp configuration.
Statistically different from WT (unpaired Student'st test, two-tailed; p < 0.002).
Kir4.1−/− oligodendrocytes exhibited a dramatic electrophysiological difference: inwardly rectifying K+currents were completely absent (Fig. 7C). The summarized data (Table 1) show that heterozygotes had an intermediate level of K+ current. The resting membrane potential in WT oligodendrocytes was −59.7 ± 7.6 mV (n = 9), but Kir4.1−/− oligodendrocytes showed significantly depolarized values of −34.3 ± 2.9 mV (n = 17;p < 0.001), and heterozygotes had an intermediate value. Whole-cell membrane conductance was reduced significantly in Kir4.1−/− (4.4 nS) compared with WT oligodendrocytes (25.5 nS;p < 0.002), again with an intermediate value for heterozygotes. These data prove that the major K+ channel in oligodendrocytes contains the Kir4.1 subunit. Kir4.1 contributes to the membrane potential by providing the dominant K+ conductance, and these properties are compromised severely in Kir4.1−/− mice.
We observed marked morphological differences of Kir4.1−/− oligodendrocytes in spinal cord cultures (P8-P12). Mature oligodendrocytes of WT cultures showed multiple extended branches and formed cell–cell interactions with astrocytes (Fig.8A,E), neurons, and other oligodendrocytes. After 1 week in culture, differentiated oligodendrocytes expressed myelin proteins and were immunopositive for MBP. Additionally at that stage, these cells elaborated large membrane sheaths that mimic the appearance of unfurled myelin sheaths (Fig.8C; Yang and Skoff, 1997). Mature oligodendrocytes formed ring structures around distal branches and partly myelinated closely located cells, indicating in vitro myelination.
Fig. 8.
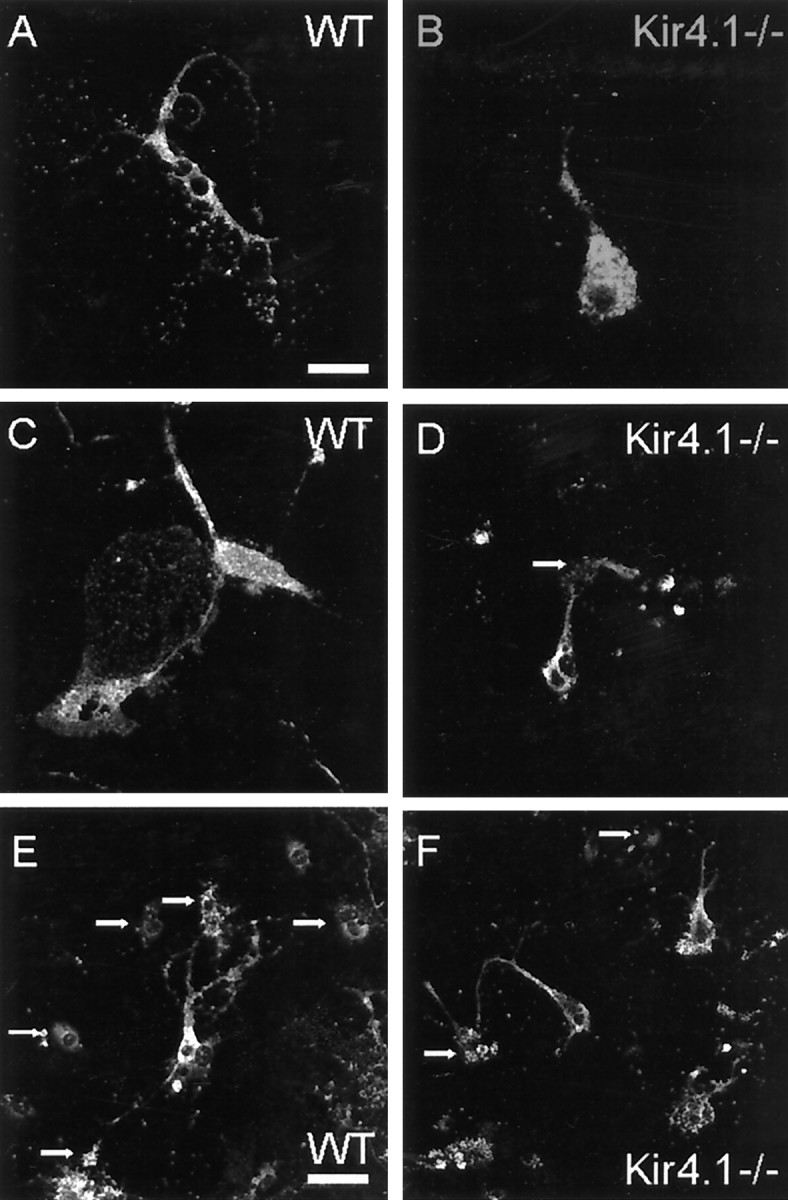
Kir4.1−/− oligodendrocytes in culture display morphological abnormalities. Shown are confocal images of cells from a WT mouse and a Kir4.1−/− littermate. A, C, E, Spinal cord cells from WT mice at P9 were cultured for 14 d. Immunostaining for proteolipid protein (PLP) reveals myelin-expressing oligodendrocytes. In WT cultures mature oligodendrocytes have multiple large processes that elaborate membrane sheaths (A, C). Note also numerous small globular structures at the tip of the branches (A) and the interaction with other, partially myelinated, cells (indicated by arrows inE). B, D, F, In Kir4.1−/− cultures oligodendrocytes show strong immunofluorescence for PLP but display a range of morphological differences from WT cells. B, Exemplar oligodendrocyte with one thick branch and a few thin processes ending in a few globular structures. No membrane sheaths are elaborated. D, Myelin-expressing Kir4.1−/− oligodendrocytes stand in contact with only a few neighboring cells (indicated by arrows in D, F).F, Several Kir4.1−/− oligodendrocytes displaying only one thick process and a few thin branches. Membrane sheaths are not elaborated. Scale bars: A–C, 25 μm;D–F, 50 μm.
In Kir4.1−/− cell cultures we observed no obvious differences in total numbers of myelin-expressing oligodendrocytes. However, Kir4.1−/− oligodendrocytes appeared less mature than WT oligodendrocytes. This point was quantified in cultures from P9 mice grown for 12 d and subsequently immunolabeled for myelin proteolipid protein (PLP) as a marker for myelin-producing oligodendrocytes. The number of oligodendrocytes elaborating membrane sheaths was reduced dramatically in Kir4.1−/− cultures (only 16% of 44 Kir4.1−/− cells had sheaths vs 74% of 73 WT cells). In contrast to WT cells, Kir4.1−/− oligodendrocytes were round (Fig.8B,D) and displayed greater somatic area than WT oligodendrocytes (566.3 ± 40 vs 254.4 ± 25.1 μm2, mean ± SEM; n= 23 and 19, respectively), displayed fewer major branches (average of 2.7 ± 0.03 vs 4.0 ± 0.04, mean ± SEM;n = 35 cells in each case), and showed fewer interactions with other cells (Fig. 8D,F).
Kir 4.1−/− neurons from spinal cord cultures are normal
Qualitative immunostaining did not show expression of Kir4.1 on neurons in the spinal cord. To address this question quantitatively, we analyzed inward rectifying potassium currents in spinal cord neurons of WT and Kir4.1−/− postnatal mice (P0-P12) cultured for up to 2 weeks. In experiments on cells with large somata from WT and Kir4.1−/− mice, a current injection produced action potentials in both cells (Fig.9E), as expected for neurons. In voltage-clamp mode these cells displayed transient inward currents and sustained outward currents in response to voltage steps from −150 to +60 mV (Fig. 9B,C). Increased extracellular K+ concentration evoked only modest increases in the inward currents (Fig. 9B,C). After 2 weeks in culture, Ba2+-sensitive K+ currents in response to 50 mm extracellular K+were upregulated substantially but were not different among WT, Kir4.1+/−, or Kir4.1−/− cells (Fig. 9D). These data confirm that the Kir4.1 channel subunit is not expressed in spinal cord neurons.
Fig. 9.
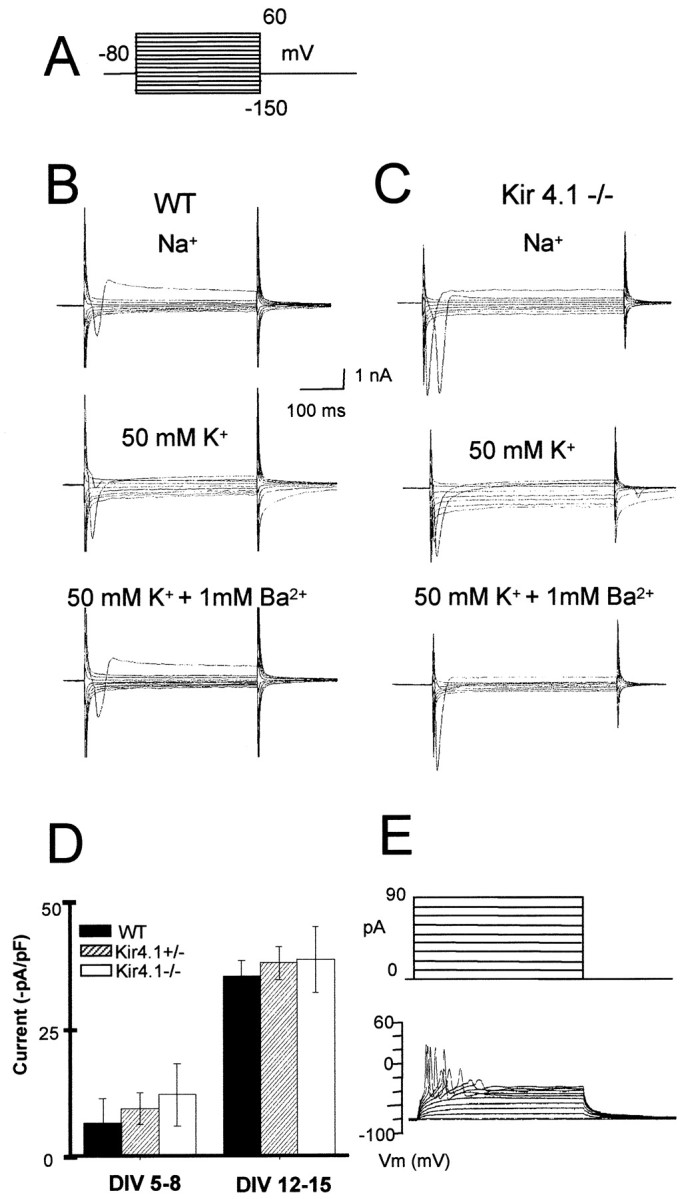
Neurons in the developing spinal cord express inwardly rectifying K+ channels other than Kir4.1.A, Voltage protocol. B, Whole-cell voltage clamp on large neuronal cell bodies of postnatal spinal cord cultures. Only traces for inward currents are depicted. A small inward current is activated with hyperpolarization in a sodium-containing solution. Inward currents augmented in 50 mm extracellular K+ solution are blocked by the addition of 1 mm Ba2+. C, Inward currents of an exemplar neuron of a Kir4.1−/− culture in Na+, and the effect of 50 mmK+ and 50 mm K+ plus 1 mm Ba2+. D, Bar graphs representing barium-sensitive inward currents induced by 50 mm external K+ to neurons from cultures prepared from P0 mice at 5–8 d in vitro (DIV 5–8; WT, n = 4; Kir4.1+/−,n = 9; Kir4.1−/−, n =15) versusDIV 12–15 (WT, n = 4; Kir4.1+/−,n = 7; Kir4.1−/−, n = 8).E, Cells were identified as neurons by injecting increasing depolarizing pulses in the current-clamp mode. These depolarizations elicited action potentials. The pattern of current pulses is indicated in the inset above the traces. Error bars represent SEM.
DISCUSSION
The Kir4.1 knock-out mouse (Kofuji et al., 2000) became the first reported genetic ablation of a constitutively active Kir channel, and the present study is the first demonstration that deletion of an ion channel affects CNS myelination, although previous reports show that mutations of connexin 32 can lead to the peripheral demyelinating disease Charcot–Marie–Tooth syndrome (Oh et al., 1997). Indeed, Kir4.1 is one of the few genes that encode neither a myelin protein, a protein of myelin synthesis/degradation, nor a protein involved in the immune response to myelin, yet still lead to CNS demyelination or dysmyelination when ablated.
The present study also outlines the pathophysiology of this process. We show that Kir4.1 underlies the principal K+ conductance in oligodendrocytes of the developing spinal cord, that Kir4.1 is probably not expressed in neurons or astrocytes in spinal cord before P12, and that ablating Kir4.1 leads to severe physiological and cellular effects, eventually disrupting the axon–glia interaction. These events lead to a hypomyelination and spongiform vacuolation in the spinal cord, predominantly affecting the white matter and neuropil with severe axonal pathology. The result is a severe neurological phenotype: motor coordination deficits and hindlimb paralysis. The additional early postnatal death of Kir4.1−/− mice may be explained by impaired feeding behavior and also by progressive dysmyelination and vacuolation of vital brainstem areas.
Although a pioneering study of Kir4.1 (Bredt et al., 1995) and a more complete recent study (Li et al., 2001) found evidence for expression in neurons, all authors agree that Kir4.1 is expressed predominantly in glial cells (Takumi et al., 1995; Kofuji et al., 2000; Poopalasundaram et al., 2000). We cannot rule out the possibility that Kir4.1 is expressed additionally in some spinal cord neurons, perhaps in adult mice, and that deletion of Kir4.1 directly causes neuronal pathology, but our study on mice younger than P12 produced no evidence to support such a process.
Implications of Kir4.1 localization in the spinal cord
Our study reveals that Kir4.1 exerts an important role in the development of the spinal cord. The restricted temporal and spatial expression pattern of Kir4.1 is appropriate for such a role: Kir4.1 was found predominantly in gray matter areas. In cross and longitudinal sections, additional labeling on oligodendrocytes within white matter areas also was detected, indicating that the channel is present in the white matter in substantially lower levels. We do not yet know the mechanism for this spatial distribution of Kir4.1 expression. First, Kir4.1 could be upregulated in a specific subset of oligodendrocytes in the gray matter or, second, downregulation could take place once oligodendrocytes reach white matter areas.
How is differential spatial Kir4.1 expression linked to its functional role? Our finding that Kir4.1 is strongly expressed in glia cells that surround motoneurons in gray matter supports the hypothesis that Kir channels of oligodendrocytes serve the second classical function, siphoning of extracellular potassium concentration away from neuronal compartments in the spinal cord in vivo (Takumi et al., 1995; Poopalasundaram et al., 2000). This role is similar to the role played by Kir4.1 in the retinal Müller cells (Kofuji et al., 2000). Our methods lack the sensitivity to decide whether Kir4.1 is expressed at the glial folds surrounding the nodes of Ranvier and therefore would participate in clearing K+away from this restricted space (Mi et al., 1996).
General roles for Kir4.1-containing channels in oligodendrocytes
The present study also provides the most direct evidence that a channel containing Kir4.1 serves the general function of setting the RMP in glial cells as well. The RMP in turn governs the transmembrane gradients of many transported molecules and therefore is required for many general processes, including signaling, differentiation, and, in particular, myelination by oligodendrocytes. Because Kir4.1 hetero-oligomerizes with other Kir subunits in vitro (Xu et al., 2000; Yang et al., 2000), we believe it likely that the channels contain Kir4.1 in association with other Kir subunits.
The present highly specific results are a satisfactory extension of previous studies that nonspecific pharmacological blockade of Kir channels, or depolarization by increasedKo, affects oligodendrocyte proliferation and development in vitro (Gallo et al., 1996;Chvatal et al., 1997). We determined that Kir4.1-containing channels are the principal inwardly rectifying potassium channels in myelin-expressing oligodendrocytes in the spinal cord: gene ablation abolishes potassium currents in Kir4.1−/− cells and leads to a depolarized membrane potential. Kir4.1−/− oligodendrocytes do not elaborate membrane sheaths, have large somatic area and display fewer branches, and rarely show interaction with other cells. Thus they reveal an immature morphology in vitro. This suggests that Kir4.1 regulates crucial steps during the maturation of oligodendrocytes. Most importantly, Kir4.1−/− oligodendrocytes fail to form compacted myelin in Kir4.1−/− mice.
Furthermore, spinal cord glial cells in Kir4.1−/− mice undergo apoptotic cell death. Whether the observed apoptosis is linked to the depolarized RMP with subsequent activation of apoptosis-inducing caspases in vivo has to be determined. However, this apoptosis is consistent with the proposed mechanism for the observed pathology in vivo.
The events that lead to recognition and ensheathment of axons by oligodendrocytes are not well understood but probably include molecular signaling in both directions (Vartanian et al., 1997; Barres and Raff, 1999; Niehaus et al., 1999). Astrocytes also appear to align oligodendrocyte processes with axons and to adhere to axons (Meyer-Franke et al., 1999). In addition, cues from actively firing axons seem to be crucial for the maturation of oligodendrocytes and for the induction of myelination in vitro (Barres and Raff, 1993; Demerens et al., 1995). Our electron microscopic findings indicate focal swellings of axons and axonal degeneration in Kir4.1−/− mice. Because Kir4.1 expression was not detected on spinal cord neurons and axonal structures, we suggest that the axonal damage seen in Kir4.1−/− mice is a secondary phenomenon caused by immature and unhealthy oligodendrocytes, disrupted oligodendrocyte-axon interactions, and the resulting hypo/dysmyelination. Hypomyelinated and dysmyelinated axons in other mouse mutants undergo severe damage and in some cases subsequently degenerate, forming vacuoles of various sizes (Kondo et al., 1995; Sanchez et al., 1996; King et al., 1997; Griffiths et al., 1998; Brady et al., 1999). Particularly marked axonal degeneration occurs in response to the double knock-out of two oligodendrocyte-specific proteins, PLP and DM-20 (Griffiths et al., 1998). However, it is a novel observation that CNS axonal degeneration occurs secondary to genetic ablation of an ion channel rather than of a protein directly involved in the structure or metabolism of myelin.
Some neuronal cell bodies are also basophilic and shrunken in the Kir4.1−/− mice (Fig. 2B), and such damage usually is not reported as a consequence of oligodendrocyte damage (Griffiths et al., 1998). However, we do not believe that this additional damage to neuronal cell bodies implies that the neurons themselves express Kir4.1; instead, the damage may arise secondary to inadequate K+ siphoning or other metabolic support by the poorly functioning Kir4.1−/− oligodendrocytes, the cell bodies of which neighbor the large neuronal cell bodies in the gray matter. Because most genetic models for hypomyelination involve myelin proteins, this point has been studied rarely. Although there are few astrocytes in spinal cord before P12 and we found no Kir4.1 expression in cultured WT astrocytes, a recent paper did report extensive Kir4.1 expression in mouse cortical astrocytes (Li et al., 2001); we cannot rule out the possibility that poorly functioning astrocytes also contribute to the motoneuron pathology in the developing spinal cord of Kir4.1−/− mice.
Implications for pathophysiology
Most genetic models for CNS dysmyelinating or demyelinating disease involve genes that directly participate in the synthesis, degradation, or immunological response to a myelin protein. Instead, Kir4.1 is crucial for the maturation of oligodendrocytes and thus for normal development of the spinal cord.
Most human demyelinating diseases have not been mapped genetically, and it is not known whether they are monogenic like the knock-out mouse studied in this paper. White matter vacuolation also occurs in a variety of human leukodystrophies, and most of these have not been mapped (van der Knaap et al., 1996; Takanashi et al., 1999; Thelle et al., 1999; Leuzzi et al., 2000). There are presumably many genes that enhance or suppress these dysmyelinating and hypomyelinating diseases. Although no known human diseases are caused directly by mutations in the Kir4.1 gene, Kir channels undergo complex trafficking, hetero-oligomerization, clustering, and other regulatory events. Mutations that disrupt such regulation might lead to decreased Kir4.1 function and therefore contribute to dysmyelination or hypomyelination.
Footnotes
This work was supported by National Institutes of Health Grants GM-29836, EY12949, DA08944, and RR13625 and the Deutsche Forschungsgemeinschaft (NE-767/1-1). We thank S. S. Velan for help with the magnetic resonance imaging, S. McKinney for help with animals, V. Santoro for help in data analysis, and B. S. Khakh for comments.
Correspondence should be addressed to Paulo Kofuji, Department of Neuroscience, University of Minnesota, 6-145 Jackson Hall, 321 Church Street SE, Minneapolis, MN 55455. E-mail: kofuj001@tc.umn.edu.
REFERENCES
- 1.Arcangeli A, Becchetti A, Mannini A, Mugnai G, De Filippi P, Tarone G, Del Bene MR, Barletta E, Wanke E, Olivotto M. Integrin-mediated neurite outgrowth in neuroblastoma cells depends on the activation of potassium channels. J Cell Biol. 1993;122:1131–1143. doi: 10.1083/jcb.122.5.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arcangeli A, Bianchi L, Becchetti A, Faravelli L, Coronnello M, Mini E, Olivotto M, Wanke E. A novel inward-rectifying K+ current with a cell-cycle dependence governs the resting potential of mammalian neuroblastoma cells. J Physiol (Lond) 1995;489:455–471. doi: 10.1113/jphysiol.1995.sp021065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barres BA, Raff MC. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature. 1993;361:258–620. doi: 10.1038/361258a0. [DOI] [PubMed] [Google Scholar]
- 4.Barres BA, Raff MC. Axonal control of oligodendrocyte development. J Cell Biol. 1999;147:1123–1128. doi: 10.1083/jcb.147.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barres BA, Koroshetz WJ, Swartz KJ, Chun LL, Corey DP. Ion channel expression by white matter glia: the O-2A glial progenitor cell. Neuron. 1990;4:507–524. doi: 10.1016/0896-6273(90)90109-s. [DOI] [PubMed] [Google Scholar]
- 6.Brady ST, Witt AS, Kirkpatrick LL, de Waegh SM, Readhead C, Tu PH, Lee VM. Formation of compact myelin is required for maturation of the axonal cytoskeleton. J Neurosci. 1999;19:7278–7288. doi: 10.1523/JNEUROSCI.19-17-07278.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bredt DS, Wang TL, Cohen NA, Guggino WB, Snyder SH. Cloning and expression of two brain-specific inwardly rectifying potassium channels. Proc Natl Acad Sci USA. 1995;92:6753–6757. doi: 10.1073/pnas.92.15.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chvatal A, Pastor A, Mauch M, Sykova E, Kettenmann H. Distinct populations of identified glial cells in the developing rat spinal cord slice: ion channel properties and cell morphology. Eur J Neurosci. 1995;7:129–142. doi: 10.1111/j.1460-9568.1995.tb01027.x. [DOI] [PubMed] [Google Scholar]
- 9.Chvatal A, Berger T, Vorisek I, Orkand RK, Kettenmann H, Sykova E. Changes in glial K+ currents with decreased extracellular volume in developing rat white matter. J Neurosci Res. 1997;49:98–106. doi: 10.1002/(sici)1097-4547(19970701)49:1<98::aid-jnr11>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 10.Chvatal A, Anderova M, Ziak D, Sykova E. Glial depolarization evokes a larger potassium accumulation around oligodendrocytes than around astrocytes in gray matter of rat spinal cord slices. J Neurosci Res. 1999;56:493–505. doi: 10.1002/(SICI)1097-4547(19990601)56:5<493::AID-JNR5>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 11.Demerens C, Stankoff B, Zalc B, Lubetzki C. Interaction between neurons and oligodendrocytes during myelination. C R Seances Soc Biol Fil. 1995;189:271–279. [PubMed] [Google Scholar]
- 12.Gallo V, Zhou JM, McBain CJ, Wright P, Knutson PL, Armstrong RC. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J Neurosci. 1996;16:2659–2670. doi: 10.1523/JNEUROSCI.16-08-02659.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- 14.Hida H, Takeda M, Soliven B. Ceramide inhibits inwardly rectifying K+ currents via a Ras- and Raf-1-dependent pathway in cultured oligodendrocytes. J Neurosci. 1998;18:8712–8719. doi: 10.1523/JNEUROSCI.18-21-08712.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hille B. Ionic channels of excitable membranes, 2nd Ed. Sinauer; Sunderland, MA: 1992. [Google Scholar]
- 16.King H, McCulloch MC, Barrie JA, Kyriakides E, Beechey CV, Cattanach BM, Griffiths IR. Hindshaker, a novel myelin mutant showing hypomyelination preferentially affecting the spinal cord. J Neurocytol. 1997;26:557–566. doi: 10.1023/a:1015486508111. [DOI] [PubMed] [Google Scholar]
- 17.Knutson P, Ghiani CA, Zhou JM, Gallo V, McBain CJ. K+ channel expression and cell proliferation are regulated by intracellular sodium and membrane depolarization in oligodendrocyte progenitor cells. J Neurosci. 1997;17:2669–2682. doi: 10.1523/JNEUROSCI.17-08-02669.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kondo A, Sendoh S, Miyata K, Takamatsu J. Spongy degeneration in the zitter rat: ultrastructural and immunohistochemical studies. J Neurocytol. 1995;24:533–544. doi: 10.1007/BF01179978. [DOI] [PubMed] [Google Scholar]
- 20.Lepple-Wienhues A, Berweck S, Bohmig M, Leo CP, Meyling B, Garbe C, Wiederholt M. K+ channels and the intracellular calcium signal in human melanoma cell proliferation. J Membr Biol. 1996;151:149–157. doi: 10.1007/s002329900066. [DOI] [PubMed] [Google Scholar]
- 21.Leuzzi V, Rinna A, Gallucci M, Di Capua M, Dionisi-Vici C, Longo D, Bertini E. Ataxia, deafness, leukodystrophy: inherited disorder of the white matter in three related patients. Neurology. 2000;54:2325–2328. doi: 10.1212/wnl.54.12.2325. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Head V, Timpe L. Identification of an inward rectifier potassium channel gene expressed in mouse cortical astrocytes. Glia. 2001;33:57–71. doi: 10.1002/1098-1136(20010101)33:1<57::aid-glia1006>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 23.Meyer-Franke A, Shen S, Barres BA. Astrocytes induce oligodendrocyte processes to align with and adhere to axons. Mol Cell Neurosci. 1999;14:385–397. doi: 10.1006/mcne.1999.0788. [DOI] [PubMed] [Google Scholar]
- 24.Mi H, Deerinck TJ, Jones M, Ellisman MH, Schwarz TL. Inwardly rectifying K+ channels that may participate in K+ buffering are localized in microvilli of Schwann cells. J Neurosci. 1996;16:2421–2429. doi: 10.1523/JNEUROSCI.16-08-02421.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morley M, Pleasure D, Kreider B. Quantification of the effects of astrocytes on oligodendroglial morphology. J Neurosci Res. 1997;49:219–228. [PubMed] [Google Scholar]
- 26.Niehaus A, Stegmuller J, Diers-Fenger M, Trotter J. Cell-surface glycoprotein of oligodendrocyte progenitors involved in migration. J Neurosci. 1999;19:4948–4961. doi: 10.1523/JNEUROSCI.19-12-04948.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oh S, Ri Y, Bennett MV, Trexler EB, Verselis VK, Bargiello TA. Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot–Marie–Tooth disease. Neuron. 1997;19:927–938. doi: 10.1016/s0896-6273(00)80973-3. [DOI] [PubMed] [Google Scholar]
- 28.Poopalasundaram S, Knott C, Shamotienko OG, Foran PG, Dolly JO, Ghiani CA, Gallo V, Wilkin GP. Glial heterogeneity in expression of the inwardly rectifying K+ channel, Kir4.1, in adult rat CNS. Glia. 2000;30:362–372. doi: 10.1002/(sici)1098-1136(200006)30:4<362::aid-glia50>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 29.Reimann F, Ashcroft FM. Inwardly rectifying potassium channels. Curr Opin Cell Biol. 1999;11:503–508. doi: 10.1016/S0955-0674(99)80073-8. [DOI] [PubMed] [Google Scholar]
- 30.Sanchez I, Hassinger L, Paskevich PA, Shine HD, Nixon RA. Oligodendroglia regulate the regional expansion of axon caliber and local accumulation of neurofilaments during development independently of myelin formation. J Neurosci. 1996;16:5095–5105. doi: 10.1523/JNEUROSCI.16-16-05095.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Signorini S, Liao YJ, Duncan SA, Jan LY, Stoffel M. Normal cerebellar development but susceptibility to seizures in mice lacking G-protein-coupled, inwardly rectifying K+ channel GIRK2. Proc Natl Acad Sci USA. 1997;94:923–927. doi: 10.1073/pnas.94.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takanashi J, Sugita K, Kohno Y. Vacuolating leukoencephalopathy with subcortical cysts with late onset athetotic movements. J Neurol Sci. 1999;165:90–93. doi: 10.1016/s0022-510x(99)00073-8. [DOI] [PubMed] [Google Scholar]
- 33.Takumi T, Ishii T, Horio Y, Morishige K, Takahashi N, Yamada M, Yamashita T, Kiyama H, Sohmiya K, Nakanishi S. A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J Biol Chem. 1995;270:16339–16346. doi: 10.1074/jbc.270.27.16339. [DOI] [PubMed] [Google Scholar]
- 34.Thelle T, Balslev T, Christensen T. van der Knaap's vacuolating leukoencephalopathy: two additional cases. Eur J Paediatr Neurol. 1999;3:83–86. doi: 10.1053/ejpn.1999.0187. [DOI] [PubMed] [Google Scholar]
- 35.van der Knaap MS, Barth PG, Vrensen GF, Valk J. Histopathology of an infantile-onset spongiform leukoencephalopathy with a discrepantly mild clinical course. Acta Neuropathol (Berl) 1996;92:206–212. doi: 10.1007/s004010050510. [DOI] [PubMed] [Google Scholar]
- 36.Vartanian T, Goodearl A, Viehover A, Fischbach G. Axonal neuregulin signals cells of the oligodendrocyte lineage through activation of HER4 and Schwann cells through HER2 and HER3. J Cell Biol. 1997;137:211–220. doi: 10.1083/jcb.137.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wickman K, Nemec J, Gendler SJ, Clapham DE. Abnormal heart rate regulation in GIRK4 knock-out mice. Neuron. 1998;20:103–114. doi: 10.1016/s0896-6273(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 38.Xu H, Cui N, Yang Z, Qu Z, Jiang C. Modulation of Kir4.1 and Kir5.1 by hypercapnia and intracellular acidosis. J Physiol (Lond) 2000;524[Pt 3]:725–735. doi: 10.1111/j.1469-7793.2000.00725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang X, Skoff RP. Proteolipid protein regulates the survival and differentiation of oligodendrocytes. J Neurosci. 1997;17:2056–2070. doi: 10.1523/JNEUROSCI.17-06-02056.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Z, Xu H, Cui N, Qu Z, Chanchevalap S, Shen W, Jiang C. Biophysical and molecular mechanisms underlying the modulation of heteromeric Kir4.1–Kir5.1 channels by CO2 and pH. J Gen Physiol. 2000;116:33–46. doi: 10.1085/jgp.116.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circ Res. 2000;87:160–166. doi: 10.1161/01.res.87.2.160. [DOI] [PubMed] [Google Scholar]