

Abstract
Parkinson's disease, a neurodegenerative movement disorder characterized by selective degeneration of nigrostriatal dopaminergic neurons, affects ∼1% of the population over 50. Because nicotinic acetylcholine receptors (nAChRs) may represent an important therapeutic target for this disorder, we performed experiments to elucidate the subtypes altered with nigrostriatal damage in parkinsonian monkeys. For this purpose we used 125I-α-conotoxin MII (CtxMII), a relatively new ligand that identifies α3 and/or α6 subunits containing nAChR subtypes. In brain from untreated monkeys, there was saturable 125I-α-CtxMII binding to a single population of high-affinity nicotinic sites (Kd = 0.9 nm), primarily localized in the visual, habenula-interpeduncular, and nigrostriatal–mesolimbic pathways. Administration of the selective dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine resulted in damage to the nigrostriatal system and parkinsonism. Autoradiographic analysis showed that 125I-α-CtxMII sites were selectively reduced (≥99%) in the basal ganglia and that the lesion-induced decreases correlated well with declines in the dopamine transporter, a marker of dopaminergic neuron integrity. These findings may indicate that most or all of 125I-α-CtxMII-labeled nAChR subtypes in the basal ganglia are present on nigrostriatal dopaminergic neurons, in contrast to 125I-epibatidine sites. These data suggest that the development of ligands directed to nAChR subtypes containing α3 and/or α6 subunits may yield a novel treatment strategy for parkinsonian patients with nigrostriatal dopaminergic degeneration.
Keywords: α-conotoxin MII, α3 nAChRs, α6 nAChRs, MPTP, monkeys, autoradiography, nigrostriatal system, dopamine, Parkinson's disease
Accumulating evidence indicates that neuronal nicotine acetylcholine receptors (nAChRs) may be important in Parkinson's disease, a movement disorder characterized by degeneration of the nigrostriatal dopaminergic system (Balfour and Fagerstrom, 1996; Quik and Jeyarasasingam, 2000). Nicotinic receptors are present in the basal ganglia (Marks and Collins, 1982; Schwartz et al., 1982, 1984; Clarke et al., 1985; Perry et al., 1987, 1995; Aubert et al., 1992; Marks et al., 1992, 1996; Gotti et al., 1997; Court et al. 2000), and nAChR stimulation evokes striatal dopamine (DA) release (Grady et al., 1992; Marshall et al., 1997; Wonnacott, 1997). Moreover, nicotine and nAChR ligands modulate motor control in rodents, monkeys, and humans (Ishikawa and Miyatake, 1993; Fagerstrom et al., 1994;Schneider et al., 1998; Domino et al., 1999; Kelton et al., 2000), possibly through activation of α4 and/or α6 containing nAChRs (Sorenson et al., 1998; Arroyo-Jiminez et al., 1999; Le Novere et al., 1999; Ross et al., 2000).
Not only does nicotinic receptor stimulation modulate locomotor activity, but nicotine has also been reported to exert a neuroprotective role against dopaminergic damage both in culture andin vivo (Janson et al., 1988; Carr and Rowell, 1990;Belluardo et al., 1998; Maggio et al., 1998; Quik and Jeyarasasingam, 2000). Moreover, over 50 epidemiological studies indicate that there is an inverse relationship between tobacco use and Parkinson's disease (Morens et al., 1995; Balfour and Fagerstrom, 1996; Gorell et al., 1999), with the risk of developing Parkinson's disease reduced from 20 to 80% in tobacco users.
Studies to investigate the nAChR subtypes that mediate the effects of nicotine are important because these receptors represent potential targets for Parkinson's disease therapy to ameliorate motor symptoms and/or protect against neurodegeneration. nAChRs form a large family of ligand-gated cation channels with diverse structures and properties (Changeux et al., 1998; Jones et al., 1999; Lukas et al., 1999; Picciotto et al., 2000). Subunit mRNAs present in basal ganglia include α2 to α7 and β2 to β4 (Gotti et al., 1997;Wonnacott, 1997; Quik et al., 2000a,b). There is thus the potential for receptors with multiple subunit combinations. However, our knowledge of the subtypes expressed in the basal ganglia has been limited in part by the small number of subtype-selective nAChR radioligands. The use of tritiated nicotine and cytisine provides some selectivity for α4- and β2-containing receptors, whereas125I-α-bungarotoxin and3H-methyllycaconitine selectively bind α7-containing receptors (Gotti et al., 1997; Lukas et al., 1999;Whiteaker et al., 1999). However, radiolabeled epibatidine is relatively nonselective for different nicotinic receptor subtypes and may bind to receptors containing α2 through α6 subunits (Perry et al., 1995; Davila-Garcia et al., 1997).
125I-α-conotoxin MII (CtxMII) is a relatively new ligand that appears to be selective for receptors containing α3 and/or α6 subunits (Cartier et al., 1996; Kulak et al., 1997; Luo et al., 1998; McIntosh et al., 1999; Vailati et al., 1999; Kuryatov et al., 2000; Whiteaker et al., 2000a). Because this represents a valuable new tool to define a subset of nicotinic receptors, we initiated experiments with 125I-α-CtxMII in normal and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned monkey brain. Monkeys were selected for study because the neuroanatomical organization of both the basal ganglia and the nAChR subunit mRNA distribution closely resemble that of humans. Moreover, monkeys treated with the neurotoxin MPTP exhibit behavioral, pathological, and neurochemical changes similar to those observed in Parkinson's disease (Langston et al., 2000; Quik et al., 2000a,b,c).
MATERIALS AND METHODS
Animals. Twenty young adult male and female squirrel monkeys (Saimiri sciureus) weighing from 0.595 to 0.752 kg were obtained from Osage Research Primates (Osage Beach, MO). All animals were housed in a 13/11 hr light/dark cycle with ad libitum access to food and water. After quarantine and testing according to standard veterinary practice, the animals were randomly assigned to the control or MPTP treatment groups. All procedures used in this study conform to the National Institutes of Health (NIH)Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee.
Behavioral testing and MPTP treatment. Quantitative activity assessment was performed using a computerized movement monitor cage containing an array of infrared sensors (Quik et al., 2000b). After an initial acclimatization period, the locomotor activity of the monkeys was monitored for a 1 hr period for 8–12 consecutive days, at the same time daily. Then, the animals were assigned randomly to treatment with MPTP (2 mg/kg, s.c.) or saline. Two to 3 weeks later, locomotor activity was measured again as described above, and animals were rated by two raters, one of whom was blinded, for motor deficits using a parkinsonian rating scale for nonhuman primates (Langston et al., 2000). The disability scores ranged from 0 to 20 in this scale, with 0 being normal and 20 very severely parkinsonian. The composite score was obtained using five clinical parameters, each having a 5 point range with 0 being normal (Langston et al., 2000). These include (1) spatial hypokinesia (reduction in use of the available cage space), (2) body bradykinesia (increased slowness in body movement), (3) manual dexterity, (4) balance, and (5) freezing. If the total Parkinson score was <3, the monkeys were given a second injection of MPTP at a lower dose (1.75 mg/kg, s.c.) than the first because our previous studies indicated that readministration of 2 mg/kg occasionally (<5%) led to animal mortality. Two to 3 weeks after this second MPTP injection treatment, locomotor motor activity and parkinsonism were determined again as described above. The monkeys were killed 4 weeks after either the first or second MPTP injection. Ketamine hydrochloride (15–20 mg/kg, i.m.) was administered to sedate the animals, followed by injection of 0.22 ml/kg intravenous euthanasia solution (390 mg sodium pentobarbital and 50 mg phenytoin sodium/ml). All procedures were performed in accordance with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association and conform to the NIH Guide for the Care and Use of Laboratory Animals.
Tissue preparation. The brains were removed, rinsed, placed in a mold, and cut into 6-mm-thick blocks that were frozen in isopentane on dry ice and stored at −80°C. The blocks were cut into 20 μm sections using a cryostat. The sections were thaw-mounted onto poly-l-lysine-coated slides, dried, and stored at −80°C. The sections were thawed and used directly for receptor and DA transporter autoradiography.
125I-α-CtxMII autoradiography. α-CtxMII was synthesized and iodinated as previously described (Whiteaker et al., 2000a). Quantitative autoradiography with125I-α-CtxMII was performed using a similar procedure as Whiteaker et al. (2000a). Sections were preincubated in buffer [binding buffer containing (in mm): 144 NaCl, 1.5 KCl, 2 CaCl2, 1 MgSO4, 20 HEPES, 0.1% BSA, pH 7.5] plus 1 mmphenylmethylsulfonyl fluoride at room temperature (RT) for 15 min. This was followed by a 2 hr incubation at room temperature in binding buffer plus 5% BSA, also containing 5 mm EDTA, 5 mm EGTA, and 10 μg/ml each of aprotinin, leupeptin, and pepstatin A, and 0.8 nm125I-α-CtxMII, unless otherwise indicated. After incubation with125I-α-CtxMII, the slides were rinsed for 30 sec in binding buffer at room temperature, followed by another 30 sec wash in ice-cold buffer (0°C), two washes for 5 sec in 0.1× binding buffer (0°C), and two 5 sec washes at 0°C in water. Nonspecific 125I-α-CtxMII binding was defined using 0.1 μm epibatidine. The sections were then air-dried and apposed to Hyperfilm β-Max (Amersham, Mt. Prospect, IL) for 2–4 d together with125I standards.
[125I]RTI-121 autoradiography.3β-(4-[125I]iodophenyl)tropane-2β-carboxylic acid isopropyl ester ([125I]RTI-121; 2200 Ci/mmol; NEN, Boston, MA) was used to identify DA transporters (Quik et al., 2000c). Thawed sections were preincubated in buffer containing (in mm): 50 Tris-HCl, pH 7.4, 120 NaCl, and 5 KCl two times for 15 min at RT. This was followed by a 2 hr incubation in the same buffer, also containing 0.025% BSA, 1 μm fluoxetine, and 50 pm[125I]RTI-121. The sections were washed four times for 15 min at 4°C in preincubation buffer, dipped in ice-cold water, air-dried, and placed against Hyperfilm β-film for 1–3 d with 125I-microscale standards. The uptake inhibitor nomifensine (100 μm) was used to define nonspecific binding. There was no binding of the radiolabel to the tissue sections in the presence of nomifensin, with blank binding being identical to the film background.
Quantitation and data analysis. The squirrel monkey (Saimiri sciureus) atlas of Emmers and Akert (1963) was used for identification of the different brain regions. Nissl-stained sections were used to identify brain areas. The area delineating the appropriate regions was quantitated using computer-assisted densitometry (ImageQuant; Molecular Dynamics, Sunnyvale, CA) under standardized light and power conditions. Optical densities of the film images were determined by subtracting background from tissue values. The optical density values were converted to femtomoles per milligram of tissue by interpolation from standard curves that were generated from 125I standards exposed with the tissue sections. The sample optical density readings for both the receptor and transporter data were within the dynamic (linear) range of the film. The saturation and competition data represent the results of three or four experiments, using one or two tissue sections per experiment for each concentration of radiolabeled125I-α-CtxMII (saturation) or competing ligand. In all other experiments, the value for the receptor binding for any brain region per monkey was obtained by taking the average value from two to four independent experiments, with one or two consecutive tissue sections used in each experiment. All values are expressed as the mean ± SEM of the indicated number of animals. For statistical analysis, data were evaluated by Student's ttest, where p ≤ 0.05 was considered significant. For saturation experiments, the Kd andBmax values were determined using GraphPad Prism (San Diego, CA). Values forKi (inhibition binding constant) were derived by the method of Cheng and Prusoff (1973) using the equationKi = IC50/1 ± (L/Kd) and the GraphPad Prism program. The data are expressed as mean ± SEM and were compared using Student'st test or one-way ANOVA followed by Newman–Keuls multiple comparison test.
RESULTS
125I-α-CtxMII binding sites are present in the visual, habenular-interpeduncular, and dopaminergic systems in control monkey brain
The autoradiographic distribution125I-α-CtxMII binding at different rostral to caudal levels of the monkey brain is illustrated in Figure1. The highest specific125I-α-CtxMII binding was obtained in the habenular-interpeduncular pathway (Table1). Prominent labeling was also observed in the supraoptic decussation, superior colliculus, and lateral geniculate nucleus, areas associated with the visual system. Moderate expression was detected in the nigrostriatal dopaminergic pathway, with greater binding in the caudate and putamen than substantia nigra. Similar levels of binding were observed in mesolimbic dopaminergic areas, including the nucleus accumbens, olfactory tubercle, and ventral tegmental area. In addition, binding was identified at low levels in various cortical areas, hippocampus, and septum (Table 1). Whiteaker et al. (2000a) obtained a similar distribution in mouse brain, although the intensity of expression in different brain areas appears to vary somewhat in the two species.
Fig. 1.
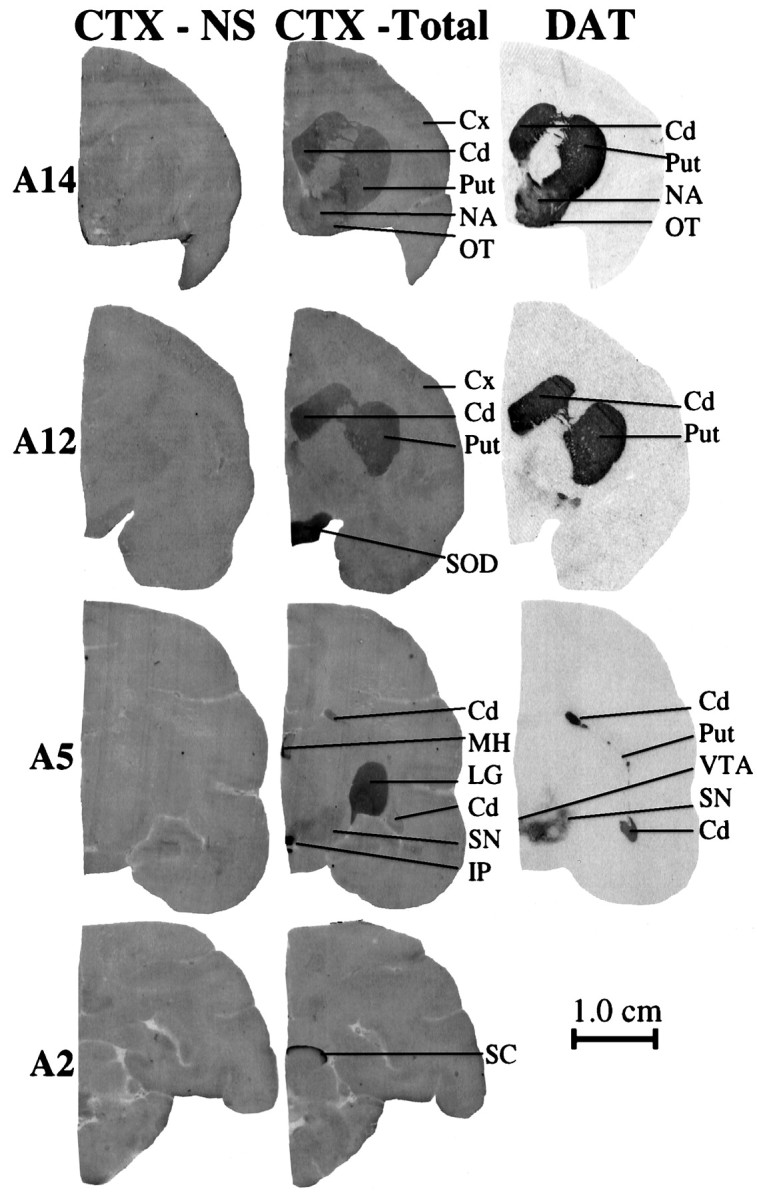
Autoradiographic distribution of125I-α-CtxMII binding in control monkey brain. Expression is observed predominantly in the nigrostriatal and visual systems, as illustrated by representative sections throughout the brain (14.0 mm anterior to the interaural line, A14.0; 12.0 mm, A12.0; 5.0 mm, A5.0; and 2.0 mm, A2.0). Nonspecific125I-α-CtxMII (CTX-NS) binding using 0.1 μm epibatidine is depicted in the set of brain sections at the left, total 125I-α-CtxMII (CTX-Total) binding is in thecenter, and dopamine transporter (DAT) binding ([125I]RTI-121) is at the right.Cd, Caudate; Cx, cortex;LG, lateral geniculate nucleus; IP, interpeduncular nucleus; MH, medial habenula;NA, nucleus accumbens; OT, olfactory tubercle; Put, putamen; SN, substantia nigra; SC, superior colliculus; SOD, supraoptic decussation; VTA, ventral tegmental area. Scale bar, 1.0 cm.
Table 1.
Quantitative assessment of the distribution of125I-α-CtxMII in control monkey brain
| Region | 125I-α-CtxMII (fmol/mg tissue) |
|---|---|
| Telencephalon | |
| Neocortex | |
| Frontal cortex | 0.20 ± 0.13 (6) |
| Parietal cortex | 0.11 ± 0.05 (4) |
| Temporal cortex | 0.12 ± 0.04 (4) |
| Rhinencephalon | |
| Hippocampus | 0.05 ± 0.03 (4) |
| Olfactory tubercles | 2.32 ± 0.24 (4) |
| Basal ganglia | |
| Nucleus accumbens | 1.94 ± 0.34 (4) |
| Caudate | 1.81 ± 0.24 (6) |
| Putamen | 1.75 ± 0.18 (6) |
| Substantia nigra | 0.55 ± 0.04 (4) |
| Ventral tegmental area | 0.51 ± 0.07 (4) |
| Septum | |
| Nucleus of stria terminalis | 0.58 ± 0.06 (3) |
| Diencephalon | |
| Dorsolateral geniculate nucleus | 3.00 ± 0.59 (4) |
| Ventrolateral geniculate nucleus | 2.01 ± 0.30 (4) |
| Medial habenula | 6.52 ± 0.56 (4) |
| Mesencephalon | |
| Interpeduncular nucleus | 9.42 ± 0.82 (4) |
| Superior colliculus | 5.78 ± 1.39 (3) |
| Fiber tracts | |
| Supraoptic decussation | 7.49 ± 0.39 (3) |
| Cerebellum | Not detected |
Coronal brain sections were prepared as described in Materials and Methods, and 125I-α-CtxMII binding was measured using a concentration of the radioligand at the Kdvalue. Each value represents the mean ± SEM of the number of animals in parentheses.
125I-α-CtxMII binds to receptors with nicotinic characteristics in control monkey brain
Because of our interest in the nigrostriatal system, we focused on the monkey caudate and putamen for the characterization work. Saturation analysis (Fig. 2) showed that the binding of 125I-α-CtxMII plateaued with a Kd of 0.93 ± 0.14 nm(n = 4) and 0.92 ± 0.14 nm(n = 4) for the caudate and putamen, respectively, and a Bmax for caudate of 2.28 ± 0.30 fmol/mg tissue (n = 4) and for putamen of 1.93 ± 0.13 fmol/mg tissue (n = 4).
Fig. 2.

Saturation binding of125I-α-CtxMII to monkey caudate and putamen. Sections were incubated with concentrations of 125I-α-CtxMII ranging from 0.1 to 6.0 nm for 2 hr. Data points are means from four experiments. For the caudate, theKd = 0.93 ± 0.14 nm (n = 4), and theBmax = 2.28 ± 0.30 fmol/mg tissue (n = 4); for the putamen, theKd = 0.92 ± 0.14 nm(n = 4), and theBmax = 1.93 ± 0.13 fmol/mg tissue (n = 4).
The potency of different drugs for inhibiting125I-α-CtxMII binding in the caudate and putamen was subsequently examined (Table2). Competition studies showed that125I-α-CtxMII binding was displaced by picomolar concentrations of epibatidine (0.08 ± 0.03 nm, n = 4), and low nanomolar concentrations of cytisine (2.01 ± 0.40 nm, n = 3) and nicotine (2.87 ± 0.21 nm, n = 4). Although the pharmacological characteristics of125I-α-CtxMII sites in the monkey brain closely resemble those in the rodent, they were not identical. TheKi for epibatidine in the monkey caudate putamen (0.08 nm) was similar to that in the rodent striatum (0.11 nm). However, theKi for cytisine (2.01 nm) and nicotine (2.87 nm) in monkey caudate putamen were very similar to each other, in contrast to rodent striatum in which the values were significantly higher (23 nm for cytisine and 348 nmfor nicotine) and also an order of magnitude different (Whiteaker et al., 2000a).
Table 2.
Inhibition of 125I-α-CtxMII binding in monkey caudate and putamen by cholinergic drugs
| Drug | Ki (nm) | |
|---|---|---|
| Caudate | Putamen | |
| Epibatidine | 0.08 ± 0.03 | 0.15 ± 0.08 |
| Conotoxin | 0.73 ± 0.18 | 1.20 ± 0.65 |
| Cytisine | 2.01 ± 0.40 | 1.80 ± 0.75 |
| Nicotine | 2.87 ± 0.21 | 1.67 ± 0.54 |
| d-tubocurarine | 263 ± 115 | 219 ± 85 |
| α-bungarotoxin | >100 | >100 |
| Atropine | >10,000 | >10,000 |
Experiments were performed using a concentration of125I-α-CtxMII at the Kd value. Values for Ki (inhibition binding constant) were derived by the method of Cheng and Prusoff (1973) using the equationKi = IC50/1 + (L/Kd) and GraphPad Prism. Values represent the mean ± SEM of three to four experiments.
The nonselective nicotinic blocker d-tubocurarine inhibited125I-α-CtxMII binding in the high nanomolar range (263 ± 115, n = 3) in monkey striatum. Thus d-tubocurarine appears to interact with a somewhat higher affinity at nicotinic receptors labeled by α-CtxMII as compared with other nicotinic receptor sites (Marks and Collins, 1982; Schwartz et al., 1982; Davila-Garcia et al., 1997; Whiteaker et al., 2000b). Alternatively, it may indicate a species difference between primates and rodents. This observation, together with the finding described above that cytisine and nicotine exhibit a similarKi, may suggest that α-CtxMII-sensitive sites in the monkey striatum represent a unique population of nicotinic receptors.
The α7 receptor antagonist α-bungarotoxin had no effect up to a concentration of 0.1 μm, nor did the muscarinic antagonist atropine (at concentrations up to 100 μm). These experiments, together with the results of the saturation analysis, suggest that 125I-α-CtxMII binds to a site(s) in monkey caudate and putamen with the characteristics of an nAChR.
MPTP treatment results in nigrostriatal damage and motor deficits
Monkeys were treated with the neurotoxin MPTP to selectively damage the dopaminergic nigrostriatal system. The animals fell into two groups, those with moderate and those with more severe nigrostriatal damage that was assessed using the dopamine transporter as a biochemical marker for the effectiveness of MPTP treatment (Figs.3, 4,5). [125I]RTI-121 binding was reduced to 28 and 23% of control in the caudate and putamen, respectively, in the moderate group and to 5% in both these regions in the severely lesioned animals (Fig. 4). The DA transporter levels in the substantia nigra were reduced to 56 and 49% of control in the moderate and severe group, respectively (Fig. 5).
Fig. 3.

Autoradiographic images showing the effect of MPTP administration on 125I-α-CtxMII binding and the DA transporter in caudate putamen. Monkeys were treated with saline (CON) or MPTP as described in Materials and Methods and killed 4 weeks later. Note the dramatic reduction in125I-α-CtxMII binding after MPTP treatment in parallel with the loss of dopamine transporter. Cd, Caudate;Put, putamen.
Fig. 4.

Quantitative assessment of the decline in125I-α-CtxMII binding and the DA transporter in monkey caudate and putamen after moderate (Mod) and severe (Sev) nigrostriatal damage. Each value represents the mean ± SEM of five to seven monkeys. Significance of difference from control: ***p ≤ 0.001.
Fig. 5.

125I-α-CtxMII binding and the DA transporter in monkey substantia nigra after moderate (Mod) and severe (Sev) nigrostriatal damage. Each value represents the mean ± SEM of four to five monkeys. The declines in 125I-α-CtxMII binding were similar to those obtained for the DA transporter. Significance of difference from control: **p ≤ 0.01; ***p < 0.001.
To evaluate the behavioral effects of MPTP treatment, animals were tested for the development of parkinsonism and alterations in baseline motor activity. The animals with moderate nigrostriatal damage exhibited very mild parkinsonism (1.57 ± 0.32). However, they showed a 39% decline in spontaneous locomotor activity (most likely reflecting bradykinesia and hypokinesia), a finding suggesting that measurement of motor activity using infrared activity monitoring may be a more sensitive index of nigrostriatal damage. The animals with more severe nigrostriatal damage had a more pronounced parkinsonian syndrome (7.75 ± 0.89, of a total score of 20) with locomotor activity diminished by 91%. The control animals did not exhibit parkinsonian features, and their locomotor activity was similar to the pretreatment saline value.
MPTP treatment decreases 125I-α-CtxMII binding in caudate, putamen, and substantia nigra
The effect of MPTP-induced nigrostriatal damage on125I-α-CtxMII binding in the caudate and putamen is depicted in the autoradiograms in Figure 3 and quantitated in Figure 4. In the severely lesioned group,125I-α-CtxMII binding is eliminated, whereas the dopamine transporter is reduced to ∼5% of control. In the animals with the moderate MPTP lesion, the DA transporter and125I-α-CtxMII binding were 28 and 5% of control, respectively, in the caudate, and 23 and 7% of control, respectively, in the putamen. As is evident, the decline in125I-α-CtxMII binding in caudate and putamen was significantly greater than the decrease in the dopamine transporter (p < 0.05), an observation suggesting that the toxin binding sites are present on a more vulnerable population of nigrostriatal dopaminergic neurons.
Consistent with previous work (Quik et al., 2000c), declines in the DA transporter were substantially less severe in the substantia nigra than in the caudate putamen after MPTP treatment (Fig. 5).125I-α-CtxMII binding sites in the substantia nigra decreased to a similar extent as the DA transporter. In the moderately lesioned animals, binding was reduced to 61% of control, and binding of the DA transporter was reduced to 56% of control. For the severely lesioned animals, binding sites were decreased to 50% of control, and the transporter was decreased to 49% of control.
The MPTP-induced decline in 125I-α-CtxMII binding occurs only in the basal ganglia
Despite an almost complete disappearance of sites in the caudate and putamen and an approximate 50% reduction in the substantia nigra, there were no declines in 125I-α-CtxMII binding sites after MPTP treatment in other brain areas at similar anatomical levels (Table 3). This includes the interpeduncular nucleus, medial habenula, and lateral geniculate nucleus, regions that are located at a similar level as the substantia nigra (A5.5 to A4.0). Similarly, there are no changes in the supraoptic decussation present at the level of the caudate putamen (A13.0 to A11.0). These results indicate that the effects of the lesion were selective for the basal ganglia.
Table 3.
MPTP treatment selectively decreases125I-α-CtxMII binding in the basal ganglia
| Region | 125I-α-CtxMII binding (fmol/mg tissue) | % control | |
|---|---|---|---|
| Control | MPTP | ||
| Caudate | 1.81 ± 0.23 | 0.047 ± 0.0313-165 | 2.2 ± 1.7 |
| Putamen | 1.76 ± 0.17 | 0.053 ± 0.0313-165 | 3.8 ± 1.8 |
| Substantia nigra | 0.60 ± 0.07 | 0.33 ± 0.023-165 | 55 ± 3.3 |
| Interpeduncular nucleus | 9.39 ± 0.64 | 8.54 ± 0.47 | 91 ± 5 |
| Medial habenula | 6.16 ± 0.39 | 6.12 ± 0.34 | 99 ± 6 |
| Supraoptic decussation | 7.49 ± 0.39 | 7.14 ± 0.50 | 95 ± 7 |
| Lateral geniculate nucleus | 2.49 ± 0.16 | 2.36 ± 0.12 | 95 ± 5 |
125I-α-CtxMII binding was quantitated for the indicated brain regions from control and all MPTP-treated animals. Each value represents the mean ± SEM of 4 to 13 animals. Significance of difference from control:
F3-165: p < 0.001.
Correlation between 125I-α-CtxMII binding sites and the DA transporter in basal ganglia in control and lesioned animals suggests a presynaptic localization
As an approach to assessing the relationship between125I-α-CtxMII binding sites and the DA transporter in the caudate, putamen, and substantia nigra, correlation analysis was performed for control and MPTP-lesioned animals (Fig.6). The results show that there was a good correlation between 125I-α-CtxMII binding sites and the DA transporter in both the caudate putamen (R2 = 0.80; p< 0.001) and substantia nigra (R2 = 0.75; p< 0.001).
Fig. 6.

Correlation between the distribution of the DA transporter and 125I-α-CtxMII binding sites in the caudate putamen (left) and the substantia nigra (right). For the caudate putamen, eachsymbol represents the mean values for 7 control and 12 MPTP-treated animals. For the substantia nigra, the symbolsrepresent the mean values for four control and nine MPTP-treated animals.
DISCUSSION
The present results are the first to investigate the localization of 125I-α-CtxMII sites in monkey brain. They show that 125I-α-CtxMII binds to an apparently homogenous population of high-affinity (Kd = 0.92 nm) nAChRs. The areas expressing125I-α-CtxMII sites are similar to those in the rodent (Whiteaker et al., 2000a), although the intensity of binding in the various regions differs between the two species. In monkey, the habenular-interpeduncular system exhibited the greatest125I-α-CtxMII binding, followed by areas associated with vision, with similar binding in the nigrostriatal and mesolimbic dopaminergic systems. In contrast, in rodents, the highest expression of 125I-α-CtxMII sites is in the visual system (Whiteaker et al., 2000a), with a similar intensity of binding in the habenular-interpeduncular pathway and the mesolimbic dopaminergic system, but a somewhat lower expression in the nigrostriatal pathway. These variations in expression between species are not uncommon (Quik et al., 2000a,b) and may reflect differences in the relative functional roles of these systems, for instance, the visual system in the rodent as compared with the monkey.
The present data show that the lesion-induced declines in125I-α-CtxMII sites parallel those in the DA transporter in both the caudate putamen and substantia nigra. These data support the contention that125I-α-CtxMII binding sites and the DA transporter share a similar cellular localization, specifically nigrostriatal dopaminergic neurons. The observation that the decline in125I-α-CtxMII sites is somewhat greater than that in the DA transporter most likely reflects a greater vulnerability to MPTP of the dopaminergic neurons on which the125I-α-CtxMII sites reside (Schneider et al., 1987).
The marked declines in 125I-α-CtxMII sites in caudate and/or putamen of moderately (93%) and severely (100%) lesioned monkeys is in contrast to results obtained with other radioligands in this brain area.125I-epibatidine binding, which recognizes α2–α6 subunits, is reduced by only 40–60% in animals with moderate to severe nigrostriatal damage (Kulak and Quik, 2000; Quik et al., 2000b), whereas 3H-cytisine, which may be selective for a subset of125I-epibatidine sites containing the α4 subunit, is reduced by only ∼30% (our unpublished observation). Furthermore, the sites labeled by125I-α-bungarotoxin, which most likely represent α7-containing nAChRs, are increased ∼100% over control in the caudate and putamen of severely lesioned animals (Kulak and Quik, 2000). These observations suggest that125I-α-CtxMII is binding to a unique nAChR population in the caudate and/or putamen that appears to be exclusively located on dopaminergic neurons, in contrast to other nAChR subtypes that show a differential localization possibly on other neurotransmitter afferents and/or postsynaptically on GABAergic or cholinergic interneurons.
A question arises in regard to the nAChR subtype(s) identified by 125I-α-CtxMII in monkey basal ganglia. Electrophysiological studies in oocytes expressing nAChRs, binding experiments in nAChR transfected cell lines, and work using rodent brain slices or synaptosomes indicate that α-CtxMII acts on α3 and/or α6-containing receptors (Cartier et al., 1996; Kulak et al., 1997; Luo et al., 1998; McIntosh et al., 1999; Vailati et al., 1999; Kuryatov et al., 2000; Whiteaker et al., 2000a). In the monkey, the α3 receptor subunit mRNA appears to be expressed at very low levels in substantia nigra (our unpublished observations) or not at all (Han et al., 2000), whereas the α6 transcript is very prominently localized primarily to the substantia nigra (Han et al., 2000; Quik et al., 2000a,b), similar to the rodent (Le Novere et al., 1996). These results suggest that the receptors identified by125I-α-CtxMII in monkey basal ganglia contain primarily the α6, rather than α3 nicotinic receptor subunit.
The β subunits with which the α6 (and/or possibly α3) subunit combine to form functional receptors may include β2 and/or β3. Evidence for this stems from rodent studies showing that β3 subunit knockout mice exhibit a decreased binding in125I-α-CtxMII, whereas animals not expressing the β2 subunit show a complete absence of125I-α-CtxMII sites (Allen et al., 1998;Whiteaker et al., 1998). These data suggest that receptors composed of α6β2 and/or α6β2β3 subunits are expressed in monkey basal ganglia. With respect to a functional role of α6-containing receptors, the recent work of Le Novere et al. (1999) indicates that intraventicular administration of α6 antisense to rats modulates locomotor activity, possibly by decreasing receptors containing the α6 nicotinic subunit. These data suggest that α6-containing nAChRs may be involved, at least in part, in motor control mediated through the nigrostriatal pathway.
In summary, this study shows that125I-α-CtxMII binds to a high-affinity nAChR population, likely containing the α6 subunit. The decline in125I-α-CtxMII binding sites after nigrostriatal damage is selective to the basal ganglia with no changes in other brain areas. The decrease in binding occurs in parallel with changes in the DA transporter, a marker of dopaminergic neuron integrity. This observation suggests that125I-α-CtxMII sites are selectively localized to dopaminergic neurons in the basal ganglia. The dopaminergic neurons with 125I-α-CtxMII binding sites may be particularly vulnerable to the effects of MPTP because the decrease in toxin binding is generally more severe than that in the DA transporter. These results suggest that receptor ligands directed to α6-containing nAChRs may be important for Parkinson's disease therapy to restore motor function and/or protect against nigrostriatal damage.
Footnotes
This work was supported by the California Tobacco Related Disease Research Program Grant TRDRP 7RT-015 (M.Q.), National Institute on Drug Abuse Grant DA12242 (J.M.M.), National Institute of Mental Health Grant MH53631 (J.M.M.), and National Institute of General Medical Sciences Grant GM48677 (J.M.M.).
Correspondence should be addressed to Dr. Maryka Quik, The Parkinson's Institute, 1170 Morse Avenue, Sunnyvale, CA 94089. E-mail:mquik@parkinsonsinstitute.org.
REFERENCES
- 1.Allen RS, Cui C, Heinemann SF. Gene targeted knock out of the β3 neuronal nicotinic acetylcholine receptor subunit. Soc Neurosci Abstr. 1998;24:1341. [Google Scholar]
- 2.Arroyo-Jiminez MM, Bourgeois JP, Marubio LM, Le Sourd AM, Ottersen OP, Rinvik E, Fairen A, Changeux JP. Ultrastructural localization of the α4-subunit of the neuronal acetylcholine nicotinic receptor in the rat substantia nigra. J Neurosci. 1999;19:6475–6487. doi: 10.1523/JNEUROSCI.19-15-06475.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aubert I, Araujo DM, Cecyre D, Robitaille Y, Gauthier S, Quirion R. Comparative alterations of nicotinic and muscarinic binding sites in Alzheimer's and Parkinson's diseases. J Neurochem. 1992;58:529–541. doi: 10.1111/j.1471-4159.1992.tb09752.x. [DOI] [PubMed] [Google Scholar]
- 4.Balfour DJ, Fagerstrom KO. Pharmacology of nicotine and its therapeutic use in smoking cessation and neurodegenerative disorders. Pharmacol Ther. 1996;72:51–81. doi: 10.1016/s0163-7258(96)00099-x. [DOI] [PubMed] [Google Scholar]
- 5.Belluardo N, Blum M, Mudo G, Andbjer B, Fuxe K. Acute intermittent nicotine treatment produces regional increases of basic fibroblast growth factor messenger RNA and protein in the tel- and diencephalon of the rat. Neuroscience. 1998;83:723–740. doi: 10.1016/s0306-4522(97)00323-0. [DOI] [PubMed] [Google Scholar]
- 6.Carr LA, Rowell PP. Attenuation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity by tobacco smoke. Neuropharmacology. 1990;29:311–314. doi: 10.1016/0028-3908(90)90019-n. [DOI] [PubMed] [Google Scholar]
- 7.Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM. A new α-conotoxin which targets α3β2 nicotinic acetylcholine receptors. J Biol Chem. 1996;271:7522–7528. doi: 10.1074/jbc.271.13.7522. [DOI] [PubMed] [Google Scholar]
- 8.Changeux JP, Bertrand D, Corringer PJ, Dehaene S, Edelstein S, Lena C, Le Novere N, Marubio L, Picciotto M, Zoli M. Brain nicotinic receptors: structure and regulation, role in learning and reinforcement. Brain Res Brain Res Rev. 1998;26:198–216. doi: 10.1016/s0165-0173(97)00040-4. [DOI] [PubMed] [Google Scholar]
- 9.Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 10.Clarke PB, Schwartz RD, Paul SM, Pert CB, Pert A. Nicotinic binding in rat brain: autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]-α-bungarotoxin. J Neurosci. 1985;5:1307–1315. doi: 10.1523/JNEUROSCI.05-05-01307.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Court JA, Piggott MA, Lloyd S, Cookson N, Ballard CG, McKeith IG, Perry RH, Perry EK. Nicotine binding in human striatum: elevation in schizophrenia and reductions in dementia with Lewy bodies, Parkinson's disease and Alzheimer's disease and in relation to neuroleptic medication. Neuroscience. 2000;98:79–87. doi: 10.1016/s0306-4522(00)00071-3. [DOI] [PubMed] [Google Scholar]
- 12.Davila-Garcia MI, Musachio JL, Perry DC, Xiao Y, Horti A, London ED, Dannals RF, Kellar KJ. [125I]IPH, an epibatidine analog, binds with high affinity to neuronal nicotinic cholinergic receptors. J Pharmacol Exp Ther. 1997;282:445–451. [PubMed] [Google Scholar]
- 13.Domino EF, Ni L, Zhang H. Nicotine alone and in combination with l-DOPA methyl ester or the D2 agonist N-0923 in MPTP-induced chronic hemiparkinsonian monkeys. Exp Neurol. 1999;158:414–421. doi: 10.1006/exnr.1999.7106. [DOI] [PubMed] [Google Scholar]
- 14.Emmers R, Akert K. A stereotaxic atlas of the brain of the squirrel monkey (Saimiri sciureus). University of Wisconsin; Madison, WI: 1963. [Google Scholar]
- 15.Fagerstrom KO, Pomerleau O, Giordani B, Stelson F. Nicotine may relieve symptoms of Parkinson's disease. Psychopharmacology (Berl) 1994;116:117–119. doi: 10.1007/BF02244882. [DOI] [PubMed] [Google Scholar]
- 16.Gorell JM, Rybicki BA, Johnson CC, Peterson EL. Smoking and Parkinson's disease: a dose-response relationship. Neurology. 1999;52:115–119. doi: 10.1212/wnl.52.1.115. [DOI] [PubMed] [Google Scholar]
- 17.Gotti C, Fornasari D, Clementi F. Human neuronal nicotinic receptors. Prog Neurobiol. 1997;53:199–237. doi: 10.1016/s0301-0082(97)00034-8. [DOI] [PubMed] [Google Scholar]
- 18.Grady S, Marks MJ, Wonnacott S, Collins AC. Characterization of nicotinic receptor-mediated [3H]dopamine release from synaptosomes prepared from mouse striatum. J Neurochem. 1992;59:848–856. doi: 10.1111/j.1471-4159.1992.tb08322.x. [DOI] [PubMed] [Google Scholar]
- 19.Han ZY, Le Novere N, Zoli M, Hill JA, Champtiaux N, Changeux JP. Localization of nAChR subunit mRNAs in the brain of Macaca mulatta. Eur J Neurosci. 2000;12:3664–3674. doi: 10.1046/j.1460-9568.2000.00262.x. [DOI] [PubMed] [Google Scholar]
- 20.Ishikawa A, Miyatake T. Effects of smoking in patients with early-onset Parkinson's disease. J Neurol Sci. 1993;117:28–32. doi: 10.1016/0022-510x(93)90150-w. [DOI] [PubMed] [Google Scholar]
- 21.Janson AM, Fuxe K, Agnati LF, Kitayama I, Harfstrand A, Andersson K, Goldstein M. Chronic nicotine treatment counteracts the disappearance of tyrosine-hydroxylase-immunoreactive nerve cell bodies, dendrites and terminals in the mesostriatal dopamine system of the male rat after partial hemitransection. Brain Res. 1988;455:332–345. doi: 10.1016/0006-8993(88)90092-3. [DOI] [PubMed] [Google Scholar]
- 22.Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555–561. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- 23.Kelton MC, Kahn HJ, Conrath CL, Newhouse PA. The effects of nicotine on Parkinson's disease. Brain Cogn. 2000;43:274–282. [PubMed] [Google Scholar]
- 24.Kulak JM, Quik M. Differential alterations in non-α7 and α7 nicotinic receptors in monkey striatum after MPTP treatment. Soc Neurosci Abstr. 2000;26:526.1. [Google Scholar]
- 25.Kulak JM, Nguyen TA, Olivera BM, McIntosh JM. α-conotoxin MII blocks nicotine-stimulated dopamine release in rat striatal synaptosomes. J Neurosci. 1997;17:5263–5270. doi: 10.1523/JNEUROSCI.17-14-05263.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuryatov A, Olale F, Cooper J, Choi C, Lindstrom J. Human α6 AChR subtypes: subunit composition, assembly, and pharmacological responses. Neuropharmacology. 2000;39:2570–2590. doi: 10.1016/s0028-3908(00)00144-1. [DOI] [PubMed] [Google Scholar]
- 27.Langston JW, Quik M, Petzinger G, Jakowec M, Di Monte DA. Investigating levodopa-induced dyskinesias in the parkinsonian primate. Ann Neurol. 2000;47:S79–89. [PubMed] [Google Scholar]
- 28.Le Novere N, Zoli M, Changeux JP. Neuronal nicotinic receptor α 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci. 1996;8:2428–2439. doi: 10.1111/j.1460-9568.1996.tb01206.x. [DOI] [PubMed] [Google Scholar]
- 29.Le Novere N, Zoli M, Lena C, Ferrari R, Picciotto MR, Merlo-Pich E, Changeux JP. Involvement of α6 nicotinic receptor subunit in nicotine-elicited locomotion, demonstrated by in vivo antisense oligonucleotide infusion. Neuroreport. 1999;10:2497–501. doi: 10.1097/00001756-199908200-00012. [DOI] [PubMed] [Google Scholar]
- 30.Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, Bertrand D, Chiappinelli VA, Clarke PB, Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW, Wonnacott S. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev. 1999;51:397–401. [PubMed] [Google Scholar]
- 31.Luo S, Kulak JM, Cartier GE, Jacobsen RB, Yoshikami D, Olivera BM, McIntosh JM. α-conotoxin AuIB selectively blocks α3 β4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J Neurosci. 1998;18:8571–8579. doi: 10.1523/JNEUROSCI.18-21-08571.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maggio R, Riva M, Vaglini F, Fornai F, Molteni R, Armogida M, Racagni G, Corsini GU. Nicotine prevents experimental parkinsonism in rodents and induces striatal increase of neurotrophic factors. J Neurochem. 1998;71:2439–2446. doi: 10.1046/j.1471-4159.1998.71062439.x. [DOI] [PubMed] [Google Scholar]
- 33.Marks MJ, Collins AC. Characterization of nicotine binding in mouse brain and comparison with the binding of α-bungarotoxin and quinuclidinyl benzilate. Mol Pharmacol. 1982;22:554–564. [PubMed] [Google Scholar]
- 34.Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci. 1992;12:2765–2784. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marks MJ, Pauly JR, Grun EU, Collins AC. ST/b and DBA/2 mice differ in brain α-bungarotoxin binding and α 7 nicotinic receptor subunit mRNA levels: a quantitative autoradiographic analysis. Brain Res Mol Brain Res. 1996;39:207–222. doi: 10.1016/0169-328x(96)00027-7. [DOI] [PubMed] [Google Scholar]
- 36.Marshall DL, Redfern PH, Wonnacott S. Presynaptic nicotinic modulation of dopamine release in the three ascending pathways studied by in vivo microdialysis: comparison of naive and chronic nicotine-treated rats. J Neurochem. 1997;68:1511–1519. doi: 10.1046/j.1471-4159.1997.68041511.x. [DOI] [PubMed] [Google Scholar]
- 37.McIntosh JM, Santos AD, Olivera BM. Conus peptides targeted to specific nicotinic acetylcholine receptor subtypes. Annu Rev Biochem. 1999;68:59–88. doi: 10.1146/annurev.biochem.68.1.59. [DOI] [PubMed] [Google Scholar]
- 38.Morens DM, Grandinetti A, Reed D, White LR, Ross GW. Cigarette smoking and protection from Parkinson's disease: false association or etiologic clue? Neurology. 1995;45:1041–1051. doi: 10.1212/wnl.45.6.1041. [DOI] [PubMed] [Google Scholar]
- 39.Perry EK, Perry RH, Smith CJ, Dick DJ, Candy JM, Edwardson JA, Fairbairn A, Blessed G. Nicotinic receptor abnormalities in Alzheimer's and Parkinson's diseases. J Neurol Neurosurg Psychiatry. 1987;50:806–809. doi: 10.1136/jnnp.50.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perry EK, Morris CM, Court JA, Cheng A, Fairbairn AF, McKeith IG, Irving D, Brown A, Perry RH. Alteration in nicotine binding sites in Parkinson's disease, Lewy body dementia and Alzheimer's disease: possible index of early neuropathology. Neuroscience. 1995;64:385–395. doi: 10.1016/0306-4522(94)00410-7. [DOI] [PubMed] [Google Scholar]
- 41.Picciotto MR, Caldarone BJ, King SL, Zachariou V. Nicotinic receptors in the brain. Links between molecular biology and behavior. Neuropsychopharmacology. 2000;22:451–465. doi: 10.1016/S0893-133X(99)00146-3. [DOI] [PubMed] [Google Scholar]
- 42.Quik M, Jeyarasasingam G. Nicotinic receptors and Parkinson's disease. Eur J Pharmacol. 2000;393:223–230. doi: 10.1016/s0014-2999(99)00888-2. [DOI] [PubMed] [Google Scholar]
- 43.Quik M, Polonskaya Y, Gillespie A, Jakowec M, Lloyd GK, Langston JW. Localization of nicotinic receptor subunit mRNAs in monkey brain by in situ hybridization. J Comp Neurol. 2000a;425:58–69. doi: 10.1002/1096-9861(20000911)425:1<58::aid-cne6>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 44.Quik M, Polonskaya Y, Gillespie A, Lloyd GK, Langston JW. Differential alterations in nicotinic receptor α6 and β3 subunit messenger RNAs in monkey substantia nigra after nigrostriatal degeneration. Neuroscience. 2000b;100:63–72. doi: 10.1016/s0306-4522(00)00244-x. [DOI] [PubMed] [Google Scholar]
- 45.Quik M, Police S, He L, Di Monte DA, Langston JW. Expression of D3 receptor messenger RNA and binding sites in monkey striatum and substantia nigra after nigrostriatal degeneration: effect of levodopa treatment. Neuroscience. 2000c;98:263–273. doi: 10.1016/s0306-4522(00)00130-5. [DOI] [PubMed] [Google Scholar]
- 46.Ross SA, Wong JY, Clifford JJ, Kinsella A, Massalas JS, Horne MK, Scheffer IE, Kola I, Waddington JL, Berkovic SF, Drago J. Phenotypic characterization of an α4 neuronal nicotinic acetylcholine receptor subunit knock-out mouse. J Neurosci. 2000;20:6431–6441. doi: 10.1523/JNEUROSCI.20-17-06431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneider JS, Yuwiler A, Markham CH. Selective loss of subpopulations of ventral mesencephalic dopaminergic neurons in the monkey following exposure to MPTP. Brain Res. 1987;411:144–150. doi: 10.1016/0006-8993(87)90691-3. [DOI] [PubMed] [Google Scholar]
- 48.Schneider JS, Pope-Coleman A, Van Velson M, Menzaghi F, Lloyd GK. Effects of SIB-1508Y, a novel neuronal nicotinic acetylcholine receptor agonist, on motor behavior in parkinsonian monkeys. Mov Disord. 1998;13:637–642. doi: 10.1002/mds.870130405. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz RD, McGee R, Jr, Kellar KJ. Nicotinic cholinergic receptors labeled by [3H]acetylcholine in rat brain. Mol Pharmacol. 1982;22:56–62. [PubMed] [Google Scholar]
- 50.Schwartz RD, Lehmann J, Kellar KJ. Presynaptic nicotinic receptors labeled by [3H]acetylcholine on catecholamine and serotonin axons in brain. J Neurochem. 1984;42:1495–1498. doi: 10.1111/j.1471-4159.1984.tb02818.x. [DOI] [PubMed] [Google Scholar]
- 51.Sorenson EM, Shiroyama T, Kitai ST. Postsynaptic nicotinic receptors on dopaminergic neurons in the substantia nigra pars compacta of the rat. Neuroscience. 1998;87:659–673. doi: 10.1016/s0306-4522(98)00064-5. [DOI] [PubMed] [Google Scholar]
- 52.Vailati S, Hanke W, Bejan A, Barabino B, Longhi R, Balestra B, Moretti M, Clementi F, Gotti C. Functional α6-containing nicotinic receptors are present in chick retina. Mol Pharmacol. 1999;56:11–19. doi: 10.1124/mol.56.1.11. [DOI] [PubMed] [Google Scholar]
- 53.Whiteaker P, Marks MJ, McIntosh JM, Piccioto MR, Changeux J-P, Collins AC. Location and composition of α-conotoxin MII binding nicotinic receptors in mouse brain. Soc Neurosci Abstr. 1998;24:836. [Google Scholar]
- 54.Whiteaker P, Davies AR, Marks MJ, Blagbrough IS, Potter BV, Wolstenholme AJ, Collins AC, Wonnacott S. An autoradiographic study of the distribution of binding sites for the novel α7-selective nicotinic radioligand [3H]-methyllycaconitine in the mouse brain. Eur J Neurosci. 1999;11:2689–2696. doi: 10.1046/j.1460-9568.1999.00685.x. [DOI] [PubMed] [Google Scholar]
- 55.Whiteaker P, McIntosh JM, Luo S, Collins AC, Marks MJ. 125I-α-conotoxin MII identifies a novel nicotinic acetylcholine receptor population in mouse brain. Mol Pharmacol. 2000a;57:913–925. [PubMed] [Google Scholar]
- 56.Whiteaker P, Jiminez M, McIntosh JM, Collins AC, Marks MJ. Identification of a novel nicotinic binding site in mouse brain using 125I-epibatidine. Br J Pharmacol. 2000b;131:729–739. doi: 10.1038/sj.bjp.0703616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]