

Abstract
Physical exercise ameliorates age-related neuronal loss and is currently recommended as a therapeutical aid in several neurodegenerative diseases. However, evidence is still lacking to firmly establish whether exercise constitutes a practical neuroprotective strategy. We now show that exercise provides a remarkable protection against brain insults of different etiology and anatomy. Laboratory rodents were submitted to treadmill running (1 km/d) either before or after neurotoxin insult of the hippocampus (domoic acid) or the brainstem (3-acetylpyridine) or along progression of inherited neurodegeneration affecting the cerebellum (Purkinje cell degeneration). In all cases, animals show recovery of behavioral performance compared with sedentary ones, i.e., intact spatial memory in hippocampal-injured mice, and normal or near to normal motor coordination in brainstem- and cerebellum-damaged animals. Furthermore, exercise blocked neuronal impairment or loss in all types of injuries. Because circulating insulin-like growth factor I (IGF-I), a potent neurotrophic hormone, mediates many of the effects of exercise on the brain, we determined whether neuroprotection by exercise is mediated by IGF-I. Indeed, subcutaneous administration of a blocking anti-IGF-I antibody to exercising animals to inhibit exercise-induced brain uptake of IGF-I abrogates the protective effects of exercise in all types of lesions; antibody-treated animals showed sedentary-like brain damage. These results indicate that exercise prevents and protects from brain damage through increased uptake of circulating IGF-I by the brain. The practice of physical exercise is thus strongly recommended as a preventive measure against neuronal demise. These findings also support the use of IGF-I as a therapeutical aid in brain diseases coursing with either acute or progressive neuronal death.
Keywords: neurodegeneration, physical exercise, insulin-like growth factor I, neurotoxins, Purkinje cell degeneration, neuroprotection
Increased prevalence of neurodegenerative illnesses in modern societies has been classically related to an increasingly aging population (Amaducci and Tesco, 1994). However, risk factors associated with modern lifestyle may also contribute to incidence of neurodegenerative diseases (Meyer et al., 1998). For example, a proposed association between vascular risk factors or diet with neurodegeneration and dementia (Blass et al., 2000; Calingasan and Gibson, 2000) suggests that factors associated with lifestyle in Western culture contribute to increased disease rates. Among these, we hypothesized that sedentary life may be a risk factor in neurodegenerative diseases because it is associated with higher risk of cerebrovascular accidents and is more pronounced in the elderly. Furthermore, several studies indicate that physical exercise may be neuroprotective. For instance, physical activity increases cognitive ability in rats and aging humans (Fordyce and Farrar, 1991;Kramer et al., 1999), attenuates motor deficits (Klintsova et al., 1998), increases new neuron formation (van Praag et al., 1999), ameliorates neurological impairments in different neurodegenerative processes (Arkin, 1999; Petajan and White, 1999; Larsen et al., 2000;Mattson, 2000), and impedes age-related neuronal loss (Larsen et al., 2000)
Our hypothesis is further supported by a different line of observations relating physical exercise to physiologically relevant neuroprotective factors, such as insulin-like growth factor I (IGF-I). We found recently that sedentary animals showed reduced brain uptake of serum IGF-I compared with exercising animals (Carro et al., 2000). Brain uptake of blood-borne IGF-I is essential for exercise-induced increases in the number of newly formed hippocampal neurons and in widespread c-Fos expression in neurons (Carro et al., 2000; Trejo et al., 2001). In addition, systemic injection of IGF-I mimics exercise-induced increase in BDNF expression in the hippocampus and is accumulated by brain cells in a pattern identical to that found after exercise (Carro et al., 2000). Other observations equaling exercise and IGF-I actions on the brain include increases in memory performance (Sonntag et al., 1997; Markowska et al., 1998; Radaka et al., 2001), in neovascularization (Black et al., 1990; Sonntag et al., 1997), and in glucose consumption (Cheng et al., 2000; Ide and Secher, 2000).
Based on these observations, we hypothesized that sedentarism increases the susceptibility to neurodegenerative processes attributable to insufficient brain uptake of serum IGF-I. To test this idea, we submitted intact, as well as brain-damaged, animals to treadmill running to stimulate brain uptake of serum IGF-I because previous observations indicated that chronic systemic administration of IGF-I resulting in increased levels of IGF-I in the brain protects against central neuronal death (Fernandez et al., 1998). We used several models of neurodegeneration affecting different brain areas because exercise-induced capture of serum IGF-I by the brain is widespread (Carro et al., 2000). We reasoned that, because IGF-I receptors are widely distributed in the brain (Bondy and Lee, 1993), neuroprotection by exercise-induced brain uptake of IGF-I should also be ample.
MATERIALS AND METHODS
Models of neurodegeneration. We used three models of experimental neurodegeneration affecting different brain areas to determine whether neuroprotection by exercise includes all types of neuronal populations or is restricted to a few. Because our aim was to induce recovery through exercise, we used partial lesions. In the first type, we injected domoic acid (0.5 mg/kg, i.p.) to adult C57BL/6 male mice (25 gm) to kill hippocampal neurons by excitotoxic damage (Stewart et al., 1990). This low dose of the excitotoxin produces partial neuronal loss (Azcoitia et al., 2001), resulting in behavioral deficits without inducing death-threatening seizures. Excitotoxic neuronal death is considered currently a major pathogenic mechanism in human neurodegenerative diseases (Ikonomidou and Turski, 1995). In the second model, we injected to adult male Wistar rats (250–300 gm) an intermediate dose of the neurotoxin 3-acetylpyridine (3AP) (40 mg/kg, i.p.), which damages a substantial proportion of inferior olive (IO) neurons in the brainstem by eliciting cellular energy imbalance (Phillips et al., 2000). Energy imbalances are currently considered to be involved in many neurodegenerative conditions (Beal, 2000). The third model, which used the pcd mouse (The Jackson Laboratory, Bar Harbor, ME), is an inherited degeneration affecting Purkinje cells in the cerebellum. Genetic neurodegeneration is also a common cause of human disease (Hardy and Gwinn-Hardy, 1998). Because the pcd mutation is still uncharacterized, we classified the animals according to their phenotype in severely and moderately ataxic, as determined in the rotarod test (see below). Because severely ataxic pcd mice do not survive for long times and run with great difficulty as a result of pronounced muscle wasting, we used mice showing moderate ataxia. Furthermore, our aim was to evaluate whether exercise prevents progression of the disease at early stages, because human genetic neurodegeneration currently can be detected early.
Behavioral evaluation. The models of neurodegeneration used display behavioral deficits that can be quantified through task-oriented tests. Spatial memory in hippocampal-lesioned mice can be evaluated with the water maze test (Petrie et al., 1992). We followed procedures described in detail previously using a water tank at 22°C with intramaze and extramaze orientation cues (Frisch et al., 2000). Briefly, after a 1 d habituation trial (day 1) in which preferences between quadrants in the different experimental groups were ruled out, for the subsequent 2–7 d, the animals learned to find a hidden platform (acquisition), followed by 1 d (day 8) of extinction trial without the platform in which swimming speed was found to be similar in all groups. At days 15 and 16, animals were tested for long-term retention (memory) with the platform placed in the original location in the water tank. On the last day (day 18), a cued version protocol was conducted to rule out possible sensorimotor and motivational differences between experimental groups. All groups found the platform faster, and the exploratory behavior was shorter than previous days, indicating a normal sensorimotor and motivational state in all of the animals. Conversely, brainstem-lesioned (3AP) and cerebellum-lesioned (pcd) animals show deficits in motor coordination that can be quantified with the rotarod test, as described in detail previously (Fernandez et al., 1998). All animals were thoroughly familiarized with the rotating rod procedure before test trials were run. Behavioral data were analyzed by ANOVA and Student's ttest.
Experimental design. We aimed to test both prevention and amelioration of neurodegeneration by exercise. Three exercise regimes were used: (1) protocol A, animals exercised before brain insult (Fig.1A); (2) protocol B, animals exercised after brain insult (Fig. 1B); and (3) protocol C, animals exercised both before and after brain insult (Fig. 1C). In protocol A, animals ran for 15 d in a treadmill apparatus and thereafter received a single injection of 3AP (rats) or domoic acid (mice). The degree of functional impairment was evaluated 5 and 7 d after 3AP and domoic acid, respectively, when maximal deleterious effects of the neurotoxins have already taken place (Fernandez et al., 1999; Azcoitia et al., 2001). Protocol B was used also in two different models: (1) Wistar rats received, on the first day of the experiment, an injection of 3AP and treadmill running for the next 25 d; and (2) moderately ataxic pcd mice underwent daily treadmill exercise for 1 month. Behavioral testing of these animals was performed every week (Fig. 1B). Protocol C was applied only in rats; animals were trained during 15 d before receiving a 3AP injection and continued training until full recovery. Motor coordination was evaluated once per week.
Fig. 1.
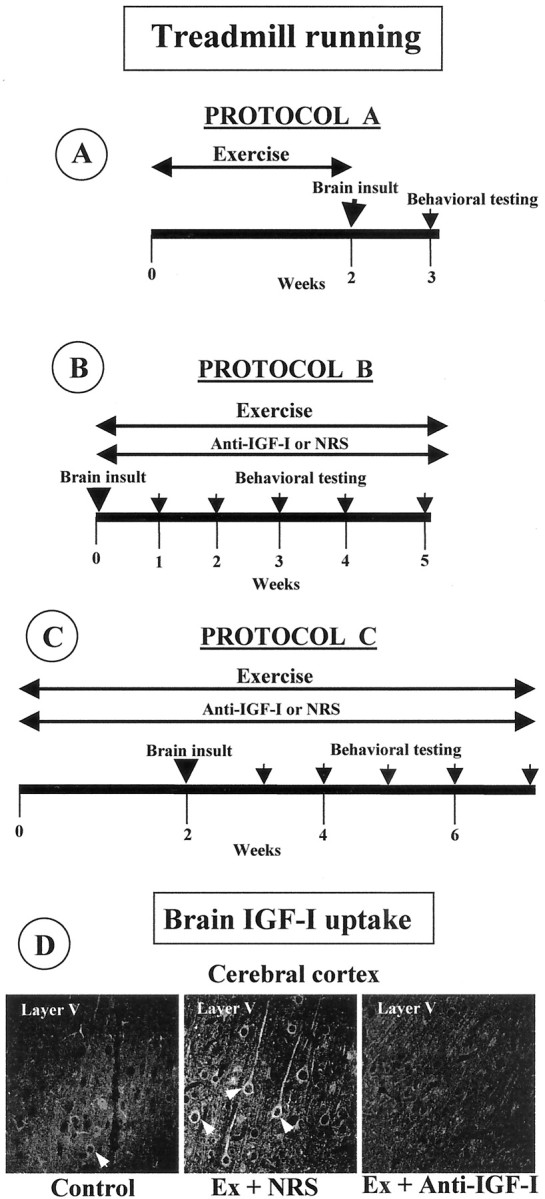
A–C, Treadmill running and anti-IGF-I delivery schedules. A, Protocol A: exercise before brain insult. Animals run during 15 d before brain insult (3AP in rats and domoic acid in mice). Behavioral testing was conducted 5–7 d later. B, Protocol B: exercise after brain insult. Brain-damaged animals ran for 4–5 weeks and were evaluated in the rotarod once per week (pcd mice and 3AP-injected rats).C, Protocol C: animals exercised both before and after brain insult (3AP). Behavioral testing was also done every week. In a parallel series of experiments, an anti-IGF-I infusion was delivered in protocols B and C to exercising animals. Control exercising animals received an NRS infusion. In all protocols, animals were killed for anatomical evaluation after the last behavioral evaluation.D, IGF-I antiserum inhibits exercise-induced brain accumulation. Control, Sedentary animals show negligible IGF-I immunostaining in the brain, whereas exercised animals receiving NRS (Ex + NRS) show a marked increase that is inhibited when an anti-IGF-I infusion is administered simultaneously (Ex + Anti-IGF-I). A representative brain area is shown.
In a second series of experiments, we determined the role of circulating IGF-I in exercise-induced neuroprotection. We administered a chronic infusion of a blocking anti-IGF-I antiserum (20% in saline) subcutaneously through an osmotic minipump (Alzet 2004 in rats and Alzet 1002 in mice; Alza, Palo Alto, CA) to animals undergoing exercise training. Infusion was maintained throughout the exercise protocol (Fig. 1B,C). We have characterized thoroughly that this procedure blocks the exercise-stimulated entrance of serum IGF-I into the brain because the anti-IGF-I antiserum blocks binding of IGF-I to its receptor (Trejo et al., 2001) (Fig. 1D). All groups of control exercising animals received an infusion of non-immune normal rabbit serum (NRS) (20% in saline), which does not impede the entrance of serum IGF-I into the brain (Fig. 1D). In separate experiments, we infused recombinant IGF-I (GroPrep) through a subcutaneous minipump (Alzet 1002; 50 μg/kg, 0.25 μl/hr) for 14 d to brain-damaged mice.
Treadmill exercise. Animals were familiarized with the treadmill apparatus (Cibertec) to minimize novelty stress and then divided in two groups: exercised and non-exercised. The procedure was described in detail previously (Carro et al., 2000). Animals that initially refused to run were encouraged by gently tapping their backs. Rats ran for 1 hr at 17 m/min every day, and mice ran for 1 hr/d at 14 m/min. These relatively low speeds were used to allow brain-damaged animals to learn to run. Training of brain-damaged animals consisted of gradual adaptation to the running schedule: the first day they ran for several minutes and, by day 5, they were able to complete 1 hr running. Control animals remained in the treadmill without running. Animals ran in the morning, 5 d/week.
Immunohistochemistry and autoradiography.Immunohistochemical procedures have been described previously (Carro et al., 2000). Brain areas were serially sectioned at 40 μm and immersed free-floating in 0.1 m phosphate buffer. A one-in-six series of sections of every animal was used. One series was used for labeling with calbindin and another series for Nissl staining with toluidine blue. Calbindin was used as a specific marker of hippocampal CA2 pyramidal cells, IO neurons, and Purkinje cells (Celio, 1990). Primary antibody used was monoclonal anti-calbindin (1:1000; Sigma, St. Louis, MO). The secondary antibody was a biotinylated donkey anti-mouse IgG (1:1000; Jackson ImmunoResearch, West Grove, PA), followed by the peroxidase-based ABC system (Vector Laboratories, Burlingame, CA). The number of neurons was determined by stereological methods, as described in detail previously (Trejo et al., 2001). Calbindin- and Nissl-positive cells were counted in a one-in-six series of sections (300 μm apart) with a 40× objective (Leica, Nussloch, Germany). The same areas and number of sections were studied for all of the animals and all of the experimental groups. A Student'st test was performed when comparing two groups.
Brain glucose uptake was determined by procedures described previously (Cheng et al., 2000). One week after injection of domoic acid to mice, 2-deoxy-d-(1-14C) glucose (1 μCi/gm, i.p.; Amersham Pharmacia Biotech, Uppsala, Sweden) was injected, and 45 min later, the animals were anesthetized. The brains were perfused with saline and snap frozen. Coronary sections (16 μm thick) were cut at −20°C, thaw mounted onto poly-l-lysine-coated slides, and dried on a 60°C plate for 15 min. Anatomically matched sections were exposed for 5 d with Kodak Biomax MR film (Eastman Kodak, Rochester, NY). Autoradiographic images were digitized by using a CanoScan FB (Canon Inc., Lake Success, NY). Signal intensity in different brain regions was analyzed with Leica Q500MC software.
Evaluation of neuronal impairment. Impairment of brainstem IO neurons after 3AP injection was assessed by determining loss of neurons expressing calbindin because it correlates well with loss of Nissl-stained IO cells (Fernandez et al., 1998) and is more accessible for stereology counts. To determine neuronal impairment in the cerebellum of moderately ataxic pcd mice, we counted calbindin-positive Purkinje cells because calbindin is a sensitive marker of neuronal impairment in ataxia and precedes cell loss (Ishikawa et al., 1995; Vig et al., 1998). Total number of Purkinje neurons in each group was also assessed by counting Nissl-stained cells in the Purkinje cell layer. In domoic-treated mice, we counted total numbers of neurons in the hippocampal hilus with Nissl. We chose this area because is severely affected by domoic acid (Stewart et al., 1990), and stereological counts are easier to perform than in other hippocampal regions. Calbindin-positive CA2 pyramidal neurons were also counted.
RESULTS
Neuroprotection by exercise
We determined whether exercise protects against brain damage by assessing behavioral performance and neuronal impairment after brain injury in sedentary and exercised animals. When laboratory rodents run 1 hr daily for 2 weeks before brain injury (Fig. 1A, protocol A), significantly better behavioral performance is observed compared with sedentary animals, regardless of the type of brain lesion (Fig. 2). Exercised rats showed significantly better motor coordination scores after 3AP injection than sedentary rats (p < 0.009) (Fig.2A). Nevertheless, exercised animals were still impaired compared with control rats (48% of the rotarod scores of intact animals; p < 0.005) (Fig.2A). Intact exercised animals show rotarod values similar to intact sedentary rats (data not shown).
Fig. 2.

Exercise prevents behavioral deficits after brain damage. A, Rats undergoing exercise training before 3AP injection, although motor-impaired compared with control animals (*p < 0.005), show significantly better motor coordination in the rotarod than sedentary 3AP rats (*p < 0.009). B, C, Mice submitted to treadmill running before injection of domoic acid have intact learning (B) and memory (C) performance, whereas sedentary mice have significantly impaired acquisition and retention scores (*p < 0.0001). Error bars are smaller than the size of the symbols at some points.
We next determined whether exercise prevents against behavioral impairment after injury of another brain area. We injected mice with domoic acid, which elicits deficits in spatial learning attributable to hippocampal lesion, and found that mice running for 15 d before receiving the neurotoxin show unimpaired acquisition and retention scores in the water maze test (Fig. 2B,C). However, sedentary domoic acid-injected mice have significantly impaired spatial learning and memory performance (Fig. 2B,C).
Because in humans neurological deficits do not usually appear until a substantial number of neurons are impaired, we determined whether physical exercise would help recover function after brain insult (protocol B). As shown in Figure3A, rats undergoing daily running after 3AP injection eventually regain full motor coordination after 5 weeks of exercise (90% of control values; p < 0.002 vs 3AP sedentary rats), whereas sedentary animals remained ataxic. When rats exercised only before but not after brain insult with 3AP, they remained 50% impaired throughout the duration of the study (data not shown). Furthermore, pcd mice with significantly impaired motor coordination in the rotarod (p < 0.0001 vs controls) before exercise training rapidly attained normal motor performance and remained normal for the duration of the training protocol (Fig. 3B). Thus, daily exercise provides continued protection against the pcd mutation. On the contrary, sedentary pcd mice remained ataxic throughout the duration of the study (Fig.3B).
Fig. 3.

Exercise induces recovery of behavioral performance in ongoing neurodegeneration. A, Rats submitted to treadmill running after 3AP insult gradually recover motor coordination and reach normal performance after 5 weeks of running (*p < 0.002 vs 3AP). B, pcd mice with moderate, albeit significantly impaired motor coordination underwent exercise training and recovered normal motor performance within 1 week. They kept normal motor coordination for the duration of the study, whereas sedentary pcd mice remained ataxic. However, exercising pcd mice simultaneously receiving an anti-IGF-I infusion did not recover limb coordination (*p < 0.001).C, Rats were submitted both before and after 3AP insult to treadmill running with simultaneous infusion of NRS and recovered full motor coordination after 5 weeks. However, rats receiving simultaneously an anti-IGF-I infusion remained severely impaired [*p < 0.01 vs 3AP and (Ex + 3AP + Ex) + Anti-IGF-I].
Exercise leads to functional recovery in different types of neurodegenerative insults, probably because target neuronal populations remain primarily unaffected. Figure4A shows that the number of calbindin-positive neurons in the inferior olive of 3AP-injected rats is almost normal in exercised animals, whereas sedentary animals show a marked reduction (p < 0.01 vs control rats). Exercise prevents neuronal impairments also in other types of injuries. Number of Nissl-stained neurons in the hilus of the hippocampus of domoic acid-injected mice was normal in exercised animals but significantly reduced in sedentary ones (p < 0.02) (Fig. 4B). Calbindin-positive CA2 pyramidal cells in domoic acid-treated mice were also reduced in number: 42,916 ± 1141 cells/mm3 after domoic acid versus 49,666 ± 1166 in controls (p < 0.05). However, exercise block the domoic acid-induced reduction: domoic acid-injected exercised mice have 50,233 ± 348 cells/mm3. Similarly, calbindin-positive Purkinje cells in pcd mice were substantially preserved in exercised animals but not in sedentary animals (p < 0.0001) (Fig. 4C). The reduction in total number of Purkinje cells, as determined by counting the number of Nissl-stained cells in the Purkinje cell layer, although less pronounced than that observed in calbindin-positive Purkinje cells, was also greater in sedentary mice, with 11% reduction compared with 4% in exercised pcd mice.
Fig. 4.

Exercise prevents neuronal loss and impairment in an IGF-I dependent manner. A, 3AP-injected rats showed profound neuronal impairment as determined by a drastic decrease in calbindin-positive cells in the IO. Exercised animals showed only a moderate, nonsignificant decrease in the number of calbindin-positive IO neurons. However, exercising animals simultaneously receiving an anti-IGF-I infusion showed sedentary-like neuronal impairment.Inset, Representative brainstem sections of the different experimental groups showing calbindin staining in the IO. Note the marked absence of calbindin-positive cell bodies in brainstem sections of sedentary 3AP and exercised plus anti-IGF-I 3AP rats. *p < 0.01. B, Domoic acid-injured mice show full protection against neuronal loss by exercise. Again, anti-IGF-I administration obliterated the protective effects of exercise. Inset, Representative Nissl-stained sections of corresponding experimental groups. Loss of neurons after domoic acid was assessed in the hippocampal hilus (Hil) by counting Nissl-stained cells. *p < 0.02 and **p < 0.01 versus respective controls.C, Sedentary pcd mice show a profound loss of calbindin staining of Purkinje cells in the cerebellar cortex. Exercising pcd mice show normal numbers of calbindin-positive Purkinje cells, whereas exercised plus anti-IGF-I-treated pcd mice have significantly reduced numbers of calbindin-positive Purkinje cells, similar to sedentary pcd mice. Inset, Representative cerebellar cortex sections of the different experimental groups. Note the marked depletion of calbindin-positive cells in the Purkinje cell layer (PC). ML, Molecular layer of the cerebellum; GL, granule cell layer. **p < 0.0001 versus respective control group.
Circulating IGF-I is necessary for exercise-induced neuroprotection
We next determined whether circulating IGF-I mediates neuroprotective effects of physical exercise because it increases levels of IGF-I in the CSF and in the brain (Carro et al., 2000; Trejo et al., 2001). Because chronic subcutaneous administration of an anti-IGF-I antibody blocks the entrance of blood-borne IGF-I into the brain parenchyma (Fig. 1D), we administered a chronic infusion of anti-IGF-I antibody to rats submitted to daily running before and after receiving 3AP (protocol C) and found that exercise-induced recovery of motor coordination was blocked (Fig.3C). We also tested whether recovery of behavioral performance after brain damage (protocol B) depends also on blood-borne IGF-I. We gave an infusion of anti-IGF-I to pcd ataxic mice undergoing exercise training and found that recovery was hindered (Fig.3B). Significantly, and as reported previously in 3AP-damaged rats (Fernandez et al., 1998), subcutaneous administration of IGF-I for 1 month to pcd ataxic mice also results in recovery of motor coordination in the rotarod test (90% of control scores; data not shown). This reinforces the notion that systemic IGF-I is the neuroprotective factor involved in the effects of exercise.
We then tested whether administration of anti-IGF-I also blocks exercise-induced neuronal protection. As shown in Figure 4, anti-IGF-I treated exercising animals show marked neuronal damage indistinguishable from that found in sedentary animals. Inhibition of exercise neuroprotection by anti-IGF-I is observed in all types of affected neuronal populations. This includes reduced brainstem calbindin-positive IO neurons after 3AP (Fig. 4A). Nissl-stained neurons of the hippocampal hilus (Fig.4B) and calbindin-positive CA2 pyramidal cells after domoic acid were also reduced (40,300 ± 602 cells/mm3 in exercise plus domoic acid plus anti-IGF-I-treated mice vs 50,233 ± 348 in exercise plus domoic acid-treated mice; p < 0.05). Finally, in pcd mice, calbindin-positive (Fig. 4C) and Nissl-stained Purkinje cells were also reduced (19,133 ± 558 cells/mm3 in controls vs 16,369 ± 1046 in exercise plus anti-IGF-I-treated mice; p < 0.01). Neuronal damage likely explains the lack of functional recovery found after anti-IGF-I treatment of exercising animals (Fig. 3).
Knowledge of the mechanisms underlying IGF-I neuroprotection are scarce, and their possible relation to exercise-induced neuroprotection is unknown. A possible mechanism involved in both IGF-I and exercise neuroprotective effects may be enhanced glucose metabolism because energy demands are increased in injured neurons (Vannucci et al., 1998) and both exercise and IGF-I increase brain glucose consumption (Cheng et al., 2000; Ide and Secher, 2000). Thus, we compared the effects of chronic subcutaneous administration of IGF-I and exercise training on brain glucose uptake in domoic acid-injured mice. As reported previously (Cheng et al., 2000), hippocampal damage with domoic acid induces an increase in glucose uptake in the hippocampus (35 ± 6% increase over controls; p < 0.0001) (Fig.5). Additional increases in hippocampal glucose uptake were found after IGF-I treatment of domoic acid-injected mice, as well as in domoic acid-injured mice submitted to exercise training (18 ± 1% increase in exercised animals,p < 0.001; 12 ± 2% increase in IGF-I-treated animals, p < 0.005; compared with domoic acid alone) (Fig. 5). In addition, both IGF-I and exercise elicited similar widespread increases in glucose consumption in other telencephalic regions (Fig. 5).
Fig. 5.
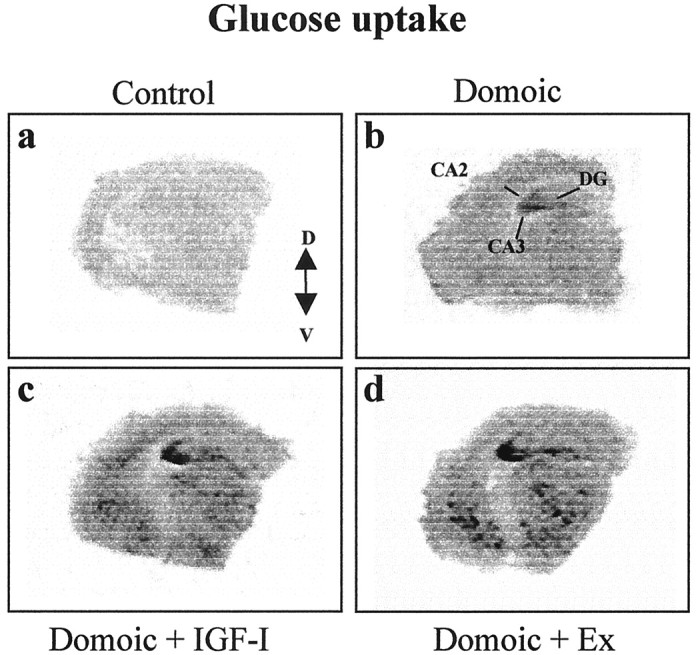
Exercise and IGF-I increase brain glucose uptake in brain-damaged animals. Domoic acid-lesioned mice (b) show increased glucose uptake in the hippocampus compared with nonlesioned mice (a). Glucose uptake is further increased by either exercise (d) or IGF-I (c) not only in the hippocampus but also in other telencephalic areas. Representative brain autoradiography hemisections are shown.CA2, CA3, Hippocampal pyramidal cell layers; DG, hippocampal dentate gyrus; V, ventral; D, dorsal.
DISCUSSION
Our results indicate that physical exercise reduces vulnerability to brain damage in models of neuronal injury involving different types of etiopathogenic mechanisms relevant to human disease. Our findings also indicate that exercise is neuroprotective because of increased passage of circulating IGF-I into the brain (Carro et al., 2000) because, when this passage is blocked, exercise is no longer neuroprotective. Additional evidence supporting that increased IGF-I input underlies exercise-induced neuroprotection is provided by the fact that systemic administration of IGF-I to brain-damaged sedentary mice (present results) or rats (Fernandez et al., 1998) is sufficient to elicit functional recovery. Based on these findings, we hypothesize that circulating IGF-I exerts a physiological neuroprotective tonic effect on the brain that is depressed in sedentary subjects. By extrapolating these observations in rodents to humans and taking into account that exercise also stimulates the growth hormone–IGF-I axis in human beings (Wallace et al., 1999), it is conceivable that changes in lifestyle associated with modern culture, and more specifically increased sedentarism, contributes to an increasing incidence of neurological diseases attributable to acute or progressive neuronal death observed in developed countries. Thus, neuronal demise could be added to the growing list of pathological conditions in which sedentarism is a risk factor.
Exercise not only attenuates the impact of a brain insult but also impedes progression of ongoing neurodegeneration. Thus, exercise reduces or abolishes (depending on the type of insult) neuronal death and decreases or entirely blocks behavioral impairment after neurotoxic insult. Sedentary rats present severe ataxia after 3AP insult compared with mild ataxia in exercised rats, and sedentary mice show memory deficits after domoic acid challenge that are not found in exercised mice. Furthermore, exercise activates endogenous homeostatic mechanisms that counteract the ongoing neurodegenerative process, as seen previously in brain-damaged or aging rats (Fernandez et al., 1998;Larsen et al., 2000). Thus, during early stages of the degenerative process, endogenous neuroprotective mechanisms (such as brain uptake of serum IGF-I) allow exercising animals to recover normal behavioral performance, whereas sedentary animals remain impaired.
Many mechanisms are likely involved in protection by exercise both after acute injury and during progress of neurodegeneration. However, our results indicate that IGF-I must be involved in both types of protective mechanisms. This agrees with previous work showing that serum IGF-I is necessary for other effects of exercise on the brain (Carro et al., 2000; Trejo et al., 2001). Conceivably, homeostatic mechanisms modulated by IGF-I, and as we now suggest, involved in exercise-induced neuroprotection, should encompass a variety of processes supporting appropriate neuronal function. These may range from those aimed to fulfill basic metabolic demands to those directed to maintain neural plasticity (Torres-Aleman, 2001). Altogether, they allow neurons to cope better with pathological threats.
Several of the neuroprotective mechanisms elicited by IGF-I have been characterized previously and include modulation of apoptosis- and neuritogenesis-related proteins (Fernandez et al., 1999). Our present findings in inherited cerebellar degeneration suggest that another mechanism likely counteracting ongoing neuronal death includes modulation of calcium homeostasis by maintaining appropriate expression of the calcium-buffering protein calbindin: exercised pcd mice show normal numbers of calbindin-positive Purkinje cells together with normal limb coordination. Indeed, a major group of proteins targeted in neurodegenerative diseases are involved in calcium homeostasis (Lin et al., 2000). Calbindin upregulation increases resistance to neuronal death (Prehn et al., 1996), and its downregulation originates an ataxic phenotype (Airaksinen et al., 1997). In addition, IGF-I is required to maintain normal levels of calbindin in the adult cerebellum (Nieto-Bona et al., 1995). An additional mechanism involved in IGF-I-mediated exercise neuroprotection is likely related to enhanced neuronal glucose metabolism. Improved glucose metabolism is essential for neurons to be able to survive to injury (Magistretti and Pellerin, 1996), and increased glucose consumption is a typical response to brain injury (Cheng et al., 2000). IGF-I enhances glucose use by neurons through upregulation of glucose transporters and modulation of glycolytic enzymes (Cheng et al., 2000), and as our present findings show, IGF-I stimulates brain glucose metabolism in brain-injured animals in a way indistinguishable of exercise.
We hypothesize that other homeostatic processes involved in IGF-I-mediated exercise neuroprotection may include increased angiogenesis and improved handling of oxygen by neurons. Although the normal adult brain do not show angiogenesis except in response to specific types of insults (Plate, 1999), exercise stimulates angiogenesis in the adult brain (Black et al., 1990), and IGF-I is involved in angiogenesis in the brain and other tissues (Sonntag et al., 1997; Dunn, 2000). Oxygen availability is also compromised in neurodegenerative conditions involving vascular derangements, and IGF-I is known to induce expression of HIF-1 (Zelzer et al., 1998), a transcription factor central in the cell response to hypoxia. Finally, modulation by IGF-I of neuronal excitability through modulation of membrane ion channels, glutamate receptors, or synapse size (Torres-Aleman, 2001) may also be instrumental in neuroprotection. At any rate, it is indeed remarkable that IGF-I exerts protection against multiple types of neuronal insults through such a diversity of mechanisms (Feldman et al., 1997). This further suggests that IGF-I is truly a physiological neuroprotectant because it appears to be specially well suited for this purpose. In this sense, physical exercise could be considered as an stimulator of a physiological neuroprotective mechanism.
A major practical consequence of our findings is that physical therapy may be an important aid in the treatment of brain diseases attributable to either acute or progressive neuronal loss. Indeed, physical therapy is currently advocated in the treatment of several neurodegenerative diseases (Grealy et al., 1999; Petajan and White, 1999; Mattson, 2000). Unfortunately, although in these studies we used a relatively moderate exercise load (i.e., 0.8–1 km/d; rodents voluntarily run much longer distances), exercise may be impractical in severely affected human patients. Administration of synthetic IGF-I could be an alternative. However, at present, this possibility is not feasible as a result of limited use of this growth factor in humans because recent observations link serum IGF-I to cancer incidence (Holly, 1998). Thus, epidemiological studies indicate that higher circulating levels of IGF-I increase the risk to suffer several types of tumors (Giovannucci, 1999). Nevertheless, recent observations by us and other laboratories indicate that many neurodegenerative diseases, including those with high incidence such as Alzheimer's disease and stroke, show significant changes in circulating IGFs (Busiguina et al., 2000). Thus, a risk–benefit assessment should be conducted for the use of IGF-I in each type of neurodegenerative disease.
In conclusion, our results points to sedentarism as a risk factor in neurological diseases showing acute or progressive neuronal death and emphasize the importance of regular exercise to prevent them. Our observations also support the use of exercise and/or IGF-I as therapeutical aids in pathological and age-related neuronal demise.
Footnotes
This study was supported by Direccion General Enseñanza Superior Investigacion y Ciencia Grant PM97-0018. E.C. and S.B. are Comunidad Autonoma de Madrid postdoctoral fellows.
Correspondence should be addressed to Ignacio Torres-Aleman, Cajal Institute, Consejo Superior de Investigaciones Científicas, Avenida Doctor Arce 37, 28002 Madrid, Spain. E-mail:torres@cajal.csic.es.
REFERENCES
- 1.Airaksinen MS, Eilers J, Garaschuk O, Thoenen H, Konnerth A, Meyer M. Ataxia and altered dendritic calcium signaling in mice carrying a targeted null mutation of the calbindin D28k gene. Proc Natl Acad Sci USA. 1997;94:1488–1493. doi: 10.1073/pnas.94.4.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amaducci L, Tesco G. Aging as a major risk for degenerative diseases of the central nervous system. Curr Opin Neurol. 1994;7:283–286. doi: 10.1097/00019052-199408000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Arkin SM. Elder rehab: a student-supervised exercise program for Alzheimer's patients. Gerontologist. 1999;39:729–735. doi: 10.1093/geront/39.6.729. [DOI] [PubMed] [Google Scholar]
- 4.Azcoitia I, Sierra A, Veiga S, Honda S, Harada N, Garcia-Segura LM. Brain aromatase is neuroprotective. J Neurobiol. 2001;47:318–329. doi: 10.1002/neu.1038. [DOI] [PubMed] [Google Scholar]
- 5.Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000;23:298–304. doi: 10.1016/s0166-2236(00)01584-8. [DOI] [PubMed] [Google Scholar]
- 6.Black JE, Isaacs KR, Anderson BJ, Alcantara AA, Greenough WT. Learning causes synaptogenesis, whereas motor activity causes angiogenesis, in cerebellar cortex of adult rats. Proc Natl Acad Sci USA. 1990;87:5568–5572. doi: 10.1073/pnas.87.14.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blass JP, Sheu RK, Gibson GE. Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann NY Acad Sci. 2000;903:204–221. doi: 10.1111/j.1749-6632.2000.tb06370.x. [DOI] [PubMed] [Google Scholar]
- 8.Bondy CA, Lee WH. Patterns of insulin-like growth factor and IGF receptor gene expression in the brain. Functional implications. Ann NY Acad Sci. 1993;692:33–43. doi: 10.1111/j.1749-6632.1993.tb26203.x. [DOI] [PubMed] [Google Scholar]
- 9.Busiguina S, Fernandez AM, Barrios V, Clark R, Tolbert DL, Berciano J, Torres-Aleman I. Neurodegeneration is associated to changes in serum insulin-like growth factors. Neurobiol Dis. 2000;7:657–665. doi: 10.1006/nbdi.2000.0311. [DOI] [PubMed] [Google Scholar]
- 10.Calingasan NY, Gibson GE. Dietary restriction attenuates the neuronal loss, induction of heme oxygenase-1 and blood-brain barrier breakdown induced by impaired oxidative metabolism. Brain Res. 2000;885:62–69. doi: 10.1016/s0006-8993(00)02933-4. [DOI] [PubMed] [Google Scholar]
- 11.Carro E, Nunez A, Busiguina S, Torres-Aleman I. Circulating insulin-like growth factor I mediates effects of exercise on the brain. J Neurosci. 2000;20:2926–2933. doi: 10.1523/JNEUROSCI.20-08-02926.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Celio MR. Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience. 1990;35:375–475. doi: 10.1016/0306-4522(90)90091-h. [DOI] [PubMed] [Google Scholar]
- 13.Cheng CM, Reinhardt RR, Lee WH, Joncas G, Patel SC, Bondy CA. Insulin-like growth factor 1 regulates developing brain glucose metabolism. Proc Natl Acad Sci USA. 2000;97:10236–10241. doi: 10.1073/pnas.170008497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn SE. Insulin-like growth factor I stimulates angiogenesis and the production of vascular endothelial growth factor. Growth Horm IGF Res. 2000;10 [Suppl A]:S41–S42. doi: 10.1016/s1096-6374(00)90020-0. [DOI] [PubMed] [Google Scholar]
- 15.Feldman EL, Sullivan KA, Kim B, Russell JW. Insulin-like growth factors regulate neuronal differentiation and survival. Neurobiol Dis. 1997;4:201–214. doi: 10.1006/nbdi.1997.0156. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez AM, de la Vega AG, Torres-Aleman I. Insulin-like growth factor I restores motor coordination in a rat model of cerebellar ataxia. Proc Natl Acad Sci USA. 1998;95:1253–1258. doi: 10.1073/pnas.95.3.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez AM, Gonzalez de la Vega AG, Planas B, Torres-Aleman I. Neuroprotective actions of peripherally administered insulin-like growth factor I in the injured olivo-cerebellar pathway. Eur J Neurosci. 1999;11:2019–2030. doi: 10.1046/j.1460-9568.1999.00623.x. [DOI] [PubMed] [Google Scholar]
- 18.Fordyce DE, Farrar RP. Enhancement of spatial learning in F344 rats by physical activity and related learning-associated alterations in hippocampal and cortical cholinergic functioning. Behav Brain Res. 1991;46:123–133. doi: 10.1016/s0166-4328(05)80105-6. [DOI] [PubMed] [Google Scholar]
- 19.Frisch C, Dere E, Silva MA, Godecke A, Schrader J, Huston JP. Superior water maze performance and increase in fear-related behavior in the endothelial nitric oxide synthase-deficient mouse together with monoamine changes in cerebellum and ventral striatum. J Neurosci. 2000;20:6694–6700. doi: 10.1523/JNEUROSCI.20-17-06694.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giovannucci E. Insulin-like growth factor-I and binding protein-3 and risk of cancer. Horm Res. 1999;51 [Suppl 3]:34–41. doi: 10.1159/000053160. [DOI] [PubMed] [Google Scholar]
- 21.Grealy MA, Johnson DA, Rushton SK. Improving cognitive function after brain injury: the use of exercise and virtual reality. Arch Phys Med Rehabil. 1999;80:661–667. doi: 10.1016/s0003-9993(99)90169-7. [DOI] [PubMed] [Google Scholar]
- 22.Hardy J, Gwinn-Hardy K. Genetic classification of primary neurodegenerative disease. Science. 1998;282:1075–1079. doi: 10.1126/science.282.5391.1075. [DOI] [PubMed] [Google Scholar]
- 23.Holly J. Insulin-like growth factor-I and new opportunities for cancer prevention. Lancet. 1998;351:1373–1375. doi: 10.1016/S0140-6736(05)79438-1. [DOI] [PubMed] [Google Scholar]
- 24.Ide K, Secher NH. Cerebral blood flow and metabolism during exercise. Prog Neurobiol. 2000;61:397–414. doi: 10.1016/s0301-0082(99)00057-x. [DOI] [PubMed] [Google Scholar]
- 25.Ikonomidou C, Turski L. Excitotoxicity and neurodegenerative diseases. Curr Opin Neurol. 1995;8:487–497. doi: 10.1097/00019052-199512000-00017. [DOI] [PubMed] [Google Scholar]
- 26.Ishikawa K, Mizusawa H, Fujita T, Ohkoshi N, Doi M, Komatsuzaki Y, Iwamoto H, Ogata T, Shoji S. Calbindin-D 28k immunoreactivity in the cerebellum of spinocerebellar degeneration. J Neurol Sci. 1995;129:179–185. doi: 10.1016/0022-510x(94)00279-w. [DOI] [PubMed] [Google Scholar]
- 27.Klintsova AY, Cowell RM, Swain RA, Napper RM, Goodlett CR, Greenough WT. Therapeutic effects of complex motor training on motor performance deficits induced by neonatal binge-like alcohol exposure in rats. I. Behavioral results. Brain Res. 1998;800:48–61. doi: 10.1016/s0006-8993(98)00495-8. [DOI] [PubMed] [Google Scholar]
- 28.Kramer AF, Hahn S, Cohen NJ, Banich MT, McAuley E, Harrison CR, Chason J, Vakil E, Bardell L, Boileau RA, Colcombe A. Ageing, fitness and neurocognitive function. Nature. 1999;400:418–419. doi: 10.1038/22682. [DOI] [PubMed] [Google Scholar]
- 29.Larsen JO, Skalicky M, Viidik A. Does long-term physical exercise counteract age-related purkinje cell loss? A stereological study of rat cerebellum. J Comp Neurol. 2000;428:213–222. [PubMed] [Google Scholar]
- 30.Lin X, Antalffy B, Kang D, Orr HT, Zoghbi HY. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci. 2000;3:157–163. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- 31.Magistretti PJ, Pellerin L. Cellular mechanisms of brain energy metabolism. Relevance to functional brain imaging and to neurodegenerative disorders. Ann NY Acad Sci. 1996;777:380–387. doi: 10.1111/j.1749-6632.1996.tb34449.x. [DOI] [PubMed] [Google Scholar]
- 32.Markowska AL, Mooney M, Sonntag WE. Insulin-like growth factor-1 ameliorates age-related behavioral deficits. Neuroscience. 1998;87:559–569. doi: 10.1016/s0306-4522(98)00143-2. [DOI] [PubMed] [Google Scholar]
- 33.Mattson MP. Neuroprotective signaling and the aging brain: take away my food and let me run. Brain Res. 2000;886:47–53. doi: 10.1016/s0006-8993(00)02790-6. [DOI] [PubMed] [Google Scholar]
- 34.Meyer JS, Terayama Y, Konno S, Akiyama H, Margishvili GM, Mortel KF. Risk factors for cerebral degenerative changes and dementia. Eur Neurol. 1998;39 [Suppl 1]:7–16. doi: 10.1159/000052064. [DOI] [PubMed] [Google Scholar]
- 35.Nieto-Bona MP, Busiguina S, Torres-Aleman I. Insulin-like growth factor I is an afferent trophic signal that modulates calbindin-28kD in adult Purkinje cells. J Neurosci Res. 1995;42:371–376. doi: 10.1002/jnr.490420311. [DOI] [PubMed] [Google Scholar]
- 36.Petajan JH, White AT. Recommendations for physical activity in patients with multiple sclerosis. Sports Med. 1999;27:179–191. doi: 10.2165/00007256-199927030-00004. [DOI] [PubMed] [Google Scholar]
- 37.Petrie BF, Pinsky C, Standish NM, Bose R, Glavin GB. Parenteral domoic acid impairs spatial learning in mice. Pharmacol Biochem Behav. 1992;41:211–214. doi: 10.1016/0091-3057(92)90084-s. [DOI] [PubMed] [Google Scholar]
- 38.Phillips RG, Lawrence MS, Ho DY, Sapolsky RM. Limitations in the neuroprotective potential of gene therapy with Bcl-2. Brain Res. 2000;859:202–206. doi: 10.1016/s0006-8993(99)02453-1. [DOI] [PubMed] [Google Scholar]
- 39.Plate KH. Mechanisms of angiogenesis in the brain. J Neuropathol Exp Neurol. 1999;58:313–320. doi: 10.1097/00005072-199904000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Prehn JH, Bindokas VP, Jordan J, Galindo MF, Ghadge GD, Roos RP, Boise LH, Thompson CB, Krajewski S, Reed JC, Miller RJ. Protective effect of transforming growth factor-beta 1 on beta-amyloid neurotoxicity in rat hippocampal neurons. Mol Pharmacol. 1996;49:319–328. [PubMed] [Google Scholar]
- 41.Radaka Z, Kanekob T, Taharab S, Nakamotoc H, Pucsokd J, Sasvarie M, Nyakase C, Gotoc S. Regular exercise improves cognitive function and decreases oxidative damage in rat brain. Neurochem Int. 2001;38:17–23. doi: 10.1016/s0197-0186(00)00063-2. [DOI] [PubMed] [Google Scholar]
- 42.Sonntag WE, Lynch CD, Cooney PT, Hutchins PM. Decreases in cerebral microvasculature with age are associated with the decline in growth hormone and insulin-like growth factor 1. Endocrinology. 1997;138:3515–3520. doi: 10.1210/endo.138.8.5330. [DOI] [PubMed] [Google Scholar]
- 43.Stewart GR, Zorumski CF, Price MT, Olney JW. Domoic acid: a dementia-inducing excitotoxic food poison with kainic acid receptor specificity. Exp Neurol. 1990;110:127–138. doi: 10.1016/0014-4886(90)90057-y. [DOI] [PubMed] [Google Scholar]
- 44.Torres-Aleman I. I Serum neurotrophic factors and neuroprotective surveillance: focus on IGF-I. Mol Neurobiol. 2001;21:153–160. doi: 10.1385/mn:21:3:153. [DOI] [PubMed] [Google Scholar]
- 45.Trejo JL, Carro E, Torres-Aleman I. Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J Neurosci. 2001;21:1628–1634. doi: 10.1523/JNEUROSCI.21-05-01628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Praag H, Kempermann G, Gage FH. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci. 1999;2:266–270. doi: 10.1038/6368. [DOI] [PubMed] [Google Scholar]
- 47.Vannucci SJ, Reinhart R, Maher F, Bondy CA, Lee WH, Vannucci RC, Simpson IA. Alterations in GLUT1 and GLUT3 glucose transporter gene expression following unilateral hypoxia-ischemia in the immature rat brain. Brain Res Dev Brain Res. 1998;107:255–264. doi: 10.1016/s0165-3806(98)00021-2. [DOI] [PubMed] [Google Scholar]
- 48.Vig PJ, Subramony SH, Burright EN, Fratkin JD, McDaniel DO, Desaiah D, Qin Z. Reduced immunoreactivity to calcium-binding proteins in Purkinje cells precedes onset of ataxia in spinocerebellar ataxia-1 transgenic mice. Neurology. 1998;50:106–113. doi: 10.1212/wnl.50.1.106. [DOI] [PubMed] [Google Scholar]
- 49.Wallace JD, Cuneo RC, Baxter R, Orskov H, Keay N, Pentecost C, Dall R, Rosen T, Jorgensen JO, Cittadini A, Longobardi S, Sacca L, Christiansen JS, Bengtsson BA, Sonksen PH. Responses of the growth hormone (GH) and insulin-like growth factor axis to exercise, GH administration, and GH withdrawal in trained adult males: a potential test for GH abuse in sport. J Clin Endocrinol Metab. 1999;84:3591–3601. doi: 10.1210/jcem.84.10.6037. [DOI] [PubMed] [Google Scholar]
- 50.Zelzer E, Levy Y, Kahana C, Shilo BZ, Rubinstein M, Cohen B. Insulin induces transcription of target genes through the hypoxia- inducible factor HIF-1alpha/ARNT. EMBO J. 1998;17:5085–5094. doi: 10.1093/emboj/17.17.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]