

Abstract
Metabotropic glutamate receptors are expressed abundantly in the spinal cord and have been shown to play important roles in the modulation of nociceptive transmission and plasticity. Most previous studies have focused on the group I metabotropic glutamate receptors (mGluR1 and mGluR5) and activation of phospholipase C signaling by these receptors in modulating nociception. Recently, it was shown that the extracellular signal-regulated kinases (ERKs)/mitogen-activated protein kinases are activated in spinal cord dorsal horn neurons in response to stimulation of nociceptors and that ERK signaling is involved in nociceptive plasticity. In the present studies, we sought to test the hypothesis that group I mGluRs modulate nociceptive transmission or plasticity via modulation of ERK signaling in dorsal horn neurons. We show that activation of mGluR1 and mGluR5 leads to activation of ERK1 and ERK2 in the spinal cord. Furthermore, we find that inflammation-evoked ERK activation, which is required for nociceptive plasticity, is downstream of mGluR1 and mGluR5. Finally, we show colocalization of group I mGluRs with activated ERK in dorsal horn neurons. These results show that mGluR1 and mGluR5 are activated in dorsal horn neurons in response to peripheral inflammation and that activation of these group I mGluRs leads to activation of ERK1 and ERK2, resulting in enhanced pain sensitivity.
Keywords: extracellular signal-regulated kinases (ERKs), mitogen-activated protein kinase (MAPK), nociception, formalin test, spinal cord, dorsal horn
The mechanisms associated with chronic inflammatory conditions, such as painful neuropathy, are not clearly understood. It is, however, thought that continuous activation of peripheral afferent fibers by noxious stimulation results in sensitization of dorsal horn neurons, which can subsequently produce aberrant activity in primary afferent fibers (Neugebauer et al., 1994;Rees et al., 1996). Together, the peripheral and central mechanisms contribute to a cycle of persistent nociception. Persistent activation of peripheral afferents may result in central changes in neurotransmitter release or receptor states, resulting in chronic nociceptive activation.
Subsequent to activation of sensory neurons, glutamate is released in the spinal dorsal horn, in which it acts via activation of ionotropic ligand-gated ion channels and G-protein-coupled metabotropic glutamate receptors (mGluRs) (Conn and Pin, 1997). The eight cloned mGluRs (mGluR1–mGluR8) are broadly classified into groups I (mGluR1 and mGluR5), II (mGluR2 and mGluR3), and III (mGluR4, mGluR6, mGluR7, and mGluR8) based on their sequence homology, pharmacology, and association with intracellular effector systems (Conn and Pin, 1997). Several mGluR subtypes are expressed in the spinal cord; in particular, there is strong expression of mGluR5 and possibly mGluR1 in dorsal horn neurons (Valerio et al., 1997a,b; Alvarez et al., 2000).
Behavioral studies show that intrathecal application of the mGluR1/5 agonist 3,5-dihydroxyphenylglycine (DHPG) induces spontaneous nociceptive behavior (Fisher and Coderre, 1996a), as well as thermal hyperalgesia and allodynia in rats, and enhances pain in the second phase of the formalin test (Fisher and Coderre, 1998; Fisher et al., 1998). DHPG also potentiates nociceptive responses of spinothalamic tract cells to cutaneous stimuli (Neugebauer et al., 1999). Furthermore, mGluR antagonists reduce sustained nociceptive inputs evoked by intraplantar formalin in rats and reduce capsaicin-induced sensitization (Fisher and Coderre, 1996b; Neugebauer et al., 1999). Finally, antisense knockdown of group I mGluRs reduces responses of dorsal horn neurons to repeated cutaneous mustard oil applications (Young et al., 1998). Together, these reports suggest a role for group I mGluRs in spinal modulation of nociception.
The cellular mechanisms by which mGluRs modulate nociception are not clear. Group I mGluRs activate phospholipase C, leading to release of calcium from intracellular stores and activation of PKC. Although most studies of group I mGluRs focus on this cascade, recent studies suggest that they can activate other downstream kinases, such as the extracellular signal-regulated kinase (ERK)/mitogen-activated protein (MAP) kinase (Peavy and Conn, 1998; Ferraguti et al., 1999). Both ERK1 and ERK2 are expressed in the spinal cord and are activated in rat dorsal horn neurons after inflammation (Thomas and Hunt, 1993;Ji et al., 1999). Inhibitors of ERK signaling reduce pain in the second phase of the formalin test, suggesting a selective role for ERKs in nociceptive sensitization (Ji et al., 1999).
The studies mentioned above suggest critical roles for ERK signaling and mGluRs in inflammatory pain plasticity. In the present study, we test the hypothesis that activation of mGluR1 and/or mGluR5 is necessary for ERK activation after peripheral inflammation and, furthermore, that this pathway is necessary for nociceptive sensitization.
MATERIALS AND METHODS
Animals. All experiments were done in accordance within the guidelines of the National Institutes of Health and The International Association for the Study of Pain and were approved by the Animal Care and Use Committee of Baylor College of Medicine. Male C57BL/6 mice weighing 20–25 gm were purchased from Baylor College of Medicine, were housed in 12 hr light/dark cycles, and were given foodad libitum.
Drug administration. The following compounds were purchased from Tocris Cookson (Ballwin, MO): (RS)-DHPG, an mGluR1 and mGluR5-selective agonist, was dissolved in 0.9% saline; 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt), a noncompetitive mGluR1-selective antagonist, was prepared as a 50 mm stock solution in an equimolar solution of NaOH and then diluted in HEPES (100 mm, pH 7.4); and 2-methyl-6-(phenylethynyl)-pyridine (MPEP), a noncompetitive mGluR5-selective antagonist, was dissolved in HEPES (100 mm, pH 7.4). 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98059), an MAP kinase kinase (MEK) inhibitor (Sigma, St. Louis, MO), was dissolved in 10% DMSO. All drugs or their appropriate vehicles were injected intrathecally in a volume of 3 μl by lumbar puncture using a Hamilton syringe and 30 gauge needles (Hylden and Wilcox, 1980).
Nociceptive testing. The total time spent in spontaneous pain behavior was recorded after intrathecal injection of (RS)-DHPG in mice for 5 min. Spontaneous pain behavior was defined as caudally oriented licking of the flanks, tail, and hindpaws after intrathecal (RS)-DHPG. In separate experiments, mice were pretreated intrathecally for 15 min with the mGluR1 or mGluR5 antagonists or vehicle before (RS)-DHPG injection. The formalin test was done as described previously (Karim et al., 1993). Mice were conditioned in a transparent Plexiglas test box (5 × 5 × 10 inches) before any drug injections for 1 hr. Mice were pretreated intrathecally for 15 min with the antagonists or appropriate vehicles. Formalin solution (2%) was injected subcutaneously into the right hindpaw, and the mouse was returned to the test box immediately. The total time spent in nociceptive behavior (licking and lifting of the injected paw) was recorded in blocks of 5 min for 1 hr. Additional experiments were done in which a combination of MPEP and CPCCOEt was administered intrathecally 15 min before hindpaw formalin injection.
Sample preparation. Mice were killed 5 min after injection of different doses of (RS)-DHPG or at different time points after hindpaw formalin injection. The spinal cords were isolated, and lumbar sections from individual mice were stored at −80°C. Lumbar spinal cord enlargements (L4–S1) were homogenized using a dounce homogenizer in ice-cold homogenization buffer (50 mm Tris HCl, pH 7.5, 50 mmNaCl, 10 mm EGTA, 5 mmEDTA, 2 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 200 μmparanitrophenylphosphate, 1 mmphenylmethylsulfonyl fluoride, 20 μg/ml leupeptin, and 4 μg/ml aprotinin; Sigma). Protein concentrations were determined by the DC assay kit (Bio-Rad, Hercules, CA).
Immunoblotting for total and phospho-ERK. Proteins (10 μg) were electrophoresed in 10% SDS polyacrylamide gels. Proteins were transferred onto protein-sensitive nitrocellulose membranes and blocked in B-TTBS [3% bovine serum albumin (BSA), 50 mmTris-HCl, pH 7.5, 150 mm NaCl, 0.02 mm Na orthovanadate, 0.05% Tween 20, and 0.01% thimerosal; Sigma] for 2 hr at room temperature. All antibody applications were done in B-TTBS. An anti-phospho-p44/42 ERK primary antibody that detects ERK phosphorylation at both Thr202 and Tyr204 (1:1000 dilution in B-TTBS; Cell Signaling Technology, Beverly, MA) was used for immunoblotting overnight at 4°C. An anti-p44/42 ERK primary antibody (1:1000 dilution in 3% BSA; Upstate Biotechnology, Lake Placid, NY) that detects total p44/42 isoforms was used for immunoblotting for 1 hr at room temperature. The blots were washed and incubated in HRP-conjugated secondary antibody for 1 hr at room temperature. Blots were developed with enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech, Arlington Heights, IL). Densitometric quantification of immunopositive bands for total or phospho-p44/42 ERK were done using NIH Image software (Scion Corp., Frederick, MD).
Immunocytochemistry for ERK and mGluR. Mice were anesthetized intraperitoneally with sodium pentobarbital (30 mg/kg). Eight minutes (determined from time course of ERK activation) after 2% subcutaneous formalin injection into the hindpaw, mice were perfused transcardially with warm saline (37°C, 0.9% NaCl), followed by 250 ml of ice-cold 4% paraformaldehyde solution. L4–S1 lumbar spinal cord sections were dissected out and post-fixed at 4°C for 4 hr with paraformaldehyde, followed by overnight cryoprotection at 4°C in 30% sucrose. Tissue sections were embedded in OCT compound (Tissue-Tek, Miles Inc., Elkhart, IN) and stored at −80°C. Coronal sections (30 μm) were cut using a freezing sliding microtome, and sections were kept in PBS (pH 7.4) for immunocytochemistry. Sections were rinsed in 10% methanol and 0.3% H2O2 in 0.1m PBS for 30 min and then blocked in 3% normal goat serum (NGS) with 0.2% Triton X-100 (NGST) two times for 10 min each. All antibodies were diluted in 1% NGST. Sections were incubated at 4°C for 36–48 hr in anti-phospho-p44/42 ERK primary antibody (1:1000 dilution in B-TTBS; Cell Signaling Technology) or an anti-total p44/42 ERK primary antibody (1:1000 dilution in 3% BSA; Upstate Biotechnology). Sections were rinsed with 1% NGST two times for 10 min each, followed by incubation in a secondary biotinylated anti-rabbit IgG antibody for 90 min (1:200; ABC kit; Vector Laboratories, Burlingame, CA). Sections were rinsed with 1% NGST two times for 10 min each and incubated in ExtraAvidin peroxidase (1:1000; Sigma) for 1 hr at room temperature. Sections were rinsed in 0.1m PBS two times for 10 min each and then in phosphate buffer (two times for 10 min each) and stained with 3,3′-diaminobenzidine tetrahydrochloride (DAB) solution (0.025% DAB in phosphate buffer containing 0.0025% H2O2; Sigma) for 5–10 min. Sections were mounted onto gelatin-coated glass slides, air-dried, dehydrated, cleared with xylene, coverslipped with DPX mounting medium, and observed for total and phospho-ERK staining. For detection of mGluR5, sections were incubated 36–48 hr at 4°C in polyclonal anti-mGluR5 primary antibody (1:2000; Upstate Biotechnology). For mGluR1a immunocytochemistry, sections were incubated 36–48 hr at 4°C in polyclonal anti-mGluR1a primary antibody (1:2000; DiaSorin, Stillwater, MN). For double-staining of phospho-ERK and mGluR5, sections were first incubated in rabbit polyclonal anti-mGluR5 antibody at 4°C overnight, rinsed, and then incubated at 4°C in mouse monoclonal phospho-ERK antibody overnight. Sections were rinsed and incubated in a mixture of anti-rabbit IgG–rhodol green or anti-rabbit IgG–Oregon green-488 and anti-mouse IgG-Cy3 (Molecular Probes, Eugene, OR) at room temperature for 1 hr. Sections were dried, mounted on slides, and viewed with a confocal microscope. In all cases in which cell counts were taken, the person performing the counts was blind to the identity of the treatments. Separate immunocytochemical experiments were done as described above 5 min (as determined from behavioral and immunoblot experiments) after intrathecal injection of (RS)-DHPG (10 nmol).
Reverse transcription-PCR. Total RNA was isolated from dorsal horn of mouse lumbar spinal cord. Tissue was homogenized with disposable plastic pestles in 300 μl of TRIZOL with 250 μg/ml glycogen (Life Technologies, Gaithersburg, MD). The homogenate was incubated at room temperature for 5 min. Chloroform (160 μl) was added to the homogenates, vortexed, and centrifuged at 14,000 × g for 15 min at 4°C. The aqueous phase was carefully pipetted to a clean tube, and 400 μl of isopropanol was added. The tubes were incubated at room temperature for 10 min and then centrifuged at 14,000 × g for 10 min at 4°C. The supernatant was decanted, and the pellet was washed with 1 ml of 75% DEPC ethanol. The pellet was air dried and resuspended in 50 μl of DEPC water. Sample RNA was quantified from a standard curve made from the RNA ladder. Reverse transcription (RT)-PCR was performed according to the manufacturer-recommended protocol (Ready-to-go RT-PCR beads) using random hexamer primers. Annealing of the mGluR-specific primers during the PCR was performed at 50°C, and extensions were 1 min each for 35 cycles.
RT-PCR was performed using primers based on rat mGluR sequences. The following C-terminal upstream and downstream primers for mGluR5 (Minakami et al., 1993) and upstream and downstream primers for mGluR1 (Soloviev et al., 1999) used were as follows: 5aRTPCRU1, 5′ tgagttgcacgttctatgcg 3′; 5aRTPCRD1, 5′ggtactcttctcattctggg 3′; 1a RTPCRU1, 5′ gctccaacaccttcctcaac 3′; and 1aRTPCRD2, 5′ acaggccgtctcattggtct 3′.
These primers will amplify all known C-terminal splice variants of mGluR1 [with the exception of mGluR1c (Soloviev et al., 1999)] and mGluR5.
Statistical analysis. Time course effects were analyzed using repeated-measure ANOVA with the Prism statistical program (Graphpad Software Inc., San Diego, CA). For comparisons of drug effects from controls, factorial ANOVA was used followed by appropriatepost hoc tests. Student's t test was used when comparisons were restricted to two means.
RESULTS
As mentioned above, previous studies had indicated that activation of spinal group I mGluRs leads to nocifensive behaviors in rats. Furthermore, inflammatory pain plasticity in rats requires ERK activation and also appears to involve activation of group I mGluRs. In the present studies, we sought to test the hypothesis that the group I mGluRs are activated after peripheral inflammation and that this mGluR activation is necessary for nociceptive-specific activation of ERKs and the resultant nociceptive plasticity. This hypothesis makes several predictions: (1) nociceptive activation should elicit an increase in ERK activation in the spinal cord; (2) activation of spinal mGluRs should activate ERKs; (3) blockade of spinal group I mGluRs should reduce the inflammation-evoked increase in ERK activation; and (4) blockade of spinal group I mGluRs should reduce nociceptive plasticity. We have tested each of these predictions using a combination of behavioral, anatomical, and biochemical approaches, described below.
Activation of Group I mGluRs in the spinal cord activates ERK signaling
Intrathecal injection of (RS)-DHPG in mice induced spontaneous nocifensive behaviors that included caudally oriented licking of the flanks, hindpaws, and tail. This increase in nocifensive behaviors was dose dependent and significantly different from vehicle-injected mice at the 1, 10, and 100 nmol doses of (RS)-DHPG (Fig.1A). Intrathecal pretreatment with the mGluR1 antagonist CPCCOEt (50 nmol) or the mGluR5 antagonist MPEP (50 nmol) significantly attenuated (RS)-DHPG-induced spontaneous nocifensive behaviors (Fig.1B). In separate experiments, mice were killed 5 min after injection of intrathecal (RS)-DHPG, and the spinal cord lumbar sections were homogenized and run on a 10% SDS-PAGE. Proteins were immunoblotted using a phospho-ERK-selective antibody, which detects activated forms of both ERK1 and ERK2. A dose-dependent activation of ERK1 and ERK2 was observed (Fig. 1C) when compared with the vehicle-injected controls. Although both ERK1 and ERK2 were activated, ERK2 was relatively more strongly activated than ERK1.
Fig. 1.
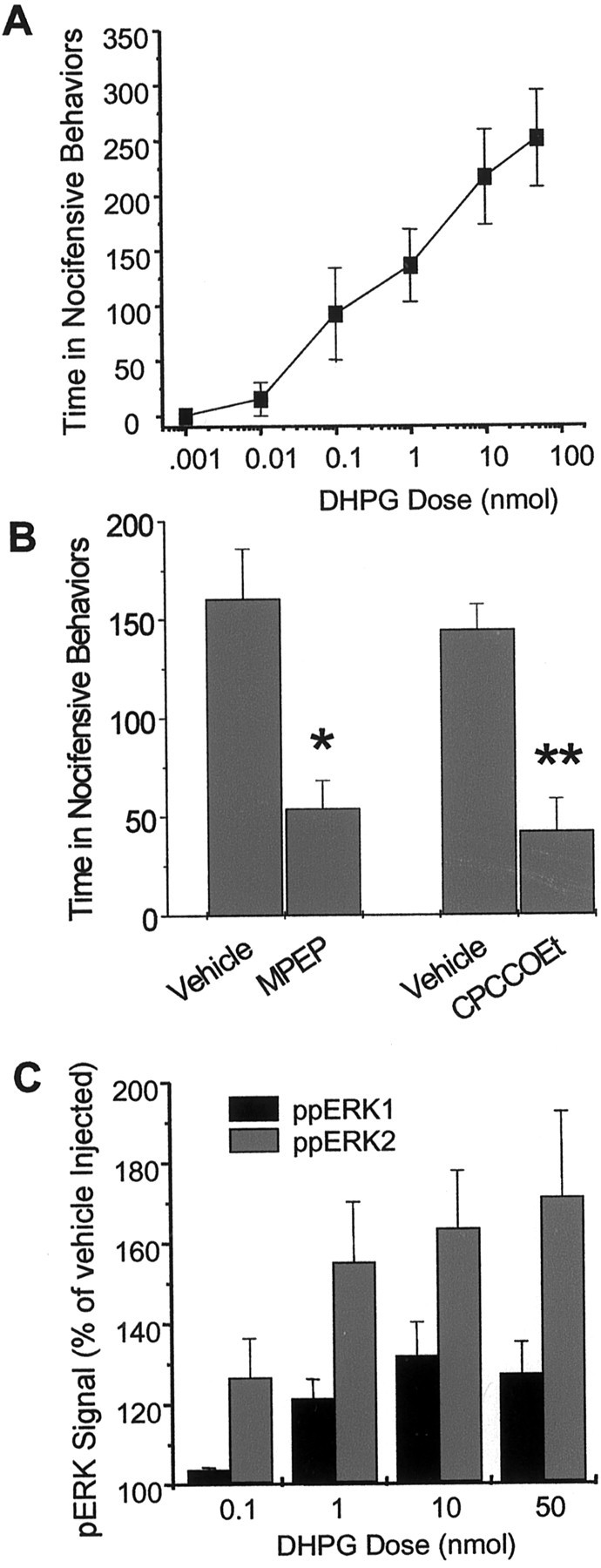
The group 1 mGluR agonist DHPG increases nociceptive behavior and activates ERK1/2 in mouse spinal cord dorsal horn. A, Total time (in seconds) spent in spontaneous nocifensive behaviors after intrathecal injection of (RS)-DHPG. Mice were given a single intrathecal injection of various doses of (RS)-DHPG, and the time spent in nocifensive behaviors was recorded for 5 min.Points represent the mean ± SEM;n = 4–5 animals per dose. B, Effect of the mGluR5 antagonist MPEP and the mGluR1 antagonist CPCCOEt on DHPG-induced spontaneous nocifensive behavior. Mice were pretreated intrathecally with 50 nmol of either antagonist 15 min before intrathecal (RS)-DHPG (1 nmol), and the time spent in nocifensive behaviors was recorded for 5 min. Pointsrepresent the mean ± SEM; n = 4 animals per dose. *p < 0.05; **p < 0.01.C, Immunoblot analysis of phosphorylated ERK1 and ERK2 bands in mouse spinal cord homogenates from mice injected intrathecally with (RS)-DHPG. Points represent the mean ± SEM densities of phospho-ERK1 and phospho-ERK2 bands normalized to total ERK for each sample from four separate experiments. In C, all points are significant atp < 0.05 compared with vehicle-injected controls, with the exception of the p44 signal at the 0.1 nmol dose.
ERK activation is required for inflammatory pain plasticity in mice
The formalin model is used frequently in the study of inflammatory pain states in rodents (Tjolsen et al., 1992). Injection of 2% formalin subcutaneously in the hindpaw of mice results in a typical biphasic nociceptive response (Hunskaar and Hole, 1987). The first phase, usually lasting <5 min, occurs a few seconds after formalin injection and is characterized by intense licking or lifting of the injected paw. This phase is attributable to acute stimulation of nociceptors. The second phase is characterized by licking and lifting of the injected paw beginning at ∼20–25 min after formalin injection and lasts until ∼45–60 min after formalin injection, although the duration and amplitude of the second phase depends on the concentration of formalin used. This second phase of inflammatory nociception is thought to involve central sensitization of dorsal horn neurons, as well as peripheral sensitization associated with the inflammation (Coderre and Melzack, 1992).
Injection of 2–5% formalin subcutaneously into the hindpaw induces activation of phospho-ERK in the lumbar spinal cord, as shown by immunoblotting using the phospho-ERK-selective primary antibody (Fig.2A,B). The phospho-ERK bands were quantitated and normalized to total ERK immunoblotted from the same samples using an anti-total ERK1/2 antibody. Compared with the contralateral cord, blots of tissue taken from the side of the cord ipsilateral to the formalin injection showed significant stimulation of both ERK1 and ERK2 at 3 min after formalin injection but not when the animals were killed immediately after formalin injection (Fig. 2B). Similarly, formalin induced ipsilateral activation of ERK that was localized to cell bodies and dendrites of dorsal horn neurons as seen in sections probed with the primary antibody for phospho-ERK (Fig. 2C; see Fig.6G). We observed no significant contralateral ERK activation in the cord. Figure 2D shows the time course of the number of dorsal horn neurons that have detectable levels of phosphorylated ERK in response to subcutaneous injection of formalin (2%). There is a time-dependent activation of ERK, which is optimal at the 3 and 8 min time points. There was no significant change in total ERK signal (Fig. 2A).
Fig. 2.

Subcutaneous injection of formalin in the hindpaw activates ERK1/2 ipsilaterally in the spinal cord.A, Representative immunoblot of mouse spinal cord homogenates using a phospho-ERK1/2 antibody (top) or total ERK1/2 antibody (bottom). Ipsilateral and contralateral lumbar spinal cord samples were taken at various time points after injection of 2% formalin. The arrows show the position of the 44 kDa (ERK1) and 42 kDa (ERK2) ERK isoforms.B, Quantitation of ERK activation after injection of formalin (2–5%). The phospho-ERK (pERK) bands were densitized and normalized to total ERK immunoblotted from the same samples using an anti-total ERK1/2 antibody and are expressed as fold stimulation of phospho-ERK on the ipsilateral side compared with the contralateral side. n = 7. *p < 0.05 compared with the contralateral side.C, Immunocytochemistry showing ipsilateral activation of ERKs 8 min after formalin injection. D, Time course of the number of dorsal horn neurons positive for phosphorylated ERK after formalin treatment. Data represent the mean ± SEM of 20 sections taken from two animals. Vehicle-injected animals showed no significant phospho-ERK staining (data not shown).
Fig. 6.

Colocalization of phospho-ERK and mGluR5 in mouse dorsal horn after intrathecal DHPG (10 nmol) (A–D) or intraplantar formalin injection (E–H). A, E, Fluorescence images showing the distribution of mGluR5 (green) and phospho-ERK (red) immunoreactivity in the lumbar spinal cord dorsal horn 5 min after DHPG (A) or 8 min after 2% subcutaneous injection of formalin in the right hindpaw (E).B and F show higher-power examples of confocal images showing the distribution of mGluR5 (green) in relation to phospho-ERK (red) in the dorsal horn. Note that some phospho-ERK cells also have apparent membrane labeling for mGluR5 (arrows), whereas other phospho-ERK-positive cells contain no detectable mGluR5 (arrowheads).C and G show higher magnifications of phospho-ERK staining of dorsal horn neurons. Note the labeling of dendritic processes (arrowheads), which are typically seen when cells are observed using a confocal microscope.D and H show higher magnification of additional example neurons with apparent membrane labeling for mGluR5 and somatic phospho-ERK. These images are representative of similar results obtained from three separate animals.
These results show that ERK1 and ERK2 are strongly activated in spinal nociceptive pathways in response to inflammation in mice and suggest that ERK activation may be involved in sensitization. To test this hypothesis, we pretreated mice with the MEK inhibitor PD98059, which inhibits ERK activation, and investigated whether this treatment attenuates nociceptive behaviors in response to formalin-induced inflammation. We found that intrathecal pretreatment with 25 nmol of PD98059 for 20 min reduced the time spent in total pain behaviors (licking and lifting of injected paw) in the second phase of the formalin test when compared with the vehicle-pretreated mice with no significant effect on the first phase response (Fig.3A). Figure 3Bshows significant dose-dependent attenuation of the total time spent in the second phase by intrathecal PD98059 when compared with vehicle-pretreated mice.
Fig. 3.

The MEK inhibitor PD98059 attenuates the second phase of formalin-induced nociceptive behavior and decreases ERK activation. A, Effect of a 20 min pretreatment with a single intrathecal injection of PD98059 (25 nmol) in the mouse formalin test. B, Effect of 5 and 25 nmol doses of PD98059 in the second phase of the formalin test (15–30 min). Each barrepresents the mean ± SEM; n = 5–7. *p < 0.05. C, Effect of PD98059 on activation of spinal ERK after formalin injection determined by cell counts of phospho-ERK-positive neurons. n = 3 each. *p < 0.05.
Consistent with our hypothesis that the effects of PD98059 are mediated by MEK inhibition, intrathecal pretreatment with 25 nmol of PD98059 for 20 min attenuated formalin-induced ERK activation as observed in sections immunostained with phospho-ERK antibody. This attenuation of ERK activation by PD98059 was statistically significant when compared with the sections stained from vehicle-pretreated mice (Fig.3C).
These results are consistent with previous studies in rat and further show that ERK1 and ERK2 are activated in response to inflammation or stimulation of group I mGluRs in mouse spinal cord dorsal horn. Because we have shown that activation of mGluR1 and/or mGluR5 induces pain and increases activation of ERKs, we wanted to test the hypothesis that mGluR1 and mGluR5 are necessary upstream activators of ERK by testing whether antagonists of mGluR1 and mGluR5 attenuate formalin-induced nociceptive behavior and ERK activation.
Blockade of spinal group I mGluRs reduces inflammation-induced ERK activation and inflammatory pain plasticity in mice
We tested whether blocking mGluR1 and mGluR5 would reduce inflammatory pain behaviors in the formalin test. We found that both the mGluR1 antagonist CPCCOEt and the mGluR5 antagonist MPEP significantly reduced pain behaviors in the second phase of the formalin test in a dose-dependent manner (Fig.4). Interestingly, we found that, at higher doses, MPEP (50 nmol) and CPCCOEt (100 nmol) also significantly attenuated the first phase, suggesting a role for mGluRs in mediation of both the acute and chronic phases of this model. Intrathecal coinjection of both CPCCOEt (50 nmol) and MPEP (50 nmol) significantly attenuated the second phase pain behaviors after subcutaneous formalin in the hindpaw. Coinjection of the antagonists reduced the second phase pain behaviors by 47% (p < 0.05) when compared with the intrathecal vehicle-treated mice. This effect was not additive, because no significant differences were observed between the effects of coinjection of MPEP and CPCCOEt versus the effects of either antagonist injected alone.
Fig. 4.
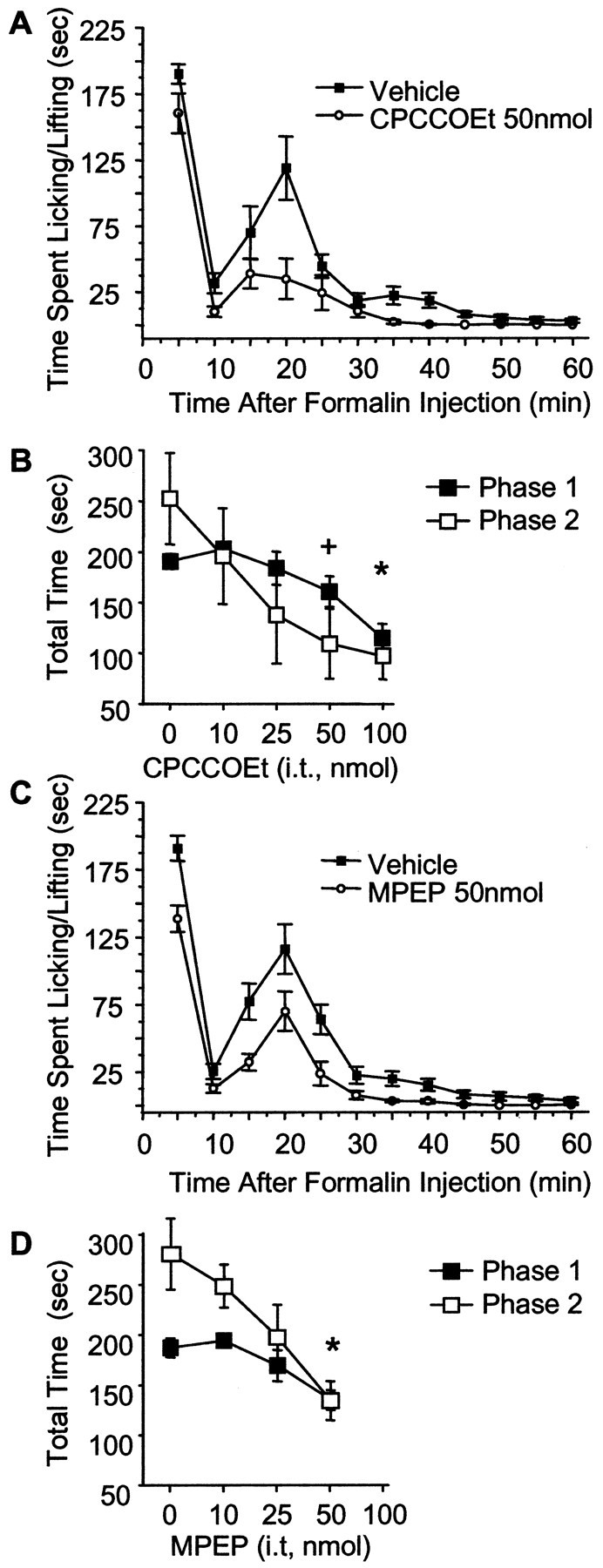
Intrathecal MPEP and CPCCOEt attenuate formalin-induced nociceptive behavior. A,C, Time course graphs of 15 min pretreatment with 50 nmol of CPCCOEt and 50 nmol of MPEP. B,D, Dose–response curves of CPCCOEt (B) and MPEP (D) on the first phase (the 5 min time point; filled squares) and the second phase (sum of 15–30 min points; open squares). Points represent the mean ± SEM;n = 5 - 11 per dose.+p < 0.05 for phase 2 only; *p < 0.05 for phase 1 and phase 2.
These results show that mGluR1 and mGluR5 are involved in nociceptive plasticity after peripheral inflammation. We therefore tested the hypothesis that this modulation of plasticity was attributable to decreased ERK activation by the mGluR1/5 antagonists. The effects of MPEP and CPCCOEt on formalin-induced activation of ERK were investigated. A 15 min pretreatment with either MPEP (50 nmol) or CPCCOEt (50 nmol) significantly attenuated formalin-induced ERK phosphorylation detected with immunocytochemistry (Fig.5).
Fig. 5.
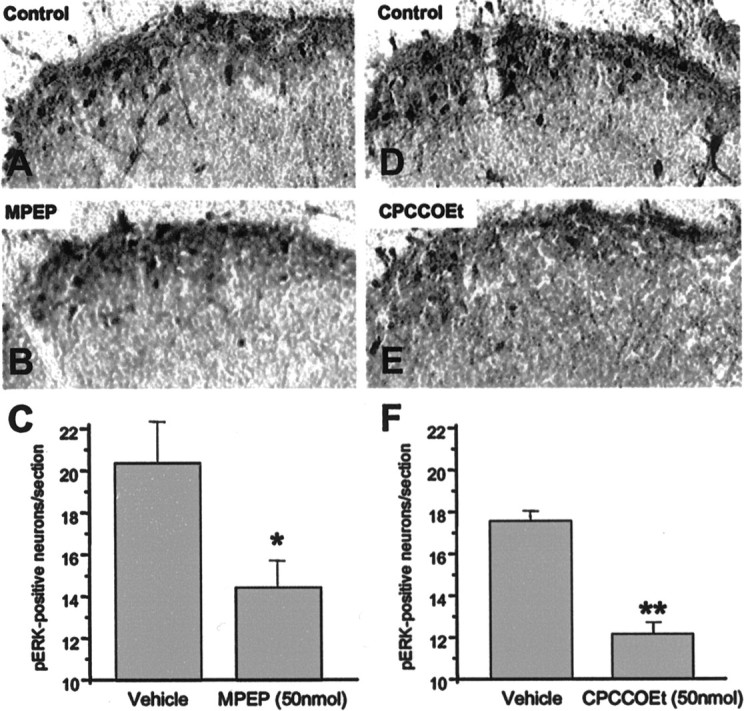
Intrathecal MPEP and CPCCOEt attenuate formalin-induced ERK activation in the spinal cord dorsal horn. Representative spinal cord lumbar sections immunostained with phospho-ERK antibody after a unilateral injection of formalin subcutaneously into the right hindpaw. Mice were pretreated with either vehicle (A) (100 mm HEPES, pH 7.4) or MPEP (B) (50 nmol). D andE show staining of spinal cords from vehicle- and CPCCOEt-pretreated mice. Quantitation of the total number of phospho-ERK-positive neurons in the dorsal horn of the lumbar spinal cord after pretreatment with either vehicle or MPEP (C) or vehicle or CPCCOEt (F) before subcutaneous formalin injection.Bars represent the mean ± SEM;n = 3–6 for each treatment. *p< 0.05; **p < 0.01.
Colocalization of inflammation-evoked ERK activation and group I mGluRs
Given that mGluR antagonists reduce ERK activation after inflammation, it is reasonable to hypothesize that mGluRs couple to ERK activation in these dorsal horn neurons. However, it is formally possible that mGluRs do not couple to ERKs but rather regulate synaptic transmission in the complex dorsal horn circuitry, leading to increased ERK activation by other receptors. If mGluRs couple directly to intracellular ERK activation rather than indirectly through modulation network activity, then a necessary condition is that the receptors should be expressed in the cells in which ERK activation occurs. We therefore investigated the localization of mGluR1 and mGluR5 in relation to the phosphorylated ERK by costaining spinal cord lumbar sections with the mGluR5 and phospho-ERK antibody after intrathecal (RS)-DHPG or subcutaneous formalin injection in the hindpaw. The pattern of phospho-ERK immunostaining in the dorsal horn neurons after intrathecal injection of (RS)-DHPG (10 nmol) (Fig.6A) was very similar to that seen after subcutaneous injection of formalin in the hindpaw (Fig.6E). Consistent with previous reports in rat, we found that mGluR5 staining is localized in the dorsal horn of the mouse spinal cord (Fig. 6A,E). Phospho-ERK immunoreactivity is seen as dense staining within cell somata and dendrites (Fig. 6B–D,F–H) of mostly the superficial dorsal horn laminas. Using confocal microscopy, we found that, in many instances, the mGluR5 staining was present as a distinct annulus surrounding phospho-ERK-positive somata, and colocalization could also be seen in processes.
Many cells positive for phosphorylated ERK showed no detectable signal for mGluR5 in the membrane or cytoplasm. It is possible that some of these mGluR5-negative cells may express mGluR1. However, spinal cord sections stained with an mGluR1a-selective antibody resulted in no detectable amounts of mGluR1a in the superficial dorsal horn of the spinal cord (data not shown). This suggests that mGluR1a may not be expressed in the superficial dorsal horn neurons, and perhaps different splice variants of mGluR1 are responsible for the mGluR1-dependent ERK activation in these dorsal horn neurons. Unfortunately, there are no commercially available antibodies at present for the other splice variants of mGluR1.
To examine the expression of other known splice variants of mGluR1, we performed RT-PCR on total RNA extracted from mouse spinal cord dorsal horn using primers that amplify mGluR1a, mGluR1b, mGluR1d, and mGluR1f (Minakami et al., 1993; Soloviev et al., 1999). Our results from the RT-PCR show that there are three RNA splice variants of mGluR1 present in the spinal cord dorsal horn: mGluR1a, mGluR1b, and mGluR1d (Fig.7A). The relative abundance of the mGluR1 splice variants in the dorsal horn rank in the order mGluR1a > mGluR1d ≫ mGluR1b. These results, combined with the results showing that CPCCOEt reduces formalin-induced nociceptive plasticity and formalin-induced activation of ERKs, suggest that mGluR1d or mGluR1b may be expressed in dorsal horn neurons and responsible for the behavioral and molecular changes observed. We also confirmed the presence of the two known splice variants of mGluR5 (mGluR5a and mGluR5b) in the mouse spinal cord dorsal horn (Fig. 7B), with mGluR5b being more abundant than the mGluR5a splice variant.
Fig. 7.

Expression of various splice variants of mGluR1 and mGluR5 in mouse spinal cord dorsal horn. RT-PCR amplification of the alternatively spliced forms of mGluR1 (A) and mGluR5 (B) from total RNA prepared from dorsal horn of the mouse spinal cord and mouse cortex. The position of predicted band sizes for mGluR1a, mGluR1b, mGluR1d, mGluR1f, mGluR5a, and mGluR5b are indicated. Molecular markers are shown in base pairs.
DISCUSSION
The present study reports several important findings. Intrathecal administration of DHPG, a group I mGluR agonist, stimulated spontaneous nociceptive behavior that included licking of the flanks, hindpaws, and the tail. Intrathecal administration of the selective mGluR1 and mGluR5 antagonists CPCCOEt and MPEP, respectively, attenuated DHPG-induced nociceptive behavior. This suggests that activation of spinal group I mGluRs generates nociceptive responses. Although a previous study showed a similar behavioral effect in rats (Fisher and Coderre, 1996b), we have extended these findings and have shown that nociceptive activation by group I mGluRs is correlated with activation of ERK1 and ERK2 in the spinal cord, identifying this cascade as a potential mediator of mGluR-induced enhancement of nociception.
Immunoblot analysis of spinal cord homogenates after intrathecal DHPG revealed a dose-dependent increase in phosphorylation of ERK1 and ERK2, with stronger activation of ERK2. Stimulation of nociceptive behavior and activation of ERKs by intrathecal DHPG occurred at similar doses, suggesting that mGluR1/5 activation in the dorsal horn may induce nociception through activation of ERKs. Furthermore, immunocytochemistry using a phospho-ERK-selective antibody localized the activation of ERKs by DHPG primarily to the superficial dorsal horn neurons. Spinal ERK activity has been shown to be induced by peripheral noxious stimuli in rats (Ji et al., 1999); however, our data are the first to report activation of spinal ERKs by selective stimulation of mGluR1/5.
We found that injection of the inflammatory agent formalin subcutaneously into the hindpaw of mice stimulates ERK activation in the ipsilateral dorsal horn. In addition, we found that intrathecal pretreatment with the MEK inhibitor PD98059 decreases the second phase of formalin-induced nociceptive behavior and decreases dorsal horn ERK activation. Similar results have been obtained in rats (Ji et al., 1999), but we have extended these findings in mice to show that inflammatory nociceptive activation correlates with quantitative increases in activation of both ERK1 and ERK2. We found a more pronounced and longer-lasting activation of ERKs in response to formalin than was observed in the previous study. This may relate to differences in formalin doses, which are difficult to compare because of differences in species, formalin concentration, and injection volume.
The mGluR5-selective antagonist MPEP and the mGluR1-selective antagonist CPCCOEt reduced the second phase of the formalin test in a dose-dependent manner, suggesting that each of these receptor subtypes contributes to the second phase nociceptive behavior. We also found that these antagonists attenuated the first phase of the formalin test at higher doses, suggesting that mGluR1 and mGluR5 are also involved in acute nociceptive transmission. This is consistent with previous studies showing a reduction of acute nociceptive transmission by intravenous MPEP and by group I mGluR antisense knockdown (Young et al., 1998; Bordi and Ugolini, 2000). It is interesting to note that, whereas the mGluR antagonists reduced the first phase, the MEK inhibitor PD98059 did not. This suggests that, although modulation of the second phase by group I mGluRs involves ERK activation, the role of group I mGluRs in acute nociception likely involves a different signaling pathway. Intrathecal coinjection of both the mGluR1 and mGluR5 antagonists decreased second phase nociceptive behavior to the same magnitude as when each antagonist was given alone. The reason for the lack of an additive effect is not clear. One possibility is that mGluR1 and mGluR5 are both required for the mGluR-mediated component of ERK activation induced by nociceptive stimulation. Indeed, we found the behavioral response to intrathecal DHPG was inhibited by >50% by each individual antagonist, suggesting functional overlap between these two receptors.
In addition to attenuating inflammatory nociceptive behavior, the group 1 mGluR antagonists also reduced formalin-induced spinal ERK activation. Attenuation of ERK induction by each antagonist was partial; this is not surprising because spinal nociceptive processing is very complex and involves interplay between several neurotransmitters or peptides that originate from descending inputs from the brain, primary inputs from the periphery, and transmission within the dorsal horn itself. Although our data show activation of ERKs by selective stimulation of spinal mGluR1/5, other glutamate receptors participate in ERK activation; for example, an NMDA receptor antagonist also decreased formalin-induced spinal ERK activation (Ji et al., 1999). There is evidence that points to interaction between mGluRs and NMDA receptors, suggesting that NMDA receptor-mediated nociceptive processing may be regulated by mGluRs (Jones and Headley, 1995). It is possible that part of the mechanism involved in mGluR antagonist-mediated reduction in ERK activation and nociceptive plasticity reflects decreased modulation of NMDA receptors. Future studies will address this possibility.
Attenuation of the inflammatory nociceptive responses by MPEP likely occurs postsynaptically in neurons of the superficial dorsal horn. Previous reports have demonstrated the expression of mGluR1 and mGluR5 in the soma and dendrites of rat superficial dorsal horn neurons, as well as in vesicle-containing profiles (Jia et al., 1999; Tao et al., 2000), suggesting that these neurons are apposed closely to nociceptive primary afferents. In addition, the mGluR5 containing neurons receive inputs from GABAergic terminals from interneurons in the spinal cord, perhaps modulating the incoming nociceptive stimuli (Tao et al. 2000). mGluR5 in these neurons may thus have a major role in spinal nociceptive processing. Our RT-PCR analysis from dorsal horn RNA, together with the immunolocalization of mGluR5, confirms the expression of mGluR5a and mGluR5b in laminas I and II of the mouse dorsal horn. We further investigated the relationship of the ERK-positive dorsal horn neurons to the distribution of the mGluR1/5 receptors in the spinal cord dorsal horn and showed that some of the lamina I neurons that show activated ERK do indeed express mGluR5.
The effects of CPCCOEt are also likely mediated in the dorsal horn neurons. However, because of lack of commercially available antibodies that detect all of the different splice variants of mGluR1, we cannot yet confirm which splice variants of mGluR1 mediate the effects of CPCCOEt. We and others have found that several commercially available antibodies for mGluR1a show cross-reactivity with mGluR5 (B. Nadin and R. W. Gereau, unpublished findings; Jia et al., 1999). There are six known splice variants of mGluR1: mGluR1a, mGluR1b, mGluR1c, mGluR1d, mGluR1e, and mGluR1f (Soloviev et al., 1999). Using RT-PCR of mouse spinal dorsal horn RNA, we confirm the presence of three splice variants in the dorsal horn of the mouse spinal cord: mGluR1a, mGluR1b, and mGluR1d. It seems likely that the superficial dorsal horn contains mGluR1b and/or mGluR1d because immunocytochemistry using an antibody selective for mGluR1a shows staining in only the deeper laminas of the dorsal horn. Thus, attenuation of the inflammatory nociceptive responses to formalin by CPCCOEt is most likely mediated via mGluR1b or mGluR1d. A recent study has reported the expression of low levels of mGluR1b in lamina II neurons (Alvarez et al., 2000). There are no reports to date of mGluR1d expression in the mouse spinal cord, and our findings are the first that show the presence of this splice variant in the spinal cord dorsal horn.
The role of ERK pathway in inflammation-induced sensitization is not clear. Activation of ERKs may regulate transcription of a variety of gene products, for example, through phosphorylation of the cAMP response element-binding protein, which has been shown to be activated in the spinal cord dorsal horn by noxious peripheral stimulation (Ji and Rupp, 1997; Impey et al., 1999). Peripheral inflammation by formalin also causes unilateral increased expression of dynorphin, enkephalin, neuropeptide Y (NPY), NPY receptor, and galanin mRNAs (Dubner and Ruda, 1992; Ji et al., 1994, 1995a,b; Donaldson et al., 1995; Woolf and Costigan, 1999; Ruda et al., 2000). Some of these could result from transcriptional activation by ERKs, because they are all expressed in the superficial dorsal horn.
Activation of spinal mGluR1 and mGluR5 may increase the excitability of spinal cord nociceptive neurons, a phenomenon referred to as central sensitization. Central sensitization may also contribute to mechanisms that cause secondary hyperalgesia. A recent electrophysiological study by Neugebauer et al. (1999) has shown that the mGluR1-selective antagonist CPCCOEt reverses capsaicin-induced central sensitization and blocks modulation by DHPG. Central sensitization may be related to windup, which is a frequency-dependent increase in excitability of spinal neurons evoked by electrical stimulation of afferents (Herrero et al., 2000). mGluRs have also been implicated in generation of windup processes in the spinal cord (Boxall et al., 1996; Budai and Larson, 1998). Although the relationship between mechanisms of windup and central sensitization are not understood, our results suggest that the mGluRs may mediate these effects partly by activation of ERK signaling.
In summary, our data suggest that mGluR1 and mGluR5 in the spinal cord are involved in functional plasticity during inflammation and that their modulatory effects involve activation of downstream ERKs. These mGluRs and the ERKs may therefore be potential targets for novel treatments for different types of inflammatory pain. Our ongoing studies include an analysis of downstream targets of ERKs in the dorsal horn, as well as electrophysiological characterization of the functional consequences of ERK activation.
Footnotes
This work was supported by National Institutes of Mental Health Grant MH60230 and The Spinal Cord Research Foundation (to R.W.G.). We thank Gautam Bhave for his assistance in this project, Brian Nadin for assistance with the confocal microscopy, and Dr. D. Sweatt for critically reading this manuscript.
Correspondence should be addressed to Robert W. Gereau IV, Baylor College of Medicine, Division of Neuroscience, One Baylor Plaza, Room S636, Houston, TX 77030. E-mail: rgereau@bcm.tmc.edu.
C.-C. Wang's present address: Fujen Catholic University, 510 Chung Cheng Road, Hsinchuang, Taipei Hsien 24205, Taiwan, Republic of China.
REFERENCES
- 1.Alvarez FJ, Villalba RM, Carr PA, Grandes P, Somohano PM. Differential distribution of metabotropic glutamate receptors 1a, 1b, and 5 in the rat spinal cord. J Comp Neurol. 2000;422:464–487. doi: 10.1002/1096-9861(20000703)422:3<464::aid-cne11>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 2.Bordi F, Ugolini A. Involvement of mGluR(5) on acute nociceptive transmission. Brain Res. 2000;871:223–233. doi: 10.1016/s0006-8993(00)02467-7. [DOI] [PubMed] [Google Scholar]
- 3.Boxall SJ, Thompson SW, Dray A, Dickenson AH, Urban L. Metabotropic glutamate receptor activation contributes to nociceptive reflex activity in the rat spinal cord in vitro. Neuroscience. 1996;74:13–20. doi: 10.1016/0306-4522(96)00101-7. [DOI] [PubMed] [Google Scholar]
- 4.Budai D, Larson AA. The involvement of metabotropic glutamate receptors in sensory transmission in dorsal horn of the rat spinal cord. Neuroscience. 1998;83:571–580. doi: 10.1016/s0306-4522(97)00390-4. [DOI] [PubMed] [Google Scholar]
- 5.Coderre TJ, Melzack R. The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury. J Neurosci. 1992;12:3665–3670. doi: 10.1523/JNEUROSCI.12-09-03665.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 7.Donaldson LF, McQueen DS, Seckl JR. Induction of transcription factor AP2 mRNA expression in rat primary afferent neurons during acute inflammation. Neurosci Lett. 1995;196:181–184. doi: 10.1016/0304-3940(95)11870-3. [DOI] [PubMed] [Google Scholar]
- 8.Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci. 1992;15:96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- 9.Ferraguti F, Baldani-Guerra B, Corsi M, Nakanishi S, Corti C. Activation of the extracellular signal-regulated kinase 2 by metabotropic glutamate receptors. Eur J Neurosci. 1999;11:2073–2082. doi: 10.1046/j.1460-9568.1999.00626.x. [DOI] [PubMed] [Google Scholar]
- 10.Fisher K, Coderre TJ. Comparison of nociceptive effects produced by intrathecal administration of mGluR agonists. NeuroReport. 1996a;7:2743–2747. doi: 10.1097/00001756-199611040-00067. [DOI] [PubMed] [Google Scholar]
- 11.Fisher K, Coderre TJ. The contribution of metabotropic glutamate receptors (mGluRs) to formalin-induced nociception. Pain. 1996b;68:255–263. doi: 10.1016/s0304-3959(96)03212-5. [DOI] [PubMed] [Google Scholar]
- 12.Fisher K, Coderre TJ. Hyperalgesia and allodynia induced by intrathecal (RS)-dihydroxyphenylglycine in rats. NeuroReport. 1998;9:1169–1172. doi: 10.1097/00001756-199804200-00038. [DOI] [PubMed] [Google Scholar]
- 13.Fisher K, Fundytus ME, Cahill CM, Coderre TJ. Intrathecal administration of the mGluR compound, (S)-4CPG, attenuates hyperalgesia and allodynia associated with sciatic nerve constriction injury in rats. Pain. 1998;77:59–66. doi: 10.1016/S0304-3959(98)00082-7. [DOI] [PubMed] [Google Scholar]
- 14.Herrero JF, Laird JM, Lopez-Garcia JA. Wind-up of spinal cord neurones and pain sensation: much ado about something? Prog Neurobiol. 2000;61:169–203. doi: 10.1016/s0301-0082(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 15.Hunskaar S, Hole K. The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. Pain. 1987;30:103–114. doi: 10.1016/0304-3959(87)90088-1. [DOI] [PubMed] [Google Scholar]
- 16.Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- 17.Impey S, Obrietan K, Storm DR. Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- 18.Ji RR, Rupp F. Phosphorylation of transcription factor CREB in rat spinal cord after formalin-induced hyperalgesia: relationship to c-fos induction. J Neurosci. 1997;17:1776–1785. doi: 10.1523/JNEUROSCI.17-05-01776.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji RR, Zhang X, Wiesenfeld-Hallin Z, Hokfelt T. Expression of neuropeptide Y and neuropeptide Y (Y1) receptor mRNA in rat spinal cord and dorsal root ganglia following peripheral tissue inflammation. J Neurosci. 1994;14:6423–6434. doi: 10.1523/JNEUROSCI.14-11-06423.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji RR, Zhang Q, Law PY, Low HH, Elde R, Hokfelt T. Expression of μ-, δ-, and κ-opioid receptor-like immunoreactivities in rat dorsal root ganglia after carrageenan-induced inflammation. J Neurosci. 1995a;15:8156–8166. doi: 10.1523/JNEUROSCI.15-12-08156.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji RR, Zhang X, Zhang Q, Dagerlind A, Nilsson S, Wiesenfeld-Hallin Z, Hokfelt T. Central and peripheral expression of galanin in response to inflammation. Neuroscience. 1995b;68:563–576. doi: 10.1016/0306-4522(95)94333-t. [DOI] [PubMed] [Google Scholar]
- 22.Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- 23.Jia H, Rustioni A, Valtschanoff JG. Metabotropic glutamate receptors in superficial laminae of the rat dorsal horn. J Comp Neurol. 1999;410:627–642. [PubMed] [Google Scholar]
- 24.Jones MW, Headley PM. Interactions between metabotropic and ionotropic glutamate receptor agonists in the rat spinal cord in vivo. Neuropharmacology. 1995;34:1025–1031. doi: 10.1016/0028-3908(95)00055-b. [DOI] [PubMed] [Google Scholar]
- 25.Karim F, Kanui TI, Mbugua S. Effects of codeine, naproxen and dexamethasone on formalin-induced pain in the naked mole-rat. NeuroReport. 1993;4:25–28. doi: 10.1097/00001756-199301000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Minakami R, Katsuki F, Yamamoto T, Nakamura K, Sugiyama H. Molecular cloning and the functional expression of two isoforms of human metabotropic glutamate receptor subtype 5. Biochem Biophys Res Commun. 1993;199:1136–1143. doi: 10.1006/bbrc.1994.1349. [DOI] [PubMed] [Google Scholar]
- 27.Neugebauer V, Lucke T, Schaible HG. Requirement of metabotropic glutamate receptors for the generation of inflammation-evoked hyperexcitability in rat spinal cord neurons. Eur J Neurosci. 1994;6:1179–1186. doi: 10.1111/j.1460-9568.1994.tb00616.x. [DOI] [PubMed] [Google Scholar]
- 28.Neugebauer V, Chen PS, Willis WD. Role of metabotropic glutamate receptor subtype mGluR1 in brief nociception and central sensitization of primate STT cells. J Neurophysiol. 1999;82:272–282. doi: 10.1152/jn.1999.82.1.272. [DOI] [PubMed] [Google Scholar]
- 29.Peavy RD, Conn PJ. Phosphorylation of mitogen-activated protein kinase in cultured rat cortical glia by stimulation of metabotropic glutamate receptors. J Neurochem. 1998;71:603–612. doi: 10.1046/j.1471-4159.1998.71020603.x. [DOI] [PubMed] [Google Scholar]
- 30.Rees H, Sluka KA, Lu Y, Westlund KN, Willis WD. Dorsal root reflexes in articular afferents occur bilaterally in a chronic model of arthritis in rats. J Neurophysiol. 1996;76:4190–4193. doi: 10.1152/jn.1996.76.6.4190. [DOI] [PubMed] [Google Scholar]
- 31.Ruda MA, Ling QD, Hohmann AG, Peng YB, Tachibana T. Altered nociceptive neuronal circuits after neonatal peripheral inflammation. Science. 2000;289:628–631. doi: 10.1126/science.289.5479.628. [DOI] [PubMed] [Google Scholar]
- 32.Soloviev MM, Ciruela F, Chan WY, McIlhinney RA. Identification, cloning and analysis of expression of a new alternatively spliced form of the metabotropic glutamate receptor mGluR1 mRNA1. Biochim Biophys Acta. 1999;1446:161–166. doi: 10.1016/s0167-4781(99)00083-4. [DOI] [PubMed] [Google Scholar]
- 33.Tao Y, Li Y, Zhao Z, Johns RA. Synaptic relationship of the neurons containing a metabotropic glutamate receptor, MGluR5, with nociceptive primary afferent and GABAergic terminals in rat spinal superficial laminae. Brain Res. 2000;875:138–143. doi: 10.1016/s0006-8993(00)02608-1. [DOI] [PubMed] [Google Scholar]
- 34.Thomas K, Hunt S. The regional distribution of extracellularly regulated kinase-1 and -2. Neuroscience. 1993;56:741–757. doi: 10.1016/0306-4522(93)90371-l. [DOI] [PubMed] [Google Scholar]
- 35.Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- 36.Valerio A, Paterlini M, Boifava M, Memo M, Spano P. Metabotropic glutamate receptor mRNA expression in rat spinal cord. NeuroReport. 1997a;8:2695–2699. doi: 10.1097/00001756-199708180-00012. [DOI] [PubMed] [Google Scholar]
- 37.Valerio A, Rizzonelli P, Paterlini M, Moretto G, Knopfel T, Kuhn R, Memo M, Spano P. mGluR5 metabotropic glutamate receptor distribution in rat and human spinal cord: a developmental study. Neurosci Res. 1997b;28:49–57. doi: 10.1016/s0168-0102(97)01175-9. [DOI] [PubMed] [Google Scholar]
- 38.Woolf CJ, Costigan M. Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci USA. 1999;96:7723–7730. doi: 10.1073/pnas.96.14.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young MR, Blackburn-Munro G, Dickinson T, Johnson MJ, Anderson H, Nakalembe I, Fleetwood-Walker SM. Antisense ablation of type I metabotropic glutamate receptor mGluR1 inhibits spinal nociceptive transmission. J Neurosci. 1998;18:10180–10188. doi: 10.1523/JNEUROSCI.18-23-10180.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]