

Abstract
Neurotransmitter release is a highly efficient secretory process exhibiting resistance to fatigue and plasticity attributable to the existence of distinct pools of synaptic vesicles (SVs), namely a readily releasable pool and a reserve pool from which vesicles can be recruited after activity. Synaptic vesicles in the reserve pool are thought to be reversibly tethered to the actin-based cytoskeleton by the synapsins, a family of synaptic vesicle-associated phosphoproteins that have been shown to play a role in the formation, maintenance, and regulation of the reserve pool of synaptic vesicles and to operate during the post-docking step of the release process. In this paper, we have investigated the physiological effects of manipulating synapsin levels in identified cholinergic synapses of Aplysia californica. When endogenous synapsin was neutralized by the injection of specific anti-synapsin antibodies, the amount of neurotransmitter released per impulse was unaffected, but marked changes in the secretory response to high-frequency stimulation were observed, including the disappearance of post-tetanic potentiation (PTP) that was substituted by post-tetanic depression (PTD), and increased rate and extent of synaptic depression. Opposite changes on post-tetanic potentiation were observed when synapsin levels were increased by injecting exogenous synapsin I. Our data demonstrate that the presence of synapsin-dependent reserve vesicles allows the nerve terminal to release neurotransmitter at rates exceeding the synaptic vesicle recycling capacity and to dynamically change the efficiency of release in response to conditioning stimuli (e.g., post-tetanic potentiation). Moreover, synapsin-dependent regulation of the fusion competence of synaptic vesicles appears to be crucial for sustaining neurotransmitter release during short periods at rates faster than the replenishment kinetics and maintaining synchronization of quanta in evoked release.
Keywords: Aplysia, synapse, exocytosis, neurotransmitter release, short-term plasticity, synaptic depression, post-tetanic potentiation
Nerve terminals have the unique property of sustaining vesicular release of neurotransmitter at high rates, although only a small fraction of synaptic vesicles (SVs) is available for immediate release. In addition, nerve terminals are able to transiently modify synaptic efficacy to give rise to various forms of short-term plasticity. These are thought to result from changes in recruitment of reserve vesicles or changes in the trafficking of vesicles between a releasable pool of cycling SVs and a reserve pool of SVs (Zucker, 1996, 1999; Brodin et al., 1997; Neher, 1998). The readily releasable pool is thought to correspond to the number of SVs docked morphologically to the presynaptic membrane that can be released (Rosenmund and Stevens, 1996; Dobrunz and Stevens, 1997). Morphological data have demonstrated that SVs in the reserve pool are organized in clusters in which SVs are reversibly linked to the actin-based cytoskeleton, possibly by a family of abundant SV-associated phosphoproteins, the synapsins (Landis et al., 1988; Bernstein and Bamburg, 1989; Hirokawa et al., 1989; Benfenati et al., 1992a, 1993;Ceccaldi et al., 1995; Pieribone et al., 1995; Kuromi and Kidokoro, 1998).
Synapsins are highly conserved, SV-associated proteins that are implicated in the formation of synaptic terminals and the regulation of neurotransmitter release. Although the vertebrate synapsin family comprises the products of at least three synapsin genes (synapsin I, II, and III) (Südhof et al., 1989; De Camilli et al., 1990;Hosaka and Südhof, 1998; Kao et al., 1998), only a single synapsin gene, which cannot yet be assigned as being synapsin I-, II- or III-like, has been found in invertebrates, includingAplysia (Klagges et al., 1996; Hilfiker et al., 1998; Angers et al., 1999; Kao et al., 1999). The synapsins have been proposed to play a major structural role in the assembly and maintenance of the reserve pool of SVs both during development and in mature neurons, as well as a dynamic role in controlling SV transitions from the reserve pool to the releasable pool during neuronal activity (Greengard et al., 1993; Brodin et al., 1997; Benfenati et al., 1999), possibly by modulation of the cross-linking of SVs to the actin-based cytoskeleton of the nerve terminal through synapsin phosphorylation (Benfenati et al., 1992a, 1993, Ceccaldi et al., 1995; Hosaka et al., 1999). More recently, a post-docking role of synapsins has been proposed (Rosahl et al., 1993, 1995; Hilfiker et al., 1998) that may involve a putative ATPase activity of synapsins (Esser et al., 1998). However, despite considerable work in the last few years and the generation of synapsin knock-out mice (Rosahl et al., 1993, 1995; Li et al., 1995; Takei et al., 1995; Ryan et al., 1996; Terada et al., 1999), the precise role of synapsins in short-term plasticity remains elusive.
In this paper, we took advantage of well characterized cholinergic synapses in the buccal ganglion of Aplysia californicaexpressing post-tetanic potentiation (PTP), facilitation, and synaptic depression (Mothet et al., 1996; Doussau et al., 1998) to analyze the role of the synapsins in neurotransmitter release and short-term plasticity. Our data demonstrate that the ability of synapses to express PTP and sustain release at a low rate of stimulation is determined by the size or maintenance, or both, of the synapsin-dependent reserve pool. Moreover, regulation by synapsin of a membrane step of the release process allows synchronization of release to be maintained and minimizes the appearance of high-frequency depression when the release rate exceeds the recycling kinetics.
MATERIALS AND METHODS
Material. Peroxidase-conjugated goat anti-rabbit secondary antibodies were from Bio-Rad (Milano, Italy), and the chemiluminescence detection system (Renaissance Western Blot Chemiluminescence Reagent Plus) was from New England Nuclear (Brussels, Belgium). Synapsin I was purified from bovine forebrain as described previously (Benfenati et al., 1992b) and stored in 200 mm NaCl, 25 mm Tris-Cl, pH 7.5. Synapsin IIa was expressed in Sf9 cells infected with the baculovirus expression vector pVLSynIIa encoding for rat synapsin IIa and purified by affinity chromatography (Siow et al., 1992; Nielander et al., 1997). Nitrocellulose membranes (0.2 μm pore size) were from Schleicher & Schuell (Milano, Italy). The antibodies used in the injection experiments were obtained in rabbits by injecting either bovine synapsin I (antibody G177) or purified rat synapsin IIa (antibody G423) with Freund's complete adjuvant. Preimmune sera were used as controls. Antisera were depleted of contaminating anti-keratin antibodies by passing through a human keratin-Sepharose 4B column as described previously (Girault et al., 1989). All other chemicals of analytical grade were obtained from Sigma (Milano, Italy). Immune rabbit serum recognizing with high-affinity vesicle associated membrane protein (VAMP)/synaptobrevin in Aplysia (Poulain et al., 1993) was a generous gift of Dr. C. Montecucco (Padova, Italy). Tetanus toxin was kindly provided by Dr. P. Boquet (Nice, France).
Immunoblotting. Freshly dissected Aplysia ganglia or rat cerebral cortices were homogenized with a Teflon–glass homogenizer in electrophoresis sample buffer (Laemmli, 1970). The homogenates were boiled for 4 min, and the insoluble material was removed by centrifugation. Proteins were separated by SDS-PAGE (Laemmli, 1970) and electrophoretically transferred to nitrocellulose membranes (Towbin et al., 1979). Immunoblotting was performed as described previously (Benfenati et al., 1992b). Membranes were blocked in Tris-buffered saline (50 mm TrisCl, 200 mm NaCl, pH 7.4) containing 5% (w/v) nonfat dry milk and incubated overnight at 4°C with preimmune, G177, or G423 serum (1:1000 dilution). After washing and incubating with horseradish peroxidase-conjugated goat anti-rabbit secondary antibodies (1:3,000–1:10,000 dilution), immunoreactivity was detected using the chemiluminescence system according to the manufacturer's instructions. To analyze the specificity of the immunostaining, the primary antibodies were preadsorbed with a molar excess of purified bovine synapsin I or a mixture of purified bovine synapsin I and rat recombinant synapsin IIa for 12 hr at 4°C before immunoblotting.
Acetylcholine release and electrical recordings atAplysia synapses. Experiments were performed at identified inhibitory cholinergic synapses in buccal ganglia ofAplysia californica (70–120 gm body weight; Marinus Inc., Long Beach, CA) (Gardner, 1971; Johannes et al., 1996; Mothet et al., 1996; Doussau et al., 1998, 2000). Detailed procedures were described previously (Doussau et al., 1998). During the experiments, two presynaptic cholinergic interneurons [B4 and B5, according toGardner (1971); 80–150 μm in diameter] and one postsynaptic neuron (B3, B6, or B8; 80–200 μm in diameter) were impaled with two glass microelectrodes (3 m KCl, Ag/AgCl2, 2–10 MΩ). Acetylcholine (ACh) release from a presynaptic neuron was monitored for the duration of the experiments (up to 24 hr continuous recording) by evoking an action potential every 40 sec (0.025 Hz). The ACh release determined at this low frequency was termed “basal release.” To avoid overlap of the postsynaptic responses originating from the two presynaptic neurons, the stimulus protocols were alternated every 20 sec. ACh release was estimated by measuring the amplitude of evoked IPSC using the conventional two-electrode voltage-clamp technique. The holding potential of the postsynaptic neuron was maintained 30 mV above ECl− as described (Doussau et al., 1998).
Post-tetanic potentiation. To initiate PTP, four trains of action potentials (1 sec each at 50 Hz) were produced at 10 sec intervals. Then, the stimulation rate was returned to control conditions. For each episode, the amplitude of PTP was determined as the maximal IPSC observed during the 5 min after the conditioning stimulus and was normalized with respect to the mean amplitude of the 10 IPSCs preceding the tetanus. In several experiments, when synapsin was neutralized, the conditioning stimulus induced a depression that preceded PTP. The amplitude of this post-tetanic depression (PTD) was determined as the minimal IPSC observed within 40 sec after conditioning and normalized as described for PTP.
Synaptic depression. In several experiments, synaptic depression was induced by stimulating presynaptic neurons at 0.5 Hz for 20 min. Plateau levels were calculated as the mean amplitude of 50 IPSCs starting from the 18th min of stimulation at 0.5 Hz and expressed as a percentage of the mean IPSC amplitude recorded under the basal rate of stimulation. To induce synaptic depression, we also used 10 and 50 Hz trains of 1 sec duration. The 10 Hz trains could be repeated as a series of four trains at 2–3 min intervals without induction of PTP, provided a 15–25 min period of rest (stimulation at 0.025 Hz) was kept between two series of stimuli. Depression in amplitude of IPSCs elicited at 20 msec interpulse time intervals was examined by analyzing the amplitude of IPSCs during 50 Hz postsynaptic responses evoked by the first of the four conditioning train stimuli used to initiate PTP (see below). However, the postsynaptic responses evoked by the second, third, and fourth trains of a conditioning protocol were not analyzed because significant PTP or PTD, or both, appears just after the first conditioning stimulus.
Calculation of IPSC amplitude during high-frequency repetitive stimulation. When the stimulation time interval is shorter than the time needed for an IPSC to return to baseline, IPSCs overlap partially. In ∼20% of the neuron pairs examined, the IPSC decay was better fitted by a bi-exponential time course (with τ1 ∼9–15 msec and τ2∼20–50 msec). Therefore, the actual amplitude of IPSC numbern in a train was calculated by deducing the extrapolated residual amplitudes of the five previous IPSCs following the equation:In ∼ Ipeak n − Σ (In−i*(a*e−i*Δt/τ1+b*e−i*Δt/τ2)), where Δt was the time interval between two subsequent IPSCs, τ1 and τ2 were the decay times of previous IPSCs, and a and b (witha + b = 1) were the respective weights of the two exponentials. τ1, τ2, a, and b were calculated in each experimental condition from several IPSCs evoked by a single action potential. The analysis was limited to the first 20 IPSCs of each 50 Hz train because of the progressive decrease of phasic release and the concomitant increase of desynchronized release in the course of the tetanic stimulation. This phenomenon can probably be attributed to the fact that during tetanic stimulation, the intraterminal Ca2+ levels approach the threshold for exocytosis.
Paired-pulse facilitation. To study paired-pulse facilitation (PPF), two brief supraliminar depolarizing pulses of 5 msec were applied at 15–8000 msec intervals to the presynaptic neuron. For each twin response, facilitation F was calculated from the equation F = (I2 −I1)/I1(Mallart and Martin, 1967), where I1and I2 are the amplitudes of the IPSC evoked by the conditioning and test stimuli, respectively. The actual amplitude of I2 was determined as described above for In in a train. Because of the high variability of paired-pulse facilitation at the cholinergic synapses studied, determination of mean paired-pulse facilitation was calculated from at least 12 paired-pulse evoked responses for the same interpulse time interval.
IPSC rise time. To avoid uncertainty in determiningt0 (IPSC foot) andtmax (IPSC peak), the time to rise from 20 to 80% of maximal IPSC amplitude was determined. Mean IPSC rise times were determined from at least 10 IPSCs recorded under the same experimental condition.
Extracellular media. Dissected buccal ganglia were maintained at 22°C using a Peltier-plate system and superfused continuously (10 ml/hr) with a physiological “control medium” containing (in mm): NaCl 460, KCl 10, CaCl2 33, MgCl2 50, MgSO4 28, Tris buffer 10, pH 7.5. Note that to minimize spontaneous neuron firing activity, the physiological medium used contained high total divalent cations, a condition known to shield plasma membrane-fixed negative charges and to alter the intramembranous electric field. When extracellular [CaCl2] needed to be modified, the respective concentrations of [CaCl2] and [MgCl2] were calculated according to the following equations: [CaCl2] (mm) =Q*(83 + [MgS04])/(Q + 1) and [MgCl2] (mm) = 83 − [CaCl2], with Q being the [Ca2+]/[Mg2+] ratio (Doussau et al., 1998); in control medium, [Ca2+]/[Mg2+] was 0.42.
Intraneuronal injection procedure. Injection electrodes were pulled from glass tubing without a capillary and contained a silver wire to allow the electrophysiological monitoring of the impalement. Samples to be injected were mixed with a vital dye (fast green FCF, 10% v/v; Sigma). The samples were air pressure-injected using a picopump PV820 (WPI Ltd.) under visual and electrophysiological monitoring. The injected volume was in the range of 1–5% of the cell body volume. After injection, the injection micropipette was removed. After intracellular injection, only neurons with membrane potentials of −60 to −45 mV and with no alterations in the action potentials were analyzed.
Data presentation. Because of the intrinsic variability of different neuronal preparations, the measurements obtained for a neuron under a given experimental condition and time range were averaged and normalized against the corresponding mean measurement made during control conditions (i.e., before the time of injection of antibodies, proteins, or buffer). Then, the normalized values obtained fromn different neurons were averaged and presented as means ± SEM. Note that because of the difficulty of applying the experimental protocols in all experiments following the same time schedule, data values were pooled for given periods of time as follows: “+300 min” refers to time range 280–320 min after injection, “+480 min” to 430–530 min, and “+1100 min” to 900–1300 min.
Statistical significance. The significance of the differences observed between groups of measurements were determined by paired or unpaired t tests when a single comparison was done. When multiple comparisons were done, the significance was tested by using either one-way or two-way ANOVA analysis depending on the number of factors considered (treatment alone, time alone, or treatment plus time); this analysis was followed by the Tukey–Kramer test. To allow multiple comparisons of the 10 or 50 Hz trains recorded under various experimental conditions, the decay of IPSC amplitude in the trains was fitted with the equation y = (100 −c)*e-t/τ+ c, where c is the plateau reached, tthe time, and τ the time constant of depression. The residual variance was analyzed and significance was determined by the Fisher–Snedecor test. For all comparisons, whenp > 0.05, the difference observed was denoted as nonsignificant (n.s.).
RESULTS
Specificity of the anti-synapsin antibodies
The ability of the antibodies directed against mammalian synapsins to recognize the Aplysia synapsin homolog is shown in Figure1. Antibody G423 (lanes 1 and3), recognizing the highly conserved domain C of synapsins (F. Benfenati, unpublished observation), and antibody G177 (lane 5), recognizing synapsins I and, to a lesser extent, synapsin II in mammals (Cibelli et al., 1996), specifically detect synapsin-like proteins in acid extracts of Aplysia ganglia. These appear as a doublet of ∼55–60 kDa molecular mass (Fig. 1). These immunoreactive species were not detectable when the corresponding preimmune sera were used in the immunoblot assays (lane 2and data not shown). The immunoreactivity was virtually abolished or markedly decreased in intensity when the antibodies had been preadsorbed with a molar excess of purified bovine synapsin I (G177 antibody; lane 6) or a mixture of purified bovine synapsin I and rat recombinant synapsin IIa (G423 antibody; lane 4).
Fig. 1.

Synapsin-like immunoreactivity inAplysia nervous tissue. Proteins from extracts of rat cerebral cortex (5 μg protein, lane 1) andAplysia ganglia (30 μg protein, lanes 2–6) were separated by SDS-PAGE on 9% polyacrylamide gels, transferred to nitrocellulose membranes, and immunoblotted with the following antibodies diluted 1:1000: lanes 1 and3, G423; lane 2, preimmune G423;lane 4, synapsin-preadsorbed G423; lane 5, G177; lane 6, synapsin preadsorbed G177. Immunoreactivity was revealed using the chemiluminescence detection system. Preadsorption of the antibodies markedly decreased or virtually abolished the immunoreactivity in the samples from both rat (data not shown) and Aplysia nervous systems. Molecular mass markers are shown on the right in kilodaltons.rSynIa,b, Rat synapsin Ia/Ib; rSynIIa, rat synapsin IIa; aSynLI, Aplysiasynapsin-like immunoreactivity; rSynIIb, rat synapsin IIb.
Effect of the intraneuronal injection of specific anti-synapsin antibodies on evoked ACh release
In each hemibuccal ganglion of Aplysia, two identified presynaptic neurons, B4 and B5, make cholinergic synapses with the same set of postsynaptic neurons (including B3, B6, and B8 cells) (Gardner, 1971). ACh release evoked by single action potentials elicited in either B4 or B5 neurons can be monitored by measuring the amplitude of IPSC recorded in one of these postsynaptic neurons. To probe the role of synapsins in neurotransmitter release, the specific anti-synapsin antibodies (either G423 or G177) were pressure injected into either B4 or B5. For all experiments described in this paper, the remaining neuron was not injected (Fig.2A) but was submitted to the same stimulation protocol and thus served as an internal control of the stability of synaptic efficacy and evoked neuroexocytosis for the whole duration of the experiment (up to 24 hr).
Fig. 2.

Effects of anti-synapsin antibody injection on ACh release. Evoked ACh release was monitored at identified synapses in the buccal ganglion of Aplysia californica. A, Schematic drawing of the neuronal connections and the recorded neurons.pre1/pre2, B4/B5 presynaptic neurons;post, B3, B6, or B8 postsynaptic neuron;Ab, antibody. B, A representative experiment is illustrated. The amplitude of IPSCs evoked every 40 sec was averaged during periods of 30 min (means of 45 measurements ± SEM) before and after the injection (arrow) of the G423 anti-synapsin antibody. ● denotes values from the antibody-injected neuron, and ○ denotes values from the control, noninjected neuron. Values were normalized with respect to the mean IPSC amplitudes recorded before the injection. C, The mean amplitude of IPSCs evoked from noninjected (n = 16, white bars), preimmune-injected (n = 4,gray bars), and antibody-injected (G423,n = 9; G177, n = 3;black bars) neurons is reported at the indicated times as a percentage (±SEM) of the mean IPSC amplitude before the injection. Note that the values reported at +1100 min after injection were obtained from only three antibody-injection experiments (G423,n = 2; G177, n = 1) and two preimmune-injection experiments in which synaptic activity could be maintained for such a long time. D, Averaged IPSCs (n = 5 for each condition) recordedbefore and +480min after the time of injection from control, noninjected (top panel) and antibody-injected (bottom panel) neurons. Averaged IPSCs have been scaled to allow comparison of their time course. E, The mean percentage changes in the IPSC rise time with respect to the mean value observed before injection are reported at the indicated times after the injection for noninjected (n = 10; white bars), preimmune-injected (n = 4;gray bars), and antibody-injected (n= 6; black bars) neurons. Means at +1100 min are from only six noninjected, three preimmune-injected, and three antibody-injected neurons. C, E, Multiple comparisons were performed by a two-way ANOVA analysis followed by Tukey–Kramer test on the relative rise time change considering two factors: treatment and time. C, n.s, Not significant. E, Effect of treatment on rise time in antibody-injected neurons versus preimmune-injected or noninjected: **p < 0.001; preimmmune-injected versus noninjected: not significant; effect of time alone on IPSC rise time of noninjected or preimmune injected neurons: not significant.F, A representative experiment of a series of four, performed as described in B, during which anti-VAMP antibodies were injected. The hatched area denotes bath application of 2 μm tetanus toxin (TeNT). G, The mean IPSC rise times were determined in antibody-injected (black bar) and noninjected (white bar) neurons 235–245 min after anti-VAMP antibody injection and 1 hr after tetanus toxin application.n.s, Not significant.
When either the B4 or the B5 presynaptic neuron was microinjected with the G423 or G177 antibodies or preimmune serum, no significant change in the amplitude of IPSC was recorded for the following 500 min as compared with IPSC evoked from the control, noninjected neuron (Fig.2B,C). These results indicate that at low rates of stimulation, the amount of ACh released per impulse remains unmodified after injection of anti-synapsin antibodies. This is consistent with observations made in the lamprey reticulospinal synapses (Pieribone et al., 1995). In several experiments, recording of IPSCs could be made with a high signal-to-noise ratio that allowed accurate analysis of IPSC kinetics. The rise time of the IPSC evoked by the stimulation of the antibody-injected neuron was significantly slowed down (Fig. 2D,E) in either G423-injected (n = 4) or G177-injected (n = 2) neurons, in the absence of significant changes in IPSC decay time. In four of the six experiments analyzed, significant slowdown of the IPSC rise time was detectable as early as 160–180 min after injection (slow by 1.22; p < 0.05; data not shown). This effect lasted for the rest of the recording, up to 1100 min after the injection (Fig. 2E). Closely similar results were observed after the injection of a conserved synapsin peptide into the squid giant presynaptic terminal (Hilfiker et al., 1998).
The above-mentioned changes in IPSC kinetics cannot be attributed to an alteration of recording conditions during these very long experiments because the IPSCs evoked by stimulation of the control, noninjected neuron and recorded from the same postsynaptic neuron (Fig.2A) were not significantly modified (Fig.2D,E). In addition, the slowdown of the IPSC rise time was specific for the presence of anti-synapsin antibodies because the IPSCs evoked by stimulation of preimmune-injected neurons were not significantly slower than those obtained from control, noninjected neurons (Fig. 2E,gray bars). Hence, the slowdown of IPSC rise time observed after synapsin neutralization indicates that synapsin may be involved in some post-docking event of exocytosis, as suggested previously (Rosahl et al., 1993, 1995; Hilfiker et al., 1998).
Because synapsins are abundant SV-associated proteins, the possibility that the IPSC slowdown was the result of steric hindrance of exocytosis caused by extensive coating of SVs by the antibody cannot be excluded. To test this possibility, we examined the consequences of the injection of antibodies directed against VAMP/synaptobrevin, an integral SV membrane protein present on SVs with a number of copies similar to or higher than those of synapsin (Walch-Solimena et al., 1995; Taubenblatt et al., 1999), which participates in the formation of the fusion core complex in exocytosis (for review, see Robinson and Martin, 1998;Benfenati et al., 1999) and the cleavage of which by tetanus toxin blocks neurotransmitter release (Schiavo et al., 1992). Figure 2shows that when anti-VAMP antibodies were injected into presynaptic neurons, they did not significantly affect the evoked ACh release (Fig.2F) and IPSC rise time (Fig. 2G) for >5 hr (the first significant effects of anti-synapsin injection appeared after 3 hr). This is consistent with previous observations obtained in PC12 cells and Aplysia synapses (Elferink et al., 1993;Poulain et al., 1993). Although no release parameter was modified after anti-VAMP injection, the ability of anti-VAMP antibodies to prevent the blocking action of tetanus toxin in all the experiments that were performed (Fig. 2F) indicates that they did indeed reach their SV target, as published previously (Poulain et al., 1993). These results show that a procedure aimed at coating SVs with antibodies does not necessarily alter ACh release kinetics and that the effects observed after injection of anti-synapsin antibodies are likely attributable to synapsin neutralization or interference with some post-docking activity of synapsin.
Effects of intraneuronal injection of anti-synapsin antibodies or of synapsin I on post-tetanic potentiation
PTP is a form of short-term synaptic plasticity that is characterized by a transient increase in synaptic strength in response to a presynaptic tetanic conditioning stimulus (for review, see Zucker, 1996, 1999). Although postsynaptic mechanisms have been reported to play some role in PTP at certain Aplysia synapses (Bao et al., 1997), PTP is mostly caused by an enhanced neurotransmitter release initiated by an accumulation of Ca2+ during tetanic stimulation (Kretz et al., 1982; Swandulla et al., 1991; Kamiya and Zucker, 1994; Bao et al., 1997) and has been proposed to involve the recruitment of SVs from the reserve pool, possibly under the control of synapsins (Greengard et al., 1993; Rosahl et al., 1995; Zucker, 1996, 1999). Hence, we investigated the possible implication of synapsin in PTP.
At the identified cholinergic synapses of the buccal ganglion, PTP is reproducibly initiated by application of four trains of impulses of 1 sec at 50 Hz. A typical PTP is illustrated in Figure3A, top panel. When PTP was elicited under control conditions, evoked ACh release increased with respect to basal values for a mean duration of 35 ± 2 min, reaching a peak increase of 48 ± 3% (means ± SEM; 41 PTPs initiated in 26 neurons). In most experiments performed under control conditions, PTP was found to reach its maximal amplitude within 40 sec after the fourth conditioning stimulus was applied (data not shown).
Fig. 3.
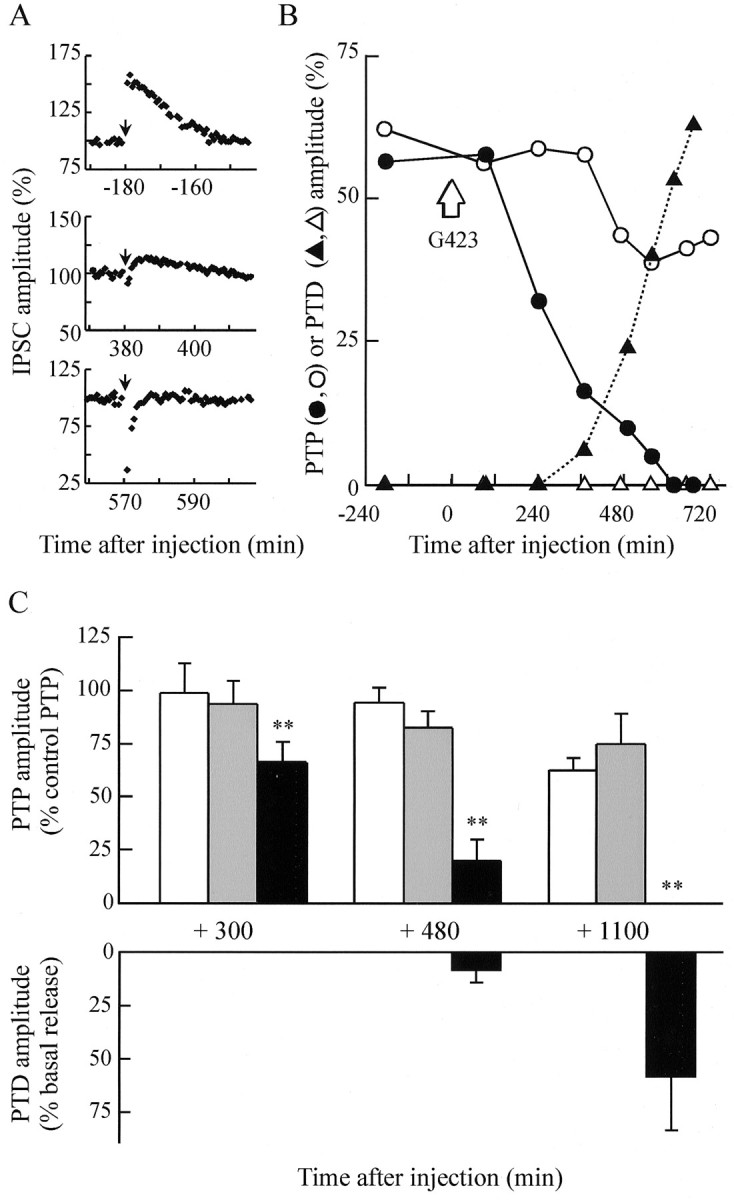
Post-tetanic potentiation (PTP) is depressed after injection of anti-synapsin antibodies.A, Four conditioning tetanic stimulations (1 sec at 50 Hz, each) were applied to initiate PTP. The expression of PTP was monitored by plotting the IPSC amplitude, evoked at 0.025 Hz, before and after the conditioning stimuli (arrow) were applied. The three PTPs illustrated were recorded at the indicated times, before (top panel) and after (middle andbottom panels) G423 antibody injection. Note the appearance of post-tetanic depression (PTD) after antibody injection. B, Representative experiment in which the amplitudes of PTP (circles) and PTD (triangles) were followed as a function of time after the injection (arrow) in G423-injected (●, ▴) and noninjected (○, Δ) neurons. PTP and PTD amplitudes were determined as described in Materials and Methods. C, The amplitudes of PTP (top panel) and PTD (bottom panel) induced in noninjected (white bars), preimmune-injected (gray bars), and antibody-injected (black bars) neurons are shown as means ± SEM. PTP amplitudes were normalized against the mean PTP amplitudes recorded in the same neurons before the injection. Average PTD amplitudes are expressed as percentage of the mean basal IPSC amplitude determined immediately before the conditioning stimulus. Number of experiments at +300 or +480 min: anti-synapsin antibodies,n = 9 (G423, n = 6; G177,n = 3); preimmune, n = 4; noninjected, n = 13. Number of experiments at +1100 min: anti-synapsin antibodies, n = 3 (G423,n = 2; G177, n = 1); preimmune,n = 2; noninjected, n = 5. Comparison between the different groups in C (top graph) was tested by using two-way ANOVA followed by the Tukey–Kramer test. PTP reduction in anti-synapsin-injected neurons wasp = 0.0045; PTP elicited in antibody-injected versus preimmune-injected or noninjected neurons wasp = 2 × 10−6 and 1.7 × 10−6, respectively. All other comparisons: n.s.
Figure 3B shows the comparative time course of PTP elicited from a presynaptic neuron injected with the G423 antibody and from the control, noninjected neuron in the same preparation. After injection (seven experiments), PTP was strongly depressed or abolished within 500 min of the antibody injection (Fig. 3C, top graph). In the three experiments performed using G177 antibodies, a similar disappearance of PTP was observed, but with slower time course, possibly caused by a lower titer or a lower affinity of the antibody. PTP recorded from noninjected neurons in the same preparations was not significantly changed (Fig. 3C,top graph, open bars), albeit a slight decrease could be observed at very long recording times, possibly because of aging of the preparations (Fig. 3C, +1100). No significant changes in PTP amplitude could be observed after injection of preimmune serum as compared with noninjected neurons (Fig.3C). The abolition of PTP observed in anti-synapsin-injected neurons was not associated with any significant change in basal ACh release, except for the effect on IPSC kinetics (Fig. 3C,top graph, compare with 2C,E). These observations suggest that the mobilization of a synapsin-dependent reserve pool of SVs is necessary for neurons to express PTP.
When conditioning protocols were induced at a time when PTP was abolished, a transient decrease in the amplitude of the postsynaptic response was observed (Fig. 3A–C). We termed it PTD. The occurrence of PTD was not restricted to a particular synapse of the network, because we observed this same phenomenon by recording IPSCs in B3, B6, or B8 postsynaptic neurons in the same hemiganglion (Fig. 4A). One of the three experiments is illustrated in Figure 4B. It is possible that when the synapsin-dependent reserve pool of SVs is depleted after action of the antibodies, the readily releasable pool becomes exhausted during the conditioning stimuli and cannot be replenished at a sufficient rate, leading to transient PTD. Such an effect should be emphasized when increasing the duration of conditioning stimuli or minimized when the release probability is reduced. Indeed, the magnitude and time course of PTD depended on the number of 50 Hz trains applied to initiate PTP (Fig. 4C), and when PTP protocols were induced in low extracellular Ca2+ (Ca/Mg = 0.14) to reduce release probability, no PTD was initiated unless the number or duration of the 50 Hz trains was increased (data not shown).
Fig. 4.

Amplitude of PTP and PTD depends on the release conditions. A, Schematic representation of the buccal ganglion of Aplysia californica.B4 and B5 denote presynaptic cholinergic neurons. B3, B6, and B8are their postsynaptic targets. B, A representative experiment of a series of three is shown. Conditioning stimuli were applied as described in Figure 3 at G423 antibody-injected (B4, arrow) and noninjected (B5) neurons. The ensuing PTDs (●) or PTPs (○) were recorded at the indicated times after the injection by impaling successively B3, B6, and B8 neurons. C, Effects of the strength of the conditioning stimulus on PTP and PTD amplitudes recorded from G423-injected (●) and noninjected (○) neurons at the indicated times after the injection. The following conditioning stimuli were applied: 1 train (1 sec at 50 Hz) (S), 4 trains (SS), 10 trains (SSS).D, The effect of anti-synapsin antibody injection on PTP amplitude depends on the release probability. Relative PTP amplitudes were determined as a percentage of the mean PTP amplitude recorded before the injection as described in Figure 3C. PTP amplitudes were determined from antibody-injected (G423,n = 2; G177, n = 1;filled bars) and noninjected (n = 3;open bars) neurons 460–515 min after antibodies were injected. Then, a low Ca2+-containing medium (Ca/Mg ratio = 0.14) was substituted for the high Ca2+medium (Ca/Mg ratio = 0.42), and the relative PTP amplitudes were determined (i.e., 520–550 min after antibody injection). *p < 0.05, **p < 0.01; Student's unpaired t test.
As shown in Figures 3A and 4C, PTD overlaps with PTP. This raises the question of whether the induction of PTD contributes to the inhibition of PTP. First, the duration of PTD was much shorter than that of PTP (Figs. 3A, 4C). Second, after PTP had been depressed by injecting G423 or G177 antibodies under normal Ca2+ conditions (Fig. 4D, compare the left open andfilled bars), a decrease in extracellular Ca2+, applied to minimize PTD, induced only small recovery of PTP as compared with the PTP recorded in noninjected neurons under the same Ca2+condition (Fig. 4D, filled bars). These data confirm that synapsin neutralization directly affects PTP mechanisms.
To further demonstrate the involvement of synapsin in the expression of PTP, we investigated the effects of an increase in the intraneuronal synapsin pool by injecting purified mammalian synapsin I into the presynaptic neurons. Purified dephosphorylated synapsin I (Fig.5, Syn) or the corresponding buffer was injected into B4 or B5 neurons. Assuming a homogenous distribution of the protein in the cytosol, the final intrasomatic concentration of synapsin was estimated to range between 0.5 and 1 μm (15 μm in the injection micropipette). In contrast to the antibody injection experiments, no change in either IPSC amplitude or kinetics could be detected up to 8 hr after the intracellular application of either synapsin or synapsin buffer as compared with the noninjected neurons (Fig. 5A–C). However, a marked and progressive enhancement in PTP amplitude was observed when ACh release was evoked from the synapsin-injected neurons (Fig.5D–F). The change in PTP amplitude started soon after injection of synapsin (Fig. 5E, compare with 3B).
Fig. 5.

Synapsin injection increases PTP without changing basal ACh release. A, Effects of synapsin I injection on basal release. A representative experiment of a series of six is illustrated. Dephosphorylated synapsin I was injected at an estimated final concentration in the soma of 0.5–1 μm. The experiments and data presentation are as described in Figure 2. ● and ○ denote IPSC amplitude from synapsin I-injected and noninjected neurons, respectively. B, C, The mean amplitude of IPSCs and the mean IPSC rise time evoked from synapsin-injected (black bars; n = 6) and buffer-injected (gray bars;n = 6) neurons after the injection are reported as a percentage of the IPSC amplitude and rise time determined before injection. D–F, Effects of synapsin I injection on PTP. D, Typical PTP recorded 300 min before (top panel) and 320 min after (bottom panel) synapsin injection. Same presentation as in Figure 3A. E, PTP amplitude was followed as a function of time after the injection (arrow) of synapsin. Presentation of data is as in Figure 3B. F, Means (±SEM) of the PTP amplitudes observed after synapsin (black bars) or buffer (gray bars) injection.n = 5 at +300 min and n = 3 at +480 min for synapsin-injected neurons, and n= 3 at both +300 min and +480 min for buffer-injected neurons. Statistical analysis was performed by using two-way ANOVA followed by Tukey–Kramer test. **p = 0.026 versus buffer injection on PTP amplitude. n.s, Not significant.
These observations indicate that synapsins play a major role in PTP mechanisms, possibly by determining the size of the reserve pool or controlling the transition of SVs between the reserve and the releasable pools, or both. A recruitment of reserve SVs has also been postulated to contribute to the maintenance of synaptic efficacy. At active mammalian synapses, the exo–endocytotic cycle of SVs takes ∼10–30 sec to complete (Stevens and Tsujimoto, 1995; Klingauf et al., 1998; Murthy and Stevens, 1999). Thus, when the rate of stimulation is increased over the recycling–refilling capacity (i.e., >0.03–0.1 Hz), synaptic efficacy cannot be maintained. However, in most synapses, the recruitment of SVs from the reserve pool contributes to the rapid replenishment of the ready releasable pool and minimizes depression (Ryan et al., 1993; Stevens and Tsujimoto, 1995;Dobrunz and Stevens, 1997). According to this model, the synaptic depression observed in lamprey synapses injected with anti-synapsin antibodies and subjected to prolonged 18–20 Hz stimulation is attributable to the exhaustion of the reserve pool (Pieribone et al., 1995). However, synapsin has also been proposed to play a role in some post-docking event of exocytosis (Rosahl et al., 1993, 1995; Hilfiker et al., 1998) (Fig. 2D,E). Hence, this raises the question of whether the inability of synapses to maintain neurotransmitter release during high-frequency stimulation is caused by exhaustion of a synapsin-dependent reserve pool or impairment of a post-docking function of synapsin, or both.
Synapsin neutralization induces fast and marked synaptic depression
The replenishment time constant of release sites by reserve SVs is on the order of several seconds (Stevens and Tsujimoto, 1995). Thus, the application of brief (1–2 sec), high-frequency stimulations may permit investigation of the effects of an altered post-docking function of synapsin induced by its neutralization by minimizing the effects caused by exhaustion of reserve pool. The amplitude of IPSCs was analyzed during 50 Hz postsynaptic responses evoked by the first of the four conditioning train stimuli used to initiate PTP and during 10 Hz trains for 1 sec that do not elicit PTP (for details, see Materials and Methods). Figures 6, A andB, and 7A show typical 50 and 10 Hz responses elicited from the same presynaptic neurons, before and after PTP had been abolished by previous injection of G423 or G177 antibodies (i.e., 500 min after injection). After anti-synapsin antibody injection, the IPSC amplitude could not be maintained during 10 and 50 Hz trains (Figs. 6C, 7B). No comparable effect was detected in control, noninjected, or preimmune injected neurons (Fig.7B). The small increase in 10 Hz-induced depression observed in control neurons indicates that aging of the preparations may also produce a small fatigue in ACh release. The time course of the increase in depression induced by the anti-synapsin antibody injection started within 2 hr after injection and developed after a time course that was closely similar to that of PTP inhibition (50 Hz train: data not shown; 10 Hz train: Fig.7C, compare with Fig. 3B). By contrast, although synapsin I injection was effective in enhancing PTP (Fig. 5), no significant change in 10 or 50 Hz-induced depression could be detected in synapsin I-injected neurons as compared with neurons injected with synapsin buffer alone (Figs. 6D, 7D). Taken together, these findings indicate that after neutralization of synapsin, ACh release cannot be maintained at high levels during a prolonged high-frequency stimulation. Interestingly, the increase in 10 and 50 Hz-induced depression was already significant at the second stimulus in the train (i.e., that elicited at a 20 or 100 msec interpulse interval) (Figs. 6 and 7, legends). This time scale is by far smaller than the mean SV residency on the plasma membrane (2–5 sec) (Klingauf et al., 1998; Murthy and Stevens, 1999). Hence, after synapsin neutralization, the inability of the synapse to sustain ACh release during short high-frequency bursts may result from alteration of a synapsin function that operates during a membrane step of the release process.
Fig. 6.

Frequency stimulation-induced depression (50 Hz) is increased after synapsin neutralization. Pairs of neurons, one injected with anti-synapsin antibodies, preimmune serum, buffer alone, or purified synapsin I and the other kept intact, were subjected to repetitive stimulation at 50 Hz. A, B, Representative postsynaptic responses evoked by 50 Hz stimulation applied before (thin line) and 500 min after G423 (A) or G177 (B) injection (thick line) (i.e., at a time when PTP was abolished). Note the different time scales in A and B. C, The mean amplitude of the 20 first IPSCs of the 50 Hz train response was determined (see Materials and Methods), expressed as a percentage of the first IPSC of the train (i.e., which amplitude is identical to IPSC amplitude recorded under basal stimulation frequency at 0.025 Hz), and normalized against the corresponding values determined before injection. Mean values (±SEM) were calculated from recordings made 430–530 min after the injection of anti-synapsin antibodies (●, 6 G423-injected neurons) or preimmune serum (gray-filled circle, 4 neurons). ○ denotes mean values from 10 control, noninjected neurons in the same preparations.D, Same kind of measurements as in Cexcept that synapsin I (▪, n = 3) or buffer alone (gray-filled square, n = 3) was injected. The comparison among the extents of 50 Hz-induced depression observed after the various treatments was performed as described in Materials and Methods (two-way ANOVA followed by a Fisher–Snedecor test). **p < 10−9 anti-synapsin versus noninjected or preimmune. Significance of the differences observed in the second response to 50 Hz trains was tested as described in Materials and Methods using the Tukey–Kramer test after one-way ANOVA:p = 0.016 and 0.004 anti-synapsin versus noninjected and preimmune, respectively.
Fig. 7.

Frequency stimulation-induced depression (10 Hz) is increased after synapsin neutralization. Representative postsynaptic responses evoked by 10 Hz (A) stimulation applied before (thin line) or 485 min after (thick line) G423 injection. B, The mean amplitude of the 10 IPSC responses evoked by the 10 Hz train 430–530 min after injection of antibodies was determined (see Materials and Methods) and normalized against the corresponding values determined before injection. Mean values (±SEM) were calculated after the injection of anti-synapsin antibodies (●, n = 6) or preimmune serum (gray-filled circle, n = 3). ○ denotes mean values from nine control, noninjected neurons in the same preparations. C, The mean amplitude of the 10th response to the 10 Hz trains recorded in the course of a representative experiment was normalized with respect to the amplitude of the first IPSC of the train (4–6 trains were averaged at each time indicated; note that SEM bars are smaller than the symbol size) and plotted as a function of time after injection (arrow) in G423-injected (●) and noninjected (○) neurons. D, Same kind of measurements as in B except that synapsin I (▪, n = 3) or its buffer (□,n = 3) was injected. Significance of the differences observed in the responses to 10 Hz trains under the various treatments was tested as described in Figure 6. Whole train: **p < 10−9anti-synapsin versus noninjected or preimmune. Second response to 10 Hz trains: **p = 0.02 and 0.03, anti-synapsin versus noninjected and preimmune, respectively.
To better characterize fast depression (and thus the underlying post-docking synapsin function), we next examined the Ca2+ dependency of 10 Hz-induced synaptic depression (three experiments). Under control conditions, no or little synaptic depression was detected when 10 Hz trains were elicited in the presence of a low Ca2+-containing medium (with Ca2+/Mg2+ = 0.14 to reduce the release probability), whereas depression could be observed when using a high Ca2+-containing medium (Ca2+/Mg2+ = 0.42 or 2.1). When similar experiments were repeated 500 min after the injection of G423 (n = 3) antibody, at a time PTP was abolished, 10 Hz-depression was dramatically enhanced when the extracellular Ca2+ concentration was raised to higher levels, whereas it was not affected at low Ca2+ (Fig.8, compare open andclosed bars). These results suggest that the synapsin-dependent regulation that operates at the membrane stage of the release process to prevent high-frequency-induced depression is limiting only when the probability of release is high.
Fig. 8.

Fast depression induced by anti-synapsin antibody injection depends on the release probability. Trains of 10 Hz were elicited in the presence of low (Ca/Mg = 0.14), normal (Ca/Mg = 0.42), or high (Ca/Mg = 2.1) Ca2+-containing medium. Mean amplitudes of IPSCs evoked by the 10 Hz stimulation of noninjected (open bars) and G423-injected (filled bars) neurons were determined as described in Materials and Methods, under the various conditions, 410–550 min after the time of antibody injection. Means (±SEM) were calculated from three independent experiments performed under each condition. Significance of the differences in the responses to 10 Hz trains between antibody-injected and control, noninjected neurons was as follows: Ca/Mg = 0.14: non-significant (ns); Ca/Mg = 0.42 or 2.1: **p < 10−9.
The time scale of the fast depression observed during high-frequency stimulation after injection of anti-synapsin antibodies overlaps with the time window (0–200 msec) for facilitation in the sameAplysia synapses (Doussau et al., 1998). To determine whether synapsin neutralization primarily enhanced depression or affected it indirectly through a decrease in facilitation, we determined the extent of IPSC facilitation induced by paired stimuli applied at time intervals ranging from 20 msec to 8 sec before and 360–480 min after (i.e., when PTP was abolished) injection of the anti-synapsin antibody G423. The injection of G423 significantly decreased the amplitude of the second response of pairs elicited at all the examined interpulse intervals (20 msec–8 sec). As shown in Figure9A, this manifested as a decrease in paired-pulse facilitation at the shorter time intervals and as paired-pulse depression at longer interpulse intervals (n = 4). Because the extent of depression was comparable with that of the decrease in facilitation (Fig.9A), this suggested that facilitation per se might be unchanged after antibody injection. To dissociate facilitation from depression, we took advantage of the observation that the effect on depression is dramatically relieved under conditions of low release probability (Fig. 8). Paired-pulse stimulation protocols were then repeated using low Ca2+-containing medium (Ca/Mg = 0.14) (Fig. 9B). Under these conditions, the amplitudes of paired-pulse facilitation determined before and after G423 injection were not significantly different (Fig. 9B) (n = 3). These results suggest that facilitation per se is not altered after neutralization of synapsin, consistent with previous observations in synapsin II or synapsin I/II knock-out mice (Rosahl et al., 1995).
Fig. 9.

PPF is not affected after synapsin neutralization under low release probability. Paired stimuli were elicited at various interpulse intervals before (○) and 360–480 min after (●) the intraneuronal injection of G423 antibody in the presence of either normal (Ca/Mg = 0.42; A) or low (Ca/Mg = 0.14;B) Ca2+-containing medium. The extent of paired-pulse facilitation (means ± SEM from 4 experiments in A and from 3 experiments in B) was calculated at the indicated interpulse intervals. Note that values reported at the 40 sec interval have been collected at the basal stimulation rate (0.025 Hz). Statistical comparison of PPF extent before and after synapsin neutralization: *p < 0.01; **p< 0.001.
Paired-stimulation experiments in anti-synapsin-injected synapses show that the time scale of paired-pulse depression extends over a 10 sec range. This time scale exceeds the mean residency time of SVs at plasma membrane but remains in the range of the replenishment kinetics of the readily releasable pool of SVs (Stevens and Tsujimoto, 1995; Klingauf et al., 1998; Murthy and Stevens, 1999). Accordingly, the inability to maintain high synaptic efficacy after synapsin neutralization in synapses subjected to twin stimulations applied at long intervals may be attributable to an altered SV trafficking between the reserve pool and the releasable pool. To investigate this possibility while minimizing the effects of an alteration of synapsin function at membrane level, we examined the effects of a prolonged increase in the stimulation rate from 0.025 to 0.5 Hz for 20 min. Under these conditions, the IPSC amplitude decreased and reached a stable level (plateau) at ∼50% of initial amplitude in ∼10–15 min (Fig.10A), as reported earlier (Mothet et al., 1996). In a series of five experiments, 0.5 Hz-induced depression was examined before and after injection of G423 antibodies and in noninjected neurons in the same preparations (a typical example is illustrated in Fig. 10A). In anti-synapsin antibody-injected neurons subjected to repeated periods of 0.5 Hz stimulation, the development of depression was significantly accelerated as compared with control recordings made before injection or in noninjected neurons (Fig.10A,C). However, the extent of depression was virtually unchanged, because the plateau level reached during 0.5 Hz stimulation was not modified significantly (Fig.10A,B). When the stimulation frequency was returned to 0.025 Hz, the initial phase of recovery from depression was significantly faster in the antibody-injected neurons (Fig. 10A,D).
Fig. 10.

Depression and recovery from 0.5 Hz stimulation periods are accelerated after synapsin neutralization.A, Representative experiment from a series of 5. Presynaptic neurons were stimulated either at basal stimulation frequency (0.025 Hz) or at 0.5 Hz for 20 min periods (horizontal bars). IPSCs were evoked in both G423-injected (●) and control, noninjected (○) neurons. Note the faster depression and recovery rates that occurred after antibody injection.B, The IPSC plateau levels (means ± SEM) reached during 0.5 Hz stimulation, determined as described in Materials and Methods, are reported for antibody-injected neurons (filled bars) and control, noninjected neurons (open bars). C, D, The amplitude of IPSCs evoked during 0.5 Hz stimulation (C) and after the stimulation frequency was returned to 0.025 Hz (D) was averaged for periods of 30 sec (0–2 min) or 60 sec (2–16 min), and the corresponding plateau values were subtracted. Means ± SEM of normalized IPSC–plateau values determined in five experiments are presented before (Δ) and +480 min after (●) the injection of G423. ○ denotes values from control, noninjected neurons. Statistical analysis was performed by using two-way ANOVA followed by the Tukey–Kramer test. *p < 0.05 antibody versus noninjected or before injection. n.s, Not significant.
DISCUSSION
Our data show that after injection of specific anti-synapsin antibodies or purified synapsin, several forms of short-term plasticity are strongly modified. This raises the questions of whether our observations can be ascribed to an effect on the synapsin-dependent reserve pool or to alteration in SV trafficking and how antibodies interfere with synapsin function.
Post-tetanic potentiation is believed to involve activity-dependent recruitment of reserve SVs (for review, see Zucker, 1996, 1999). The implication of synapsin in PTP has remained an unclear issue: decreased PTP, concomitant with long-lasting depression, has been found in synapsin II or synapsin I/synapsin II but not synapsin I mutant mice (Rosahl et al., 1995). Because we found that PTP amplitude increases after synapsin injection and is virtually abolished after synapsin neutralization, our findings unambiguously implicate the synapsins as a major determinant for this form of synaptic plasticity. The expression of PTP is generally believed to involve a fast recruitment of SVs from the reserve pool to the releasable pool (Zucker, 1996, 1999) that can be achieved, at least in part, through Ca2+-dependent phosphorylation of synapsin (Fig. 11, step 5) (Greengard et al., 1993). However, the enhancement of PTP amplitude after injection of dephosphorylated synapsin and the relative long delay for the appearance of the changes in PTP expression suggest that antibody- or synapsin-dependent variations in the size of SV pools are involved. In fact, in recent years the synapsins have been suggested to have a structural role by favoring the assembly of the reserve pool of SVs during synaptogenesis and regulating its size in mature neurons (Lu et al., 1992; Li et al., 1995; Pieribone et al., 1995; Rosahl et al., 1995; Takei et al., 1995; Valtorta et al., 1995; Ryan et al., 1996;Hilfiker et al., 1998; Fiumara et al., 2001). Thus, it is possible that the synapsin-induced enhancement of PTP results from an increase in the size of the reserve pool at steady state (Fig. 11, box A, balance between steps 5 and 6) that, in turn, will increase the supply of reserve SVs to the readily releasable pool in response to a conditioning stimulus (Fig. 11, step5), whereas the antibody would elicit the opposite effect by inducing depletion of the synapsin-dependent reserve pool of SVs. Moreover, the ability of exogenous synapsin to increase PTP indicates that the mechanism by which synapsins recruit SVs to the reserve pool is not saturated at physiological synapsin concentrations.
Fig. 11.

Synapsin-dependent regulation of SV pools and trafficking during short-term plasticity. Numbers refer to the following steps in the release process: 1, recycling of fused SVs; 2, refilling of free release sites or docking; 3, priming of docked SVs;4, SV fusion; 5, SV trafficking between synapsin-dependent reserve pool and membrane-bound pool;6, sequestration of recycled SV in the synapsin-dependent reserve pool. Synapsin molecules are shown asblack ribbons; open arrows denote proposed synapsin-dependent steps. Box A highlights the putative mechanisms involved in the expression of PTP and depression at low rate of stimulation. Box B highlights the putative mechanisms involved in the occurrence of depression at a high rate of stimulation and synchronization of release. For more details, see Discussion.
Several of our data show that after synapsin neutralization, synaptic efficacy cannot be maintained during and after repetitive stimulations elicited at time intervals ranging from the millisecond to the second time scale. This manifests by the appearance of increased depression, decreased facilitation, and PTD. As detailed below, depending on the stimulation rate, the underlying synapsin-dependent mechanisms altered by antibody injection seem to be different.
An experimental situation to be considered is the depression induced by sustained 0.5 Hz stimulation (Fig. 10). After synapsin neutralization, it is characterized by both accelerated kinetics and no change in the plateau level as compared with controls. At active synapses, when the rate of stimulation is increased over the SV cycle rate (Fig. 11, steps1-4) (∼10–30 sec), synaptic efficacy depresses and reaches a level that is determined both by the refilling–recycling capacity and by the concomitant recruitment of reserve SVs (Ryan et al., 1993; Stevens and Tsujimoto, 1995; Dobrunz and Stevens, 1997). Because the reserve pool is limited, when repetitive stimulations are prolonged, transmitter release is likely to reach a level determined entirely by the recycling capacity. In view of this model, the unchanged plateau level that characterizes 0.5 Hz depression indicates that at moderate stimulation frequency, synapsin does not affect the rate of SV cycling (Fig. 11, steps1-4). This is consistent with the unchanged endocytosis and repriming rates observed in synapsin knock-out mice (Ryan et al., 1996). The marked effects of synapsin neutralization during the early phase of 0.5 Hz-depression, as well as the appearance of PTD, are compatible with the notion that synapsin controls either the size of the reserve pool (Fig. 11, step6) or the supply of reserve SVs to the releasable pool to minimize depression (Fig. 11, step 5). However, we cannot exclude the possibility that 0.5 Hz-depression is also contributed to by the alteration of a synapsin function that operates at the membrane level, as discussed below for fast depression (Fig. 11, step 3). After synapsin neutralization, we found that ACh release returns faster to its initial level when the stimulation rate is slowed from 0.5 to 0.025 Hz. This is reminiscent of the faster replenishment observed at inhibitory synapses in mice lacking synapsin I (Terada et al., 1999). A likely possibility is that when the stimulation rate is slowed down, the amount of recycled SVs (determined by the preceding higher rate stimulation period) exceeds the replenishment need during a time window in the order of the recycling time. In intact neurons, these extra SVs are sequestered in the reserve pool (Fig. 11, step 6). After synapsin neutralization, this process is no longer possible, and thereby all recycled SVs may directly enter the releasable pool (Fig. 11, step2), leading to faster recovery of synaptic efficacy to its initial level. Obviously, the synapsin-dependent mechanisms involved in minimizing the depression induced by moderate stimulation rates share several similarities with PTP mechanisms (Fig. 11, box A).
A different experimental situation is represented by the fast synaptic depression induced by synapsin neutralization in Aplysiasynapses subjected to short trains of high-frequency (10–50 Hz) stimulation. A disruption of the reserve pool and the subsequent impaired refilling of the releasable pool is unlikely to be involved because the fast kinetics of depression indicates that the SVs involved should be made available for exocytosis in a time scale (10–100 msec) that is significantly shorter than the estimated time (several seconds) needed for reserve SVs to replenish the releasable pool (Stevens and Tsujimoto, 1995). Taking into account an average residency time of SVs at the presynaptic membrane of 2–5 sec (Klingauf et al., 1998; Murthy and Stevens, 1999), this suggests that fast depression results from disruption of a synapsin action that operates during a membrane step of the release process (Fig. 11, box B). At variance with previous deductions made in mice lacking synapsin I (Rosahl et al., 1995), our data do not support the possibility that this synapsin-dependent membrane action is related to facilitation mechanisms. Indeed, although paired-pulse facilitation is depressed after synapsin neutralization under control conditions (Fig. 9), it is not affected under low Ca2+ conditions that maximize facilitation and minimize depression. Most likely, synapsin plays a modulatory role in the mechanism by which membrane-bound SVs attain fusion competence or fuse after stimulation (Fig. 11, steps 3-4). Because fast depression develops only when the release probability and stimulation rates are high (Fig. 8), the membrane action of synapsin becomes limiting under conditions of exhaustion of the releasable pool. The fact that these regulatory actions of synapsin are not enhanced after injection of exogenous synapsin suggests that, at variance with PTP, the involved molecular event(s) may already be saturated by the levels of endogenous synapsin.
The implication of synapsin at a plasma membrane step of the exocytotic process is also supported by the slower IPSC rise time induced by anti-synapsin antibodies (Fig. 2). This effect is similar to that observed after injection of a conserved synapsin domain into the squid giant presynaptic terminal (Hilfiker et al., 1998). The slower rise time of IPSC observed after synapsin neutralization in the absence of a significant synaptic delay suggests that the release process has been desynchronized. This is compatible with a facilitatory action of synapsin on the priming–fusion steps (Fig. 11, steps3-4). However, this regulatory action appears to be dispensable for the release process, because the changes in IPSC rise time were observed in the absence of a significant effect on IPSC amplitude. As mentioned in Materials and Methods, during 50 Hz trains the IPSCs are superimposed on progressively increasing desynchronized release, and this effect prevents accurate analysis of IPSC amplitude during the whole trains. Further work is needed to determine whether the reduction of IPSC amplitude during the course of 50 Hz responses elicited after synapsin neutralization (Fig.6A) is caused by increased desynchronization during the train, thereby revealing a role of synapsin in maintaining the phasic character of evoked release during high-frequency stimuli.
Regarding the mechanisms of synapsin “neutralization” by the antibodies, it seems unlikely that the antibodies are able to enter the thick clusters of vesicles and actin filaments and directly compete with the binding of synapsin to vesicles and actin. Neutralization of synapsin most likely consists of a slow sequestration of the few synapsin molecules that are released into the cytosol during the exo–endocytotic cycle of SVs (Torri Tarelli et al., 1992; Greengard et al., 1993) and of the subsequent shift from the bound to the free form attributable to the mass action law (Schiebler et al., 1986; Benfenati et al., 1991). The relatively long delay (>2 hr) needed to neutralize endogenous synapsin may be attributable to the time needed to bring enough IgG to the nerve terminals to inactivate a substantial fraction of the synapsin molecules. It should be pointed out that because synapsins are present in >10 copies per SV (Schiebler et al., 1986) and several hundreds of SVs are present in Aplysia nerve terminals (∼1.25 μm diameter) (Bailey et al., 1979), each terminal is likely to contain >1000 synapsin molecules.
In conclusion, our data demonstrate that synapsin controls multiple steps in the release process and participates in multiple forms of short-term plasticity. A major regulation concerns the maintenance of a reserve pool that confers the nerve terminal the ability to release neurotransmitter at firing rates exceeding the maximum SV recycling capacity and to dynamically change the efficiency of release in response to conditioning stimuli (e.g., the PTP) by transiently shifting the equilibrium between the reserve and the releasable SV pools (Fig. 11,box A). Moreover, our data support the notion that synapsin regulates the fusion competence or priming of SVs. In allowing faster entry of releasable SV pools in the fusion mechanisms, the synapsins control the ability of nerve terminals to maintain phasic and efficient release, albeit for a brief period of time (Fig. 11, box B).
Footnotes
This work was supported by grants from the Association Française contre les Myopathies (B.P.), the Fisher Center for Alzheimer's Disease Research, the Associazione Italiana Ricerca sul Cancro, Consiglio Nazionale delle Ricerche, Ministero dell'Universitá e della Ricerca Scientifica e Tecnologica Cofin 1998 and 2000 (F.B.), and United States Public Health Service Grants MH39327 and AG15072 (P.G.). The financial support of Telethon–Italy (Grant 1131 to F.B.) is gratefully acknowledged. We thank Drs. Eric Kandel (Columbia University, New York, NY), Pietro De Camilli (Yale University, New Haven, CT), Giampietro Schiavo (Imperial Cancer Research Fund, London, UK), and Nancy Grant-Takeda (UPR 2356 Strasbourg, France) for critical reading of this manuscript, Dr. Jean-Luc Rodeau (Université Louis Pasteur/Centre National de la Recherche Scientifique UMR 7519, Strasbourg) for help in data analysis, and Drs. Andrew J. Czernik and Chris Siow (The Rockefeller University, New York, NY) for their contribution to the preparation of the G423 antibody.
Correspondence should be addressed to Bernard Poulain, Neurotransmission et Sécrétion Neuroendocrine, UnitéPropre de Recherche 2356 Centre National de la Recherche Scientifique, 5 rue Blaise Pascal, F-67084 Strasbourg Cedex, France. E-mail:poulain@neurochem.u-strasbg.fr.
F. Vitiello's present address: Dipartimento di Farmacologia e Fisiologia Umana, Facoltà di Medicina e Chirurgia, Università di Bari, Bari, Italy.
REFERENCES
- 1.Angers A, Bean AJ, Byrne JH. Cloning and molecular characterization of Aplysia synaptic vesicle protein synapsin. Soc Neurosci Abstr. 1999;25:1749. [Google Scholar]
- 2.Bailey CH, Thompson EB, Castellucci VF, Kandel ER. Ultrastructure of the synapses of sensory neurons that mediate the gill-withdrawal reflex in Aplysia. J Neurocytol. 1979;8:415–444. doi: 10.1007/BF01214801. [DOI] [PubMed] [Google Scholar]
- 3.Bao JX, Kandel ER, Hawkins RD. Involvement of pre- and postsynaptic mechanisms in post-tetanic potentiation at Aplysia synapses. Science. 1997;275:969–973. doi: 10.1126/science.275.5302.969. [DOI] [PubMed] [Google Scholar]
- 4.Benfenati F, Valtorta F, Greengard P. Computer modeling of synapsin I binding to synaptic vesicles and F-actin: implications for the regulation of neurotransmitter release. Proc Natl Acad Sci USA. 1991;88:575–579. doi: 10.1073/pnas.88.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benfenati F, Valtorta F, Chieregatti E, Greengard P. Interaction of free and synaptic vesicle-bound synapsin I with F-actin. Neuron. 1992a;8:377–386. doi: 10.1016/0896-6273(92)90303-u. [DOI] [PubMed] [Google Scholar]
- 6.Benfenati F, Valtorta F, Rubenstein JR, Gorelick FS, Greengard P, Czernik AJ. Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature. 1992b;359:417–420. doi: 10.1038/359417a0. [DOI] [PubMed] [Google Scholar]
- 7.Benfenati F, Valtorta F, Rossi MC, Onofri F, Sihra T, Greengard P. Interactions of synapsin I with phospholipids: possible role in synaptic vesicle clustering and in the maintenance of bilayer structures. J Cell Biol. 1993;123:1845–1855. doi: 10.1083/jcb.123.6.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benfenati F, Onofri F, Giovedi S. Protein-protein interactions and protein modules in the control of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:243–257. doi: 10.1098/rstb.1999.0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernstein BW, Bamburg JR. Cycling of actin assembly in synaptosomes and neurotransmitter release. Neuron. 1989;3:257–265. doi: 10.1016/0896-6273(89)90039-1. [DOI] [PubMed] [Google Scholar]
- 10.Brodin L, Low P, Gad H, Gustafsson J, Pieribone VA, Shupliakov O. Sustained neurotransmitter release: new molecular clues. Eur J Neurosci. 1997;9:2503–2511. doi: 10.1111/j.1460-9568.1997.tb01679.x. [DOI] [PubMed] [Google Scholar]
- 11.Ceccaldi PE, Grohovaz F, Benfenati F, Chieregatti E, Greengard P, Valtorta F. Dephosphorylated synapsin I anchors synaptic vesicles to actin cytoskeleton: an analysis by videomicroscopy. J Cell Biol. 1995;128:905–912. doi: 10.1083/jcb.128.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cibelli G, Ghirardi M, Onofri F, Casadio A, Benfenati F, Montarolo PG, Vitiello F. Synapsin-like molecules in Aplysia punctata and Helix pomatia: identification and distribution in the nervous system and during the formation of synaptic contacts in vitro. Eur J Neurosci. 1996;8:2530–2543. doi: 10.1111/j.1460-9568.1996.tb01547.x. [DOI] [PubMed] [Google Scholar]
- 13.De Camilli P, Benfenati F, Valtorta F, Greengard P. The synapsins. Annu Rev Cell Biol. 1990;6:433–460. doi: 10.1146/annurev.cb.06.110190.002245. [DOI] [PubMed] [Google Scholar]
- 14.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 15.Doussau F, Clabecq A, Henry J-P, Darchen F, Poulain B. Calcium-dependent regulation of Rab3 in short-term plasticity. J Neurosci. 1998;18:3147–3157. doi: 10.1523/JNEUROSCI.18-09-03147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doussau F, Gasman S, Humeau Y, Vitiello F, Popoff M, Boquet P, Bader MF, Poulain B. A Rho-related GTPase is involved in Ca2+-dependent neurotransmitter exocytosis. J Biol Chem. 2000;275:7764–7770. doi: 10.1074/jbc.275.11.7764. [DOI] [PubMed] [Google Scholar]
- 17.Elferink LA, Peterson MR, Scheller RH. A role for synaptotagmin (p65) in regulated exocytosis. Cell. 1993;72:153–159. doi: 10.1016/0092-8674(93)90059-y. [DOI] [PubMed] [Google Scholar]
- 18.Esser L, Wang CR, Hosaka M, Smagula CS, Südhof TC, Deisenhofer J. Synapsin I is structurally similar to ATP-utilizing enzymes. EMBO J. 1998;17:977–984. doi: 10.1093/emboj/17.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fiumara F, Ghirardi M, Onofri F, Benfenati F, Montarolo PG (2001) Intracellular injection of synapsin I induces neurotransmitter release in C1 neurons of Helix pomatiacontacting a wrong target. Neuroscience, in press. [DOI] [PubMed]
- 20.Gardner D. Bilateral symmetry and interneuronal organization in the buccal ganglia of Aplysia. Science. 1971;173:550–553. doi: 10.1126/science.173.3996.550. [DOI] [PubMed] [Google Scholar]
- 21.Girault J-A, Gorelik FS, Greengard P. Improving the quality of immunoblots by chromatography of polyclonal antisera on keratin affinity columns. Anal Biochem. 1989;182:193–198. doi: 10.1016/0003-2697(89)90741-0. [DOI] [PubMed] [Google Scholar]
- 22.Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 23.Hilfiker S, Schweizer FE, Kao H-T, Czernik AJ, Greengard P, Augustine GJ. Two sites of action for synapsin domain E in regulating neurotransmitter release. Nat Neurosci. 1998;1:29–35. doi: 10.1038/229. [DOI] [PubMed] [Google Scholar]
- 24.Hirokawa N, Sobue K, Kanda K, Harada A, Yorifuji H. The cytoskeletal architecture of the presynaptic terminal and molecular structure of synapsin I. J Cell Biol. 1989;108:111–126. doi: 10.1083/jcb.108.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosaka M, Südhof TC. Synapsin III, a novel synapsin with an unusual regulation by Ca2+. J Biol Chem. 1998;273:13371–13374. doi: 10.1074/jbc.273.22.13371. [DOI] [PubMed] [Google Scholar]
- 26.Hosaka M, Hammer RE, Südhof TC. A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron. 1999;24:377–387. doi: 10.1016/s0896-6273(00)80851-x. [DOI] [PubMed] [Google Scholar]
- 27.Johannes L, Doussau F, Clabecq A, Henry JP, Darchen F, Poulain B. Evidence for a functional link between Rab3 and the SNARE complex. J Cell Sci. 1996;109:2875–2884. doi: 10.1242/jcs.109.12.2875. [DOI] [PubMed] [Google Scholar]
- 28.Kamiya H, Zucker RS. Residual Ca2+ and short-term synaptic plasticity. Nature. 1994;371:603–606. doi: 10.1038/371603a0. [DOI] [PubMed] [Google Scholar]
- 29.Kao HT, Porton B, Czernik AJ, Feng J, Yiu G, Haring M, Benfenati F, Greengard P. A third member of the synapsin gene family. Proc Natl Acad Sci USA. 1998;95:4667–4672. doi: 10.1073/pnas.95.8.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kao HT, Porton B, Hilfiker S, Stefani G, Pieribone VA, DeSalle R, Greengard P. Molecular evolution of the synapsin gene family. J Exp Zool. 1999;285:360–377. [PubMed] [Google Scholar]
- 31.Klagges BRE, Heimbeck G, Godenschwege TA, Hofbauer A, Pflugfelder GO, Reifegerste R, Reisch D, Schaupp M, Buchner S, Buchner E. Invertebrate synapsins: a single gene codes for several isoforms in Drosophila. J Neurosci. 1996;16:3154–3165. doi: 10.1523/JNEUROSCI.16-10-03154.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klingauf J, Kavalali ET, Tsien RW. Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature. 1998;394:581–585. doi: 10.1038/29079. [DOI] [PubMed] [Google Scholar]
- 33.Kretz R, Shapiro E, Kandel ER. Post-tetanic potentiation at an identified synapse in Aplysia is correlated with a Ca2+-activated K+ current in the presynaptic neuron: evidence for Ca2+ accumulation. Proc Natl Acad Sci USA. 1982;79:5430–5434. doi: 10.1073/pnas.79.17.5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuromi H, Kidokoro Y. Two distinct pools of synaptic vesicles in single presynaptic boutons in a temperature-sensitive Drosophila mutant, shibire. Neuron. 1998;20:917–925. doi: 10.1016/s0896-6273(00)80473-0. [DOI] [PubMed] [Google Scholar]
- 35.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 36.Landis DMD, Hall AK, Weinstein LA, Reese TS. The organization of cytoplasm at the presynaptic active zone of a central nervous system synapse. Neuron. 1988;1:201–209. doi: 10.1016/0896-6273(88)90140-7. [DOI] [PubMed] [Google Scholar]
- 37.Li L, Chin L-S, Shupliakov O, Brodin L, Sihra TS, Hvalby Ø, Jensen V, Zheng D, McNamara JO, Greengard P, Andersen P. Impairment of synaptic vesicle clustering and of synaptic transmission and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci USA. 1995;92:9235–9239. doi: 10.1073/pnas.92.20.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu B, Greengard P, Poo MM. Exogenous synapsin I promotes functional maturation of developing neuromuscular synapses. Neuron. 1992;8:521–529. doi: 10.1016/0896-6273(92)90280-q. [DOI] [PubMed] [Google Scholar]
- 39.Mallart A, Martin AR. An analysis of facilitation of transmitter release at the neuromuscular junction of the frog. J Physiol (Lond) 1967;193:679–697. doi: 10.1113/jphysiol.1967.sp008388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mothet JP, Fossier P, Tauc L, Baux G. Opposite actions of nitric oxide on cholinergic synapses: which pathways? Proc Natl Acad Sci USA. 1996;93:8721–8726. doi: 10.1073/pnas.93.16.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murthy VN, Stevens CF. Reversal of synaptic vesicle docking at central synapses. Nat Neurosci. 1999;2:503–507. doi: 10.1038/9149. [DOI] [PubMed] [Google Scholar]
- 42.Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 43.Nielander HB, Onofri F, Schaeffer E, Menegon A, Fesce R, Valtorta F, Greengard P, Benfenati F. Phosphorylation-dependent effects of synapsin IIa on actin polymerization and network formation. Eur J Neurosci. 1997;9:2712–2722. doi: 10.1111/j.1460-9568.1997.tb01700.x. [DOI] [PubMed] [Google Scholar]
- 44.Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P. Distinct pools of synaptic vesicles in neurotransmitter release. Nature. 1995;375:493–497. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- 45.Poulain B, Rossetto O, Deloye F, Schiavo G, Tauc L, Montecucco C. Antibodies against rat brain vesicle-associated membrane protein (synaptobrevin) prevent inhibition of acetylcholine release by tetanus toxin or botulinum neurotoxin type B. J Neurochem. 1993;61:1175–1178. doi: 10.1111/j.1471-4159.1993.tb03640.x. [DOI] [PubMed] [Google Scholar]
- 46.Robinson LJ, Martin TF. Docking and fusion in neurosecretion. Curr Opin Cell Biol. 1998;10:483–492. doi: 10.1016/s0955-0674(98)80063-x. [DOI] [PubMed] [Google Scholar]
- 47.Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC, Sudhof TC. Short-term synaptic plasticity is altered in mice lacking synapsin I. Cell. 1993;75:661–670. doi: 10.1016/0092-8674(93)90487-b. [DOI] [PubMed] [Google Scholar]
- 48.Rosahl TW, Spillane D, Missler M, Herz J, Seling OK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsin I and II in synaptic vesicles regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- 49.Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 50.Ryan TA, Reuter H, Wendland B, Schweizer E, Tsien RW, Smith SJ. The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- 51.Ryan TA, Li L, Chin L-S, Greengard P, Smith SJ. Synaptic vesicle recycling in synapsin I knock-out mice. J Cell Biol. 1996;134:1219–1227. doi: 10.1083/jcb.134.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature. 1992;359:832–835. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- 53.Schiebler W, Jahn R, Doucet JP, Rothlein J, Greengard P. Characterization of synapsin I binding to small synaptic vesicles. J Biol Chem. 1986;261:8383–8390. [PubMed] [Google Scholar]
- 54.Siow C, Chilcote T, Benfenati F, Greengard P, Thiel G. Synapsin IIa: expression in insect cells, purification and characterization. Biochemistry. 1992;31:4268–4275. doi: 10.1021/bi00132a017. [DOI] [PubMed] [Google Scholar]
- 55.Stevens CF, Tsujimoto T. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc Natl Acad Sci USA. 1995;92:846–849. doi: 10.1073/pnas.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Südhof TC, Czernik AJ, Kao HT, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS, DeCamilli P. Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science. 1989;245:1474–1480. doi: 10.1126/science.2506642. [DOI] [PubMed] [Google Scholar]
- 57.Swandulla D, Hans M, Zipser K, Augustine G. Role of residual calcium in synaptic depression and posttetanic potentiation: fast and slow calcium signaling in nerve terminals. Neuron. 1991;7:915–926. doi: 10.1016/0896-6273(91)90337-y. [DOI] [PubMed] [Google Scholar]
- 58.Takei Y, Harada A, Takeda S, Kobayashi K, Terada S, Noda T, Takahashi T, Hirokawa N. Synapsin I deficiency results in structural changes in the presynaptic terminals in the murine nervous system. J Cell Biol. 1995;131:1789–1800. doi: 10.1083/jcb.131.6.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taubenblatt P, Dedieu JC, Gulik-Krzywicki T, Morel N. VAMP (synaptobrevin) is present in the plasma membrane of nerve terminals. J Cell Sci. 1999;112:3559–3567. doi: 10.1242/jcs.112.20.3559. [DOI] [PubMed] [Google Scholar]
- 60.Terada S, Tsujimoto T, Takei Y, Takahashi T, Hirokawa N. Impairment of inhibitory synaptic transmission in mice lacking synapsin I. J Cell Biol. 1999;145:1039–1048. doi: 10.1083/jcb.145.5.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Torri Tarelli F, Bossi M, Fesce R, Greengard P, Valtorta F. Synapsin I partially dissociates from synaptic vesicles during exocytosis induced by electrical stimulation. Neuron. 1992;9:1143–1153. doi: 10.1016/0896-6273(92)90072-l. [DOI] [PubMed] [Google Scholar]
- 62.Towbin L, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedures and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Valtorta F, Iezzi N, Benfenati F, Lu B, Poo MM, Greengard P. Accelerated structural maturation induced by synapsin I at developing neuromuscular synapses of Xenopus laevis. Eur J Neurosci. 1995;7:261–270. doi: 10.1111/j.1460-9568.1995.tb01062.x. [DOI] [PubMed] [Google Scholar]
- 64.Walch-Solimena C, Blasi J, Edelmann L, Chapman ER, von Mollard GF, Jahn R. The t-SNAREs syntaxin 1 and SNAP-25 are present on organelles that participate in synaptic vesicle recycling. J Cell Biol. 1995;128:637–645. doi: 10.1083/jcb.128.4.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zucker RS. Exocytosis: a molecular and physiological perspective. Neuron. 1996;17:1049–1055. doi: 10.1016/s0896-6273(00)80238-x. [DOI] [PubMed] [Google Scholar]
- 66.Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol. 1999;9:305–313. doi: 10.1016/s0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]