

Abstract
Oxidative stress is a key feature in the Alzheimer's disease (AD) brain and manifests as lipid peroxidation (LPO). Isoprostanes (iPs) are specific and sensitive markers of in vivo LPO. To determine whether amyloid β (Aβ) deposition in vivois associated with increased LPO, we examined iP levels in a transgenic mouse model (Tg2576) of AD amyloidosis. Urine, plasma, and brain tissues were collected from Tg2576 and littermate wild-type (WT) animals at different time points starting at 4 months of age and continuing until 18 months of age. Levels of urinary 8,12-iso-iPF2α-VI were higher in Tg2576 than in WT animals as early as 8 months of age and remained this high for the rest of the study. A similar pattern was observed for plasma levels of 8,12-iso-iPF2α-VI. Homogenates from the cerebral cortex and hippocampus of Tg2576 mice had higher levels of 8,12-iso-iPF2α-VI than those from WT mice starting at 8 months of age. In contrast, a surge of Aβ 1–40 and 1–42 levels as well as Aβ deposits in Tg2576 mouse brains occurred later, at 12 months of age. A direct correlation was observed between brain 8,12-iso-iPF2α-VI and Aβ 1–40 and 1–42. Because LPO precedes amyloid plaque formation in Tg2576 mice, this suggests that brain oxidative damage contributes to AD pathogenesis before Aβ accumulation in the AD brain.
Keywords: Alzheimer's disease, Tg2576 transgenic animal model, lipid peroxidation, isoprostanes, amyloid β protein, plasma, urine
Increasing evidence suggests that oxidative stress plays an important role in the pathogenesis of Alzheimer's disease (AD) (Markesbery and Carney, 1999; Praticòand Delanty, 2000). The CNS is particularly vulnerable to oxidative damage because it has a high energy requirement, a high oxygen consumption rate, and a relative deficit of antioxidant defense systems compared with other organs (Floyd, 1999). Oxidative damage to the CNS predominantly manifests as lipid peroxidation (LPO) because of the high content of polyunsaturated fatty acids that are particularly susceptible to oxidation. Isoprostanes (iPs) are specific and sensitive markers of in vivo LPO (Praticò, 1999). We have shown recently that levels of a major iP, 8,12-iso-iPF2α-VI, are increased not only in specific AD brain regions (Praticò et al., 1998) but also in the urine, plasma, and CSF of patients with a clinical diagnosis of AD (Praticò et al., 2000). The specific increase of 8,12-iso-iPF2α-VI in urine and plasma raised the possibility that it may be a useful peripheral biomarker for progression of AD. However, the mechanisms underlying the specific increase in 8,12-iso-iPF2α-VI in AD brain and bodily fluids are unknown, and it is unclear whether oxidative damage is a cause or a consequence of amyloid (A)β peptide (Aβ) accumulation or whether they are two independent processes.
To address this, we examined iP levels in a transgenic (Tg) mouse model (Tg2576 mice) of AD amyloidosis, which develops characteristic AD-like Aβ brain deposits because of overexpression of a human amyloid precursor protein (APP) transgene with a double mutation found in a Swedish (APP swe) family with early onset AD (Hsiao et al., 1996). Urine, plasma, and brain tissues were collected from these animals and from wild-type (WT) littermate controls at different time points, starting at 4 months of age and continuing until 18 months of age. Biochemical analyses revealed that urine, plasma, and CNS levels of 8,12-iso-iPF2α-VI are increased as early as 8 months of age in Tg2576 mice compared with WT mice; this preceded the onset of Aβ deposition and a surge in CNS levels (Aβ 1–40 and 1–42 levels) in the Tg2576 mice. These results suggest that LPO may play an earlier role than previously anticipated in the pathogenesis of AD.
MATERIALS AND METHODS
Animals. The genotype and phenotypic features of the heterozygote APP swe (K670N, M671L) transgenic mice (Tg2576) and WT littermates studied here have been described previously (Hsiao et al., 1995, 1996). A total of 20 Tg2576 and 20 WT animals were studied. Mice were weaned at 4 weeks of age and kept on a chow diet; males were always separated from females for the entire study. Starting at 4 months of age, a 24 hr urine collection was performed every month in metabolic cages (Nalgene, Rochester, NY), and blood was collected retro-orbitally. Brain tissue was obtained from cohorts of these animals at 4, 8, 12, and 15 months of age (three Tg mice and three WT mice for each time point). Finally, brain collection for similar studies was performed at 18 months of age (five Tg mice and five WT mice).
Isoprostane analysis. Urine was collected in plastic tubes containing 0.01% of the antioxidant butylated hydroxytoluene (BHT). Samples were spiked with a fixed amount of internal standard (d4-8,12-iso-iPF2α-VI) extracted on a C18 cartridge column. The eluate was purified by thin-layer chromatography and finally assayed by negative ion chemical ionization gas chromatography–mass spectrometry as described previously (Praticò et al., 1998, 1999, 2000). A urine aliquot (0.1 ml) was used for measurement of creatinine levels by a commercially available, standardized, automated colorimetric assay (Sigma, St. Louis, MO). Urine levels were expressed as nanograms per milligram of creatinine. Blood was anticoagulated with trisodium citrate (3.8%) and centrifuged at 3000 rpm for 15 min at 4°C to obtain plasma. Plasma was spiked and treated as described above for urine. Levels were expressed as picograms per milliliter. All of the assays were performed without knowledge of the genotype or age.
Tissue preparation. Animals were anesthetized and killed according to the recommendation of the Panel on Euthanasia of the American Veterinary Medical Association. They were perfused intracardially for 30 min with ice-cold 0.9% PBS containing 2 mm/l EDTA and 20 mm/l BHT, pH 7.4. Brains were removed, and one hemisphere was fixed by immersion in 4% paraformaldehyde in 0.1 m PBS, pH 7.4, at 4°C overnight, blocked in the coronal plane, and embedded in paraffin as described previously for immunohistochemistry (Murai et al., 1998;Nakagawa et al., 2000). The other hemisphere was gently rinsed in cold 0.9% PBS and then immediately dissected in three anatomical regions (cerebral cortex, cerebellum, and hippocampus) for isoprostane and Aβ measurements.
For isoprostane analysis, tissue was homogenized, and total lipids were extracted using Folch solution (chloroform/methanol 2:1 vol) (Praticò et al., 1999). Next, base hydrolysis was performed using 15% KOH at 45°C for 1 hr, and the total 8,12-iso-iPF2α-VI levels were measured as described above. Brain tissues were always analyzed in a coded manner.
Aβ sandwich ELISA. Brain tissues were homogenized in 1 ml of 70% formic acid and centrifuged at 100,000 × g for 1 hr. The supernatant was recovered and neutralized by a 20-fold dilution in 1 m Tris base buffer. Samples were mixed with buffer EC [0.02 m sodium phosphate, 0.2 mm EDTA, 0.4m NaCl, 0.2% BSA, 0.05% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 0.4% Block-ace (Dainippon Suita, Osaka, Japan), and 0.05% sodium azide, pH 7.0] and analyzed directly using the Ban50–BA27 (for Aβ 1–40) or Ban50–BC-05 (for Aβ 1–42/43) sandwich ELISA systems as described previously (Gravina et al., 1995). The values were calculated by comparison with a standard curve of synthetic Aβ 1–40 and Aβ 1–42 (Bachem, King of Prussia, PA) as described previously (Turner et al., 1996). Absorbance values were calculated for dilution and initial weight. Results were expressed as picomoles per gram of tissue. Analyses were always performed in duplicate and in a coded manner.
Immunohistochemistry. Serial 6-μm-thick paraffin sections were cut throughout each brain and mounted on 3-aminopropyltriethoxysilane-coated slides. Sections were deparaffinized, hydrated, rinsed with PBS, and pretreated with formic acid (88%) for 10 min for antigen retrieval; with 3% H2O2 in methanol for 30 min to eliminate endogenous peroxidase activity in the tissue; and with the blocking solution (5% normal horse serum in Tris buffer, pH 7.6). Subsequently, sections were incubated with a biotinylated antibody against Aβ (4G8) (1:10,000 dilution) at 4°C overnight (Nakagawa et al., 2000). Sections were then incubated with secondary antibody for 1 hr (dilution 1:1000) and reacted with horseradish peroxidase–avidin–biotin complex (Vector Laboratories, Burlingame, CA); immunocomplexes were visualized by using 3,3′-diaminobenzidine as the chromogen. Finally, the sections were dehydrated with ethanol, cleared with xylene, and coverslipped with Cytoseal. As a control, sections from the same group of animals were treated in the same manner, except for the primary antibody. Analyses were always performed in a coded manner.
Statistical analysis. Data for 8,12-iso-iPF2α-VI and Aβ 1–40 and Aβ 1–42 were expressed as the mean ± SEM. Isoprostane and Aβ levels were assessed by ANOVA and subsequently by a Student's unpaired two-tailed t test. Significance was set at p < 0.05. Correlations between parameters were tested by linear regression analysis.
RESULTS
Starting at 4 months of age, 24 hr urine collections were performed monthly until the mice were 18 months old. Urinary 8,12-iso-iPF2α-VI levels in Tg2576 mice were not different from the levels observed in WT mice until 6 months of age (Fig. 1); thereafter, these levels increased in Tg mice compared with WT mice, and this increase became statistically significant (p < 0.01) by 8 months (Fig. 1). Urinary 8,12-iso-iPF2α-VI levels continued to increase, reaching a plateau by ∼12 months of age (Fig. 1). A similar pattern was observed for plasma 8,12-iso-iPF2α-VI levels in Tg mice, with significantly higher levels at 8 months that reached a maximum by 9–10 months of age compared with WT mice (Fig.2). Indeed, there were no significant changes in urine or plasma 8,12-iso-iPF2α-VI levels with advancing age in the WT mice. Because previous studies documented the onset of Aβ deposition in Tg2576 mice at ∼10–12 months (Hsiao et al., 1995, 1996), these data and ours suggest that elevated 8,12-iso-iPF2α-VI levels beginning at ∼7–8 months of age precede Aβ deposition in Tg2576 mice. In addition, the direct correlation between urine and plasma 8,12-iso-iPF2α-VI levels (r2 = 0.81; p< 0.01) suggests a common mechanism for LPO in Tg2576 mice.
Fig. 1.

Increased urinary levels of 8,12-iso-iPF2α-VI in Tg2576 mice. Urinary levels of 8,12-iso-iPF2α-VI in Tg2576 mice (closed circles) and wild-type littermates (closed squares) from 4 until 18 months of age are shown (*p < 0.01; **p < 0.001).
Fig. 2.

Increased plasma levels of 8,12-iso-iPF2α-VI in Tg2576 mice. Plasma levels of 8,12-iso-iPF2α-VI in Tg2576 mice (closed circles) and wild-type littermates (closed squares) from 4 until 18 months of age are shown (*p < 0.01; **p < 0.001).
To determine whether urine and plasma reflect brain iP levels, Tg and WT mice at 4, 8, 12, and 15 months of age (n = 3 Tg and 3 WT mice for each time point) as well as at 18 months of age (n = 5 Tg and 5 WT mice) were killed, and the cerebral cortex, hippocampus, and cerebellum were immediately dissected and homogenized to measure total 8,12-iso-iPF2α-VI levels. No difference was observed between Tg and WT animals at 4 months of age in all regions examined, but by 8 months of age, the cerebral cortex of Tg mice had significantly higher 8,12-iso-iPF2α-VI levels than that of WT mice, and this difference became increasingly significant at ≥12 months of age (Fig. 3A). These values also directly correlated with plasma and urinary levels of 8,12-iso-iPF2α-VI in the Tg mice (r2 = 0.70, p< 0.01; r2 = 0.68,p < 0.001, respectively). A similar pattern was observed for the hippocampus (Fig. 3B), but cortical 8,12-iso-iPF2α-VI levels were higher than those in the hippocampus (Fig. 3, compare A withB) of the Tg mice, whereas 8,12-iso-iPF2α-VI levels in the cerebellum of Tg mice did not differ from those in WT mice (Fig.3C).
Fig. 3.

Tg2567 mice have elevated 8,12-iso-iPF2α-VI levels in brain homogenates. Total brain cortex (A), hippocampus (B), and cerebellum (C) levels of 8,12-iso-iPF2α-VI in Tg2576 mice (filled bars) and wild-type littermates (open bars) at different ages (*p < 0.01; **p < 0.003).
To compare these data with brain levels of Aβ 1–40 and 1–42, we measured both species of Aβ using a sensitive sandwich ELISA at the same time points and in the same brain regions as for 8,12-iso-iPF2α-VI. Although WT mice showed undetectable (at 4, 8, and 12 months) or negligible (at 15 and 18 months) levels of Aβ 1–40 and 1–42 (Table1), the Tg2576 mice showed low cortical levels of both peptides at 4 and 8 months but dramatically increased cortical levels of Aβ 1–40 and 1–42 by 12 months of age, which continued to increase at 15 and 18 months of age (Table 1). A similar pattern was observed for the hippocampus, although lower levels of Aβ 1–40 and 1–42 were detected in the hippocampus compared with the cortex, and the cerebellum showed no significant increase in Aβ 1–40 and 1–42 with aging in these Tg mice (Table 1). Interestingly, increased levels of 8,12-iso-iPF2α-VI were found only in Tg mouse brain regions with high levels of Aβ 1–40 and 1–42 (i.e., the cortex and hippocampus, but not the cerebellum), whereas cortical levels of Aβ 1–40 and 1–42 were highly correlated with cortical levels of 8,12-iso-iPF2α-VI (r2 = 0.77, p < 0.001; r2= 0.64, p < 0.001, respectively) in the Tg mice (Fig.4A,B).
Table 1.
Aβ 1–40 and Aβ 1–42 levels in the cerebral cortex, hippocampus, and cerebellum of Tg and WT littermates at different ages
| Age (months) | Cortex | Hippocampus | Cerebellum | ||||
|---|---|---|---|---|---|---|---|
| Aβ 1–40 (pmol/g) | Aβ 1–42 (pmol/g) | Aβ 1–40 (pmol/g) | Aβ 1–42 (pmol/g) | Aβ 1–40 (pmol/g) | Aβ 1–42 (pmol/g) | ||
| Tg | 4 | 23 ± 17 | 42 ± 16 | 173 ± 23 | 179 ± 18 | 133 ± 22 | 156 ± 24 |
| 8 | 39 ± 25 | 52 ± 25 | 179 ± 23 | 221 ± 36 | 159 ± 24 | 148 ± 26 | |
| 12 | 5041 ± 688 | 2353 ± 706 | 1688 ± 89 | 1221 ± 53 | 520 ± 205 | 622 ± 185 | |
| 15 | 7840 ± 1413 | 3910 ± 706 | 4365 ± 580 | 2024 ± 497 | 628 ± 307 | 700 ± 299 | |
| 18 | 19,181 ± 2437 | 5646 ± 718 | 13,604 ± 1317 | 4784 ± 263 | 1030 ± 180 | 1034 ± 161 | |
| WT | 4 | <5 | <5 | <5 | <5 | <5 | <5 |
| 8 | <5 | <5 | <5 | <5 | <5 | <5 | |
| 12 | <5 | <5 | <5 | <5 | <5 | <5 | |
| 15 | 11 ± 3 | 9 ± 3 | 11.5 ± 3 | 12 ± 4 | 10 ± 3 | 9 ± 2 | |
| 18 | 11 ± 3 | 9.6 ± 3 | 9 ± 2 | 9 ± 2 | 11 ± 2 | 10 ± 3 | |
Results are expressed as mean ± SEM. Cortex, Cerebral cortex.
Fig. 4.
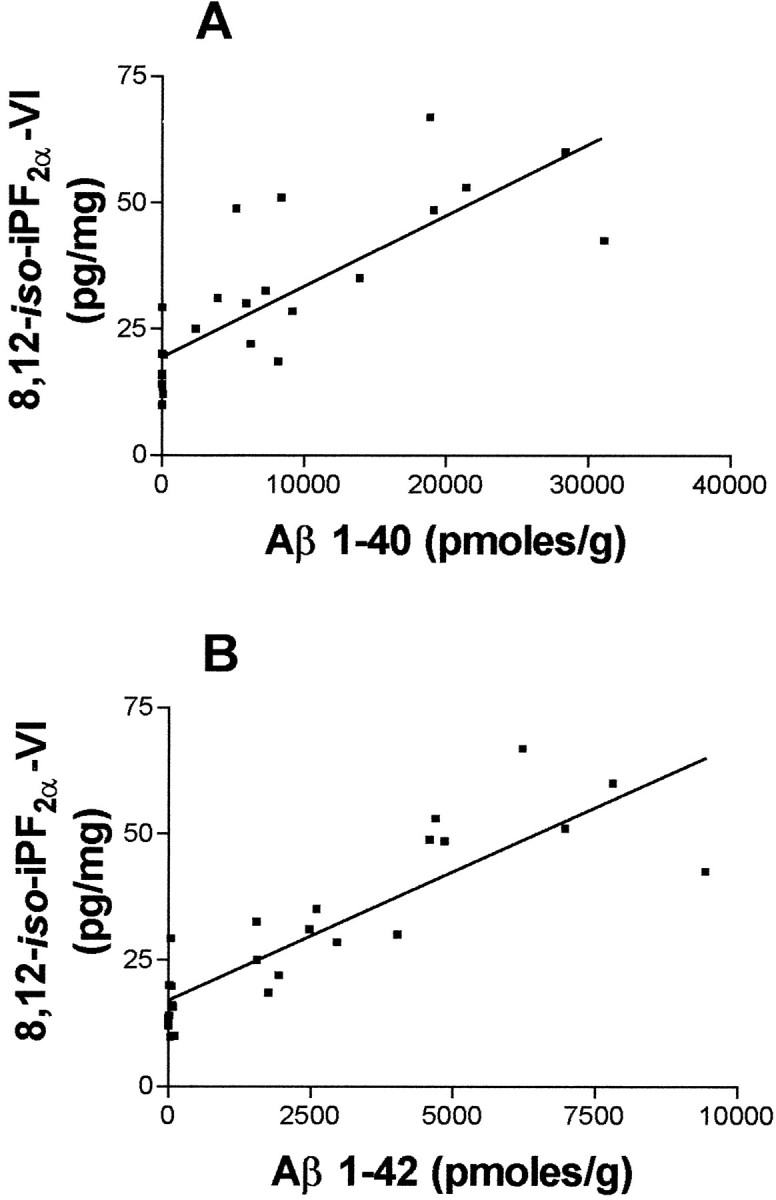
Cortex Aβ 1–40 and 1–42 highly correlate with cortex 8,12-iso-iPF2α-VI levels in Tg2576 mice. A correlation between the total cerebral cortex levels of 8,12-iso-iPF2α-VI and Aβ 1–40 (A) and Aβ 1–42 (B) in Tg2576 mice is apparent.
Because a surge in brain Aβ 1–40 and 1–42 levels correlates with onset of Aβ deposition (Hsiao et al., 1995, 1996), we monitored the brains of the Tg mice for Aβ plaques using immunohistochemistry, which revealed no Aβ deposits at 8 months of age (Fig.5A) but scattered Aβ deposits in the cerebral cortex and the hippocampus at 12 months of age (Fig. 5B) and more abundant Aβ deposits throughout the neocortex and hippocampus by 18 months of age (Fig. 5C). No detectable immunoreactive Aβ deposits were observed in any WT brains (data not shown).
Fig. 5.

Tg2576 mice have an age-dependent increase in Aβ immunostaining. Immunohistochemical staining of a Tg2576 mouse brain at 8, 12, and 18 months of age is shown; the 4G8 antibody was used for staining. Immunostaining was conducted on the same Tg2576 mice as for brain Aβ 1–40, Aβ 1–42, and 8,12-iso-iPF2α-VI measurements. All of the panels are at the same magnification. Scale bar, 0.5 mm.
DISCUSSION
Our study provides compelling evidence that Tg2576, a mouse model of AD amyloidosis, shows evidence of brain oxidative damage reflected by an age-dependent brain and a systemic increase in LPO compared with WT littermates. Significantly, we also observed that increased LPO, as indicated by elevated levels of 8,12-iso-iPF2α-VI, preceded onset of significant Aβ deposition in the Tg2576 mice. Thus, these data provide important and novel insights into the pathogenesis of AD.
First, our observation that the Tg2576 mice showed an age-dependent increase in LPO that correlated with subsequent elevations in brain Aβ levels suggests an early mechanistic role for brain oxidative stress in AD pathogenesis. Although oxidative stress has been primarily implicated in mechanisms of AD brain degeneration (Markesbery and Carney, 1999; Praticò and Delanty, 2000), it has been difficult to fully evaluate the contribution of oxidative processes to the neuropathology of AD for several reasons. For example, only the recent development of Tg mouse models of AD amyloidosis has enabled testing of the oxidative stress and Aβ hypotheses of AD pathogenesis (Guenette and Tanzi, 1999; Janus et al., 2000; Takeuchi et al., 2000). The Tg2576 mice are well characterized models of AD amyloidosis (Hsiao et al., 1996) because these mice develop extensive extracellular Aβ deposits and some associated AD-like neuropathology (Irizarry et al., 1997). Indeed, previous immunohistochemical studies of brains from 20-month-old Tg2576 mice demonstrated evidence of oxidative stress (Pappolla et al., 1998; Smith et al., 1998). However, it is difficult to determine from these studies of older Tg mice whether oxidant stress is important in the early evolution of AD amyloidosis, or a consequence thereof. Thus, our study is novel and highly significant because it provides the first quantitative analysis of oxidant stress and LPO in an animal model of AD amyloidosis from onset to terminal stages of this neuropathology. In addition, assessment of LPO in vivo has been hampered by the use of assays with unsatisfactory specificity and/or sensitivity (Gutteridge and Halliwell, 1990). iPs, a recently described new family of lipids, are chemically stable isomers of prostaglandins formed by a free radical peroxidation of polyunsaturated fatty acids and are sensitive and specific markers of LPO in vivo (Praticò, 1999). Thus, using this highly sensitive marker enabled us to demonstrate increased biosynthesis of 8,12-iso-iPF2α-VI in the urine, plasma, and brain of Tg2576 mice as a function of advancing age.
Second, although signatures of oxidant damage have been detected in AD brains, it remained unclear whether they reflected causes or consequences of AD neuropathology. However, the early and continued increase of cortical and hippocampal 8,12-iso-iPF2α-VI levels in the Tg2576 mice, and their correlation with subsequent elevations of Aβ and amyloid plaque formation in the same brain regions, would support the hypothesis that LPO is not a consequence of AD amyloidosis. However, our observation that there is increased production of 8,12-iso-iPF2α-VI in the absence of significant Aβ deposition in Tg2576 mice does not completely eliminate the possibility that immunohistochemically undetectable microaggregates of extracellular or intracytoplasmic Aβ could initiate LPO in these mice. Indeed, the recent demonstration of rare Aβ plaques as well as scant amounts of formic acid-extractable Aβ in 8-month-old Tg2576 mouse brains prompts cautious interpretation of our data with respect to whether LPO precedes or is linked to the initial stages of AD-like amyloidosis in Tg2576 mice (Kawarabayashi et al., 2001). For example, the shift in Aβ from SDS to formic acid-soluble pools may reflect early evidence of Aβ fibrillation, including β-pleated sheet formation (Kawarabayashi et al., 2001), and it is possible that these subtle changes augment LPO as reflected in the urine and plasma of the Tg2576 mice.
Third, the results presented here support emerging data from studies of AD patients that indicate that increased levels of 8,12-iso-iPF2α-VI in blood or urine appear to herald the onset of AD (Praticò et al., 2000) and that oxidative damage is integral to mechanisms of AD progression (Yan et al., 1995; Zhang et al., 1997; Iadecola et al., 1999; Markesbery and Carney, 1999; Praticò and Delanty, 2000). Indeed, our ability to demonstrate a significant increase in LPO, as indicated by a rise in 8,12-iso-iPF2α-VI levels without similar increases Aβ 1–40 and 1–42 as well as without appreciable Aβ deposition in 7- to 8-month-old Tg2576 mice, suggests that monitoring this peripheral biomarker may be useful for confirming the onset and following the progression of AD. Indeed, before the recent demonstration of elevated 8,12-iso-iPF2α-VI levels in the urine and plasma of AD patients, there were no convenient peripheral biomarkers for AD (Praticò et al., 2000). Thus, the early detection of this iP in the Tg2576 mice provides additional evidence for its utility as an AD biomarker and suggests that 8,12-iso-iPF2α-VI levels may be informative for assessing the response of AD patients to novel therapies for this disorder. Nonetheless, additional studies are needed to clarify the utility of measuring iPs for early diagnosis for AD, as well as to assess the potential of antioxidant therapy to treat living AD patients.
Footnotes
This work was supported in part by grants from the American Heart Association, the National Institutes of Health (AG11542), and the Oxford Foundation. V.M.-Y.L. is the John M. Ware Professor of Alzheimer's disease. We thank Dr. Karen Hsiao for the Tg2576 mice.
Correspondence should be addressed to Domenico Praticò, Center for Experimental Therapeutics, University of Pennsylvania, BRB II/III, Room 812, 421 Curie Boulevard, Philadelphia, PA 19104. E-mail:domenico@spirit.gcrc.upenn.edu.
REFERENCES
- 1.Floyd RA. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc Soc Exp Biol Med. 1999;222:236–245. doi: 10.1046/j.1525-1373.1999.d01-140.x. [DOI] [PubMed] [Google Scholar]
- 2.Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Jr, Younkin LH, Suzuki N, Younkin SG. Amyloid β protein (Aβ) in Alzheimer's disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ40 or Aβ42(43). J Biol Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 3.Guenette SY, Tanzi RE. Progress toward valid transgenic mouse model for Alzheimer's disease. Neurobiol Aging. 1999;20:201–211. doi: 10.1016/s0197-4580(99)00042-1. [DOI] [PubMed] [Google Scholar]
- 4.Gutteridge JMC, Halliwell B. The measurement and mechanism of lipid peroxidation in biological systems. Trends Biochem Sci. 1990;15:129–135. doi: 10.1016/0968-0004(90)90206-q. [DOI] [PubMed] [Google Scholar]
- 5.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficit, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 6.Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, Iadecola C, Clark HB, Carlson G. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor protein. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- 7.Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao K, Carlson GA. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- 8.Irizarry MC, McNamara M, Fedorchack K, Hsiao K, Hyman BT. APPsw transgenic mice develop age-related Aβ deposits and neuropil abnormalities, but not neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Janus C, Chishti MA, Westaway D. Transgenic mouse models of Alzheimer's disease. Biochim Biophys Acta. 2000;1502:63–75. doi: 10.1016/s0925-4439(00)00033-8. [DOI] [PubMed] [Google Scholar]
- 10.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Hsiao-Ashe K, Younkin S. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Markesbery WR, Carney JM. Oxidative alterations in Alzheimer's disease. Brain Pathol. 1999;9:133–146. doi: 10.1111/j.1750-3639.1999.tb00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murai H, Pierce JES, Raghupathi R, Smith DH, Saatman KE, Trojanowski JQ, Lee VM-Y, Loring JF, Eckman C, Younkin S, McIntosh TK. Two-fold over expression of human β-amyloid precursor proteins in transgenic mice does not affect the neuromotor, cognitive, or neurodegenerative sequelae following experimental brain injury. J Comp Neurol. 1998;392:428–438. doi: 10.1002/(sici)1096-9861(19980323)392:4<428::aid-cne2>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 13.Nakagawa Y, Reed L, Nakamura M, McIntosh T, Smith DH, Saatman KE, Raghupathi R, Clemens J, Saido TC, Lee VM-Y, Trojanowski JQ. Brain trauma in aged transgenic mice induces regression of established Aβ deposits. Exp Neurol. 2000;163:244–252. doi: 10.1006/exnr.2000.7375. [DOI] [PubMed] [Google Scholar]
- 14.Pappolla MA, Chyan Y-J, Omar RA, Hsiao K, Perry G, Smith MA, Bozner P. Evidence of oxidative stress and in vivo neurotoxicity of β-amyloid in a transgenic mouse model of Alzheimer's disease. Am J Pathol. 1998;152:871–877. [PMC free article] [PubMed] [Google Scholar]
- 15.Praticò D. F2-isoprostanes: sensitive and specific non-invasive indices of lipid peroxidation in vivo. Atherosclerosis. 1999;147:1–10. doi: 10.1016/s0021-9150(99)00257-9. [DOI] [PubMed] [Google Scholar]
- 16.Praticò D, Delanty N. Oxidative injury in diseases of the central nervous system: focus on Alzheimer's disease. Am J Med. 2000;109:577–585. doi: 10.1016/s0002-9343(00)00547-7. [DOI] [PubMed] [Google Scholar]
- 17.Praticò D, Lee VM-Y, Trojanowski JQ, Rokach J, FitzGerald GA. Increased F2-isoprostanes in Alzheimer's disease: evidence of enhanced lipid peroxidation in vivo. FASEB J. 1998;12:1777–1783. doi: 10.1096/fasebj.12.15.1777. [DOI] [PubMed] [Google Scholar]
- 18.Praticò D, Rokach J, Tangirala RK. Brains of aged apolipoprotein E-deficient mice have increased levels of F2-isoprostanes, in vivo markers of lipid peroxidation. J Neurochem. 1999;73:736–741. doi: 10.1046/j.1471-4159.1999.0730736.x. [DOI] [PubMed] [Google Scholar]
- 19.Praticò D, Clark CM, Lee VM-Y, Trojanowski JQ, Rokach J, FitzGerald GA. Increased 8,12-iso-iPF2α-VI in Alzheimer's disease: correlation of a non-invasive index of lipid peroxidation with disease severity. Ann Neurol. 2000;48:809–812. [PubMed] [Google Scholar]
- 20.Smith MA, Hirai K, Hsiao K, Pappolla MA, Harris PLR, Siedlak SL, Tabaton M, Perry G. Amyloid β deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem. 1998;70:2212–2215. doi: 10.1046/j.1471-4159.1998.70052212.x. [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M, Mann DMA, Hyman BT, Iwatsubo T. Age-related amyloid β deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid β precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol. 2000;157:331–339. doi: 10.1016/s0002-9440(10)64544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turner SR, Suzuki N, Chyung ASC, Younkin SG, Lee VM-Y. Amyloids β40 and β42 are generated intracellularly in cultured human neurons and their secretion increases with maturation. J Biol Chem. 1996;271:8966–8970. doi: 10.1074/jbc.271.15.8966. [DOI] [PubMed] [Google Scholar]
- 23.Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P, Zweier JL, Stern D. Non-enzymatically glycated τ in Alzheimer's disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid β-peptide. Nat Med. 1995;1:693–699. doi: 10.1038/nm0795-693. [DOI] [PubMed] [Google Scholar]
- 24.Zhang L, Zhao B, Yew DT, Kusiak JW, Roth GS. Processing of Alzheimer's amyloid precursor protein during H2O2-induced apoptosis in human neuronal cells. Biochem Biophys Res Commun. 1997;235:845–848. doi: 10.1006/bbrc.1997.6698. [DOI] [PubMed] [Google Scholar]