

Abstract
Brain-derived neurotrophic factor (BDNF) is synthesized by small neuron cell bodies in the dorsal root ganglia (DRG) and is anterogradely transported to primary afferent terminals in the dorsal horn where it is involved in the modulation of painful stimuli. Here we show that BDNF is released in the rat isolated dorsal horn after chemical stimulation by capsaicin or electrical stimulation of dorsal roots. Capsaicin superfusion (1–100 μm) induced a dose-dependent release of BDNF, measured using ELISA. The highest dose of capsaicin also induced a depletion of BDNF protein in the dorsal horn. BDNF release was also seen after electrical stimulation of the dorsal roots at C-fiber strength. This release was encoded by specific patterns of afferent fiber stimulation. Neither continuous low-frequency (480 pulses, 1 Hz) nor tetanic high-frequency (300 pulses in 3 trains, 100 Hz) stimulation evoked release of BDNF, although substance P (SP) release was observed under both of these conditions. However, BDNF was released after short bursts of high-frequency stimulation (300 pulses in 75 trains, 100 Hz) along with SP and glutamate. The NMDA antagonist d-AP-5 inhibited electrically evoked BDNF release. BDNF release was also measured after systemic or intrathecal NGF treatment. This upregulated BDNF content in the DRG and increased the capsaicin-evoked release of BDNF. Similarly, the amount of BDNF released by burst stimulation was increased after NGF treatment. This activity-dependent release continued to be encoded solely by this stimulation pattern. These experiments demonstrate that BDNF release in the dorsal horn is encoded by specific patterns of afferent fiber stimulation and is mediated by NMDA receptor activation.
Keywords: neurotrophin release, sensory neurons, capsaicin, burst stimulation, NMDA receptor, nociception
There is now good evidence that neuronally derived brain-derived neurotrophic factor (BDNF) contributes to the process of hippocampal synaptic plasticity via its activity-dependent release (Canossa et al., 1997; Schuman, 1999). It has also been proposed recently that this neurotrophin might regulate synaptic efficacy in the dorsal horn of the spinal cord (Snider and McMahon, 1998; Woolf and Salter, 2000). BDNF is synthesized by a subpopulation of unmyelinated primary afferents (primarily nociceptors) that contain substance P (SP) and glutamate (De Biasi and Rustioni, 1988; Apfel et al., 1996; Zhou and Rush, 1996; Conner et al., 1997;Michael et al., 1997). These neurons use glutamate (stored in clear vesicles) as a fast neurotransmitter, but some of them also release SP from dense-core vesicles when they fire repetitively (Duggan et al., 1995; Marvizón et al., 1997). Like SP, BDNF is packaged in large dense-core vesicles in dorsal root ganglion (DRG) cells, and it is anterogradely transported to axon terminals in the dorsal horn (Zhou and Rush, 1996; Conner et al., 1997; Michael et al., 1997). BDNF is found in laminas I and II, the known termination area for SP-containing C fibers that express receptors for capsaicin (VR1) and nerve growth factor (NGF) (TrkA) (Averill et al., 1995; Tominaga et al., 1998). Small fibers are thought to be the major source for BDNF in the superficial laminas of the dorsal horn because after axotomy both small DRG cells and axon terminals in the superficial laminas downregulate BDNF expression (Cho et al., 1998; Michael et al., 1999;Zhou et al., 1999). However, BDNF immunoreactive cells are found in the ventral spinal cord white matter (Dreyfus et al., 1999), and descending noradrenergic fiber terminals might contribute to BDNF spinal cord pools as reported for some brain areas (Fawcett et al., 1998). The receptor for BDNF, TrkB, is found throughout the dorsal horn with particularly high density in the superficial laminas (Zhou et al., 1993) where it is associated with dorsal horn cells but not with afferent terminals (Bradbury et al., 1998). BDNF present in C-fiber terminals may exert positive modulation of spinal glutamatergic NMDA-mediated nociceptive signaling, sharing this role with SP (Kerr et al., 1999; Woolf and Salter, 2000). Differently, in the brainstem, BDNF can negatively modulate glutamatergic AMPA-mediated currents in second-order sensory neurons (Balkowiec et al., 2000). Because BDNF is released with activity and in a frequency-dependent manner in cranial ganglion cells in culture (Balkowiec and Katz, 2000), this neurotrophin may play a role in the modulation of visceral sensory information in the brainstem.
Until now, signaling through the TrkB receptor in the dorsal horn was accepted as evidence of BDNF release. We have used an in vitro preparation consisting of dorsal horn slices with attached dorsal roots (Malcangio and Bowery, 1993; Malcangio et al., 1997) to make direct measurement of BDNF release evoked by electrical stimulation of the roots, as well as chemical stimulation. Concomitant release of glutamate and SP has also been evaluated.
MATERIALS AND METHODS
NGF administration. Adult male Wistar rats (250 gm body weight) were used and killed by decapitation. All procedures were in accordance with United Kingdom Home Office regulations. Human recombinant NGF (Genentech, South San Francisco, CA) was administered to adult rats either systemically (1 mg/kg, s.c., three times per week for 1–2 weeks) (Malcangio et al., 1997) or intrathecally (Michael et al., 1997) for 14 d (12 μg/d) or 10 d (24 μg/d). Control animals received vehicle (saline containing 0.1% rat serum albumin; Sigma, Poole, UK).
Release of BDNF, SP, and glutamate from dorsal horn slices.Horizontal dorsal horn slices (400 μm thick) with dorsal roots attached were obtained from the lumbar spinal cord of adult rats as described previously (Malcangio and Bowery, 1993; Malcangio et al., 1997). Only one slice was obtained from each rat, mounted in the central compartment of a three-compartment chamber, and continuously superfused (1 ml/min) with oxygenated (95% O2and 5% CO2) Krebs' solution containing 0.05–0.1% bovine serum albumin (BSA), 1 μg/ml aprotinin, 0.03 μg/ml cystatin, 0.2 μg/ml bestatin, 0.1 mmbenzethonium chloride, 1 mm benzamidine, 10 μg/ml leupeptin, 1 μm phosphoramidon, 100 μm captopril, 20 μg/ml bacitracin, and 6 μm dithiothreitol (Sigma). Experiments were performed at room temperature because peptide release was shown previously to be measurable but submaximal under these conditions and thus susceptible to pharmacological manipulation (Malcangio et al., 1997).
BSA and protease inhibitors were added to minimize loss of detectable BDNF-like immunoreactivity (LI) and SP-LI through surface adhesion and to prevent degradation. The dorsal roots were placed in the lateral compartments on bipolar platinum electrodes and covered in mineral oil to avoid dehydration. Before, during, and after dorsal root stimulation, fractions of 3 or 8 ml of the superfusates were collected in ice-cooled siliconized tubes (Sigmacote; Sigma) to minimize BDNF-LI loss or in acetic acid (0.1N) to stabilize SP. Three different stimulation protocols were used: (1) continuous stimulation (CS) for 8 min at 1 Hz (480 pulses) or 1 min at 30 Hz (1800 pulses); (2) tetanic stimulation (TS), which was three or six trains of 100 pulses at 100 Hz separated by 10 sec intervals; and (3) burst stimulation (BS), which was 75 trains of four pulses at 100 Hz separated by 0.2 sec intervals. Square pulses of 0.5 msec duration and 20 V [corresponding to 10–14 mA of current in our conditions (Malcangio and Bowery, 1993)] or 10 mA were used to recruit C fibers. To selectively stimulate A fibers, pulses of 0.1 msec duration and 0.1–2 mA were used (Malcangio et al., 2000). In another set of experiments, BDNF release was induced by superfusing the slices with capsaicin (1–100 μm) at 1 ml/min for 3 min (Malcangio et al., 1998). Capsaicin was dissolved in ethanol (1 mm) and then diluted with modified Krebs' solution. Capsaicin was also superfused at the end of some experiments after electrical stimulation had been applied to check the viability of the preparations.
Processing of collected samples. To quantify BDNF-LI and glutamate contents in the same superfusates, samples were desalted and concentrated by Ultrafree-15 centrifugal device 10K (Waters Associates, Watford, UK). Retentates were freeze-dried and reconstituted in 300 μl of block and sample buffer (Promega, Southampton, UK) and 100 μl assayed for BDNF-LI content by ELISA (see below). Filtrates were used for determining glutamate concentration by HPLC coupled with o-phthalaldehyde precolumns derivatization and fluorometric detection (Neal et al., 1994). SP-LI content could not be measured in the filtrates because the recovery of the peptide through the Ultrafree-15 dialysis membrane was <30%. Separate experiments were performed, and superfusates were used for measuring SP and glutamate contents. Samples were desalted and partially purified by using Sep-Pak C18 reverse-phase silica gel cartridges (Waters Associates) (Malcangio et al., 1997). The cartridges were conditioned with acetonitrile (100%; HPLC grade; BDH Chemicals, Poole, UK) and trifluoroacetic acid (TFA) (0.1%; HPLC grade; BDH Chemicals). Samples were then loaded into the columns, and the peptide was eluted using acetonitrile/TFA (80:20) solution. The eluates were dried by evaporation under nitrogen (recovery not <85%). Dried samples were reconstituted in 300 μl of phosphate buffer and 100 μl assayed by radioimmunoassay in duplicate (1.3 pg/100 μl assay sensitivity) using scintillation proximity assay (Amersham Pharmacia Biotech, Buckinghamshire, UK) as described previously (Malcangio and Bowery 1993; Malcangio et al., 1997). The remaining 100 μl of reconstituted samples were used for glutamate content determination by HPLC (see above).
BDNF tissue extraction. DRG were harvested from both naïve and NGF-treated rats and processed to extract detectable levels of BDNF-LI using ELISA (see below). Several methods were used to maximize BDNF recovery from the tissue. A cocktail of protease inhibitors was added to the homogenization buffer to reduce enzymatic breakdown of BDNF [137 mm NaCl, 20 mm Tris-HCl, 1% NP-40, 10% glycerol, 0.1% BSA, 1 mm α-toluenesulfonil fluoride, 10 μg/ml aprotinin, 0.5 mm sodium vanadate (Sigma)]. Samples were kept at 4°C to slow down proteolytic activity. Non-ionic detergents (NP-40) were added to the buffer, to both aid tissue dissociation and prevent nonspecific adsorption of BDNF. BSA (1 mg/ml) was added in the homogenization buffer to provide soluble binding sites for BDNF. Tissue samples were acidified for 15 min because this has been reported to aid dissociation of bound trophic factors from their receptors (Okragly and Hakk-Frendscho, 1997). In our conditions, BDNF-LI content in DRG (four per rat) increased from 20.9 ± 5.6 to 55.0 ± 5.6 pg/mg protein (n = 4 rats per group) after acidification. Samples were neutralized and centrifuged to remove particulates and then diluted 1:10 with assay buffer to prevent interference with the ELISA (see below). Protein content was determined by the Bradford assay.
BDNF ELISA. Nunc (Roskilde, Denmark) MaxiSorp 96-well plates were used. BDNF standards (100 μl of 4–500 pg/ml solutions) and 100 μl of unknown samples were run in duplicate and triplicate, respectively, following a protocol slightly modified from the instructions of the manufacturer (Emax ImmunoAssay kit; Promega) (Son et al., 1999; Cejas et al., 2000). The plates were incubated for 48 hr with the monoclonal primary antibody. A standard curve was run for each plate, and this was linear at all levels of detection (r2 = 0.994 ± 0.002;n = 10). In each plate, some wells were spiked and DRG extracts were also run because they would contain more BDNF and prove that the assay was adequate to distinguish among different BDNF levels: from zero, to modest (>4 pg/ml), moderately high (20 pg/ml), and high (DRG extracts, 100 pg/ml). Samples were considered BDNF positive when their signal was higher than the background signal (modified Krebs' solution) and within the range of the standard curve.
The assay showed <3% cross-reactivity with human recombinant NGF. Human recombinant BDNF standard was used to assess BDNF recovery. This was determined by adding 5–20 pg/3 ml BDNF to modified Krebs' solution (see above) that had not been superfused through dorsal horn tissue and then processing under the same conditions as experimental samples. Recovery was 60% for 10 pg/3 ml BDNF.
Data calculation and statistical analysis of release data.The data are presented as mean ± SEM. ANOVA, followed by Tukey's or Dunnett's tests, and Student's t test were used when appropriate.
Neurokinin-11 receptor internalization. Neurokinin-1 (NK1) receptor internalization in lumbar spinal cord slices obtained from 14- to 30-d-old rats was assessed in transverse slices (400 μm) with one dorsal root attached (Marvizón et al., 1997, 1999). After electrical stimulation at the root (300 pulses, BS; see above), slices were fixed and cryoprotected. Sections (25 μm) were immunostained for NK1 receptor (Marvizón et al., 1997, 1999). Internalization was quantified as the percentage of NK1 receptor-IR neuronal somas showing internalization (Marvizón et al., 1997, 1999). All NK1 receptor-IR somas in at least three sections per slice were counted by a person blinded to the treatment. Confocal images were acquired with a Leica (Nussloch, Germany) TCS-SP confocal microscope.
BDNF immunostaining in the dorsal horn. After the release experiments, tissue was post-fixed in 4% paraformaldehyde (2 hr at 4°C), cryopreserved in 20% sucrose (overnight at 4°C), and blocked in OCT embedding compound (BDH Chemicals), and transverse sections (20 μm) were cryostat cut. Sections were immunostained for BDNF using indirect tyramide signal amplification (DuPont NEN, Boston, MA) (Michael et al., 1997). Sections were incubated with the following (all reagents were diluted in PBS containing 0.2% Triton X-100): normal goat serum (10%, 1 hr), rabbit polyclonal anti-BDNF (0.4 μg/ml, 12 hr; a gift from Amgen, Thousand Oaks, CA), biotin-conjugated goat anti-rabbit antibody (1:400, 2 hr; Vector Laboratories, Burlingame, CA), avidin–biotin complex (1:5 with no Triton X-100, 30 min; Vector Laboratories), biotinyl tyramide (1:75, 10 min; DuPont NEN), and FITC-conjugated extra-avidin (1:500, 2 hr; Sigma, St Louis, MO). Slides were coverslipped in Vectashield mounting medium (Vector Laboratories) and visualized under a Leitz (Wetzlar, Germany) fluorescent microscope. For quantification, images of BDNF staining in the dorsal horn of naïve and capsaicin-treated rats (n = 4 per group) were captured using a Hamamatsu (Bridgewater, NJ) digital camera. The intensity of BDNF immunostaining in the superficial laminas from four sections per animal was quantified using SigmaScan image analysis software (SPSS, Chicago, IL).
Electrophysiology. The tibial nerve of anesthetized adult male Sprague Dawley rats (280 gm body weight) was exposed, and the uncut nerve was placed on a bipolar tungsten stimulating electrode immediately proximal to the ankle in a pool of warm mineral oil. A bipolar platinum wire (diameter of 30 μm) recording electrode was positioned above the nerve 5–7 mm proximal to the stimulating electrode. A noxious heat stimulus was used to locate nociceptive fibers having receptive fields in the footpad. Successive fine nerve filaments (10–15 μm diameter) were dissected from the nerve until one was obtained containing a unit responsive to heating of the footpad to 47–48°C using a Peltier-effect thermal stimulator (1 cm square element; courtesy of Dr. Bruce Naliboff, University of California, Los Angeles, CA). The response to noxious heating was tested, and the plantar surface was explored for mechanosensitive receptive fields using a blunt probe and calibrated von Frey type stimulators (Stoelting Inc., Kiel, WI). At least 10 min after the final stimulus, capsaicin (100 mg/100 ml) was injected subdermally into the footpad, and the response was recorded. At the end of experiments, the tibial nerve was cut proximal to the recording electrode, and the nerve was stimulated to determine conduction velocities for the units recorded. Single-unit records were obtained via sorting of multiunit activity (one to six units typically) using Spike2 software (Cambridge Electronic Design, Cambridge, UK). For one fiber, a reliable conduction velocity could not be determined and its identity as a C fiber was inferred from its spike waveform.
Receptor antagonists.d(−)2-Amino-5-phosphonopentanoate (d-AP-5) and 6-cyano-7-nitro-quinoxaline-2,3-dione (CNQX) were from Research Biochemicals (Natick, MA). Naloxone was from Sigma. (−)Bicuculline methobromide was from Tocris Cookson (Bristol, UK).
RESULTS
Electrically evoked release of BDNF in the dorsal horn
Because BDNF and nociceptive peptides are colocalized in some C fibers (Michael et al., 1997), BDNF-LI release in dorsal horn was first examined using a pattern of sensory neuron electrical stimulation known to induce SP-LI release, an indication of high threshold fiber recruitment (Malcangio et al., 2000). Glutamate release was also evaluated in the same samples, although both A and C fibers contribute to its release (Kangrga and Randiç, 1991; Teoh et al., 1996).
As shown previously (Malcangio and Bowery, 1993; Malcangio et al., 1997), CS of the dorsal roots at C-fiber strength and low frequency (1 Hz, 480 pulses in 8 min) was effective at releasing SP-LI but did not lead to detectable release of BDNF-LI or glutamate (Fig.1A). The lack of glutamate detection in this set of experiments may be explained by fast uptake of the amino acid by both neurons and glia. The absence of BDNF-LI release could have been the consequence of poor tissue penetrability and/or loss of detectable BDNF-LI through surface adhesion (Leibrock et al., 1989; Anderson et al., 1995). However, in this study, BSA addition in the superfusion buffer and sample collection in siliconized tubes resulted in BDNF-LI basal outflow values that were in the range of those found in hippocampal slice perfusates (Canossa et al., 1997).
Fig. 1.
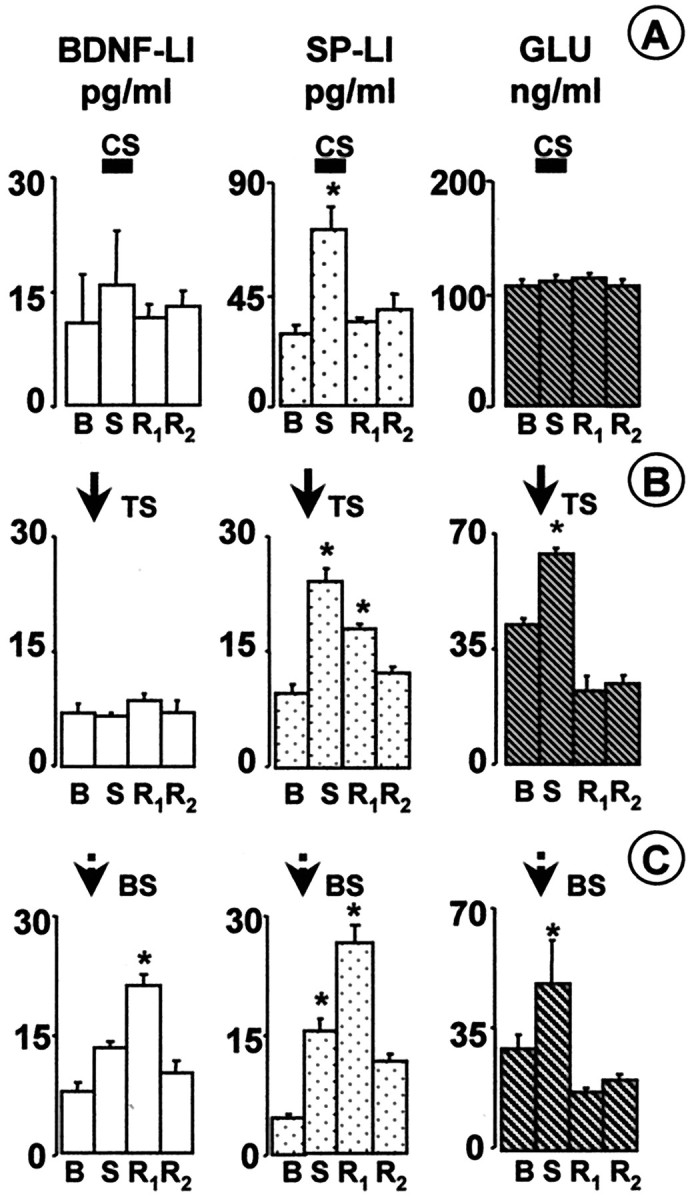
BDNF-LI, SP-LI, and glutamate (GLU) were released in the dorsal horn by different patterns of afferent fiber stimulation. Basal (B), high-threshold fiber strength-stimulated (S) and recovery (R1 andR2) fractions were collected at a rate of 1 ml/min for 8 (A) or 3 (B,C) min. Basal values (B) represent the mean of BDNF content in three fractions collected before the stimulated fraction (S).R2 values represent content in two fractions collected after the first recovery fraction (R1). Dorsal root stimulation was at high-threshold fiber strength (20 V or 5 mA, 0.5 msec) and was started at the beginning of the collection of the S fraction.A, CS of the dorsal roots at low frequency (1 Hz, 480 pulses, 8 min) induced SP-LI (n = 4 slices), but not BDNF-LI (n = 4 slices) or glutamate, release (n = 4 slices). B, TS (300 pulses in 3 trains, 100 Hz) evoked the release of SP-LI (n = 6 slices) and glutamate (n= 4 slices) but not of BDNF-LI (n = 7 slices).C, BS (300 pulses in 75 trains, 100 Hz) induced release of BDNF-LI (n = 6 slices), SP-LI (n = 7 slices), and glutamate (n = 4 slices). Values (means ± SE) express concentrations in reconstituted samples. *p < 0.05 versus both B and R2fraction values (ANOVA, followed by Tukey's test).
High-frequency stimulation (100 Hz, three trains of 100 pulses) of primary afferent fibers evokes long-term potentiation (LTP) of their synapses with dorsal horn neurons (Randiç et al., 1993; Liu and Sandkühler, 1998). The same pattern of stimulation was very effective in evoking SP-LI release in the dorsal horn (Marvizón et al., 1997), which may participate in this form of LTP (Liu and Sandkühler, 1997). We applied the same stimulation pattern (TS, 300 pulses at 100 Hz in three trains separated 10 sec) to the dorsal roots to test whether it was able to evoke BDNF release. Significant release of SP-LI and glutamate was detected (Fig.1B). BDNF-LI release was not detected even after 600 pulses were delivered at 100 Hz in six trains (basal outflow was 8.0 ± 0.8 pg/ml; release was 8.3 ± 0.2 pg/ml;n = 3; p > 0.05). Likewise, 1800 pulses delivered at 30 Hz as a single 1 min train did not produce any significant BDNF-LI release (11.2 ± 0.5 pg/ml) over basal (10.7 ± 0.01 pg/ml; n = 3; p > 0.05). In vivo, C fibers tend to fire in short bursts of high frequency in response to several forms of noxious stimulation (see below) (Puig and Sorkin, 1995; Adelson et al., 1996, 1997). Therefore, we stimulated the dorsal roots with the same number (300) of high-intensity pulses used for TS but delivered in 75 trains (bursts) of four pulses at 100 Hz, separated by a 0.2 sec interburst interval (BS). As shown in Figure 1C, BS induced significant release of BDNF-LI, SP-LI, and glutamate. Interestingly, whereas the release of glutamate peaked during the BS period, SP-LI release persisted after stimulation (first recovery, R1) and BDNF release was significant in the first recovery fraction (Fig.1C) and then returned to basal values. BDNF-LI release evoked by BS was intensity dependent (Fig.2A) and occurred only above the intensity threshold to recruit C fibers (5–10 mA), remaining constant at higher stimulation strengths. Electrically evoked BDNF-LI release (basal outflow increased to 240 ± 37.9% at 10 mA intensity) was comparable with glutamate-induced BDNF-LI release in hippocampal slices (Canossa et al., 1997). However, activity-induced BDNF-LI release in the hippocampus was not dependent on extracellular Ca2+ (Canossa et al., 1997), whereas BDNF-LI release in the dorsal horn was abolished in Ca2+-free medium (Fig.2B). To further ascertain that BDNF release was attributable to nerve impulse conduction, experiments were performed in the presence of tetrodotoxin (TTX), which abolished electrically evoked BDNF-LI release but did not modify basal outflow (Fig. 2C).
Fig. 2.

Intensity dependence of BS-induced BDNF release (A). BS was applied at intensities and durations ranging from A-fiber (0.1–1 mA, 0.1 msec) to C-fiber (5–10 mA, 0.5 msec) strength. Numbers inside columnsindicate the number of slices. *p < 0.05 versus basal values (B) (ANOVA, followed by Dunnett's test). B, Calcium dependence of BS-induced BDNF release. BS induced the release of BDNF-LI in the presence of Ca2+ ions (▴) but not when slices were superfused with Ca2+-free Krebs' solution containing 10 mm EDTA (▪). BS (300 stimuli; 10 mA, 0.5 msec, 100 Hz) was applied (arrow) after collection of two basal outflow fractions. Points represent means ± SE of four slices. *p < 0.05 versus analog fractions collected in the presence of Ca2+ (ANOVA, followed by Tukey's test). C, TTX superfusion blocked BS-induced BDNF release without changing basal outflow. TTX (2 μm) was superfused during the last 2 min of the third fraction (basal) and the first minute of the fourth fraction (stimulated) (■;n = 4 slices). Four slices were run in the absence of TTX as controls (▴). BS (300 stimuli; 10 mA, 0.5 msec, 100 Hz) was applied (arrow) after collection of three basal outflow fractions. Points represent means ± SEM. *p < 0.05 versus analog fractions collected in the presence of TTX (ANOVA, followed by Tukey's test).
To confirm that BS of the dorsal roots recruited some C fibers, SP release was indirectly assessed via NK1 receptor activation in spinal cord transverse slices (Marvizón et al., 1997). BS of the dorsal root produced NK1receptor internalization in the majority (Fig.3B,C) of the NK1 receptor-immunoreactive neurons in laminas I and IIo of the stimulated side of the slices but not in the contralateral side or in deeper laminas (Fig.3A,C).
Fig. 3.

BS produces NK1 receptor internalization in the dorsal horn. Confocal images of the contralateral (A; 10 optical sections) and the stimulated (B; 3 optical sections) side of a representative slice. Insets, Higher magnification of the neurons in the rectangles (8 optical sections inA; 5 optical sections in B). Scale bar: 50 and 16 μm for low- and high-magnification, respectively.I, II, and IIIindicate the location of Rexed's laminas. C, Percentage of NK1 receptor-immunoreactive (NK1R-ir) neuronal somata in laminas I-IIo with internalization after BS; mean ± SEM of four slices obtained from two rats. ***p < 0.0001 (t test).
Electrically evoked release of BDNF in the dorsal horn after NGF treatment
Primary afferent fibers expressing TrkA receptor can retrogradely transport NGF produced by their target cells to the DRG cells in which NGF can stimulate the expression of BDNF (Apfel et al., 1996; Michael et al., 1997). Thus, we investigated whether NGF induced alterations in BDNF content, and these were reflected in altered release in the dorsal horn.
Figure 4A shows that both systemic and intrathecal NGF treatments increased BDNF-LI content in the DRG of approximately threefold. In addition, when cords from NGF-treated rats that upregulated BDNF were used, enhanced amount of BDNF-LI was released in the dorsal horn after BS (Fig.4B). BDNF-LI basal outflow was unchanged, and TS was still ineffective in eliciting BDNF-LI release in both NGF-treated and nontreated rats (Fig. 4B). Thus, TS effectiveness in evoking BDNF release was not increased by increasing the amount of BDNF available.
Fig. 4.

A, NGF treatment upregulated BDNF-LI content in DRG. NGF and control vehicle (Con) (saline plus 0.1% rat serum albumin) were administered either systemically (NGF-s; 1 mg/kg 3 times per week for 1 or 2 weeks) or intrathecally (NGF-it; 12 μg/d for 14 d or 24 μg/d for 10 d), and BDNF content in the DRG was measured by ELISA (see Materials and Methods). B, NGF treatment increased the amount of BDNF-LI released over basal outflow (white columns) evoked by BS of the dorsal roots (striped columns). TS was still ineffective after NGF treatment (white and black columnsindicate basal and stimulated fractions, respectively). Both BS and TS consisted of 300 pulses of 10 mA or 20 V, 0.5 msec, given at 100 Hz in 75 (BS) or three (TS) trains. *p < 0.05 versus basal values; #p < 0.05 versus BS-evoked release in controls (ANOVA, followed by Tukey's test).Numbers inside columns indicate number of animals per slice.
Capsaicin-induced release of BDNF in the dorsal horn in naïve and NGF-treated rats
Peripheral noxious stimuli can cause bursting activity in fine afferent fibers (Puig and Sorkin, 1995; Adelson et al., 1996, 1997). Thus, we evaluated whether the chemical irritant capsaicin injected into the paw would induce a similar pattern of activity in C fibers. Using a thermal moderately noxious heat search stimulus (ramp to 47–48°C via a Peltier-effect thermal stimulator, 1 cm square element) we surveyed dissected thin filaments (10–15 μm diameter) of the tibial nerve until we isolated one containing a heat-sensitive C unit having a receptive field in the footpad. We tested the responses to the heating ramp several times (Fig.5B) and explored the mechanosensitivity of the units. Capsaicin (20–25 μl, 100 mg/ml) injected into the footpad adjacent to the heat-sensitive receptive field produced bursting activity (Fig.5A,C,D) in three of three experiments. Capsaicin can stimulate polymodal nociceptors to release their contents and, when superfused through the dorsal horn preparation, can dose dependently release SP-LI (EC50 of 100 nm) (Malcangio et al., 1993, 1998). Thus, in this study, the ability of capsaicin to release BDNF-LI from the dorsal horn was evaluated. Figure6A shows that capsaicin dose dependently released BDNF-LI but at higher doses than those effective in releasing SP. The highest dose (100 μm) induced a significant release of BDNF-LI during superfusion (fourth fraction) compared with basal outflow (first three fractions) (Fig. 6B). The release persisted in the following (fifth) fraction, and then BDNF-LI values recovered to basal (Fig. 6B). Capsaicin-induced BDNF-LI release was abolished by cosuperfusion of capsazepine (100 μm) (Fig. 6B). The doses of capsaicin necessary to release BDNF were unexpectedly high, so we tested whether capsaicin would be more effective if BDNF contents were increased by NGF treatment, which would have also increased nociceptor sensitivity to capsaicin (Winter et al., 1988). Figure 6Cshows that capsaicin (1 μm) significantly released BDNF in spinal cords of rats treated with NGF but was ineffective in controls. We then examined slices after high-dose capsaicin superfusion to determine whether this noxious stimulus led to BDNF depletion. Superfusion of capsaicin (300 μm) produced a significant loss of BDNF immunoreactivity throughout the dorsal horn (Fig.7B) compared with control slices that displayed the expected dense BDNF immunostaining in laminas I and II (Fig. 7A). Simultaneous staining of control and experimental slices revealed a 40% reduction in BDNF labeling after capsaicin treatment (Fig. 7C). To check that this decrease in BDNF immunoreactivity was not attributable to nonspecific terminal damage caused by capsaicin superfusion, we stained adjacent sections for a primary afferent marker. Staining of the isolectin B4 revealed no difference between capsaicin and control sections, indicating that binding sites were still present in the tissue (data not shown).
Fig. 5.

Intraplantar injection of capsaicin induces bursting discharge in nociceptors. A, Instantaneous frequency plot of burst discharge of a heat-sensitive C nociceptor in response to subdermal injection of 100 μg/100 μl capsaicin (CAPS; rectangleindicates the duration of the injection). B, Response of the same unit as in A to repeated heating to 47.6°C (bottom trace indicates the temperature ramp).C, A different type of bursting discharge in another heat-sensitive C nociceptor evoked by subdermal injection of 100 μg/100 μl capsaicin (first rectangleindicates the duration of the injection). D, Detail of period of discharge indicated by second(bold) rectangle inC.
Fig. 6.

Superfusion of dorsal horn slices with capsaicin induced BDNF release. A, Capsaicin (Caps; superfused for 3 min) dose dependently released BDNF. Values are obtained from at least four slices for each dose. *p < 0.05 versus basal outflow (B) (ANOVA, followed by Dunnett's test).B, Superfusion of dorsal horn slices with capsaicin (Caps; 100 μm, black horizontal bar) during collection of the fourth fraction (3 min) caused significant release of BDNF-LI in the fourth and fifth fractions (n = 6 slices). BDNF release returned to basal afterward. Superfusion of capsazepine (Cpz; 100 μm; white horizontal bar;n = 4 slices) one fraction before and during capaicin superfusion blocked BDNF release. C, Capsaicin (Caps; 1 μm)-induced release of BDNF-LI over basal outflow (B) was increased after NGF pretreatment (n = 10 slices). *p < 0.05 (Student's t test versus basal values); #p < 0.05 versus capsaicin in controls (n = 6 slices).
Fig. 7.

Capsaicin superfusion induced depletion of BDNF immunostaining in the dorsal horn. BDNF-IR in the dorsal horn of a naïve dorsal horn slice (A) and a slice superfused with 300 μm capsaicin for 3 min (B). In control slices (A), BDNF is present in fibers (probably primary afferent terminals) in laminas I and II, being particularly abundant in the medial portion. After capsaicin treatment (B), there is a marked reduction in BDNF immunoreactivity in the superficial dorsal horn (arrows in B). C, Quantitative analysis (see Materials and Methods) confirms a significant reduction in BDNF immunoreactivity after capsaicin treatment. *p < 0.05 (Student's ttest; n = 4 dorsal horn slices in each group).
Modulation of electrically evoked release of BDNF in the dorsal horn
It has been shown previously that SP and glutamate release from primary afferent terminals in the dorsal horn are modulated by glutamate acting on NMDA receptors located presynaptically (Liu et al., 1997; Marvizón et al., 1997; Malcangio et al., 1998). The possibility that glutamate released after BS of the dorsal roots controlled the release of BDNF was tested by assessing the effect of antagonists for NMDA and non-NMDA receptors on evoked BDNF release. Figure 8A shows that, in control dorsal horn slices, BS of primary afferent fibers induced a significant and reversible release of BDNF-LI (9–12 min time interval) over basal outflow (3–9 min time interval). However, superfusion of the NMDA receptor antagonist d-AP-5 significantly inhibited BS-induced release of BDNF-LI (Fig. 8A). In contrast, the presence of the non-NMDA antagonist CNQX did not inhibit BS-induced release of BDNF-LI (Fig. 8A). These data indicate that BDNF release, like SP release, is likely to be modulated by glutamate activating NMDA receptors, whereas non-NMDA receptors are not involved.
Fig. 8.

Pharmacological modulation of BS-induced release of BDNF-LI. Burst stimulation (300 stimuli; 10 mA, 0.5 msec, 100 Hz) was applied (arrow) during collection of the fourth fraction. In control slices (▴; n = 9), BS induced significant and reversible release of BDNF over basal outflow (first 3 fractions) (A, B). Superfusion of d-AP-5 (50 μm; n = 5) in the fractions before, during, and after stimulation (horizontal white bar) inhibited evoked release of BDNF-LI (A). Superfusion of CNQX (A; 5 μm; n = 4), naloxone (B, NAL; 1 μm;n = 7), or (−) bicuculline methobromide (B, BIC; 100 μm;n = 5) did not modify BDNF-LI release. *p < 0.05, stimulated (fourth fraction) versus correspondent basal outflow fractions (first plus second or third fractions); ANOVA, followed by Tukey's test.
Importantly, SP release from primary afferent terminals in the dorsal horn is also under the inhibitory control of endogenous opioid and GABAergic systems that counteract NMDA-evoked facilitation (Jessell and Iversen, 1977; Malcangio et al., 1993; Teoh et al., 1996). Thus, we tested the effect of naloxone and bicuculline (antagonists for μ and GABAA receptors, respectively) on BS-induced BDNF release and found that neither antagonist could modify the evoked release (Fig. 8B). These findings fail to support the possibility that BS of primary afferent fibers activates endogenous dorsal horn inhibitory systems sufficiently to affect BDNF release.
DISCUSSION
BDNF may act as a modulator of synaptic transmission in the spinal dorsal horn (Snider and McMahon, 1998; Woolf and Salter, 2000), as found previously in the hippocampus (Canossa et al., 1997; Schuman, 1999). To understand the action of BDNF in the cord, it is important to establish what type of stimuli are effective at releasing BDNF. Here we report that BDNF-LI release from the dorsal horn is evoked by capsaicin and electrical stimulation of the dorsal roots. We have used several protocols of electrical stimulation, and we found that BDNF release was dependent on the pattern of stimulation of primary afferents. Thus, dorsal root stimulation with short bursts of high-frequency pulses (BS) evoked BDNF release, whereas sustained stimulation at low frequency (1 Hz, CS) or high frequency (100 Hz, TS) failed to produce any measurable release. Remarkably, CS or TS were unable to evoke any BDNF release, even when the number of pulses was increased well above those used for BS. Moreover, TS was ineffective even when the BDNF content was increased by NGF treatment, indicating that the pattern of stimulation of primary afferent fibers is the main determinant of BDNF release. BS-induced BDNF release was greatly increased after NGF treatment, probably because of an increased BDNF content in C fibers expressing the TrkA receptor. However, it cannot be excluded that prolonged treatment with NGF might have enhanced afferent fiber excitability, for example by sodium channel induction (Toledo-Aral et al., 1995).
The physiological relevance of these findings is underscored by the transition of C fibers to a bursting firing pattern when the intensity of nociceptive stimuli increases over a certain threshold (Puig and Sorkin, 1995; Adelson et al., 1996, 1997). We show that C fibers in the tibial nerve develop bursting activity after an injection of capsaicin into the footpad. Other studies have shown C-fiber bursting activity in visceral nerves in response to hydrogen peroxide (Adelson et al., 1996) or bradykinin (Adelson et al., 1997). Therefore, certain noxious stimuli may produce bursting activity in C fibers, which in turn elicits BDNF release in the dorsal horn.
BS of the dorsal roots was also able to evoke the release of SP in the dorsal horn preparation. Previous studies (Go and Yaksh, 1987; Duggan et al., 1995; Marvizón et al., 1997) have shown that SP release is facilitated at high frequencies of stimulation, although it also occurs if enough pulses are given at low frequency (Malcangio and Bowery, 1993; Allen et al., 1999). Interestingly, unlike BDNF, approximately the same amount of SP-LI release was elicited by TS and BS. Confirming this, BS (Fig. 3) and TS (Marvizón et al., 1997) produced similar amounts of NK1 internalization in the superficial dorsal horn, an indicator of NK1 receptor activation by released tachykinins. CS also produced a substantial amount of SP release. Hence, SP appears to be released by a wider range of firing patterns than BDNF. This observation is somewhat surprising given the fact that SP and BDNF are likely to be present in the same population of primary afferent fibers (Michael et al., 1997) and may be both packaged in large, dense-core synaptic vesicles. Alternatively, BDNF may be stored in a different population of dense-core vesicles that undergo exocytosis after BS, which might lead to a substantial buildup of calcium concentrations in presynaptic terminals. (Muschol and Salzberg, 2000) Indeed, BS-evoked release of BDNF was Ca2+ dependent and returned to basal levels after stimulation, suggesting that it was produced by a synaptic mechanism and was not attributable to electropermeabilization of neuronal membranes. Moreover, electrically evoked release of BDNF was inhibited by TTX perfusion, indicating that the release was a consequence of neuronal activity involving voltage-dependent Na+ channels. In contrast, BDNF basal outflow was neither calcium dependent nor modified by TTX superfusion and was likely to be unrelated to neuronal firing. The observation that NGF treatment did not increase BDNF basal outflow further supports this idea.
Importantly, BDNF release evoked by BS of the dorsal roots was associated with glutamate release, inhibited by the NMDA receptor antagonist d-AP-5 but not by the non-NMDA antagonist CNQX, indicating that glutamate stimulates BDNF release by activating NMDA receptors in the dorsal horn. However, TS did not produce significant BDNF release but increased extracellular glutamate content, suggesting that glutamate alone is not sufficient to induce BDNF release. Glutamate also stimulates SP release from primary afferent terminals by putatively activating presynaptically located NMDA receptors (Liu et al., 1997; Marvizón et al., 1997; Malcangio et al., 1998). Thus, one explanation for our findings would be that BDNF release is modulated by presynaptic NMDA receptors, which have been found in the central terminals of primary afferents (Liu et al., 1994). Alternatively, postsynaptic NMDA receptors may stimulate BDNF release by triggering the release of diffusible retrograde messengers (such as nitric oxide) from dorsal horn cells. Synaptic transmission between primary afferents and dorsal horn neurons is primarily driven by AMPA receptors (Gerber and Randiç, 1989; Yoshimura and Jessell, 1990;Randiç et al., 1993; Yoshimura and Nishi, 1993), with NMDA receptors contributing only a small component of the EPSP. Therefore, our observation that the AMPA receptor antagonist CNQX did not inhibit BS-evoked BDNF release suggests that BDNF is likely to be mainly released from primary afferent terminals, although we cannot rule out entirely that BDNF was released from dorsal horn neurons. Although GABAA and opioid receptors play an important role in modulating nociceptive signals in the dorsal horn (Millan, 1999), these inhibitory mechanisms do not appear to modulate BDNF release, because it was not altered by the GABAAantagonist bicuculline or the opioid antagonist naloxone.
Our findings suggest that different patterns of stimulation of primary afferents may encode the release of different transmitters. Notably, neuropeptide release is likely to depend on stimulation frequency (Bartfai et al., 1986), whereas neurotrophin release appears to depend on the pattern of stimulation, which may be critical for the activation of specific transmission pathways to evoke BDNF release. One consequence may be that different transmitters–modulators are released in different pain states, which would have important implications for analgesic therapy.
Our data are consistent with a recent report (Balkowiec and Katz, 2000) showing that BDNF is released from cultured neonatal nodose and petrosal ganglia (NPG) neurons by high-frequency burst field stimulation but not short-term KCl depolarization. These authors suggested that the magnitude of BDNF release in NPG neurons depended on the pattern and frequency of stimulation. Likewise, we show that stimulus pattern-dependent release of BDNF occurs in the dorsal horn of the spinal cord, under conditions that resemble nociceptive sensory neuron activation.
BDNF release was also evoked by capsaicin, and BDNF immunoreactivity measured in situ in the dorsal horn was substantially reduced after capsaicin application, indicating that primary afferent BDNF pools can be readily depleted. Concentrations of capsaicin necessary to stimulate BDNF release (1–100 μm) were higher than those that release SP under the same experimental conditions (EC50 of 100 nm) (Malcangio et al., 1997), which raised the issue of specificity of the action of capsaicin (Bevan and Szolcsanyi, 1990). Although VR1 and SP have been shown to colocalize (Tominaga et al., 1998), no such data exist for BDNF, and the effect of capsaicin might be indirect, for example, mediated by glutamate release and nitric oxide production. Capsazepine prevented BDNF release, however, suggesting that the effect of capsaicin was attributable to VR1 activation. Moreover, after NGF treatment, capsaicin significantly released BDNF at a dose (1 μm) that is close to its affinity for VR1 receptors (Caterina et al., 1997).
The release of BDNF in the dorsal horn may have important physiological consequences. Existing evidence suggests that BDNF participates in central sensitization and inflammatory pain states (Kerr et al., 1999;Mannion et al., 1999). Moreover, BDNF increases the excitability of nociceptive spinal reflexes and potentiates NMDA receptor-mediated responses in the dorsal horn (Kerr et al., 1999). However, there is evidence suggesting that chronic spinal delivery of BDNF alleviates allodynia and hyperalgesia in neuropathic rats (Cejas et al., 2000). Furthermore, BDNF is antinociceptive in normal and inflamed rats when injected into the brainstem (Siuciak et al., 1994, 1995) where this neurotrophin can strongly inhibit AMPA-mediated currents via TrkB receptor activation (Balkowiec et al., 2000). Thus, the action of BDNF is likely to be complex and dependent on the specific site within nociceptive pathways.
In summary, this study shows that BDNF is released by specific patterns of stimulation of afferent fibers and is mediated by NMDA receptor activation. These data support the idea that endogenous BDNF plays a role in the modulation of nociceptive stimuli in the dorsal horn.
Footnotes
This work was supported by a Wellcome Trust Career Development Fellowship (to M.M.). S.B.M. and E.J.B. thank the Wellcome Trust for support. We thank Drs. Reg Docherty, Tim Boucher, and Greg Michael for helpful discussion, Drs. Emeran A. Mayer and Yvette Taché for their support, and Viv Cheah for technical assistance.
Correspondence should be addressed to Marzia Malcangio, Sensory Function, Center for Neuroscience, Hodgkin Building, King's College London, Guy's Campus, London Bridge, London SE1 1UL, UK. E-mail: marzia.malcangio@kcl.ac.uk.
REFERENCES
- 1.Adelson DW, Wei JY, Kruger L. H2O2 sensitivity of afferent splanchnic C fiber units in vitro. J Neurophysiol. 1996;76:371–380. doi: 10.1152/jn.1996.76.1.371. [DOI] [PubMed] [Google Scholar]
- 2.Adelson DW, Wei JY, Kruger L. Warm-sensitive afferent splanchnic C-fiber units in vitro. J Neurophysiol. 1997;77:2989–3002. doi: 10.1152/jn.1997.77.6.2989. [DOI] [PubMed] [Google Scholar]
- 3.Allen BJ, Li J, Menning PM, Rogers SD, Ghilardi J, Mantyh PW, Simone DA. Primary afferent fibers that contribute to increased substance P receptor internalization in the spinal cord after injury. J Neurophysiol. 1999;81:1379–1390. doi: 10.1152/jn.1999.81.3.1379. [DOI] [PubMed] [Google Scholar]
- 4.Anderson KD, Alderson RF, Altar CA, DiStefano PS, Corcoran TL, Lindsay RM, Wiengand SJ. Differential distribution of exogenous BDNF, NGF and NT-3 in the brain corresponds to the relative abundance and distribution of the high affinity and low affinity neurotrophin receptors. J Comp Neurol. 1995;357:296–317. doi: 10.1002/cne.903570209. [DOI] [PubMed] [Google Scholar]
- 5.Apfel SC, Wright DE, Wiideman AM, Dormia C, Snider WD, Kessler JA. Nerve growth factor regulates the expression of brain-derived neurotrophic factor mRNA in the peripheral nervous system. Mol Cell Neurosci. 1996;7:134–142. doi: 10.1006/mcne.1996.0010. [DOI] [PubMed] [Google Scholar]
- 6.Averill S, McMahon SB, Clary SB, Reichart LF, Priestley JV. Immunocytochemical localisation of trkA receptors in chemically identified subgroups of adult rat sensory neurons. Eur J Neurosci. 1995;7:1484–1494. doi: 10.1111/j.1460-9568.1995.tb01143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balkowiec A, Kunze DL, Katz DM. Brain-derived neurotrophic factor acutely inhibits AMPA-mediated currents in developing sensory relay neurons. J Neurosci. 2000;20:1904–1911. doi: 10.1523/JNEUROSCI.20-05-01904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartfai T, Iverfeldt K, Brodin E, Ögren S-O. Functional consequences of coexistence of classical and peptide neurotransmitters. Prog Brain Res. 1986;68:321–330. doi: 10.1016/s0079-6123(08)60247-2. [DOI] [PubMed] [Google Scholar]
- 10.Bevan S, Szolcsanyi J. Sensory neuron-specific actions of capsaicin: mechanisms and application. Trends Pharmacol Sci. 1990;11:330–333. doi: 10.1016/0165-6147(90)90237-3. [DOI] [PubMed] [Google Scholar]
- 11.Bradbury EJ, King V, Simmons LJ, Priestley JV, McMahon SB. NT-3 but not BDNF prevents atrophy and death of axotomised spinal cord projection neurons. Eur J Neurosci. 1998;10:3058–3068. doi: 10.1046/j.1460-9568.1998.00307.x. [DOI] [PubMed] [Google Scholar]
- 12.Canossa M, Griesbeck O, Berninger B, Campana G, Kolbeck R, Thönen H. Neurotrophin release of neurotrophins: implications for activity-dependent neuronal plasticity. Proc Natl Acad Sci USA. 1997;94:13279–13286. doi: 10.1073/pnas.94.24.13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caterina MJ, Scumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 14.Cejas PJ, Martinez M, Karmally S, McKillop M, McKillop J, Plunkett JA, Oudega M, Eaton MJ. Lumbar transplant of neurons genetically modified to secrete brain-derived neurotrophic factor attenuates allodynia and hyperalgesia after sciatic nerve constriction. Pain. 2000;86:195–210. doi: 10.1016/s0304-3959(00)00245-1. [DOI] [PubMed] [Google Scholar]
- 15.Cho H-J, Kim J-K, Park H-C, Kim J-K, Kim D-S, HA S-O, Hong H-S. Changes in brain-derived neurotrophic factor immunoreactivity in rat dorsal root ganglia, spinal cord, and gracile nuclei following cut or crush injuries. Exp Neurol. 1998;154:224–230. doi: 10.1006/exnr.1998.6936. [DOI] [PubMed] [Google Scholar]
- 16.Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Biasi S, Rustioni A. Glutamate and substance P coexist in primary afferent terminals in the superficial laminae of spinal cord. Proc Natl Acad Sci USA. 1988;85:7820–7824. doi: 10.1073/pnas.85.20.7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dreyfus CF, Dai X, Lercher LD, Racey BR, Friedman WJ, Black IB. Expression of neurotrophins in the adult spinal cord in vivo. J Neurosci Res. 1999;56:1–7. doi: 10.1002/(SICI)1097-4547(19990401)56:1<1::AID-JNR1>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 19.Duggan AW, Riley RC, Mark MA, Macmillan SJA, Schaible H-G. Afferent volley patterns and the spinal release of immunoreactive substance P in the dorsal horn of the anaesthetized spinal cat. Neuroscience. 1995;65:849–858. doi: 10.1016/0306-4522(94)00541-c. [DOI] [PubMed] [Google Scholar]
- 20.Fawcett JP, Bamji SX, Causing CG, Aloyz R, Ase AR, Reader TA, McLean JH, Miller FD. Functional evidence that BDNF is an anterograde neuronal trophic factor in the CNS. J Neurosci. 1998;18:2802–2821. doi: 10.1523/JNEUROSCI.18-08-02808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerber G, Randiç M. Excitatory amino acid-mediated components of synaptically evoked input from dorsal roots to deep dorsal horn neurons in the rat spinal cord slice. Neurosci Lett. 1989;106:211–219. doi: 10.1016/0304-3940(89)90228-0. [DOI] [PubMed] [Google Scholar]
- 22.Go VLW, Yaksh TL. Release of substance P from the cat spinal cord. J Physiol (Lond) 1987;391:141–167. doi: 10.1113/jphysiol.1987.sp016731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jessell TM, Iversen LL. Opiate analgesics inhibit substance P release from rat trigeminal nucleus. Nature. 1977;268:549–551. doi: 10.1038/268549a0. [DOI] [PubMed] [Google Scholar]
- 24.Kangrga I, Randiç M. Outflow of endogenous amino acids from the rat spinal dorsal horn in vitro by activation of low- and high-threshold primary afferent fibers-modulation by μ opioids. Brain Res. 1991;553:342–352. doi: 10.1016/0006-8993(91)90848-p. [DOI] [PubMed] [Google Scholar]
- 25.Kerr BJ, Bradbury EJ, Bennett DLH, Trivedi PM, Dassan P, French J, Shelton DB, McMahon SB, Thompson SWN. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci. 1999;19:5138–5148. doi: 10.1523/JNEUROSCI.19-12-05138.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, Thonen H, Barde Y-A. Molecular cloning and expression of brain-derived neurotrophic factor. Nature. 1989;341:149–152. doi: 10.1038/341149a0. [DOI] [PubMed] [Google Scholar]
- 27.Liu H, Wang H, Sheng M, Jan LY, Jan YN, Basbaum AI. Evidence for presynaptic N-methyl-d-aspartate autoreceptors in the spinal cord dorsal horn. Proc Natl Acad Sci USA. 1994;91:8383–8387. doi: 10.1073/pnas.91.18.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, Mantyh PW, Basbaum AI. NMDA-receptor regulation of substance P release from primary afferent nociceptors. Nature. 1997;386:721–724. doi: 10.1038/386721a0. [DOI] [PubMed] [Google Scholar]
- 29.Liu X-G, Sandkühler J. Characterization of long-term potentiation of C-fiber-evoked potentials in spinal dorsal horn of adult rat: essential role of NK1 and NK2 receptors. J Neurophysiol. 1997;78:1973–1982. doi: 10.1152/jn.1997.78.4.1973. [DOI] [PubMed] [Google Scholar]
- 30.Liu X-G, Sandkühler J. Activation of spinal N-methyl-d-aspartate or neurokinin receptors induces long-term potentiation of spinal C-fiber-evoked potentials. Neuroscience. 1998;86:1209–1216. doi: 10.1016/s0306-4522(98)00107-9. [DOI] [PubMed] [Google Scholar]
- 31.Malcangio M, Bowery NG. Gamma-aminobutyric acidB, but not γ-aminobutyric acidA receptor activation, inhibits electrically evoked substance P-like immunoreactivity release from the rat spinal cord in vitro. J Pharmacol Exp Ther. 1993;266:1490–1496. [PubMed] [Google Scholar]
- 32.Malcangio M, Garrett NE, Cruwys S, Tomlinson DR. Nerve growth factor- and neurotrophin-3-induced changes in nociceptive threshold and the release of substance P from the rat isolated spinal cord. J Neurosci. 1997;17:8459–8467. doi: 10.1523/JNEUROSCI.17-21-08459.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malcangio M, Fernandes K, Tomlinson DR. NMDA receptor activation modulates evoked release of substance P from rat spinal cord. Br J Pharmacol. 1998;125:1625–1626. doi: 10.1038/sj.bjp.0702260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malcangio M, Ramer MS, Jones MG, McMahon SB. Abnormal substance P release from the spinal cord following injury to primary sensory neurons. Eur J Neurosci. 2000;12:397–399. doi: 10.1046/j.1460-9568.2000.00946.x. [DOI] [PubMed] [Google Scholar]
- 35.Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma Q-P, Holstege JC, Ji R-R, Acheson A, Lindsay RM, Wilkinson GA, Woolf CJ. Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci USA. 1999;96:9385–9390. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marvizón JCG, Martínez V, Grady EF, Bunnett NW, Mayer EA. Neurokinin receptor internalization in spinal cord slices induced by dorsal root stimulation is mediated by NMDA receptors. J Neurosci. 1997;17:8129–8136. doi: 10.1523/JNEUROSCI.17-21-08129.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marvizón JCG, Grady EF, Stefani E, Bunnett NW, Mayer EA. Substance P release in the dorsal horn assessed by receptor internalization: NMDA receptors counteract a tonic inhibition by GABAB receptors. Eur J Neurosci. 1999;11:417–426. doi: 10.1046/j.1460-9568.1999.00445.x. [DOI] [PubMed] [Google Scholar]
- 38.Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DHL, Yan Q, Priestley JV. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in tyrosine-kinase-A expressing dorsal root ganglion cells and their central terminations within the spinal cord. J Neurosci. 1997;17:8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michael GJ, Averill S, Shortland PJ, Yan Q, Priestley JV. Axotomy results in major changes in BDNF expression by dorsal root ganglion cells: BDNF expression in large trkB and trkC cells, in pericellular baskets, and in projections to deep dorsal horn and dorsal column nuclei. Eur J Neurosci. 1999;11:3539–3551. doi: 10.1046/j.1460-9568.1999.00767.x. [DOI] [PubMed] [Google Scholar]
- 40.Millan MJ. The induction of pain: an integrative review. Prog Neurobiol. 1999;57:1–164. doi: 10.1016/s0301-0082(98)00048-3. [DOI] [PubMed] [Google Scholar]
- 41.Muschol M, Salzberg BM. Dependence of transient and residual calcium dynamics on action-potential patterning during neuropeptide secretion. J Neurosci. 2000;20:6773–6780. doi: 10.1523/JNEUROSCI.20-18-06773.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neal MJ, Cunningham JR, Hutson PH, Hogg J. Effects of ischaemia on neurotransmitter release from the isolated retina. J Neurochem. 1994;62:1025–1033. doi: 10.1046/j.1471-4159.1994.62031025.x. [DOI] [PubMed] [Google Scholar]
- 43.Okragly AJ, Hakk-Frendscho M. An acid-treatment method for the enhanced detection of GDNF in biological samples. Exp Neurol. 1997;145:592–596. doi: 10.1006/exnr.1997.6500. [DOI] [PubMed] [Google Scholar]
- 44.Puig S, Sorkin LS. Formalin-evoked activity in identified primary afferent fibers: systemic lidocaine suppresses phase-2 activity. Pain. 1995;64:345–355. doi: 10.1016/0304-3959(95)00121-2. [DOI] [PubMed] [Google Scholar]
- 45.Randiç M, Jiang MC, Cerne R. Long-term potentiation and long-term depression of primary afferent neurotransmission in the rat spinal cord. J Neurosci. 1993;13:5228–5241. doi: 10.1523/JNEUROSCI.13-12-05228.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schuman EM. Neurotrophin regulation of synaptic transmission. Curr Opin Neurobiol. 1999;9:105–109. doi: 10.1016/s0959-4388(99)80013-0. [DOI] [PubMed] [Google Scholar]
- 47.Siuciak JA, Altar CA, Wiengand SJ, Lindsay RM. Antinociceptive effect of brain-derived neurotrophic factor and neurotrophin-3. Brain Res. 1994;633:326–330. doi: 10.1016/0006-8993(94)91556-3. [DOI] [PubMed] [Google Scholar]
- 48.Siuciak JA, Wong V, Pearsall D, Wiegand SJ, Lindsay RM. BDNF produces analgesia in the formalin test and modifies neuropeptide levels in rat brain and spinal cord areas associated with nociception. Eur J Neurosci. 1995;7:663–670. doi: 10.1111/j.1460-9568.1995.tb00670.x. [DOI] [PubMed] [Google Scholar]
- 49.Snider WD, McMahon SB. Tackling pain at the source: new ideas about nociceptors. Neuron. 1998;20:629–632. doi: 10.1016/s0896-6273(00)81003-x. [DOI] [PubMed] [Google Scholar]
- 50.Son JH, Chun HS, Joh TH, Cho S, Conti B, Lee JW. Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J Neurosci. 1999;19:10–20. doi: 10.1523/JNEUROSCI.19-01-00010.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teoh H, Malcangio M, Bowery NG. GABA, glutamate and substance-P like immunoreactivity release: effects of novel GABAB antagonists. Br J Pharmacol. 1996;118:1153–1160. doi: 10.1111/j.1476-5381.1996.tb15518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Toledo-Aral JJ, Brehm P, Halegoua A, Mandel G. A single pulse of nerve growth factor triggers long term neuronal excitability through sodium channels induction. Neuron. 1995;14:607–611. doi: 10.1016/0896-6273(95)90317-8. [DOI] [PubMed] [Google Scholar]
- 53.Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 54.Winter J, Forbes CA, Sternberg J, Lindsay RM. Nerve growth factor (NGF) regulates adult rat cultured dorsal root ganglion neuron responses to the excitotoxin capsaicin. Neuron. 1988;1:973–981. doi: 10.1016/0896-6273(88)90154-7. [DOI] [PubMed] [Google Scholar]
- 55.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1768. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 56.Yoshimura M, Jessell T. Amino acid-mediated EPSPs at primary afferent synapses with substantia gelatinosa neurones in the rat spinal cord. J Physiol (Lond) 1990;430:315–335. doi: 10.1113/jphysiol.1990.sp018293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshimura M, Nishi S. Blind patch-clamp recordings from substantia gelatinosa neurons in adult rat spinal cord slices: pharmacological properties of synaptic currents. Neuroscience. 1993;53:519–526. doi: 10.1016/0306-4522(93)90216-3. [DOI] [PubMed] [Google Scholar]
- 58.Zhou X-F, Rush RA. Endogenous brain derived neurotrophic factor is anterogradely transported in primary sensory neurons. Neuroscience. 1996;74:945–951. doi: 10.1016/0306-4522(96)00237-0. [DOI] [PubMed] [Google Scholar]
- 59.Zhou X-F, Parada LF, Soppet D, Rush RA. Distribution of TrkB tyrosine kinase immunoreactivity in the rat central nervous system. Brain Res. 1993;622:63–70. doi: 10.1016/0006-8993(93)90802-t. [DOI] [PubMed] [Google Scholar]
- 60.Zhou X-F, Chie ET, Deng Y-S, Zhong J-H, Xue Q, Rush RA, Xian CJ. Injured primary sensory neurons switch phenotype for brain-derived neurotrophic factor in the rat. Neuroscience. 1999;92:841–853. doi: 10.1016/s0306-4522(99)00027-5. [DOI] [PubMed] [Google Scholar]