

Abstract
The generation of high-frequency spike bursts (“complex spikes”), either spontaneously or in response to depolarizing stimuli applied to the soma, is a notable feature in intracellular recordings from hippocampal CA1 pyramidal cells (PCs) in vivo. There is compelling evidence that the bursts are intrinsically generated by summation of large spike afterdepolarizations (ADPs). Using intracellular recordings in adult rat hippocampal slices, we show that intrinsic burst-firing in CA1 PCs is strongly dependent on the extracellular concentration of Ca2+([Ca2+]o). Thus, lowering [Ca2+]o (by equimolar substitution with Mn2+ or Mg2+) induced intrinsic bursting in nonbursters, whereas raising [Ca2+]o suppressed intrinsic bursting in native bursters. The induction of intrinsic bursting by low [Ca2+]o was associated with enlargement of the spike ADP. Low [Ca2+]o-induced intrinsic bursts and their underlying ADPs were suppressed by drugs that reduce the persistent Na+ current (INaP), indicating that this current mediates the slow burst depolarization. Blocking Ca2+-activated K+ currents with extracellular Ni2+ or intracellular chelation of Ca2+ did not induce intrinsic bursting. This and other evidence suggest that lowering [Ca2+]o may induce intrinsic bursting by augmenting INaP. Because repetitive neuronal activity in the hippocampus is associated with marked decreases in [Ca2+]o, the regulation of intrinsic bursting by extracellular Ca2+ may provide a mechanism for preferential recruitment of this firing mode during certain forms of hippocampal activation.
Keywords: intrinsic bursting, calcium, persistent sodium current, pyramidal cell, hippocampus, phenytoin, gap junction, PKC, rat
The discharge of many hippocampal CA1 pyramidal cells (PCs) in vivo is a mixture of single spikes and high-frequency bursts of several spikes (Kandel and Spencer, 1961; Fujita, 1975; Nuñez et al., 1990). There is convincing evidence that the spike bursts (or “complex spikes”) are generated intrinsically by slow, voltage-gated membrane currents (Kandel and Spencer, 1961; Fujita, 1975; Nuñez et al., 1990; Kamondi et al., 1998). Such bursts are thought to play important roles in electrical signaling, in neuronal synchronization, and in the induction of long-term synaptic plasticity (for review, see Lisman, 1997).
In isolated hippocampal slices perfused with standard saline, only a small fraction (∼20%) of CA1 PCs manifest an intrinsic tendency to burst-fire; most of these neurons are regular firing cells (Schwartzkroin, 1975; Masukawa et al., 1982; Jensen et al., 1994). The scarcity of intrinsic bursters in CA1 in vitro may reflect the fact that the firing pattern of an individual PC is not an invariable attribute. Rather, it is strongly modulated by the ionic composition of the extracellular fluid. Thus, modest increases in extracellular pH (Church and Baimbridge, 1991) or concentration of K+([K+]o) (Jensen et al., 1994), or modest decreases in extracellular osmolality (Azouz et al., 1997), can convert regular firing PCs to intrinsic bursters. Because these factors vary as a function of ongoing neuronal activity in the brain, it is possible that the intrinsic firing pattern of CA1 PCs in vivo may alternate between nonbursting and bursting in different states of hippocampal activation.
The baseline concentration of free extracellular Ca2+(Ca2+o) is 1.2–1.5 mm, but neuronal activity can cause Ca2+o concentration ([Ca2+]o) to decrease considerably (Heinemann et al., 1977). Therefore, it is important to establish how changes in [Ca2+]o affect the firing pattern of hippocampal neurons. In a previous study we concluded that somatic bursting in CA1 PCs is neither driven by Ca2+ currents nor terminated by Ca2+-activated K+ currents (Azouz et al., 1996). However, we noted that lowering [Ca2+]o increases the propensity of these neurons to burst in response to depolarization. In this study we describe this effect of low [Ca2+]o in more detail and examine three putative hypotheses regarding its underlying mechanism: (1) suppression of repolarizing Ca2+-activated K+ currents, (2) upmodulation of persistent Na+ current (INaP), and (3) increase in electrotonic coupling through gap junctions. Our data are consistent only with the second hypothesis.
A preliminary report of these findings has been published previously in a recent abstract (Su et al., 1999).
MATERIALS AND METHODS
Slice preparation. All experimental protocols were approved by the Hebrew University Animal Care and Use Committee. Transverse hippocampal slices were prepared from adult Sabra rats (150–200 gm). Animals were anesthetized with ether and decapitated with a guillotine. The brain was removed and immediately immersed in ice-cold, oxygenated (95% O2, 5% CO2) dissection saline. The caudal two-thirds of one hemisphere (containing one hippocampus) were glued to the stage of a Vibratome (Campden Instruments). Transverse slices (400 μm thick) were cut from the region of the hemisphere containing the anterior hippocampus. The hippocampal portion was dissected out of each slice and transferred to an incubation chamber containing oxygenated saline at room temperature (21–24°C). For experimental recordings, the slices were transferred to an interface slice chamber and perfused from below with oxygenated (95% O2, 5% CO2) saline at 33.5°C. The upper surface of the slices was exposed to the humidified gas mixture. The slices were allowed to recover at least 1 hr before the experiment was started.
Solutions and drugs. The standard saline solution contained (in mm): NaCl 124, KCl 3.5, MgSO4 2, CaCl2 2, NaHCO3 26, and d-glucose 10, pH 7.3. Low-Ca2+ salines were prepared by equimolar substitution of CaCl2 with MnCl2. In some experiments, where indicated, CaCl2 was replaced with MgCl2. High-Ca2+saline was prepared by replacing MgSO4 with CaCl2.
All of the salines also contained the glutamate receptor antagonists 6-cyano-7-nitro-quinoxaline-2,3-dione (CNQX; 15 μm) and 2-amino-5-phosphono-valeric acid (50 μm) to block fast EPSPs and the GABAA receptor antagonist bicuculline methiodide (10 μm) to block fast IPSPs.
Salts and drugs were purchased from Sigma (St. Louis, MO), with the exception of CNQX (RBI, Natick, MA) and doxyl-stearic acid (Aldrich, Milwaukee, WI). Phenytoin was dissolved in 1N NaOH. The phorbol esters 4β-phorbol 12,13-dibutyrate (PDB) and 4α-phorbol 12,13-didecanoate (PDC) were dissolved in dimethyl sulfoxide (DMSO), and doxyl-stearic acid was dissolved in ethanol before it was added to the saline. Control salines contained equivalent concentrations of DMSO or ethanol (not >0.1%), which had no effects on the measured parameters.
Intracellular recordings. Current-clamp recordings from the somata of PCs in the CA1 pyramidal layer were made using sharp, K+-acetate-filled (4m) glass microelectrodes (60–90 MΩ). In some experiments, where indicated, 200 mm BAPTA, or 2% biocytin, were included in the filling solution. An active bridge circuit in the amplifier (Axoclamp 2A, Axon Instruments) allowed simultaneous injection of current and measurement of membrane potential. The bridge balance was carefully monitored and adjusted before each measurement. Bipolar platinum electrodes (50 μm) connected to a stimulator by an isolation unit were used for focal stimulation (1–20 V, 50–70 μsec) of pyramidal axons in the alveus. Neurons were identified as PCs if they responded with short latency spikes to antidromic stimulation and manifested strong spike frequency adaptation during a sustained depolarization. The PCs accepted for this study had stable resting potentials of at least −55 mV and overshooting action potentials.
Cell staining. For assessing the incidence of gap junctions among CA1 PCs, some neurons were injected with biocytin. After the experiment, the slices were fixed overnight in 4% paraformaldehyde, cut into thin (120 μm) sections, and incubated with avidin–biotin complex (Vectastain ABC elite kit, Vector Laboratories, Burlingame, CA). The stained cells were photographed at 200× magnification.
Data measurement and analysis. The intracellular signals were digitized and stored on a personal computer using a data acquisition system (TL-1, Axon Instruments). Off-line data analyses were performed using pCLAMP software (Axon Instruments).
To measure passive membrane properties, the PCs were injected with small (0.1–0.5 nA) 200 msec negative current pulses. The input resistance was provided by the slope of the linear regression line fitted through the linear portion of the steady-state voltage versus current amplitude plot. The apparent membrane time constant (τm) was taken as the slowest component (τ0) of multiexponential function fitted to the charging curve produced by application of a small negative current step (from onset to steady state), as suggested by Rall (1977). The fast spike afterhyperpolarization (AHP) was measured as the potential attained at the end of fast spike repolarization. A measure for the size of the spike and subthreshold afterdepolarizations (ADPs) was provided by the area delimited by the ADP waveform and resting membrane potential. This measure was sensitive to both ADP amplitude and duration. Inward rectification was evaluated from the nonlinear voltage responses to slow (0.9 sec long) depolarizing current ramps. The membrane potential at which the membrane began to rectify was determined by extrapolating a line fitted to the linear portion of the voltage–current relation. The initial and mean firing rates during a burst were calculated from the first interspike interval and from the mean interspike interval, respectively.
Averaged data are expressed as mean ± SD. The significance of the differences between the measured spike parameters was evaluated using Student's paired t test or Wilcoxon's paired-sample test with a significance level of 0.05. Correlation between the firing pattern and the incidence of dye coupling of the PCs was tested with the Spearman rank order correlation.
Classification of PC firing patterns. In this study we defined a burst as a cluster of three or more closely spaced action potentials, riding on a distinct slow depolarizing envelope. As suggested previously (Jensen et al., 1994, 1996; Azouz et al., 1997), PCs were classified into three groups according to their response to long (150–200 msec) depolarizing current pulses of increasing intensity (in steps of 30–100 pA). The variant firing patterns observed in CA1 PCs perfused with nominally Ca2+-free saline are portrayed in Figure1. Nonbursters generated accommodating trains of independent action potentials in response to all suprathreshold stimuli (Fig. 1A). High-threshold bursters (HTBs) generated burst-like responses only when subjected to strong suprathreshold stimuli (Fig. 1B), whereas low-threshold bursters (LTBs) fired in burst mode in response to threshold-straddling stimuli. To quantify the propensity to generate a burst, the latter heterogeneous group was further divided into three subgroups of increasing tendency for burst generation. Grade I LTBs fired only one spike in response to a brief (3–5 msec) depolarizing current pulse (Fig. 1C); grade II LTBs generated a burst also in response to a brief stimulus (Fig. 1D); and grade III LTBs also fired spontaneously in burst mode (Fig.1E). It should be stressed that this classification of CA1 PCs into five different subgroups does not imply a fundamental difference in mechanisms and is used only for descriptive purposes.
Fig. 1.

Variant firing patterns of CA1 PCs in low [Ca2+]o. Intracellular recordings were made in five PCs in slices perfused with nominally Ca2+-free saline. The PCs were injected with long (200 msec) and brief (3–5 msec) positive current pulses of increasing intensity (in steps of 50–100 pA). The top panels(a) in A–Dillustrate the firing patterns of four different PCs evoked by two long current pulses. The bottom panels(b) depict the respective responses to brief stimuli. The injected current pulses are indicated below the voltage traces. A, A nonburster (NB). This neuron generated nonclustered spike trains in response to long current pulses (a) and a single spike in response to brief current pulses (b). B, A high-threshold burster (HTB). This neuron generated an initial burst only in response to strong long current pulses (a) and a single spike in response to brief current pulses (b). C, Grade I low-threshold burster (LTB I). This neuron generated bursts in response to threshold and suprathreshold long current pulses (a), but a single spike in response to brief current pulses (b).D, Grade II LTB (LTB II). This neuron fired bursts in response to both long (a) and brief (b) current pulses. E, Grade III LTB (LTB III). In addition to firing bursts in response to any threshold stimuli (data not shown), this neuron displayed spontaneous, rhythmic burst firing. The spontaneous burst marked with an asterisk in the top trace of E is shown in the bottom trace on an expanded time scale. In this and the following Figures, dashed lines indicate truncated spikes. Resting potentials are provided to the left of topmost panels.
RESULTS
Effects of lowering [Ca2+]o on the firing patterns of CA1 PCs
In standard saline, the proportion of intrinsically bursting CA1 PCs was small and the bursting threshold in these bursters was high, as described previously (Jensen et al., 1994). Of 136 neurons examined, 112 (82%) were nonbursters and 24 (17%) were bursters (of which 20 were HTBs and 4 grade I LTBs). Lowering [Ca2+]o markedly increased the incidence of, and decreased the threshold for, intrinsic bursting in most PCs. In the representative experiment illustrated in Figure 2, changing from standard to 1.2 mm Ca2+ saline converted the nonbursting PC (Fig. 2A) into an HTB (Fig.2B). Further reduction of [Ca2+]o to 0.5 mm converted the PC into a grade I LTB (Fig.2C). Finally, in nominally Ca2+-free saline the PC changed into a grade II LTB (Fig. 2D).
Fig. 2.

Effect of lowering [Ca2+]o on the intrinsic firing pattern of a CA1 PC. Recordings were made in a slice perfused with salines containing different concentrations of Ca2+.A, In standard saline, the PC was a nonburster, as determined from its responses to 200 msec (a) and 3 msec (b) depolarizing current pulses.B, After a 30 min wash in 1.2 mmCa2+-saline, the PC became an HTB, firing a burst in response to a strong 200 msec depolarization (a).C, After a 30 min wash in 0.5 mmCa2+-saline, the PC became a grade I LTB, firing a burst also in response to a threshold 200 msec depolarization (a). D, Finally, after a 30 min wash in Ca2+-free saline, the PC became a grade II LTB, firing a burst in response to any threshold depolarization (a, b). E, The transition from regular firing to bursting was associated with a progressive decrease in the fast AHP and an increase in size of the spike ADP, as shown in the overlay of the expanded traces from b inA–D.
Similar concentration-dependent effects were seen in 15 of 20 PCs exposed to graded reductions in [Ca2+]o, although the final grade of bursting attained in Ca2+-free saline varied among the neurons. As summarized in Figure 3, lowering [Ca2+]o from 2 to 1.2 mm converted 8 of 17 nonbursters to HTBs. Further reduction to 0.5 mm converted many of the HTBs to LTBs. Subsequent exposure to Ca2-free saline further augmented the bursting propensity of these LTBs. Spontaneous intrinsic bursters (grade III LTBs) were encountered only in nominally Ca2+-free saline in 5 of the 20 PCs (25%) in this sample.
Fig. 3.

The firing patterns of CA1 PCs in different Ca2+o concentrations. The firing patterns of 20 CA1 PCs was monitored while [Ca2+]o was reduced in a stepwise manner from 2 mm (standard saline) to 0 mm(Ca2+-free saline). In control conditions, 17 PCs were nonbursters, 2 were HTBs, and only 1 was a grade I LTB. Thelines in the graph depict the direction of change in firing pattern within each subclass of PCs when [Ca2+]o is lowered to the next concentration. Thus, of the 17 native nonbursters, 8 PCs became HTBs in 1.2 mm Ca2+ and LTBs in 0.5 mm Ca2+, another 4 PCs became HTBs in 0.5 mm Ca2+, and the remaining 5 PCs did not change at all.
The waveform of intrinsic bursts in low [Ca2+]o was similar to that of native intrinsic bursts (Jensen et al., 1994, 1996). The number of intraburst spikes varied between three and seven across different cells (averaging 3.5 ± 1.1; n = 40) but was rather constant in any given cell. The initial and mean firing rates during a burst were 178.7 ± 28.8 and 134.7 ± 28.2 Hz (n = 40), respectively.
Effects of lowering [Ca2+]o on the spike ADP
Lowering [Ca2+]o did not significantly affect resting membrane potential, input resistance, or time constant, nor did it affect spike threshold or amplitude (Table1). However, it strongly modulated the spike afterpotentials, namely the fast AHP and ADP, in most PCs. As illustrated in Figure 2E, lowering [Ca2+]o reduced the fast AHP and augmented the size of the ADP in a concentration-dependent manner. On average, changing from standard to Ca2+-free saline reduced the fast AHP by 5.8 ± 4.0 mV and variably augmented the spike ADP by 63.2 ± 54.8% (n = 40) (Table 1).
Table 1.
Effects of low [Ca2+]o on intrinsic membrane properties of CA1 PCs
| 2 Ca2+ | 0 Ca2+ | |
|---|---|---|
| Resting VM, mV | −64.3 ± 3.8 | −64.6 ± 3.8 |
| RN, MΩ | 28.4 ± 9.1 | 30.2 ± 6.7 |
| τm, msec | 13.7 ± 5.2 | 14.6 ± 5.5 |
| Spike threshold, mV | −52.3 ± 3.8 | −52.5 ± 4.0 |
| Spike amplitude, mV | 87.0 ± 5.9 | 87.5 ± 6.6 |
| Fast AHP, mV | −56.0 ± 4.8 | −50.1 ± 5.0* |
| ADP, mV · msec | 171.7 ± 46.3 | 261.5 ± 117.1* |
Recordings from each PC (n = 40) were made first in standard saline (2 Ca2+) and then after ∼30 min of perfusion with nominally Ca2+-free saline (0 Ca2+). All entries are expressed as mean ±sd. Resting VM andRN are resting membrane potential and input resistance, respectively. τm is the passive time constant of the membrane measured at resting VM. Fast AHP is the membrane potential reached at the end of fast spike repolarization. ADP is the slow depolarizing potential that follows the fast AHP (see Materials and Methods).
Significantly different (p < 0.05) from control.
During the lowering of [Ca2+]o, the size of the spike ADP and the propensity to burst increased in parallel (Fig. 2). Accordingly, the final size of the spike ADP in Ca2+-free saline was significantly larger in bursters (275.9 ± 103.7 mV · msec;n = 33) than in nonbursters (196.4 ± 54.2 mV · msec; n = 7). These observations are consistent with the notion that intrinsic bursts are triggered by suprathreshold spike ADPs (Kandel and Spencer, 1961; Jensen and Yaari, 1996).
Effects of INaP blockers on low [Ca2+]o-induced intrinsic bursting
In pharmacologically untreated CA1 PCs, somatic intrinsic bursting is driven by INaP (Azouz et al., 1996). We examined whether a similar ionic mechanism underlies intrinsic bursting in low [Ca2+]o by testing its sensitivity to three drugs previously shown to reduceINaP in CA1 PCs, namely tetrodotoxin (TTX), the anti-epileptic drug phenytoin, and the protein kinase C (PKC) activator PDB. Representative results are shown in Figure4.
Fig. 4.
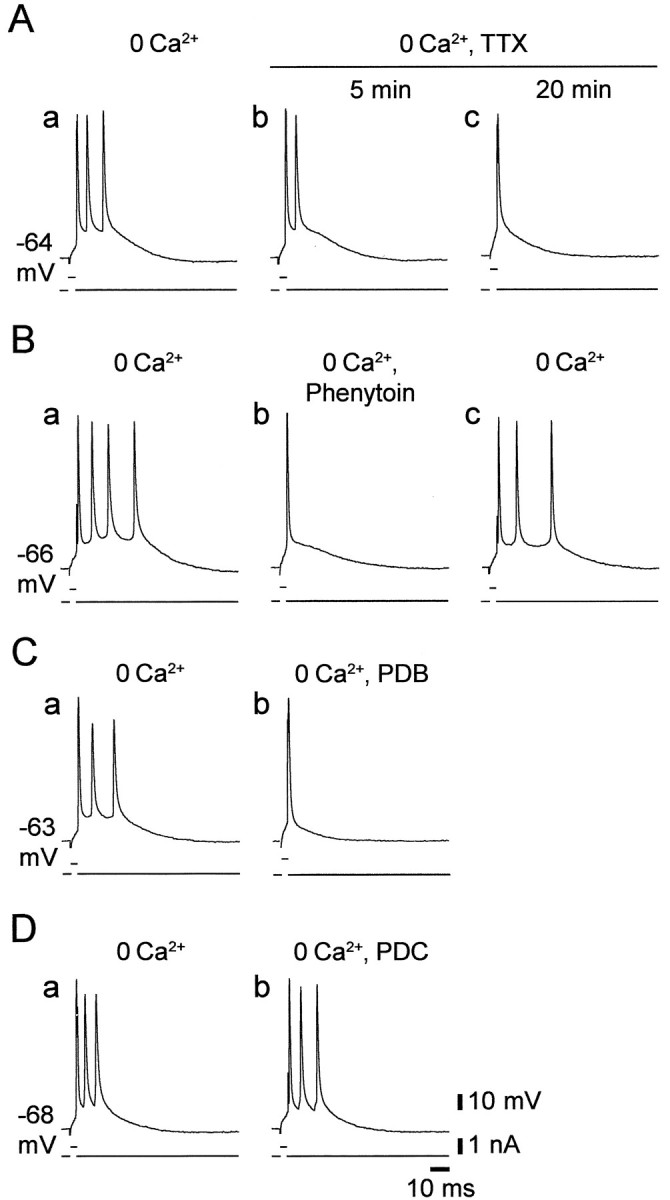
Blockers of INaPsuppress intrinsic bursting in low [Ca2+]o. Recordings were made from four grade II LTBs in Ca2+-free saline. Burst firing was evoked by 3 msec depolarizing stimuli.A, The responses of the neuron are shown before (a) and 5 and 20 min after (b andc, respectively) adding 0.1 μm TTX to the Ca2+-free saline, which caused a gradual suppression of intrinsic bursting. B, The responses of the neuron are shown before (a), 30 min after adding 50 μm phenytoin to the Ca2+-free saline (b), and 30 min after wash of phenytoin (c). C, The responses of the neuron are shown before (a) and 30 min after (b) adding 5 μm PDB to the Ca2+-free saline (b). Both phenytoin and PDB blocked the burst response completely but affected the first spike very little. D, The responses of the neuron are shown before (a) and 45 min after (b) adding 50 μm PDC to the Ca2+-free saline. No effect of this drug was noted.
Tetrodotoxin
This agent blocks both transient Na+current and INaP in CA1 PCs (French et al., 1990). As illustrated in Figure 4A, adding 0.1 μm TTX to the saline caused a progressive block of the burst responses concurrent with an increase in spike threshold. Longer exposures to TTX caused complete suppression of spike responses (data not shown). Similar results were consistently obtained in seven PCs.
Phenytoin
Phenytoin was shown to preferentially reduceINaP in CA1 PCs (Chao and Alzheimer, 1995; Segal and Douglas, 1997). We tested in nine PCs the effects of 50–100 μm phenytoin on low Ca2+-induced intrinsic bursting. In all cases, phenytoin reversibly suppressed intrinsic bursting, as illustrated in Figure 4B. Concurrently, phenytoin caused a small (3.36 ± 3.26 mV) but significant increase in spike threshold (from −54.0 ± 2.6 to −50.8 ± 2.9 mV;n = 9) but did not significantly affect spike amplitude. Unlike TTX, phenytoin did not suppress the initiation of single spikes even after a prolonged (>1 hr) exposure.
Phorbol esters
In CA1 PCs, activation of PKC suppressesINaP (Alroy et al., 1999). Application of 2–10 μm PDB, a phorbol ester that potently activates PKC (Castagna et al., 1982), suppressed low Ca2+-induced intrinsic bursting within 30–45 min (Fig. 4C). The effect was irreversible with 1 hr wash, but similar effects were seen in all nine bursters tested with this drug. Like phenytoin, PDB also caused a small (∼4 mV) increase in spike threshold but did not significantly affect spike amplitude. To control for unspecific phorbol ester effects, we exposed slices for up to 1 hr to 50 μm PDC, a phorbol ester that does not activate PKC (Castagna et al., 1982). In all cases (n = 4), PDC did not affect intrinsic bursting (Fig.4D).
Effects of INaP blockers on low [Ca2+]o-enhanced spike ADPs
In a fraction of the PCs, changing to Ca2+-free saline enhanced the spike ADP, but not sufficiently to trigger a burst in response to a brief stimulus (i.e., HTBs and grade I LTBs) (Fig.1B,C). We tested in these neurons the effects of the three INaP blockers on the enhanced spike ADPs. As illustrated in Figure5, the spike ADP was consistently suppressed by 0.1–0.2 μm TTX (Fig.5A) (n = 4), 50–100 μm phenytoin (Fig. 5B) (n = 9), and 2–10 μm PDB (Fig.5C) (n = 9), but not by 50 μm PDC (n = 3; data not shown). These results are consistent with the notion that the enhanced spike ADPs in low [Ca2+]o are driven primarily by INaP.
Fig. 5.
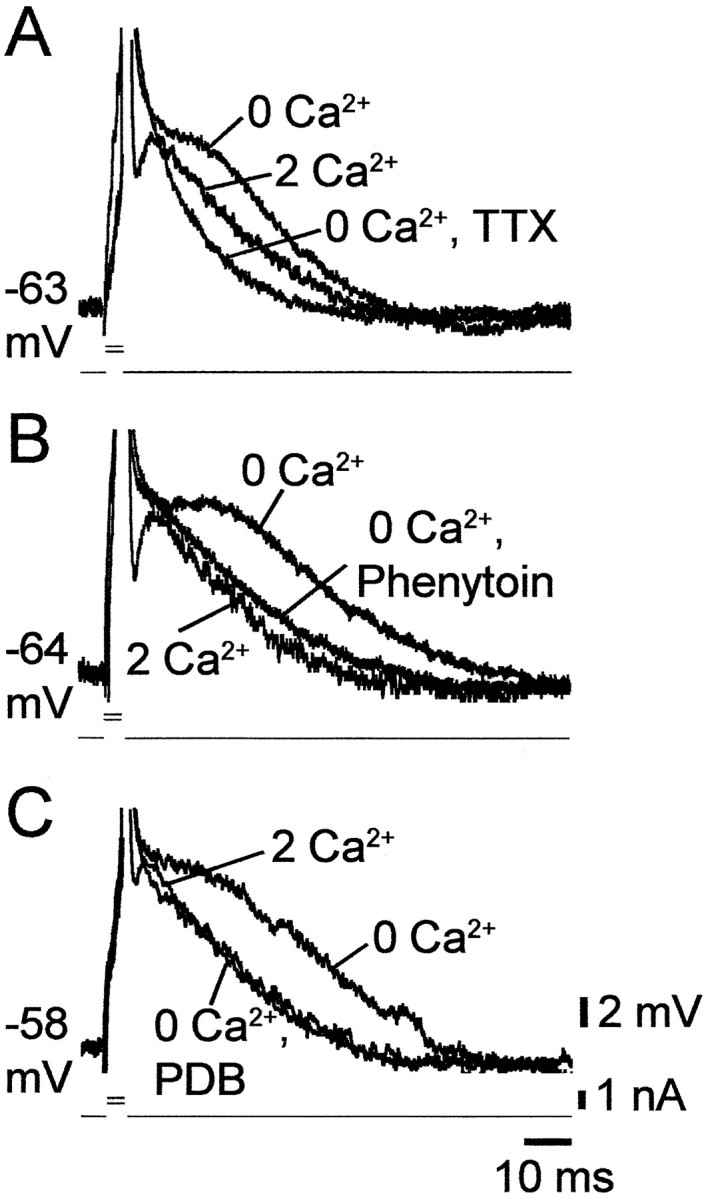
Blockers of INaPsuppress spike ADPs in low [Ca2+]o.A–C, Recordings of single spikes evoked by 3 msec stimuli were made in three CA1 PCs in slices perfused sequentially with standard saline (2 Ca2+), Ca2+-free saline (0 Ca2+), and Ca2+-free saline containing a drug known to suppressINaP. In the three PCs the native spike was followed by an active ADP. Lowering [Ca2+]o caused spike broadening, blocked the fast AHP, and enhanced the active ADP. The latter potential was completely depressed after 20 min exposure to 0.2 μm TTX (A). Likewise, it was markedly reduced after 30 min exposure to 50 μmphenytoin (B) or 5 μm PDB (C).
Effects of INaP blockers on low [Ca2+]o-enhanced subthreshold ADPs
When CA1 PCs are depolarized with a brief current pulse to near threshold potential, the membrane potential decays back to resting potential more slowly than expected from the membrane time constant. These subthreshold ADPs are readily blocked by TTX, but not by Ca2+ channel blockers (Azouz et al., 1996), suggesting that they are generated byINaP, the activation threshold of which is slightly more negative than spike threshold (French et al., 1990). We tested the effects of lowering [Ca2+]o on these potentials. In all cases (n = 22), as illustrated in Figure 6, deleting Ca2+o markedly enhanced the subthreshold ADPs. Thus, the waveform of these potentials increased significantly from 134.9 ± 50.3 to 333.5 ± 71.2 mV · msec after changing from standard to Ca2+-free saline. Consistent with the notion that they are generated byINaP, these potentials were readily blocked by 0.1–0.2 μm TTX (Fig.6A) (n = 4), 50–100 μm phenytoin (Fig. 6B) (n = 9), and 2–10 μm PDB (Fig.6C) (n = 9).
Fig. 6.
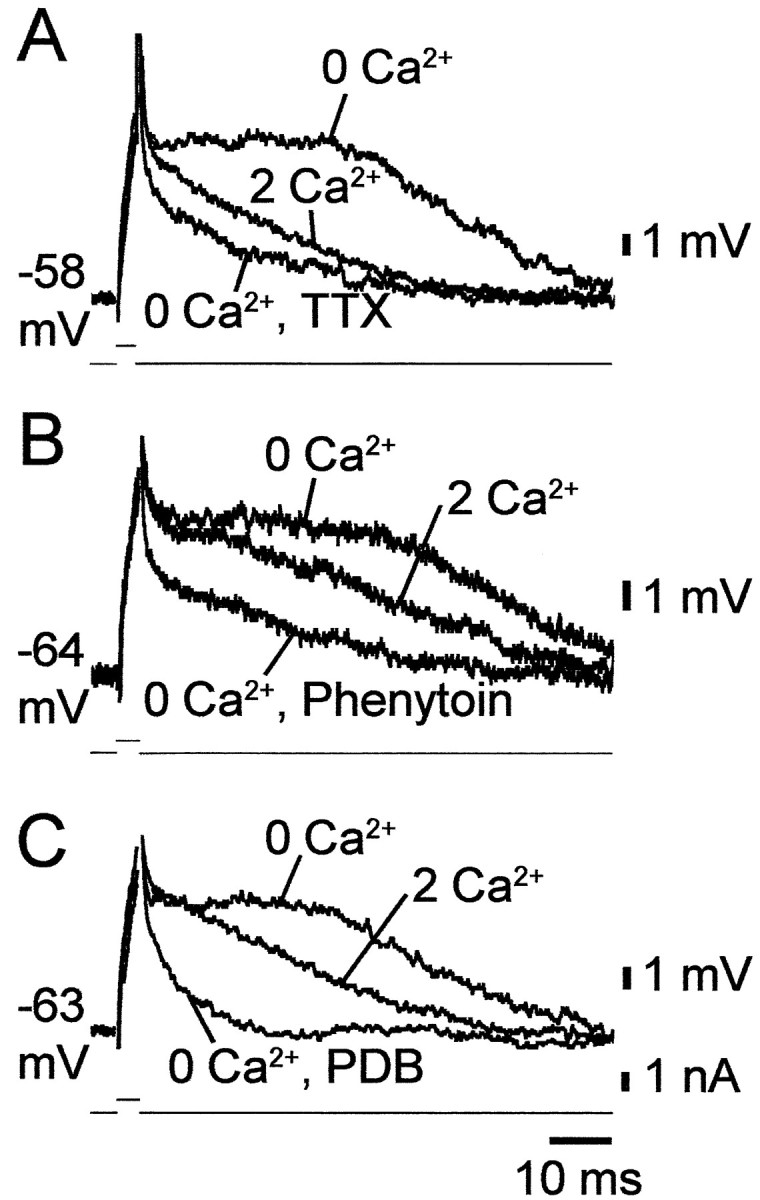
Blockers of INaPsuppress subthreshold ADPs in low [Ca2+]o. A–C, Recordings of subthreshold ADPs evoked by 3 msec threshold-straddling stimuli were made in three CA1 PCs in slices perfused sequentially with standard saline (2 Ca2+), Ca2+-free saline (0 Ca2+), and Ca2+-free saline containing a drug known to suppressINaP. In the three PCs, when the stimulus failed to trigger a spike, it evoked a slow depolarizing potential that declined more slowly to resting potential than expected from passive membrane charging. Lowering [Ca2+]o induced a subthreshold ADP in these PCs. The latter potential was suppressed by 0.2 μm TTX (20 min; A), 50 μm phenytoin (30 min; B), and 2 μm PDB (40 min; C).
Effects of Ca2+ current block with Ni2+
The results described above established that low [Ca2+]o-induced intrinsic bursting is most likely driven byINaP. However, they did not indicate the mechanism by which lowering [Ca2+]o augments the slow potentials that trigger the burst discharge. One possibility is that lowering [Ca2+]o reduces outward Ca2+-activated K+ currents, thereby increasing the depolarizing impact of INaP (Azouz et al., 1996). Alternatively, lowering [Ca2+]o may upmodulate INaP itself. The first hypothesis predicts that blocking Ca2+channels pharmacologically also would induce intrinsic bursting. We tested this notion in four nonbursters by adding 1 mm Ni2+ (in replacement of 1 mmMg2+) to the standard saline that perfused the slices. Consistent results were obtained in all cases and are illustrated in Figure 7. Adding Ni2+ suppressed the fast AHP (Fig. 7, compare a in A and B), as expected from block of the fast Ca2+-activated K+ current (Ic) (Storm, 1987). Likewise, it suppressed the slow AHP that follows a train of several spikes, which is generated by the slow Ca2+-activated K+ current (IAHP) (Madison and Nicoll, 1984), as well as the associated spike frequency accommodation (Fig. 7, compareb in A and B). Despite the apparent block of these K+ currents, the nonbursting pattern of the PC did not change (Fig. 7, comparec in A and B). Yet, bursting was readily induced in the Ni2+-containing saline by deleting Ca2+ from the solution (Fig. 7C, c).
Fig. 7.

Block of Ca2+-activated K+ currents with Ni2+ does not induce intrinsic bursting. Recordings were obtained from a native nonbursting CA1 PC. Shown are the responses of the neuron to 4 msec (a), 400 msec (b), and 200 msec depolarizing stimuli (c) in three conditions.A, In standard saline, the solitary spike was followed by distinct fast AHP and ADP (a), and repetitive firing was followed by a medium AHP and slow AHPs (b). B, After the change to standard saline containing 1 mm Ni2+, the fast and slow AHPs were suppressed, whereas the medium AHP was preserved (a, b). Despite the reduction in spike frequency accommodation (b), the nonbursting firing pattern of the PCs was unaltered (c). C, Changing to Ca2+-free saline containing 1 mm Ni2+(b) converted the PC to a grade I LTB (c).
Effects of buffering intracellular Ca2+with BAPTA
As an additional test for the role of Ca2+-activated currents in shaping the firing mode of CA1 PCs, we injected seven neurons with the Ca2+ chelator BAPTA by applying negative current pulses (up to 0.5 nA; 10–30 min). The effects of BAPTA were similar in all cases. As illustrated in Figure8, BAPTA injection expectedly suppressed the fast AHP (Fig. 8, compare a in A andB), the slow AHP, and the associated spike frequency accommodation (Fig. 8, compare b in A andB). Despite these effects, BAPTA injection did not induce intrinsic bursting (Fig. 8, compare c in A andB). However, bursting was readily induced in the BAPTA-injected neuron by removing Ca2+o (Fig.8C, c).
Fig. 8.

Buffering of intracellular Ca2+concentration with BAPTA does not induce intrinsic bursting. Recordings were obtained from a native nonbursting CA1 PC. Shown are the responses of the neuron to 4 msec (a), 400 msec (b), and 200 msec depolarizing stimuli (c) in three conditions.A, In standard saline, the solitary spike was followed by distinct fast AHP and ADP (a), and repetitive firing was followed by a medium and slow AHPs (b). B, Injecting BAPTA into the neuron suppressed the fast and slow AHPs and reduced spike frequency accommodation (a, b), but the nonbursting firing pattern of the PC was unaltered (c).C, Changing to Ca2+-free saline converted the PC to a grade I LTB (c).
Effect of low [Ca2+]o on slow spikes
Perfusing CA1 PCs with standard saline containing 10 mm TEA, which blocks various voltage-gated K+ currents in these neurons (e.g., delayed rectifier and M-current) (Storm, 1990), induces compound slow (duration 100–400 msec) and fast spike responses to brief depolarizing stimuli. Deleting Ca2+o converts the slow spikes to “plateau” potentials lasting 1–2 sec (García-Muñoz et al., 1993; Jensen et al., 1996). These potentials are blocked by TTX at low concentrations that do not affect the fast spikes, indicating that they are generated byINaP. Accordingly, they are also blocked by phenytoin (H. Su and Y. Yaari, unpublished observations) and PKC activation (Alroy et al., 1999).
We tested whether blocking Ca2+-activated K+ currents with Ni2+ also induces plateau potentials in TEA-containing saline. A representative experiment is illustrated in Figure 9A. In standard saline containing 10 mm TEA, a brief depolarizing pulse evoked a complex of several fast spikes riding on a slow spike, followed by a slow AHP (Fig. 9A, a). Adding 1 mm Ni2+ abolished the slow AHP and slightly enhanced the slow spike, but did not induce a plateau potential (Fig. 9A, b). However, deleting Ca2+ from the Ni2+-containing saline induced a prolonged (∼1.8 sec) plateau potential (Fig. 9A, c). This slow potential was blocked entirely by 0.2 μmTTX before the primary fast spike was affected (Fig. 9A,d). Similar results were obtained in all experiments (n = 4).
Fig. 9.

Lowering of [Ca2+]o, but not block of Ca2+-activated K+ currents, induces TTX-sensitive plateau potentials. A, In this experiment the slice was perfused with standard saline containing 10 mm TEA to block several voltage-gated K+currents. A constant negative current was injected into the cell to maintain the native resting membrane potential (−63 mV). Suprathreshold responses were evoked by 5 msec positive current pulses. Salines were exchanged every 30 min. In standard saline containing TEA, the PC generated a compound response, consisting of fast spikes riding on a slow spike, and followed by a slow AHP (a). Adding 1 mm Ni2+ to the saline suppressed the slow AHP completely and slightly prolonged the slow spike (b). Deleting Ca2+ from the saline induced a prolonged plateau potential lasting ∼1.8 sec (c), which was suppressed by 0.2 μmTTX within 10 min of exposure (d).B, In another slice perfused with salines containing 10 mm TEA, [Ca2+]o was reduced in a stepwise manner every 30 min. The responses of the PC to 5 msec pulse stimulation are shown in standard saline (a), 1.2 mm Ca2+saline (b), 0.5 mmCa2+ saline (c), and Ca2+-free saline (d). The control response comprised fast and slow spikes (a). The duration of the plateau potential increased as [Ca2+]o decreased (b–d).
These results indicated that blocking Ca2+-activated K+ currents itself is not sufficient to induce plateau potentials in TEA-containing saline. Rather, Ca2+o removal per se is required for this effect. Indeed, we found that even partial Ca2+o replacement is effective in prolonging the slow spike component in TEA-containing saline (n = 4). As illustrated in Figure 9B, reducing [Ca2+]oin a stepwise manner caused a concentration-dependent growth of the plateau potential (Fig. 9B, a–d).
Effect of low [Ca2+]o on inward rectification
CA1 PCs display inward rectification, manifested as an apparent increase in input resistance near action-potential threshold (Hotson et al., 1979). A component of inward rectification persists after suppression of Ca2+ currents and is blocked by TTX, indicating that it is caused by activation ofINaP (Benardo et al., 1982). If low [Ca2+]oupmodulates INaP, then it should augment TTX-sensitive inward rectification. We tested this notion in five slices perfused with saline containing 1 mmNi2+ by injecting slow current ramps (duration 0.9 sec) of increasing magnitude into the PCs. In the example illustrated in Figure 10, the current was linearly increased from 0 to 160 pA, causing the neuron to depolarize from −72 mV to spike threshold potential (approximately −60 mV). In standard saline containing Ni2+, inward rectification was seen at membrane potentials more positive than −63.8 mV (Fig.10A, a). After changing to Ni2+-containing Ca2+-free saline, inward rectification occurred already at −67.1 mV (Fig. 10, trace b). Adding 1 μm TTX completely blocked the inward rectification (Fig. 10, trace c). Similar results were obtained in all five experiments. On average, the membrane potential at which the membrane began to rectify shifted from −62.0 ± 2.9 to −67.2 ± 2.0 mV after removal of Ca2+o (a significant difference of −5.2 ± 1.4 mV).
Fig. 10.

Lowering [Ca2+]oenhances TTX-sensitive inward rectification. Recordings were obtained from a native nonbursting CA1 PC. The neuron was injected with current ramps from 0 to 160 pA (duration 0.9 sec). Plotted are the subthreshold voltage responses of the neuron to these ramps. Each trace is an average of four consecutive responses. The slice was first perfused with standard saline containing 1 mmNi2+ to block voltage-sensitive Ca2+ currents. In these conditions (trace a), the membrane potential first depolarized linearly from its resting level (−72 mV) until −63.8 mV, after which it depolarized exponentially until spike threshold was attained (at −60 mV). After 30 min of perfusion with Ni2+-containing Ca2+-free saline (trace b), the nonlinear response began already at −67.1 mV and was steeper than in control. Addition of 0.1 μm TTX (trace c) completely suppressed the nonlinear subthreshold response of the neuron in 10 min.
Effects of increasing [Ca2+]o on native intrinsic bursting and slow spikes
In three experiments we tested how elevating [Ca2+]o affects native intrinsic bursting. A representative experiment is illustrated in Figure 11A. In standard saline, strong positive current pulses evoked an initial burst response of three spikes (Fig. 11A, a). Raising [Ca2+]o to 4 mm (by substituting 2 mmMg2+ with 2 mmCa2+) blocked the burst response at all stimulation intensities (Fig. 11A, b). This effect reversed after wash with standard saline (Fig.11A, c). Similar results were obtained in two additional PCs. The suppression of intrinsic bursting by doubling [Ca2+]o was associated with a small (∼3 mV) increase in spike threshold.
Fig. 11.

Raising [Ca2+]oblocks native intrinsic bursting and slow spikes. A, In this experiment recordings were obtained from a native HTB. Shown are the responses of the neuron to 200 msec depolarizing stimuli in standard saline (a), 30 min after switching to saline containing 4 mm Ca2+(b), and 30 min after changing back to standard saline (c). Raising [Ca2+]o reversibly increased the threshold for evoking spikes and suppressed the initial burst response to the depolarizing pulses. B, In another slice perfused with salines containing 10 mm TEA, [Ca2+]o was increased to 4 mm. The responses of the PC to 5 msec pulse stimulation are shown in standard saline (a), after 20 min in 4 mm Ca2+ saline (b), and after 30 min wash back to standard saline (c). The slow spike was reversibly suppressed by doubling [Ca2+]o(a–c).
We also tested the effects of raising [Ca2+]o on the slow spikes in TEA-containing saline (n = 3). As shown in Figure 11B, raising [Ca2+]o to 4 mm selectively and reversibly blocked the slow spike component but spared the fast spike (Fig. 11B,a–c). Consequently, the slow AHP also was reversibly blocked by doubling [Ca2+]o. Similar results were obtained in all three experiments.
Dye coupling and firing patterns of CA1 PCs in low [Ca2+]o
Previous work in CA1 PCs suggested that bursters are more likely to be electrically coupled to each other via gap junctions than nonbursters (Church and Baimbridge, 1991). It was suggested that there may be a causal relationship between electrical coupling and intrinsic bursting. Presumably, the spread of spikes within a small network of electrically coupled neurons can generate a burst response in each neuron, which would appear as an “intrinsic” burst. Because lowering [Ca2+]oreportedly increases gap junctional communication and, by implication, electrotonic coupling between CA1 PCs (Perez Velazquez et al., 1994), we examined whether this effect may contribute to low [Ca2+]o-induced intrinsic bursting. To this end, we injected 46 PCs with the dye tracer biocytin during perfusion with nominally Ca2+-free saline. Of these, nine neurons (21.4%) were found to be dye coupled to one, two, or three other PCs (mean number of PCs in a coupled cluster 2.6 ± 0.9). However, we found no significant correlation between the firing pattern of the neuron and the incidence of dye coupling (Table2). Figure12 illustrates two exemplary biocytin-injected neurons, a grade II LTB (Fig. 12A,a and b), which was not coupled to other neurons (Fig. 12A, c), and a regular firing cell (Fig. 12B, a and b), which was coupled to two other PCs (Fig. 12B,c).
Table 2.
Relation between firing pattern and dye coupling in CA1 PCs perfused with Ca2+-free saline
| PC type | n | Number of dye-coupled clusters | Incidence of dye coupling (%) |
|---|---|---|---|
| NB | 10 | 2 | 20.0 |
| HTB | 10 | 3 | 30.0 |
| Grade I LTB | 9 | 2 | 22.2 |
| Grade II LTB | 8 | 2 | 25.0 |
| Grade III LTB | 9 | 1 | 11.1 |
| Total | 46 | 10 | 21.7 |
The biocytin-stained CA1 PCs (n = 46) were grouped according to their firing mode in nominally Ca2+-free saline. For each subset of PCs, the number of dye-coupled clusters observed and the overall incidence of dye coupling are provided. NB, Nonburster; HTB, high-threshold burster; LTB, low-threshold burster.
Fig. 12.
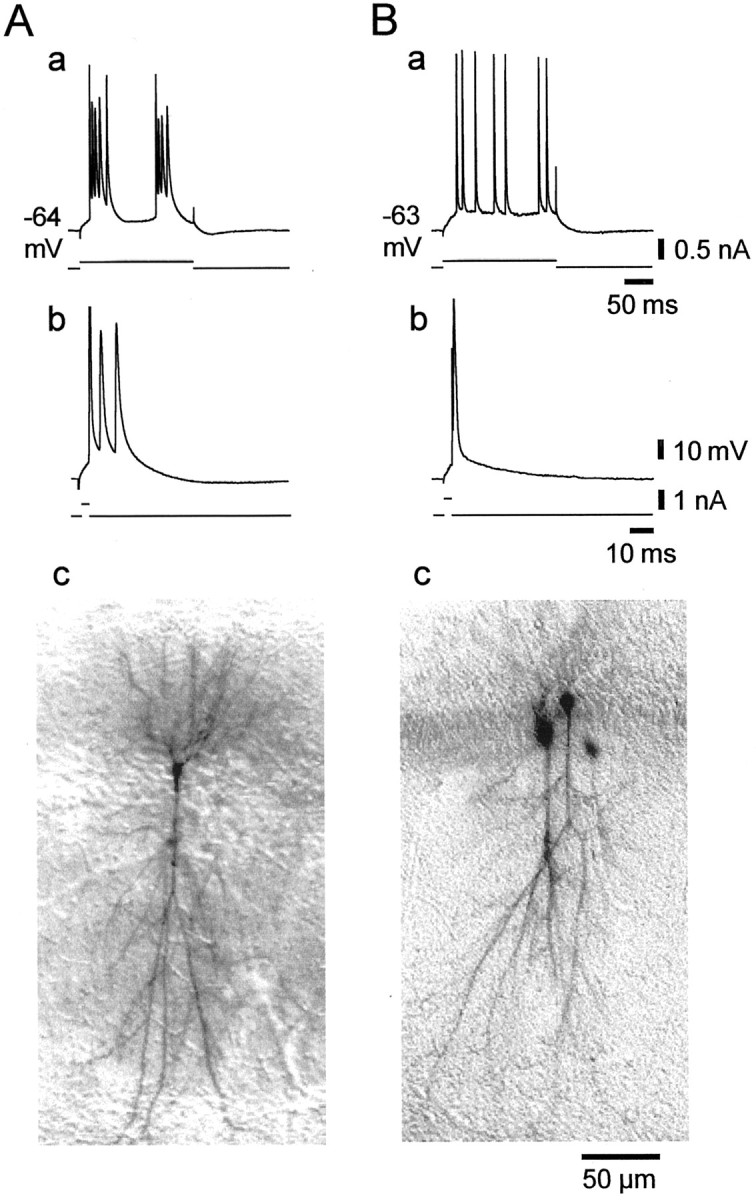
Dissociation between dye coupling and intrinsic bursting in low [Ca2+]o.A, B, Recordings from two native nonbursters 30 min after changing to Ca2+-free saline. Both PCs were stained with biocytin to detect dye coupling. A, This neuron became a grade II LTB in low [Ca2+]o, as is evident from its burst responses to 200 msec (a) and 3 msec depolarizing current pulses (b). As shown in the photomicrograph of the neuron (c), it was not dye coupled to other cells. B, This neuron remained nonbursting in low [Ca2+]o (a,b). Biocytin injection into this cell resulted in the staining of three PCs (c), suggesting that the neuron formed gap junctions with one or two other PCs.
It has been argued recently that in some neurons dye coupling may not be a reliable indicator for the presence of electrical coupling via gap junctions (Gibson et al., 1999). Therefore, we also tested the effect of the gap junctional blocker doxyl-stearic acid (Strata et al., 1997;Zahs and Newman, 1997) on low [Ca2+]o-induced intrinsic bursting. This drug was chosen because unlike other commonly used gap junction blockers tested (propionate, octanol, halothane, and carbenoxolone), it exerted no deleterious effects on intrinsic neuronal properties at concentrations that block neuronal gap junctions (Su and Yaari, unpublished observations). Addition of 50 μmdoxyl-stearic acid to the Ca2+-free saline did not affect intrinsic bursting during 45–60 min of perfusion in all cases (n = 6; data not shown).
Taken together, these data do not support a role for electrical coupling in the generation of intrinsic bursting in low [Ca2+]o.
DISCUSSION
The main finding in this study is that Ca2+o regulates the intrinsic firing mode of CA1 PCs. In standard saline containing 2 mm Ca2+, only a small fraction of these neurons burst-fire in response to somatic depolarization. Lowering [Ca2+]oincreases, whereas elevating [Ca2+]o decreases, the propensity of these neurons to generate bursts. We also show that intrinsic bursts induced by lowering [Ca2+]o are driven by INaP. The simplest hypothesis that accounts for our findings is that Ca2+acting extracellularly exerts a depressant effect on noninactivating Na+ channels that generateINaP. Accordingly, decreases in [Ca2+]o enhance intrinsic bursting via increases inINaP.
Ionic mechanism of intrinsic bursts in low [Ca2+]o
Several slow inward currents have been implicated in the generation of somatic bursts in cortical neurons, including voltage-sensitive Ca2+ (Wong and Prince, 1981; Foehring and Waters, 1991; Deisz, 1996) and Na+ currents (Franceschetti et al., 1995;Azouz et al., 1996; Mattia et al., 1997), and a Ca2+-activated cationic current (Kang et al., 1998). In CA1 PCs in vitro, native somatic bursting, as well as that induced by elevating [K+]o (Jensen et al., 1994), are insensitive to block of Ca2+ currents. Rather, they are suppressed by blockers of INaP, implicating the latter current in their generation (Azouz et al., 1996;Alroy et al., 1999). Somatic bursting can be induced in these neurons also by exposure to millimolar concentrations of 4-aminopyridine (4-AP), which blocks several K+conductances (Magee and Carruth, 1999). Unlike native bursting, 4-AP-induced bursts are suppressed by Ca2+channel blockers, consistent with the view that they are generated by dendritic Ca2+ spikes (Magee and Carruth, 1999).
Two lines of evidence suggest that somatic bursting in low [Ca2+]o is driven by INaP. First, in most experiments bursting was readily induced in nominally Ca2+-free saline containing 2 mm Mn2+. Blocking Ca2+ channels with Mn2+ in addition to deleting Ca2+ ensured that neither Ca2+ currents nor Na+ currents through Ca2+ channels (Lux et al., 1990) were involved in bursting. Thus, the only slow inward current available in this saline was INaP. Second, intrinsic bursting induced by low [Ca2+]o was readily suppressed by TTX, phenytoin, and PDB. Although these drugs interact with voltage-gated Na+ channels in different ways (Catterall, 1999), they share a common depressant effect on INaP.
We have examined three hypotheses that explain whyINaP becomes more effective in producing intrinsic bursting as [Ca2+]o is lowered: (1) suppression of opposing Ca2+-activated K+ currents, (2) upmodulation ofINaP itself, and (3) increase in electrotonic coupling through gap junctions. These hypotheses will be assessed in the following sections.
Suppression of Ca2+-activated K+ currents
In CA1 PCs, IC activates during the action potential and contributes largely to the late phase of spike repolarization (Storm, 1987). Thus, the density ofIC influences the waveform of the spike ADP and may determine whether it will progress to a burst. We compared the effects of three experimental manipulations that causeIC suppression, namely, lowering [Ca2+ ]o, blocking Ca2+ currents with extracellular Ni2+, and preventing intracellular Ca2+ accumulation with BAPTA. As expected, all of these manipulations reduced theIC-dependent fast AHP. However, only low [Ca2+ ]oinduced intrinsic bursting, indicating that reduction inIC (or any other Ca2+-activated current) by itself is not sufficient to invoke this firing pattern in CA1 PCs. Rather, lowering [Ca2+ ]o exerts additional effects that are crucial for the induction of bursting.
Upmodulation of INaP
An attractive possibility is that low [Ca2+ ]oupmodulates INaP. We tested this possibility by monitoring the effects of Ca2+o removal on TTX-sensitive plateau potentials and inward rectification that are generated by INaP. Both the plateau potentials and inward rectification were enhanced in this condition, supporting the view that lowering [Ca2+]o upmodulatesINaP.
In a previous study in supraoptic hypothalamic neurons, lowering [Ca2+ ]o enhancedINaP and the associated spontaneous intrinsic bursting (Li and Hatton, 1996). However, replacement of Ca2+ with other divalent cations prevented these effects, suggesting that they are caused by a net decrease in extracellular divalent ion concentration (which would shiftINaP activation to more positive potentials by unscreening the external negative membrane surface potential) rather than by a specific decrease in [Ca2+ ]o. In our experiments, the concentrations of divalent cations were maintained constant at 4 mm in all salines, and the induction of intrinsic bursting occurred regardless of the species of divalent cations used to replace Ca2+o. These data suggest that in CA1 PCs, Ca2+o decreasesINaP by a mechanism that involves its selective binding to membrane receptors. The receptors that bind Ca2+o may be the Na+ channels themselves (Armstrong and Cota, 1991) or G-protein-coupled Ca2+-sensing receptors (Yamaguchi et al., 2000), such as metabotropic glutamate receptors (Kubo et al., 1998), that may modulate INaP via second messenger cascades. The latter mechanism is less likely, because 100 μm neomycin (n = 3) and 10 nm gadolinium (n = 3), which activate Ca2+-sensing receptors (Xiong and MacDonald, 1999), failed to reverse the induction of intrinsic bursting by low [Ca2+ ]o(Su and Yaari, unpublished observations).
Increase in electrical coupling through gap junctions
We also examined whether augmentation of electrotonic coupling via gap junctions contributes to the appearance of intrinsic bursting in low [Ca2+]o. Both experimental (Getting and Willows, 1974) and theoretical (Sherman and Rinzel, 1992) data suggest that electrotonic coupling can cause neurons to burst. Indeed, it was shown that raising extracellular pH enhances both dye coupling (and by implication, electrotonic coupling) and intrinsic bursting in CA1 PCs and that only PCs coupled to other neurons display bursting behavior (Church and Baimbridge, 1991). Although we found significant dye coupling between CA1 PCs bathed in low [Ca2+]o, we could not substantiate a role for electrotonic coupling in the bursting behavior. First, the incidence of dye coupling among bursters and nonbursters was the same. Second, the gap junction blocker doxyl-stearic acid did not affect intrinsic bursting. It should be noted that both the overall incidence of dye coupling (21.4%) and the size of a coupled aggregate (two to four cells) in Ca2+-free saline were much lower than in high pH saline (88% and two to nine cells, respectively) (Church and Baimbridge, 1991). Thus, if electrical coupling via gap junctions and intrinsic bursting are causally associated, this mechanism may be more important for intrinsic bursting in high extracellular pH than in low [Ca2+]o.
Functional implications
Despite the strict homeostatic control of [Ca2+]o in the brain, neuronal activity can cause substantial decreases in [Ca2+]o (Heinemann et al., 1977). In hippocampal slices perfused with 2 mmCa2+ saline, repetitive orthodromic activation of CA1 PCs at θ frequencies (5–10 Hz) decreases [Ca2+]o down to 1.4 mm (Benninger et al., 1980). This decrease, in combination with an associated increase in [K+]o (up to 12 mm), would convert many nonbursting PCs into intrinsic bursters. This may explain why θ frequency orthodromic activation of CA1 PCs also leads to the development of postsynaptic bursting (Thomas et al., 1998).
The induction of intrinsic bursting by activity-dependent decreases in [Ca2+]o also may be germane to the genesis of epileptic seizures. In vivorecordings of [Ca2+]o in experimental models of epilepsy have shown dramatic decreases in [Ca2+]o (down to 0.2 mm) during seizure activity (Pumain et al., 1985). These decreases would be expected to enhance intrinsic bursting, which in turn would contribute to the explosive development and spread of seizure activity (Jensen and Yaari, 1997). Consistent with this view is the finding that lowering [Ca2+]o to 0.2 mm or less induces spontaneous population bursts in rat hippocampal slices despite the block of chemical synaptic transmission (Jefferys and Haas, 1982; Taylor and Dudek, 1982; Yaari et al., 1983).
Footnotes
This work was supported by the Bundesministerium für Bildung Wissenschaft (BMBF), the Israel Ministry of Science (MOS), and the Israel Science Foundation (ISF) administered by the Israel Academy of Sciences and Humanities. E.D.K. was supported by a fellowship from Teva Pharmaceuticals Inc.
Correspondence should be addressed to Dr. Yoel Yaari, Department of Physiology, Hebrew University School of Medicine, P.O. Box 12272, Jerusalem 91121, Israel. E-mail:yaari@md2.huji.ac.il.
REFERENCES
- 1.Alroy G, Su H, Yaari Y. Protein kinase C mediates muscarinic block of intrinsic bursting in rat hippocampal neurons. J Physiol (Lond) 1999;517:71–79. doi: 10.1111/j.1469-7793.1999.0071r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong CM, Cota G. Calcium ion as a cofactor in Na channel gating. Proc Natl Acad Sci USA. 1991;88:6528–6531. doi: 10.1073/pnas.88.15.6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azouz R, Jensen MS, Yaari Y. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol (Lond) 1996;492:211–223. doi: 10.1113/jphysiol.1996.sp021302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azouz R, Alroy G, Yaari Y. Modulation of endogenous firing patterns by osmolarity in rat hippocampal neurones. J Physiol (Lond) 1997;502:175–177. doi: 10.1111/j.1469-7793.1997.175bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benardo LS, Masukawa LM, Prince DA. Electrophysiology of isolated hippocampal pyramidal dendrites. J Neurosci. 1982;2:1614–1622. doi: 10.1523/JNEUROSCI.02-11-01614.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benninger C, Kadis J, Prince DA. Extracellular calcium and potassium changes in hippocampal slices. Brain Res. 1980;177:165–172. doi: 10.1016/0006-8993(80)90502-8. [DOI] [PubMed] [Google Scholar]
- 7.Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. 1982;257:7847–7851. [PubMed] [Google Scholar]
- 8.Catterall WA. Molecular properties of brain sodium channels: an important target for anticonvulsant drugs. Adv Neurol. 1999;79:441–456. [PubMed] [Google Scholar]
- 9.Chao TI, Alzheimer C. Effects of phenytoin on the persistent Na+ current of mammalian CNS neurones. NeuroReport. 1995;6:1778–1780. doi: 10.1097/00001756-199509000-00017. [DOI] [PubMed] [Google Scholar]
- 10.Church J, Baimbridge KG. Exposure to high-pH medium increases the incidence and extent of dye coupling between rat hippocampal CA1 pyramidal neurons in vitro. J Neurosci. 1991;11:3289–3295. doi: 10.1523/JNEUROSCI.11-10-03289.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deisz RA. A tetrodotoxin-insensitive sodium current initiates burst firing of neocortical neurons. Neuroscience. 1996;70:341–351. doi: 10.1016/0306-4522(95)00362-2. [DOI] [PubMed] [Google Scholar]
- 12.Foehring RC, Waters RS. Contributions of low-threshold calcium current and anomalous rectifier (Ih) to slow depolarizations underlying burst firing in human neocortical neurons in vitro. Neurosci Lett. 1991;124:17–21. doi: 10.1016/0304-3940(91)90812-8. [DOI] [PubMed] [Google Scholar]
- 13.Franceschetti S, Guatteo E, Panzica F, Sancini G, Wanke E, Avanzini G. Ionic mechanisms underlying burst firing in pyramidal neurons: intracellular study in rat sensorimotor cortex. Brain Res. 1995;696:127–139. doi: 10.1016/0006-8993(95)00807-3. [DOI] [PubMed] [Google Scholar]
- 14.French CR, Sah P, Buckett KJ, Gage PW. A voltage-dependent persistent sodium current in mammalian hippocampal neurons. J Gen Physiol. 1990;95:1139–1157. doi: 10.1085/jgp.95.6.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujita Y. Two types of depolarizing after-potentials in hippocampal pyramidal cells of rabbits. Brain Res. 1975;94:435–446. doi: 10.1016/0006-8993(75)90227-9. [DOI] [PubMed] [Google Scholar]
- 16.García-Muñoz A, Barrio LC, Buño W. Membrane potential oscillations in CA1 hippocampal pyramidal neurons in vitro: intrinsic rhythms and fluctuations entrained by sinusoidal injected current. Exp Brain Res. 1993;97:325–333. doi: 10.1007/BF00228702. [DOI] [PubMed] [Google Scholar]
- 17.Getting PA, Willows AOD. Modifications of neuron properties by electrotonic synapses. II. Burst formation by electrotonic synapses. J Neurophysiol. 1974;37:858–868. doi: 10.1152/jn.1974.37.5.858. [DOI] [PubMed] [Google Scholar]
- 18.Gibson JR, Beierlein M, Connors BW. Two networks of electrically coupled inhibitory neurons in neocortex. Science. 1999;402:75–79. doi: 10.1038/47035. [DOI] [PubMed] [Google Scholar]
- 19.Heinemann U, Lux HD, Gutnick MJ. Extracellular free calcium and potassium during paroxysmal activity in the cerebral cortex of the cat. Exp Brain Res. 1977;27:237–243. doi: 10.1007/BF00235500. [DOI] [PubMed] [Google Scholar]
- 20.Hotson JR, Prince DA, Schwartzkroin PA. Anomalous inward rectification in hippocampal neurons. J Neurophysiol. 1979;42:889–895. doi: 10.1152/jn.1979.42.3.889. [DOI] [PubMed] [Google Scholar]
- 21.Jefferys JGR, Haas HL. Synchronized bursting of CA1 hippocampal pyramidal cells in the absence of synaptic transmission. Nature. 1982;300:448–450. doi: 10.1038/300448a0. [DOI] [PubMed] [Google Scholar]
- 22.Jensen MS, Yaari Y. Role of intrinsic burst firing, potassium accumulation, and electrical coupling in the elevated potassium model of hippocampal epilepsy. J Neurophysiol. 1997;77:1224–1233. doi: 10.1152/jn.1997.77.3.1224. [DOI] [PubMed] [Google Scholar]
- 23.Jensen MS, Azouz R, Yaari Y. Variant firing patterns in rat hippocampal pyramidal cells modulated by extracellular potassium. J Neurophysiol. 1994;71:831–839. doi: 10.1152/jn.1994.71.3.831. [DOI] [PubMed] [Google Scholar]
- 24.Jensen MS, Azouz R, Yaari Y. Spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol (Lond) 1996;492:199–210. doi: 10.1113/jphysiol.1996.sp021301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamondi A, Acsady L, Buzsaki G. Dendritic spikes are enhanced by cooperative network activity in the intact hippocampus. J Neurosci. 1998;18:3919–3928. doi: 10.1523/JNEUROSCI.18-10-03919.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kandel ER, Spencer WA. Electrophysiology of hippocampal neurones: II. After-potentials and repetitive firing. J Neurophysiol. 1961;24:243–259. doi: 10.1152/jn.1961.24.3.243. [DOI] [PubMed] [Google Scholar]
- 27.Kang Y, Okada T, Ohmori H. A phenytoin-sensitive cationic current participates in generating the afterdepolarization and burst afterdischarge in rat neocortical pyramidal cells. Eur J Neurosci. 1998;10:1363–1375. doi: 10.1046/j.1460-9568.1998.00155.x. [DOI] [PubMed] [Google Scholar]
- 28.Kubo Y, Miyashita T, Murata Y. Structural basis for a Ca2+-sensing function of the metabotropic glutamate receptors. Science. 1998;279:1722–1725. doi: 10.1126/science.279.5357.1722. [DOI] [PubMed] [Google Scholar]
- 29.Lux HD, Carbone E, Zucker H. Na+ currents through low-voltage-activated Ca2+ channels of chick sensory neurones: block by external Ca2+ and Mg2+. J Physiol (Lond) 1990;430:159–188. doi: 10.1113/jphysiol.1990.sp018287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z, Hatton GI. Oscillatory bursting of phasically firing rat supraoptic neurones in low-Ca2+ medium: Na+ influx, cytosolic Ca2+ and gap junctions. J Physiol (Lond) 1996;496:379–394. doi: 10.1113/jphysiol.1996.sp021692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- 32.Madison DV, Nicoll RA. Control of the repetitive discharge of rat CA1 pyramidal neurones in vitro. J Physiol (Lond) 1984;354:319–331. doi: 10.1113/jphysiol.1984.sp015378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Magee JC, Carruth M. Dendritic voltage-gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1999;82:1795–1901. doi: 10.1152/jn.1999.82.4.1895. [DOI] [PubMed] [Google Scholar]
- 34.Masukawa LM, Benardo LS, Prince DA. Variations in electrophysiological properties of hippocampal neurons in different subfields. Brain Res. 1982;242:341–344. doi: 10.1016/0006-8993(82)90320-1. [DOI] [PubMed] [Google Scholar]
- 35.Mattia D, Kawasaki H, Avoli M. Repetitive firing and oscillatory activity of pyramidal-like bursting neurons in the rat subiculum. Exp Brain Res. 1997;114:507–517. doi: 10.1007/pl00005660. [DOI] [PubMed] [Google Scholar]
- 36.Nuñez A, Garcia-Austt E, Buño W. Slow intrinsic spikes recorded in vivo in rat CA1-CA3 hippocampal pyramidal neurons. Exp Neurol. 1990;109:294–299. doi: 10.1016/s0014-4886(05)80020-2. [DOI] [PubMed] [Google Scholar]
- 37.Perez-Velazquez JL, Valiante TA, Carlen PL. Modulation of gap junctional mechanisms during calcium-free induced field burst activity: a possible role for electronic coupling in epileptogenesis. J Neurosci. 1994;14:4308–4317. doi: 10.1523/JNEUROSCI.14-07-04308.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pumain R, Menini C, Heinemann U, Louvel J, Silva Barrat C. Chemical synaptic transmission is not necessary for epileptic seizures to persist in the baboon Papio papio. Exp Neurol. 1985;89:250–258. doi: 10.1016/0014-4886(85)90280-8. [DOI] [PubMed] [Google Scholar]
- 39.Rall W. Handbook of physiology. The nervous system. American Physiology Society; Bethesda, MD: 1977. Core conductor theory and cable properties of neurones. [Google Scholar]
- 40.Schwartzkroin PA. Characteristics of CA1 neurons recorded intracellularly in the hippocampal in vitro slice preparation. Brain Res. 1975;85:423–436. doi: 10.1016/0006-8993(75)90817-3. [DOI] [PubMed] [Google Scholar]
- 41.Segal MM, Douglas AF. Late sodium channel openings underlying epileptiform activity are preferentially diminished by the anticonvulsant phenytoin. J Neurophysiol. 1997;77:3021–3304. doi: 10.1152/jn.1997.77.6.3021. [DOI] [PubMed] [Google Scholar]
- 42.Sherman A, Rinzel J. Rhythmogenic effects of weak electrotonic coupling in neuronal models. Proc Natl Acad Sci USA. 1992;89:2471–2474. doi: 10.1073/pnas.89.6.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol (Lond) 1987;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- 45.Strata F, Atzori M, Molnar M, Ugolini G, Tempia F, Cherubini E. A pacemaker current in dye coupled hilar interneurons contributes to the generation of giant GABAergic potentials in developing hippocampus. J Neurosci. 1997;17:1435–1446. doi: 10.1523/JNEUROSCI.17-04-01435.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su H, Alroy G, Kirson ED, Yaari Y. Intrinsic bursting induced by low extracellular calcium in rat hippocampal neurons: role of persistent sodium current. Neurosci Lett. 1999;54:S40. [Google Scholar]
- 47.Taylor CP, Dudek FE. Synchronous neural afterdischarges in rat hippocampal slices without active chemical synapses. Science. 1982;217:810–812. doi: 10.1126/science.7134978. [DOI] [PubMed] [Google Scholar]
- 48.Thomas MJ, Watabe AM, Moody TD, Makhinson M, O'Dell TJ. Postsynaptic complex spike bursting enables the induction of LTP by theta frequency synaptic stimulation. J Neurosci. 1998;17:7117–7126. doi: 10.1523/JNEUROSCI.18-18-07118.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong RK, Prince DA. Afterpotential generation in hippocampal pyramidal cells. J Neurophysiol. 1981;45:86–97. doi: 10.1152/jn.1981.45.1.86. [DOI] [PubMed] [Google Scholar]
- 50.Xiong ZG, MacDonald JF. Sensing of extracellular calcium by neurones. Can J Physiol Pharmacol. 1999;77:715–721. [PubMed] [Google Scholar]
- 51.Yaari Y, Konnerth A, Heinemann U. Spontaneous epileptiform activity of CA1 hippocampal neurons in low extracellular calcium solutions. Exp Brain Res. 1983;51:153–156. doi: 10.1007/BF00236813. [DOI] [PubMed] [Google Scholar]
- 52.Yamaguchi T, Chattopadhyay N, Brown EM. G protein-coupled extracellular Ca2+ (Ca2+o)-sensing receptor (CaR): roles in cell signaling and control of diverse cellular functions. Adv Pharmacol. 2000;2000:209–253. doi: 10.1016/s1054-3589(08)60113-1. [DOI] [PubMed] [Google Scholar]
- 53.Zahs KR, Newman EA. Asymmetric gap junctional coupling between glial cells in the rat retina. Glia. 1997;20:10–22. [PubMed] [Google Scholar]