

Abstract
Alzheimer's Disease (AD) is the most common of the senile dementias, the prevalence of which is increasing rapidly, with a projected 14 million affected worldwide by 2025. The signal transduction mechanisms that underlie the learning and memory derangements in AD are poorly understood. β-Amyloid (Aβ) peptides are elevated in brain tissue of AD patients and are the principal component of amyloid plaques, a major criterion for postmortem diagnosis of the disease. Using acute and organotypic hippocampal slice preparations, we demonstrate that Aβ peptide 1-42 (Aβ42) couples to the mitogen-activated protein kinase (MAPK) cascade via α7 nicotinic acetylcholine receptors (nAChRs). In vivo elevation of Aβ, such as that exhibited in an animal model for AD, leads to the upregulation of α7 nAChR protein. α7 nAChR upregulation occurs concomitantly with the downregulation of the 42 kDa isoform of extracellular signal-regulated kinase (ERK2) MAPK in hippocampi of aged animals. The phosphorylation state of a transcriptional mediator of long-term potentiation and a downstream target of the ERK MAPK cascade, the cAMP-regulatory element binding (CREB) protein, were affected also. These findings support the model that derangement of hippocampus signal transduction cascades in AD arises as a consequence of increased Aβ burden and chronic activation of the ERK MAPK cascade in an α7 nAChR-dependent manner that eventually leads to the downregulation of ERK2 MAPK and decreased phosphorylation of CREB protein.
Keywords: Alzheimer's disease, MAPK, nicotinic receptor, amyloid, kinase, hippocampus, learning, memory
Alzheimer's Disease (AD) clinically presents itself as impaired memory formation, yet despite intensive study the mechanisms underlying AD-related memory dysfunction remain mysterious. The discovery that soluble β-amyloid (Aβ) peptides are elevated in the brains of AD patients raises the issue of whether these molecules play a causative role in AD (Kuo et al., 1996). Aβ peptides are generated from amyloid precursor protein (APP) via endo-proteolytic cleavage by β- and γ-secretases (Selkoe, 1998). In normal individuals Aβ40 comprises the majority of the Aβ population; a far smaller fraction is made up of Aβ42 (Kuo et al., 1996). Aβ42 is highly fibrillogenic and exhibits trophic and toxic effects on neurons (Lambert et al., 1998; Hartley et al., 1999; Dodart et al., 2000).
Early onset AD is associated with several risk factors, the best correlated being age and the inheritance of specific genes that result in the increased production of Aβ peptides. Thus several laboratories are seeking to gain insights into AD by studying the effects of Aβin vitro and in vivo (Hsiao, 1998; Dodart et al., 2000). Studies on transgenic animals that express the human genes linked to early onset familial AD (FAD) have shown that aberrant Aβ production leads to many pathological features of AD (Borchelt, 1998;Hsiao, 1998). One transgenic strain (Tg2576) exhibits several pathological features of AD, including elevated Aβ production with subsequent hippocampus-dependent learning and memory deficits, age-dependent accumulation of Aβ fibrils, and plaque formation (Hsiao et al., 1996; Irizarry et al., 1997a; Chapman et al., 1999). The Tg2576 mouse strain at 9 months of age exhibits learning deficits without neuronal cell loss or Aβ deposition into plaques (Irizarry et al., 1997b). Therefore, the impairments leading to this phenotype are likely in the normal cellular signaling cascades involved in learning and memory.
Using this rationale, we investigated Aβ42 activation of the extracellular signal-regulated kinase (ERK2) isoform of the ERK mitogen-activated protein kinase (MAPK) cascade, because activation of this kinase in hippocampus is required for contextual and spatial memory formation in mammals (Atkins et al., 1998; Blum et al., 1999;Schafe et al., 1999; Selcher et al., 1999). In addition, we evaluated the phosphorylation state of a downstream target of ERK MAPK in hippocampus, the cAMP-regulatory element binding (CREB) protein, also a necessary component for hippocampus-dependent memory formation in mammals (Bourtchuladze et al., 1994).
The α7 nicotinic acetylcholine receptor (nAChR) is expressed in brain regions particularly susceptible to the ravages of AD, and the functional location of α7 nAChRs in hippocampus indicates a role for these receptors in memory formation (Perry et al., 1995; Frazier et al., 1998; McQuiston and Madison, 1999). Recent studies have shown that Aβ42 coimmunoprecipitates with the α7 nAChR in samples from postmortem AD hippocampus, and α7 nAChR antagonists compete for Aβ42 peptide binding to heterologously expressed α7 nAChRs (Wang et al., 2000). Furthermore, preincubation with Aβ42 peptide antagonizes the activation of α7 nAChR-like currents in hippocampal interneurons (Pettit et al., 2001). Therefore, we tested the hypothesis that the α7 nAChR functions as a receptor for Aβ42 in the hippocampus, coupling Aβ42 to the ERK MAPK cascade. Furthermore, in an animal model of AD we evaluated the effects of chronic elevated Aβ on this signal transduction cascade.
MATERIALS AND METHODS
Materials. Unless otherwise stated, chemicals and drugs were purchased from Sigma (St. Louis, MO). Synthetic rat Aβ42 was purchased from Calbiochem (La Jolla, CA). Aβ42 stock solutions were prepared at 100 μm in 100 mm HEPES, pH 8.5, aliquoted, and stored frozen. Anti-α7 nAChR antibody was purchased from Babco (Richmond, CA). Anti-ERK1/2 and anti-ERK1/2 dually phosphorylated at Thr202/Tyr204 antibodies were purchased from Cell Signaling Technologies. Anti-CREB antibody also was purchased from Cell Signaling Technologies. Anti-CREB phosphorylated at Ser133 antibody was developed and characterized in the laboratory of J. D. Sweatt. Tissue culture mediums and buffers were prepared with supplies from Life Technologies (Gaithersburg, MD).
Animal subjects. Acute hippocampal slices were obtained from mice heterozygous for the L250T α7 nAChR transgene (Orr-Urtreger et al., 2000). Morris water maze testing was performed on Tg2576 mice carrying a human APP transgene with the K670N-M671L mutation (Hsiao et al., 1996). This was followed by a longitudinal biochemical analysis of ERK, CREB, and α7 nAChR levels at 4, 13, and ∼20 months of age. Postnatal day 7 (P7), P10, or P13 Sprague Dawley rat pups (Harlan Sprague Dawley, Indianapolis, IN) were used for hippocampal explant cultures. All animal experiments were performed in accordance with the Baylor College of Medicine Institutional Animal Care and Use Committee and with national regulations and policies.
Hippocampus dissection. Animals were decapitated, and both hippocampi were removed and placed into ice-cold cutting solution [containing (in mm) 1.25 NaH2PO4, 28 NaHCO3, 60 NaCl, 3 KCl, 110 sucrose, 0.5 CaCl2, 7 MgCl2, 5 glucose, and 0.6 ascorbate]. For experiments that used α7 nAChR L250T heterozygote animals, 400 μm transverse slices were prepared (see below). For quantitative immunoblot that used samples from Tg2576 mice and age-matched control animals (most often littermates), area CA1 and dentate gyrus (DG) were subdissected from each hippocampus and prepared for quantitative immunoblot as described previously (Roberson et al., 1999). The following numbers of test subjects were used: 4 month Tg2576 animals, n = 10; 13 month animals, n = 8; 18–22 (∼20) month animals,n = 20.
Acute hippocampal slice preparation and drug treatments.Transverse hippocampal slices (400 μm) were prepared as described previously (Roberson et al., 1999) from mice heterozygote for the L250T mutation in the α7 nAChR (Orr-Urtreger et al., 2000). Slices were maintained in aCSF [containing (in mm) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, and 25 glucose] for drug treatments. Slices were incubated in 500 μm nicotine for 10 min with or without pretreatment (for 30 min) with 1 μm MLA. Basal samples represent identical slices that were left untreated. Samples were subjected to quantitative immunoblot as described previously. All experiments included a 10 μm PDA treatment (for 10 min) as a positive control for ERK MAPK activation. Data that are normalized to basal samples are reported as average ± SEM. Experimental results represent replicates of three to five slices per treatment.
Hippocampal slice culture and treatments. Hippocampal slice cultures were prepared from P7, P10, or P13 Sprague Dawley rat pups and maintained in culture according to the method of Stoppini et al. (1991). Then 24 hr before assay the cultures were rinsed and switched into serum-free culture medium (Neurobasal medium, pH 7.2, supplemented with B-27, 1% penicillin–streptomycin, 1.4% 1.8 mglucose, 0.5% 200 mm glutamine, and 0.25 μg/ml Fungizone). After 5–11 d in vitro the slices were assayed for ERK MAPK activation after various treatments. Assays were performed on at least eight slices per treatment in triplicate or greater for each experiment. Basal samples represent identical cultures that were left untreated. Samples were subjected to quantitative immunoblot as described previously. Data that are normalized to basal samples are reported as average ± SEM. Results represent at least three experimental replicates. Treatments were performed in serum-free culture medium and included (1) 500 μm nicotine (for 10 min) with or without pretreatment (for 30 min) with 1 μm MLA; (2) 100 nm Aβ42 for 2, 5, 10, 30, or 120 min; (3) 5 min incubation with 0.01, 0.1, 1.0, 10, or 100 nm Aβ42; (4) 5 min incubation with 100 nm Aβ42 with or without pretreatment (for 30 min) with 1 μm MLA or (for 2 hr) with 100 μm α-BTX; (5) 500 μmnicotine (for 10 min) with or without pretreatment (for 2 hr) with 100 nm Aβ42; (6) 10 μm PDA (for 10 min) with or without pretreatment (for 2 hr) with 100 nm Aβ42; (7) 100 nmAβ42 with or without pretreatment (for 1 hr) with 1 μm TTX; (8) 100 nm Aβ42 with or without preincubation (for 1 hr) in culture medium containing 5 mm EGTA; (9) 100 pm Aβ42 treatment for 144 hr. As a positive control for ERK MAPK activation, where relevant, the experiments included a 10 μm PDA treatment (for 10 min).
Quantitative immunoblotting. Harvested brain tissue was sonicated in sonication buffer [containing (in mm) 10 HEPES, pH 7.4, 150 NaCl, 50 NaF, 1 ED/EGTA, 10 Na4P2O7, and 1 Na3VO4 plus 200 nm calyculin A, 10 μg/ml leupeptin, 2 μg/ml aprotinin, and 1 μm microcystin-LR], and protein concentration was determined with BCA (Pierce, Rockford, IL). Samples were subjected to SDS-PAGE and transferred to Immobilon-P (Millipore, Bedford, MA), followed by immunoblotting with the appropriate primary and secondary antibodies and chemiluminescence (ECL, Amersham/Pharmacia Biotech, Piscataway, NJ). Band intensity was quantified with Scion Image software (NIH Image, Bethesda, MD) from film exposures (BioMax, Kodak, Rochester, NY) in the linear range for each antibody and normalized to basal/control level. Normalized basal/control values were determined for each immunoblot by averaging basal/control values, dividing each basal/control and test/transgenic sample density by the average of the basal/control set, and then determining the average and SEM for basal/control and test/transgenic samples.
Spatial learning task. Initially, Tg2576 mice and age-matched control animals underwent cued training for 3 d consecutively (8 trials/d), swimming to a raised black platform marked with a black and white striped pole. The platform location and start quadrant were varied pseudo-randomly in each trial. Hidden platform training was performed over 9 d consecutively (4 trials/d), wherein mice were required to locate within 60 sec a platform submerged 1.5 cm beneath the surface of opaque water. Once the platform was reached in hidden platform training, the mice were allowed to remain on the platform for 30 sec. Mice who failed to reach the platform within 60 sec were led to the platform with a retrieval scoop. At 16–24 hr after the 12th, 24th, and 36th training trials, probe trials were run in which mice swam for 60 sec in the pool with no platform. Trials were monitored by a ceiling-mounted camera above the pool and analyzed with the HVS tracking system (HVS Image Ltd., Hampton, UK). Further analysis was done with Wintrack (kindly provided by Dr. David Wolfer, University of Zurich, Switzerland).
Exclusion criteria omitted mice with obvious swimming difficulties, such as persistent floating, sinking, or abnormal swimming patterns. Mice exhibiting an escape latency >2 SD longer than the age-matched Tg2576 mean during the last four cued trials and mice that consistently failed to orient to or follow the retrieval scoop by the end of the testing period also were excluded from data analysis.
Statistical methods. Student's t test or one-way ANOVA was performed on quantitative immunoblot results, followed bypost hoc analysis with the method of Tukey.
RESULTS
We first tested whether α7 nAChRs could mediate activation of the ERK2 MAPK cascade by treating hippocampal slices with nicotine and assaying for a resultant increase in ERK2 MAPK phosphorylation, a direct indicator of kinase activation (Sturgill et al., 1988; Payne et al., 1991). Because the wild-type α7 nAChR receptor rapidly desensitizes, the slices in these experiments were prepared from the hippocampi of heterozygote transgenic (L250T) animals that contain a targeted mutation in the α7 nAChR gene that renders the expressed α7 nAChRs resistant to desensitization (Revah et al., 1991;Orr-Urtreger et al., 2000). Nicotine activates the 42 kDa ERK2 isoform of MAPK in hippocampi from L250T animals (Fig.1a). Stimulation of ERK2 activity is dependent on α7 nAChR function because an α7-selective antagonist, methyllycaconitine (MLA), attenuates nicotine-induced activation of ERK2 MAPK in the L250T hippocampal slices. In addition, wild-type rat hippocampal slices maintained in culture exhibit activation of ERK2 MAPK with nicotine treatment that is blocked by MLA pretreatment (Fig. 1b). These results demonstrate that α7 nAChR activation is capable of stimulating ERK2 MAPK in hippocampus.
Fig. 1.

Nicotine activation of ERK2 in hippocampal slices is mediated by α7 nAChRs. a, Quantitative immunoblot demonstrates that 500 μm nicotine stimulated ERK2 MAPK activity, which is antagonized by 1 μm MLA in hippocampal slices from mice heterozygous for the L250T α7 nAChR transgene. Basal, 1.00 ± 0.22; nicotine (nic), 2.33 ± 0.13; MLA + nicotine, 1.34 ± 0.14. *Significant difference from basal level; p < 0.01, post hocTukey multiple comparison test. Representative immunoblot results are depicted below the response histogram. ERK1 (topband) activation paralleled that of ERK2; however, absolute levels of phospho-ERK1 were far below ERK2 and were not quantified. Slices from wild-type animals did not exhibit significant ERK2 MAPK activation with nicotine treatment, likely because of the rapid desensitization kinetics of wild-type α7 nAChRs precluding biochemical detection of ERK2 MAPK activation (Seguela et al., 1995). b, Quantitative immunoblot demonstrates that ERK2 MAPK activation by nicotine in cultured rat hippocampal slices is blocked by 1 μm MLA. Basal, 1.01 ± 0.08; nicotine, 1.66 ± 0.13; MLA + nicotine, 1.22 ± 0.09. *Denotes nicotine stimulation is significantly different from basal level;p < 0.001, post hoc Tukey multiple comparison test. Representative immunoblot results are depictedbelow the response histogram.
Hippocampal slice culture technique was used to evaluate whether α7 nAChRs mediate Aβ42 stimulation of the ERK MAPK cascade. Aβ activates several kinase systems in cultured cells (Saitoh et al., 1993; Kosik et al., 1996), including the ERK MAPK cascade in cultured primary cortical and hippocampal neurons (Ekinci et al., 1999; Rapoport and Ferreira, 2000). The Aβ preparations used in these previous studies varied vis-à-vis the aggregation state and length of the synthetic peptides that were used. Our work used Aβ42 peptide that was prepared under conditions to promote solubility and to retard aggregation (Burdick et al., 1992; Garzon-Rodriguez et al., 1997). We tested our Aβ42 preparations for aggregation in a Congo red assay. At the concentrations used and under the incubation conditions tested, none of the Aβ42 preparations significantly pelleted out of solution with Congo red dye at 2.5 or 25 μm (Brining, 1997) (data not shown). We found that synthetic Aβ42 activates ERK2 MAPK in cultured hippocampal slices at concentrations typically found in the CSF and brains of AD patients and in animal models of AD (Hsiao et al., 1996; Andreasen et al., 1999a,b; Tapiola et al., 2000) (Fig.2a). Furthermore, ERK2 MAPK activation occurs rapidly and decreases with prolonged exposure to Aβ42, indicative of receptor or MAPK cascade desensitization (Fig.2b).
Fig. 2.
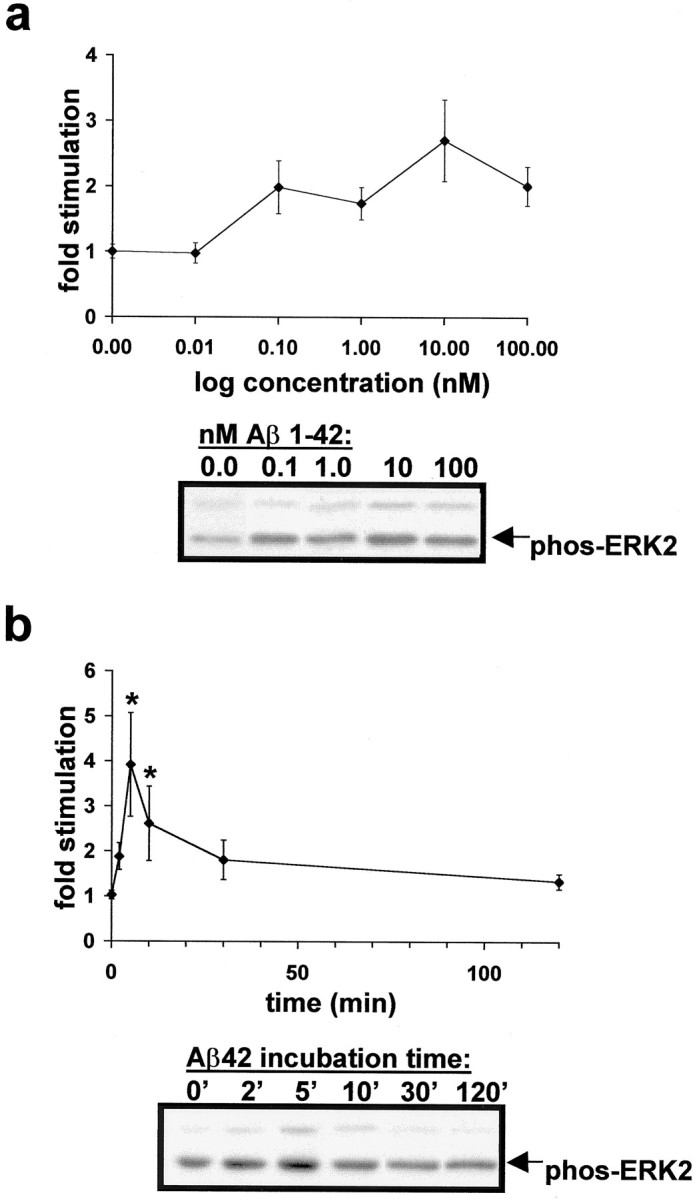
Time course and concentration dependence of Aβ42 activation of ERK2 MAPK in cultured rat hippocampal slices.a, Aβ42 activates ERK MAPK in the picomolar to nanomolar range. Data points are as follows: 0 nm(1.00 ± 0.15), 0.01 (0.98 ± 0.16), 0.1 (1.98 ± 0.48), 1.0 (1.74 ± 0.25), 10 (2.70 ± 0.62), and 100 (2.01 ± 0.30) nm Aβ42 for 5 min. Representative immunoblot results are depicted below the response curve.b, The 100 nm Aβ42 rapidly activates ERK2 MAPK in rat hippocampal slice cultures. Peak response occurs at 5 min Aβ42 and returns to baseline within 2 hr. Data points are as follows: 2 (1.88 ± 0.30), 5 (3.92 ± 1.15), 10 (2.61 ± 0.83), 30 (1.81 ± 0.44), and 120 (1.34 ± 0.17) min. *Significant difference from basal level (1.06 ± 0.12); p< 0.05, Student's t test with Welch's correction because variances differ significantly according to Bartlett's test. Representative immunoblot results are depicted below the response curve.
Stimulation of ERK2 MAPK activity with Aβ42 in cultured rat hippocampal slices is blocked by the α7 nAChR antagonists MLA and α-bungarotoxin (BTX), demonstrating that α7 nAChR function is necessary for Aβ42 coupling to the ERK MAPK cascade (Fig.3a). This was also the case for acute hippocampal slices prepared from L250T animals and exposed to identical drug treatments (data not shown). Cross-desensitization is one way to test the possibility that two different agonists couple via the same receptor type. Pretreatment of cultured hippocampal slices with Aβ42 blocked nicotine stimulation of ERK MAPK activity, evidence that nicotine and Aβ42 mediate their effects on ERK2 MAPK via the same receptor type (Fig. 3b). Aβ42 pretreatment of cultured slices did not interfere with the ability of phorbol 12,13-dibutyrate (PDA) to activate ERK MAPK, demonstrating that the MAPK cascade was not desensitized by Aβ42 treatment, evidence in support of α7 nAChRs mediating this Aβ42 effect (Fig.3c). We tested whether action potential generation is necessary for the ERK2 MAPK activation by Aβ42 by including tetrodotoxin (TTX) in the assay medium. TTX alone decreased the basal level of ERK MAPK activation, indicating that endogenous synaptic transmission in the hippocampal slice cultures contributes to basal ERK2 MAPK activity level. Aβ42 in the presence of TTX results in significant ERK2 MAPK activation as compared with cultures treated with TTX alone (Fig. 3d). Because α7 nAChRs are highly permeable to Ca2+ (Seguela et al., 1993), we tested the Ca2+ dependency of Aβ42-induced ERK MAPK activation. Depleting the assay system of external Ca2+ by including EGTA in the culture medium blocks Aβ42-induced activation of ERK2 MAPK (Fig.3d). Overall, our experiments show that Aβ42 rapidly activates ERK2 MAPK in hippocampus for which α7 nAChR function is necessary. This activation uses extracellular Ca2+ (likely via α7 nAChRs directly and α7 nAChR-dependent depolarization) and is action potential-independent; the ability of Aβ42 to activate ERK2 MAPK exhibits desensitization without inhibiting phorbol ester activation of the cascade. Furthermore, ERK MAPK activation occurs in response to picomolar and nanomolar concentrations of Aβ42.
Fig. 3.

α7 couples Aβ42 activation of ERK2 MAPK in cultured rat hippocampal slices. a, Quantitative immunoblot demonstrates that ERK2 MAPK activation by 100 nm Aβ42 is blocked by 1 μm MLA and 100 μm BTX. Basal, 1.00 ± 0.04; Aβ42 (light-shaded bar), 1.58 ± 0.09; MLA + Aβ42, 1.19 ± 0.07; Aβ42 (dark-shaded bar), 1.67 ± 0.11; BTX + Aβ42, 0.94 ± 0.14. *Significant difference from basal ERK2 MAPK activity; p < 0.0001, Student'st test with Welch's correction because variances differ significantly according to Bartlett's test. Representative immunoblot results are depicted below the response histogram.b, Aβ42 (100 nm) desensitizes the nicotine-induced (500 μm) activation of ERK2 MAPK. Basal, 1.05 ± 0.14; nicotine (nic), 1.94 ± 0.22; Aβ42 (at 2 hr), 1.14 ± 0.24; Aβ42 + nicotine (at 2 hr), 1.03 ± 0.13. *Significant difference from basal ERK2 MAPK activity; p < 0.001, post hoc Tukey multiple comparison test. Representative immunoblot results are depicted below the response histogram. c, Aβ42 (100 nm) does not desensitize the PDA-induced (10 μm) activation of ERK2 MAPK. Basal, 1.00 ± 0.08; Aβ42 (at 2 hr), 0.78 ± 0.11; Aβ42 + PDA (at 2 hr), 2.15 ± 0.54; PDA, 2.25 ± 0.13. *Significant difference from basal ERK2 MAPK activity; p < 0.01, post hoc Tukey multiple comparison test. Representative immunoblot results are depicted below the response histogram.d, The 100 nm Aβ42 activation of ERK2 MAPK is not blocked by TTX and exhibits Ca2+ dependency. Basal, 1.00 ± 0.06; Aβ42, 1.30 ± 0.07; TTX, 0.67 ± 0.10; Aβ42 + TTX, 1.02 ± 0.05; EGTA, 0.59 ± 0.04; Aβ42 + EGTA, 0.65 ± 0.05. *Significant difference from basal-induced ERK2 MAPK activity. #, Significant difference from TTX-induced ERK2 MAPK activity; p < 0.001, post hocTukey multiple comparison test.
As a complement to these in vitro experiments, we investigated the effects of elevated Aβ in vivo. We used Tg2576 animals to evaluate the long-term effects of elevated Aβ on hippocampal α7 nAChR expression level and ERK2 MAPK activity. A typical consequence of chronic exposure to nAChR agonist is nAChR upregulation (Marks et al., 1983; Fenster et al., 1999). We therefore hypothesized that α7 nAChRs would be upregulated in Tg2576 hippocampus as a consequence of chronic exposure to Aβ. We found an age-dependent increase in α7 nAChR protein in the hippocampi of these animals as compared with age-matched controls (Fig.4a,b). This increase is detected as early as 4 months of age in dentate gyrus (DG). With age, α7 nAChR protein continues to increase in the DG as well as in area CA1. As a control, we measured the level of another subtype of nAChR subunit, the α4 subunit, and found no significant increase in α4 nAChR protein in the hippocampi of Tg2576 animals (data not shown). Thus our data demonstrate a selective age-dependent upregulation of α7 nAChR in the hippocampus of Tg2576 mice. An explanation for these data in light of our findings in vitro is that α7 nAChRs in the hippocampi of Tg2576 animals are stimulated chronically by Aβ, leading to desensitization and upregulation of α7 nAChRs.
Fig. 4.

α7 nAChR is upregulated in hippocampus and DG of Tg2576 mice. a, Quantitative immunoblot of area CA1 and DG from 4, 13, and ∼20 month Tg2576 hippocampus reveals upregulation of α7 nAChR protein as early as 4 months. *Significantly higher α7 nAChR level than age-matched control animals (p < 0.03) by Student's ttest. Dashed line represents normalized control animal level; note the change in scale for ∼20 month animals. Filled bar, CA1; shaded bar, DG. b, Representative immunoblot for α7 nAChR level in Tg2576 and age-matched control animals.
Given the necessity for ERK2 MAPK activity in certain forms of learning and memory, the finding that Aβ42 activates ERK MAPK via α7 nAChRs, and the observation that α7 nAChRs are upregulated in Aβ-overexpressing mice, we determined whether the ERK MAPK cascade is disrupted in Tg2576 hippocampus. Quantitative immunoblot reveals that ERK2 phosphorylation is increased significantly in Tg2576 hippocampus as compared with controls (Fig.5a,c, Table1). In area CA1 at 13 months and in DG at 4 and 13 months, ERK2 MAPK hyperactivation is detected. These results correlate with elevated α7 nAChR protein in these brain regions as was described above. By 20 months of age ERK2 MAPK is downregulated; the hyperactivation detected at earlier ages is absent in DG and is below control animal levels in area CA1. At this age total ERK2 MAPK protein is unchanged in DG, whereas in area CA1 both total ERK2 MAPK protein and activity are downregulated by 22 and 27%, respectively. Previous evidence suggests that this level of reduction in ERK2 MAPK activity in hippocampus can lead to learning and memory impairments. For example, Selcher et al. (1999) have shown that partial inhibition of ERK2 MAPK activity in hippocampus blocks two forms of hippocampus-dependent learning in the mouse.
Fig. 5.

Downstream targets of α7 nAChR activation in Tg2576 hippocampus exhibit dysregulation. ERK2 and CREB proteins undergo hyperactivation, followed by downregulation, as compared with age-matched control animals. Differences between Tg2576 and age-matched control animals were detected by quantitative immunoblot of area CA1 and DG from 4, 13, and ∼20 month Tg2576 hippocampus. Samples were evaluated for total and phospho-ERK2 MAPK (a, c) and total and phospho-CREB (b, d) levels in CA1 and DG, respectively. *Significant difference from age-matched control animal level (p ≤ 0.05) by Student'st test. Dashed line represents normalized control animal level. Filled bar, Total; shaded bar, phospho.
Table 1.
Data summary for the parameters measured in hippocampi of Tg2576 mice at different ages as compared with age-matched control animals
| Protein detected | Age (months) | ||
|---|---|---|---|
| 4 | 13 | ∼20 | |
| CA1 | |||
| ERK2 MAPK | 0.95 ± 0.06 | 1.10 ± 0.07 | 0.78 ± 0.06 |
| (ns) | (ns) | (0.017) | |
| Phospho-ERK2 MAPK | 0.85 ± 0.09 | 1.15 ± 0.06 | 0.73 ± 0.06 |
| (ns) | (0.03) | (0.001) | |
| CREB | 1.02 ± 0.11 | 0.93 ± 0.13 | 1.03 ± 0.14 |
| (ns) | (ns) | (ns) | |
| Phospho-CREB | 1.27 ± 0.16 | 1.50 ± 0.25 | 0.82 ± 0.07 |
| (ns) | (0.041) | (0.015) | |
| α7 nAChR | 1.15 ± 0.17 | 1.45 ± 0.33 | 19.17 ± 5.89 |
| (ns) | (ns) | (0.002) | |
| DG | |||
| ERK2 MAPK | 0.97 ± 0.09 | 1.06 ± 0.09 | 1.01 ± 0.08 |
| (ns) | (ns) | (ns) | |
| Phospho-ERK2 MAPK | 1.40 ± 0.23 | 1.56 ± 0.13 | 1.13 ± 0.10 |
| (0.05) | (0.001) | (ns) | |
| CREB | 1.17 ± 0.10 | 1.78 ± 0.35 | 1.35 ± 0.16 |
| (ns) | (0.025) | (0.03) | |
| Phospho-CREB | 1.64 ± 0.29 | 0.90 ± 0.17 | 1.19 ± 0.16 |
| (0.015) | (ns) | (ns) | |
| α7 nAChR | 1.57 ± 0.16 | 1.82 ± 0.40 | 10.00 ± 2.08 |
| (0.005) | (0.028) | (<0.001) | |
Relative densities are measured from immunoblots as described. Mean normalized densities are reported ± SEM. In parentheses below each mean value are Student's t test results. ns, Not significant.
Because ERK MAPK is altered in the hippocampi of Tg2576 animals, it is of interest to learn whether downstream targets of this kinase cascade also are affected. CREB is phosphorylated at Ser133 by rsk2, a kinase that is activated by ERK MAPK phosphorylation (Xing et al., 1996). We evaluated the phosphorylation state and total protein level of CREB protein in CA1 and DG of Tg2576 hippocampi by quantitative immunoblotting (Fig. 5b,d, Table 1). In Tg2576 hippocampus, CREB phosphorylation is elevated at 13 months, followed by downregulation by 20 months of age. In area CA1, CREB phosphorylation fluctuates with ERK2 MAPK activity, consistent with CREB phosphorylation being coupled to the ERK MAPK cascade (Roberson and Sweatt, 1996; Impey et al., 1998; Watabe et al., 2000). CREB protein level was not altered significantly in area CA1. These data demonstrate that, in area CA1, derangements of the ERK MAPK cascade occur in conjunction with dysregulation of a downstream target, the transcriptional regulator CREB. In DG, however, CREB protein level is elevated at 13 and 20 months of age, indicative of differential regulation of CREB in DG versus area CA1. Furthermore, the correlated CREB phosphorylation with ERK2 activation was not observed in DG, although coupling between ERK MAPK activity and CREB phosphorylation at Ser133 in this region has been demonstrated (Davis et al., 2000).
Tg2576 animals exhibit long-term potentiation (LTP) and working memory deficits by 9 months of age and a spatial learning impairment that is evident at 6 months, which deteriorates further at 20–26 months of age (M. Westerman and K. H. Ashe, personal communication). We tested whether the increased α7 nAChR protein we observed correlated with behavioral learning defects in these animals. Before biochemical analysis, Tg2576 and control animals were trained and tested in the Morris water maze. A scatter plot of the α7 nAChR levels of individual animals versus the percentage of time spent in the target quadrant during a third probe trial in the Morris water maze illustrates a negative correlation (R2 = −0.283 and −0.193;p ≤ 0.05, for CA1 and DG, respectively) between α7 nAChR level and Morris water maze performance (Fig.6). These data do not indicate a simple linear correlation between α7 nAChR protein levels and Morris water maze performance. However, these observations are consistent with the idea that α7 nAChR upregulation in hippocampus may serve as a biochemical marker for the synaptic plasticity derangement and learning and memory deficits in Tg2576 animals.
Fig. 6.

Animals with elevated α7 nAChR level fail to perform to criteria in the Morris water maze. Scatter plot of the third probe trial performance of individual ∼20-month-old Tg2576 and control animals versus α7 nAChR protein levels in CA1 and DG.Dashed line indicates performance criterion.Filled squares, Control; filled diamonds, Tg.
We further tested the hypothesis that chronic exposure to Aβ42 leads to α7 nAChR upregulation in hippocampus by incubating cultured hippocampal slices with 100 pm Aβ42 for 6 d. After 144 hr of Aβ exposure, α7 nAChR protein was increased by over twofold (Fig. 7). Treated slices had the same appearance as control slices after 9 d in culture. These data demonstrate that prolonged exposure in vitro to a concentration of Aβ found in the brains of AD patients and Tg2576 animals leads to α7 nAChR upregulation in hippocampal tissue.
Fig. 7.

Chronic exposure to Aβ42 leads to increased α7 nAChR protein in hippocampal slice cultures. Cultured rat hippocampal slices were exposed to 100 pm Aβ42 for 144 hr, and α7 nAChR protein was quantified by immunoblot. The data that are expressed are normalized to the α7 nAChR protein level in cultures that were left untreated. Representative immunoblot results are shownbelow the histogram. *Significant difference from control level (p < 0.0001) by Student'st test. Basal, 1.00 ± 0.11; Aβ42-treated, 2.35 ± 0.13.
DISCUSSION
We have demonstrated that the α7 nAChR couples Aβ42 to the ERK2 MAPK cascade in hippocampus, uses Ca2+, and is independent of action potential propagation. We found that, in vitro, concentrations of Aβ42 that occur in the CSF and brain of AD patients rapidly activate hippocampal ERK2 MAPK and lead to α7 nAChR upregulation with chronic exposure. The experiments performed in this study do not address directly whether the α7 nAChR is a receptor for Aβ42. However, the following observations are consistent with this idea: (1) extended exposure of cultured rat hippocampal slices to Aβ42 blocks subsequent α7 nAChR agonist activation of ERK2 MAPK (under these conditions the ERK MAPK cascade is not desensitized to activation by phorbol ester); (2) both MLA and BTX block Aβ42 activation of ERK2 MAPK; (3) TTX does not block Aβ42 activation of ERK2 MAPK. Furthermore, ERK2 MAPK activation by Aβ42 requires Ca2+ influx, which is consistent with the model that Ca2+ influx via α7 nAChRs and α7 nAChR-dependent depolarization-induced Ca2+ influx leads to ERK2 MAPK activation. We favor the interpretation that Aβ42 is an agonist for α7 nAChRs in hippocampus and that prolonged exposure to Aβ can elicit α7 nAChR upregulation as a result of chronic receptor stimulation, a typical phenomenon after chronic nAChR agonist exposure (Marks et al., 1983; Fenster et al., 1999). In turn, we hypothesize that prolonged exposure of α7 nAChRs to Aβ leads to chronic stimulation of ERK2 MAPK.
As predicted by our in vitro findings, we observed effects on α7 nAChR and ERK2 MAPK in the hippocampus of a transgenic mouse line that overproduces Aβ peptides (Hsiao et al., 1996). In the hippocampi of these animals α7 nAChR protein increases in an age-dependent manner, whereas the basal activation state of ERK2 MAPK exhibits an age-dependent hyperactivation followed by downregulation. ERK2 MAPK hyperactivation is coincident with α7 nAChR upregulation at young ages; one interpretation of these data is that, in these animals, Aβ chronically stimulates the ERK MAPK cascade via α7 nAChRs. Likewise, the phosphorylation state of CREB, a downstream effector of the ERK MAPK cascade in area CA1, parallels the changes measured in ERK2 MAPK in this region. Thus, in Tg2576 animals and, we hypothesize in AD as well, elevated Aβ has profound effects on an Aβ receptor (α7 nAChR) and a signal transduction cascade (ERK MAPK) to which it is coupled. In older Tg2576 animals the pronounced upregulation of α7 nAChRs is associated with the downregulation of ERK2 MAPK.
Exposure of hippocampal slices to Aβ42 triggers signal transduction events that are important for hippocampal synaptic plasticity and learning and memory; activation of the ERK2 MAPK cascade in vitro by Aβ42 requires α7 nAChR function. These findings are consistent with the concept that α7 nAChR is an Aβ42 receptor. There is an emerging literature implicating the α7 nAChR in AD pathophysiology and memory loss. Evidence complementing our data that Aβ42 signals via the α7 nAChR includes the previous observations that (1) α7 nAChRs coimmunoprecipitate from postmortem human AD hippocampus; (2) Aβ peptides compete α7 agonist and antagonist binding to SK-N-MC cells; (3) 20-fold more α7 nAChR protein immunoprecipitates with Aβ42 from AD hippocampus as compared with control; (4) preincubation with Aβ42 antagonizes an α7 nAChR-like current in hippocampal interneurons; and (5) nicotine protects SK-N-MC cells from neurotoxicity induced by prolonged exposure to Aβ42 (Kihara et al., 1997; Wang et al., 2000). These last two observations are akin to the cross-desensitization of Aβ42 and nicotine demonstrated in this study. It must be emphasized, however, that additional Aβ receptors that play important roles in AD etiology are likely (Yan et al., 1996).
What then are the consequences of α7 nAChR coupling Aβ42 to the ERK MAPK cascade in AD? Our findings suggest that early AD etiology involves chronic activation of the ERK MAPK cascade as a consequence of elevated Aβ binding to and activating this signaling cascade via α7 nAChRs and that these events are concomitant with α7 nAChR upregulation and derangement of ERK2 MAPK signaling. Aβ burden is emerging as an early indicator of cognitive decline in AD in that total Aβ (nonaggregate and aggregates in diffuse or mature senile plaques combined) in the brains of elderly patients correlates with recent premorbid Clinical Dementia Rating scale values, even in the absence of severe dementia (Cummings and Cotman, 1995; Cummings et al., 1996;Naslund et al., 2000). Moreover, CSF Aβ is elevated to nanomolar level in patients with short disease duration and mild cognitive impairment (Nakamura et al., 1994; Tapiola et al., 2000). These findings support the idea that extracellular Aβ levels are elevated in at-risk individuals before gross plaque deposition and severe cognitive impairment. According to our findings, one consequence of nanomolar Aβ in the extracellular milieu of AD brain is the (chronic) activation of the ERK2 MAPK cascade via α7 nAChRs, leading to upregulation of this receptor type. In fact, we have demonstratedin vitro that chronic exposure to Aβ42 leads to increased α7 nAChR protein in hippocampus. An interesting implication of this hypothesis is that α7 nAChR-selective radioimaging techniques may be useful as a diagnostic test for early AD or risk for AD (Nordberg, 1999; Scheffel et al., 2000).
The ERK2 MAPK cascade is known to play a critical role in hippocampus synaptic plasticity and learning. In area CA1 of the rodent hippocampus ERK2 MAPK is necessary for the expression of a late phase of LTP (English and Sweatt, 1997; Impey et al., 1998; Selcher et al., 1999) and is an important pathway through which neurotransmitters modulate LTP induction (Roberson et al., 1999; Winder et al., 1999; Watabe et al., 2000). Furthermore, ERK2 MAPK activation is necessary for both contextual fear conditioning and escape training in the Morris water maze, both hippocampus-dependent associative learning paradigms (Atkins et al., 1998; Blum et al., 1999; Selcher et al., 1999). Tg2576 animals, in which Aβ production is elevated, exhibit deficits in hippocampus LTP, working memory, and escape training in the Morris water maze (Hsiao et al., 1996; Chapman et al., 1999). On the basis of our findings, we propose a mechanism linking Aβ overproduction to memory dysfunction; specifically, our data suggest that memory deficits occur in part via Aβ42 eliciting downstream derangements in ERK MAPK signaling. Aβ42 impinging on the ERK MAPK cascade suggests a molecular basis for the disruptions in memory formation accompanying AD, because the proper functioning of the ERK MAPK cascade is critical for certain types of memory formation.
Our model for the signal transduction events underlying AD etiology posits that elevated Aβ, such as occurs in humans genetically predisposed to the disease, chronically stimulates the ERK MAPK cascade via α7 nAChRs. Chronic exposure to Aβ leads to upregulation of α7 nAChRs and hyperactivation, followed by downregulation of ERK2 MAPK as well as perturbed functionality of downstream targets of this kinase. This derangement of the ERK MAPK signaling cascade may underlie the learning and memory deficits attributed to hippocampal dysfunction in AD. An additional site of action is likely to be disruption of the normal function of the hippocampal circuit, specifically the altered excitability of pyramidal neurons because α7 nAChRs located on GABAergic interneurons can regulate hippocampal pyramidal neuron excitability (Freund et al., 1988, 1990; Frazier et al., 1998). Modifications of GABAergic signaling to area CA1 pyramidal neurons in the hippocampi of animals in which Aβ production is either enhanced or genetically knocked-out have been demonstrated (Fitzjohn et al., 2000; Zaman et al., 2000). Overall, these findings highlight a potential therapeutic target for AD: the α7 nAChR.
In addition to assuaging the MAPK signaling derangement we have described, α7 nAChR antagonist therapy might benefit other aspects of AD etiology such as the selective loss of cholinergic inputs to the hippocampus and cortex as well as effects on Aβ production itself. The cholinergic input to the hippocampus and cortex is a particularly vulnerable neural circuit in AD, and α7 nAChRs are expressed on these projection neurons from the basal forebrain (Arendt et al., 1985;Breese et al., 1997). Our data suggest the possibility that Aβ42 binding to presynaptic α7 nAChRs may elicit cholinergic fiber loss, and α7 nAChR antagonism might delay this aspect of neurodegeneration in AD.
Finally, although the molecular basis for Aβ overproduction that is a consequence of FAD-linked gene inheritance is not understood, the ERK MAPK cascade has been implicated in regulating Aβ production in neurons (Mills et al., 1997). One of the effects of downregulated ERK2 MAPK activity may be the setting up of a positive feedback loop for Aβ accumulation; blocking the derangement of MAPK signaling via the α7 nAChR thus may alleviate one stimulant of Aβ production. Overall, the findings presented here provide insights into the molecular basis of Aβ-induced pathology and advance the possibilities for treatment targets in AD.
Footnotes
This work was supported by awards to J.D.S. from the National Institute on Aging (NIA), National Institute of Mental Health, National Alliance for Research on Schizophrenia and Depression, and the Texas Advanced Technology Program; an NIA National Research Service Award to K.T.D.; and a National Institute of Neurological Disorders and Stroke award to J.W.P. We thank Drs. James W. Patrick and Daniel H. S. Lee for helpful discussions during the preparation of this manuscript.
Correspondence should be addressed to Kelly T. Dineley or J. David Sweat, Division of Neuroscience, Room S603, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030. E-mail: kdineley@cns.bcm.tmc.eduor david@cns.bcm.tmc.edu.
REFERENCES
- 1.Andreasen N, Hesse C, Davidsson P, Minthon L, Wallin A, Winblad B, Vanderstichele H, Vanmechelen E, Blennow K. Cerebrospinal fluid β-amyloid (1-42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease [see comments]. Arch Neurol. 1999a;56:673–680. doi: 10.1001/archneur.56.6.673. [DOI] [PubMed] [Google Scholar]
- 2.Andreasen N, Minthon L, Vanmechelen E, Vanderstichele H, Davidsson P, Winblad B, Blennow K. Cerebrospinal fluid tau and Aβ42 as predictors of development of Alzheimer's disease in patients with mild cognitive impairment. Neurosci Lett. 1999b;273:5–8. doi: 10.1016/s0304-3940(99)00617-5. [DOI] [PubMed] [Google Scholar]
- 3.Arendt T, Bigl V, Tennstedt A, Arendt A. Neuronal loss in different parts of the nucleus basalis is related to neuritic plaque formation in cortical target areas in Alzheimer's disease. Neuroscience. 1985;14:1–14. doi: 10.1016/0306-4522(85)90160-5. [DOI] [PubMed] [Google Scholar]
- 4.Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- 5.Blum S, Moore AN, Adams F, Dash PK. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J Neurosci. 1999;19:3535–3544. doi: 10.1523/JNEUROSCI.19-09-03535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borchelt DR. Inherited neurodegenerative diseases and transgenic models. Lab Anim Sci. 1998;48:604–610. [PubMed] [Google Scholar]
- 7.Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 8.Breese CR, Adams C, Logel J, Drebing C, Rollins Y, Barnhart M, Sullivan B, Demasters BK, Freedman R, Leonard S. Comparison of the regional expression of nicotinic acetylcholine receptor α7 mRNA and [125I]-α-bungarotoxin binding in human postmortem brain. J Comp Neurol. 1997;387:385–398. doi: 10.1002/(sici)1096-9861(19971027)387:3<385::aid-cne5>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 9.Brining SK. Predicting the in vitro toxicity of synthetic β-amyloid (1-40). Neurobiol Aging. 1997;18:581–589. doi: 10.1016/s0197-4580(97)00153-x. [DOI] [PubMed] [Google Scholar]
- 10.Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C. Assembly and aggregation properties of synthetic Alzheimer's α4/β-amyloid peptide analogs. J Biol Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- 11.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 12.Cummings BJ, Cotman CW. Image analysis of β-amyloid load in Alzheimer's disease and relation to dementia severity. Lancet. 1995;346:1524–1528. doi: 10.1016/s0140-6736(95)92053-6. [DOI] [PubMed] [Google Scholar]
- 13.Cummings BJ, Pike CJ, Shankle R, Cotman CW. β-Amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer's disease [see comments]. Neurobiol Aging. 1996;17:921–933. doi: 10.1016/s0197-4580(96)00170-4. [DOI] [PubMed] [Google Scholar]
- 14.Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dodart JC, Mathis C, Ungerer A. The β-amyloid precursor protein and its derivatives: from biology to learning and memory processes. Rev Neurosci. 2000;11:75–93. doi: 10.1515/revneuro.2000.11.2-3.75. [DOI] [PubMed] [Google Scholar]
- 16.Ekinci FJ, Malik KU, Shea TB. Activation of the L voltage-sensitive calcium channel by mitogen-activated protein (MAP) kinase following exposure of neuronal cells to β-amyloid. MAP kinase mediates β-amyloid-induced neurodegeneration. J Biol Chem. 1999;274:30322–30327. doi: 10.1074/jbc.274.42.30322. [DOI] [PubMed] [Google Scholar]
- 17.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long-term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 18.Fenster CP, Whitworth TL, Sheffield EB, Quick MW, Lester RA. Upregulation of surface α4β2 nicotinic receptors is initiated by receptor desensitization after chronic exposure to nicotine. J Neurosci. 1999;19:4804–4814. doi: 10.1523/JNEUROSCI.19-12-04804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fitzjohn SM, Morton RA, Kuenzi F, Davies CH, Seabrook GR, Collingridge GL. Similar levels of long-term potentiation in amyloid precursor protein: null and wild-type mice in the CA1 region of picrotoxin-treated slices. Neurosci Lett. 2000;288:9–12. doi: 10.1016/s0304-3940(00)01204-0. [DOI] [PubMed] [Google Scholar]
- 20.Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV. Acetylcholine activates an α-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci. 1998;18:1187–1195. doi: 10.1523/JNEUROSCI.18-04-01187.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freund RK, Jungschaffer DA, Collins AC, Wehner JM. Evidence for modulation of GABAergic neurotransmission by nicotine. Brain Res. 1988;453:215–220. doi: 10.1016/0006-8993(88)90160-6. [DOI] [PubMed] [Google Scholar]
- 22.Freund RK, Luntz-Leybman V, Collins AC. Nicotine interferes with GABA-mediated inhibitory processes in mouse hippocampus. Brain Res. 1990;527:286–291. doi: 10.1016/0006-8993(90)91148-a. [DOI] [PubMed] [Google Scholar]
- 23.Garzon-Rodriguez W, Sepulveda-Becerra M, Milton S, Glabe CG. Soluble amyloid Aβ (1-40) exists as a stable dimer at low concentrations. J Biol Chem. 1997;272:21037–21044. doi: 10.1074/jbc.272.34.21037. [DOI] [PubMed] [Google Scholar]
- 24.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid-β protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsiao K. Transgenic mice expressing Alzheimer amyloid precursor proteins. Exp Gerontol. 1998;33:883–889. doi: 10.1016/s0531-5565(98)00045-x. [DOI] [PubMed] [Google Scholar]
- 26.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice [see comments]. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 27.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 28.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related Aβ deposits and neuropil abnormalities, but no neuronal loss in CA1 [see comments]. J Neuropathol Exp Neurol. 1997a;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Aβ deposition is associated with neuropil changes, but not with overt neuronal loss, in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997b;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kihara T, Shimohama S, Sawada H, Kimura J, Kume T, Kochiyama H, Maeda T, Akaike A. Nicotinic receptor stimulation protects neurons against β-amyloid toxicity. Ann Neurol. 1997;42:159–163. doi: 10.1002/ana.410420205. [DOI] [PubMed] [Google Scholar]
- 31.Kosik KS, Qiu WQ, Greenberg S. Cellular signaling pathways and cytoskeletal organization. Ann NY Acad Sci. 1996;777:114–120. doi: 10.1111/j.1749-6632.1996.tb34409.x. [DOI] [PubMed] [Google Scholar]
- 32.Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE. Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271:4077–4081. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- 33.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marks MJ, Burch JB, Collins AC. Effects of chronic nicotine infusion on tolerance development and nicotinic receptors. J Pharmacol Exp Ther. 1983;226:817–825. [PubMed] [Google Scholar]
- 35.McQuiston AR, Madison DV. Nicotinic receptor activation excites distinct subtypes of interneurons in the rat hippocampus. J Neurosci. 1999;19:2887–2896. doi: 10.1523/JNEUROSCI.19-08-02887.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mills J, Laurent Charest D, Lam F, Beyreuther K, Ida N, Pelech SL, Reiner PB. Regulation of amyloid precursor protein catabolism involves the mitogen-activated protein kinase signal transduction pathway. J Neurosci. 1997;17:9415–9422. doi: 10.1523/JNEUROSCI.17-24-09415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura T, Shoji M, Harigaya Y, Watanabe M, Hosoda K, Cheung TT, Shaffer LM, Golde TE, Younkin LH, Younkin SG. Amyloid-β protein levels in cerebrospinal fluid are elevated in early-onset Alzheimer's disease. Ann Neurol. 1994;36:903–911. doi: 10.1002/ana.410360616. [DOI] [PubMed] [Google Scholar]
- 38.Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline [see comments]. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 39.Nordberg A. PET studies and cholinergic therapy in Alzheimer's disease. Rev Neurol (Paris) 1999;155:S53–S63. [PubMed] [Google Scholar]
- 40.Orr-Urtreger A, Broide RS, Kasten MR, Dang H, Dani JA, Beaudet AL, Patrick JW. Mice homozygous for the L250T mutation in the α7 nicotinic acetylcholine receptor show increased neuronal apoptosis and die within 1 day of birth. J Neurochem. 2000;74:2154–2166. doi: 10.1046/j.1471-4159.2000.0742154.x. [DOI] [PubMed] [Google Scholar]
- 41.Payne DM, Rossomando AJ, Martino P, Erickson AK, Her JH, Shabanowitz J, Hunt DF, Weber MJ, Sturgill TW. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J. 1991;10:885–892. doi: 10.1002/j.1460-2075.1991.tb08021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perry EK, Morris CM, Court JA, Cheng A, Fairbairn AF, McKeith IG, Irving D, Brown A, Perry RH. Alteration in nicotine binding sites in Parkinson's disease, Lewy body dementia, and Alzheimer's disease: possible index of early neuropathology. Neuroscience. 1995;64:385–395. doi: 10.1016/0306-4522(94)00410-7. [DOI] [PubMed] [Google Scholar]
- 43.Pettit DL, Shao Z, Yakel JL. β-Amyloid 1-42 peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci. 2001;21:RC120. doi: 10.1523/JNEUROSCI.21-01-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rapoport M, Ferreira A. PD98059 prevents neurite degeneration induced by fibrillar β-amyloid in mature hippocampal neurons. J Neurochem. 2000;74:125–133. doi: 10.1046/j.1471-4159.2000.0740125.x. [DOI] [PubMed] [Google Scholar]
- 45.Revah F, Bertrand D, Galzi JL, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux JP. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- 46.Roberson ED, Sweatt JD. Transient activation of cyclic AMP-dependent protein kinase during hippocampal long-term potentiation. J Biol Chem. 1996;271:30436–30441. doi: 10.1074/jbc.271.48.30436. [DOI] [PubMed] [Google Scholar]
- 47.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saitoh T, Horsburgh K, Masliah E. Hyperactivation of signal transduction systems in Alzheimer's disease. Ann NY Acad Sci. 1993;695:34–41. doi: 10.1111/j.1749-6632.1993.tb23023.x. [DOI] [PubMed] [Google Scholar]
- 49.Schafe GE, Nadel NV, Sullivan GM, Harris A, LeDoux JE. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem. 1999;6:97–110. [PMC free article] [PubMed] [Google Scholar]
- 50.Scheffel U, Horti AG, Koren AO, Ravert HT, Banta JP, Finley PA, London ED, Dannals RF. 6-[18F]fluoro-A-85380: an in vivo tracer for the nicotinic acetylcholine receptor. Nucl Med Biol. 2000;27:51–56. doi: 10.1016/s0969-8051(99)00082-7. [DOI] [PubMed] [Google Scholar]
- 51.Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Selcher JC, Atkins CM, Trzaskos JM, Paylor R, Sweatt JD. A necessity for MAP kinase activation in mammalian spatial learning. Learn Mem. 1999;6:478–490. doi: 10.1101/lm.6.5.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selkoe DJ. The cell biology of β-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 54.Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 55.Sturgill TW, Ray LB, Erikson E, Maller JL. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature. 1988;334:715–718. doi: 10.1038/334715a0. [DOI] [PubMed] [Google Scholar]
- 56.Tapiola T, Pirttila T, Mikkonen M, Mehta PD, Alafuzoff I, Koivisto K, Soininen H. Three-year follow-up of cerebrospinal fluid tau, β-amyloid 42 and 40 concentrations in Alzheimer's disease. Neurosci Lett. 2000;280:119–122. doi: 10.1016/s0304-3940(00)00767-9. [DOI] [PubMed] [Google Scholar]
- 57.Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. β-Amyloid (1-42) binds to α7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- 58.Watabe AM, Zaki PA, O'Dell TJ. Coactivation of β-adrenergic and cholinergic receptors enhances the induction of long-term potentiation and synergistically activates mitogen-activated protein kinase in the hippocampal CA1 region. J Neurosci. 2000;20:5924–5931. doi: 10.1523/JNEUROSCI.20-16-05924.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by β-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- 60.Xing J, Ginty DD, Greenberg ME. Coupling of the RAS–MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 61.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease [see comments]. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 62.Zaman SH, Parent A, Laskey A, Lee MK, Borchelt DR, Sisodia SS, Malinow R. Enhanced synaptic potentiation in transgenic mice expressing presenilin 1 familial Alzheimer's disease mutation is normalized with a benzodiazepine. Neurobiol Dis. 2000;7:54–63. doi: 10.1006/nbdi.1999.0271. [DOI] [PubMed] [Google Scholar]