

Abstract
Excessive consumption of ethanol during pregnancy can produce teratogenic effects in offspring and is the leading cause of mental deficiency in the Western world. The objective of this study was to examine the effects of chronic prenatal ethanol exposure on the number of GABAA receptors and relative protein levels for GABAA receptor α1 and β2/3 subunits in the adult guinea pig cerebral cortex. Timed pregnant Dunkin-Hartley strain guinea pigs were given one of the following oral treatments daily throughout gestation: 4 gm of ethanol per kilogram of maternal body weight, isocaloric-sucrose with pair feeding, or isovolumetric water withad libitum access to food. The ethanol treatment resulted in a peak maternal blood ethanol concentration of 328 ± 55 mg/dl (71.3 ± 12.0 mm) on gestational day 57 (term, ∼68 d). Chronic prenatal exposure to ethanol resulted in increased spontaneous locomotor activity throughout development and decreased cerebral cortical weight in adult offspring. The number of cerebral cortical [3H]muscimol binding sites was increased in adult offspring from the ethanol treatment group, and there was a corresponding increase in the amount of GABAAreceptor α1 and β2/3 subunit proteins in these same animals. For individual offspring, there were correlations between locomotor activity and cerebral cortical weight, as well as between cerebral cortical weight and GABAA receptor neurochemistry. There was no effect of chronic prenatal ethanol exposure on [3H]MK-801 binding in this tissue. These data demonstrate that chronic prenatal ethanol exposure has long-term consequences on the regulation of GABAA receptor expression in the cerebral cortex.
Keywords: GABAA receptor, NMDA receptor, prenatal ethanol exposure, cerebral cortex, guinea pig, radioligand binding, Western blot
Excessive consumption of ethanol during pregnancy produces teratogenic effects in the fetus that can manifest postnatally as fetal alcohol syndrome (FAS) (Jones and Smith, 1973). The most debilitating and persistent characteristic of FAS is CNS dysfunction that can present as impairments in emotion management, attention, learning, memory, and problem solving. (Streissguth et al., 1991; Mattson et al., 1996; Steinhausen and Spohr, 1998). FAS is recognized as the leading cause of mental deficiency in the Western world.
The GABAA receptor is the major mediator of inhibitory neurotransmission in the CNS and is a heteromeric ion channel complex composed of five subunits. CNS GABAA receptor subunits are organized into six distinct families based on sequence homology (α, β, γ, δ, ε, and θ), and some families contain subunit subtypes (e.g., α1-α6). Generally, a combination of α, β, and γ subunits forms fully functional receptors. The GABAA receptor is an important site of action for benzodiazepines, barbiturates, neuroactive steroids, convulsants, and ethanol (for review, see Rabow et al., 1995;Mehta and Ticku, 1999). Different GABAA receptor isoforms result from the combination of different subunit subtypes, producing GABAA receptors with distinct pharmacological characteristics (Barnard et al., 1998). GABAA receptor subunit subtype expression varies both developmentally and regionally throughout the CNS. The expression of GABAA receptor subunit subtypes also may be altered significantly by chronic exposure to xenobiotics, including ethanol (Mhatre et al., 1993; Mhatre and Ticku, 1994; Devaud et al., 1995, 1997).
The effect of chronic exposure to ethanol in utero on GABAA receptor number or subunit subtype protein expression has not been elucidated, although certain GABAA receptor-mediated behavioral (Zimmerberg et al., 1995; Osborn et al., 1998) and neurochemical (Allan et al., 1998;Hsiao et al., 1998, 1999) dysfunctions have been identified. Data from our laboratory demonstrate that chronic prenatal ethanol exposure increases the number of cerebral cortical benzodiazepine-binding GABAA receptors and decreases [3H]flunitrazepam affinity for these GABAA receptors in adult offspring (Bailey et al., 1999). Using the guinea pig as an experimental animal model, the objectives of this study were to determine (1) whether chronic prenatal ethanol exposure increases the number of all GABAA receptors in the adult cerebral cortex using [3H]muscimol as the ligand for the GABA binding site and (2) whether this increase in GABAA receptor number occurs concurrently with an increase in the relative protein content of the GABAA receptor α1 and β2/3 subunits, each of which is the most abundant subtype or subtypes of their subunit families in the adult cerebral cortex (Laurie et al., 1992). The effect of chronic prenatal ethanol exposure on the number of NMDA receptors in the adult guinea pig cerebral cortex also was determined, because the function of this excitatory neurotransmitter receptor also has been demonstrated to be altered by chronic prenatal exposure to ethanol (Morrisett et al., 1989; Costa et al., 2000).
MATERIALS AND METHODS
Experimental animals. Nulliparous female Dunkin-Hartley strain guinea pigs (550–600 gm body weight; Charles River Canada, St. Constant, Quebec, Canada) were bred with male guinea pigs of the same strain (650–1000 gm body weight) according to an established procedure (Elvidge, 1972). The last day of full vaginal membrane opening was identified as gestational day 0 (G0) (term, ∼G68). On G1, the pregnant animals were housed individually at an ambient temperature of 23°C with a 12 hr light/dark cycle with lights on at 8:00 A.M.. Vaginal membrane status and general health of the pregnant animals were monitored, and body weight was measured daily throughout gestation. All guinea pigs were cared for according to the principles and guidelines of the Canadian Council on Animal Care. The experimental protocol was approved by the Queen's University Animal Care Committee.
Animal treatment regimens. On G2, pregnant guinea pigs were randomly assigned to receive oral administration of one of the following treatment regimens up to and including G67: (1) 4 gm of ethanol per kilogram of maternal body weight per day with ad libitum access to pellet food (PMI Nutrition International guinea pig diet 5025) and water; (2) isocaloric-sucrose and pair feeding withad libitum access to water; or (3) isovolumetric water withad libitum access to food and water. The pair-feeding regimen consisted of each sucrose-treated guinea pig being paired to an ethanol-treated guinea pig, receiving sucrose that was isocaloric and isovolumetric to the daily ethanol dose and receiving food in an amount that was equal to that consumed by the ethanol-treated guinea pig on each day of gestation. The daily treatments were given via oral intubation into the mouth and were administered as two equally divided doses 2 hr apart commencing between 9:30 and 11:00 A.M.. The ethanol (30% v/v) and sucrose (42% w/v) solutions were prepared with tap water. On G57, 200 μl of blood was taken from an ear blood vessel of the ethanol-treated pregnant animals at 1 hr after the second divided dose of ethanol for the determination of ethanol concentration by an established gas-liquid chromatographic method (Steenaart et al., 1985). Blood was collected from animals of the isocaloric-sucrose and water treatment groups at 1 hr after the second daily dose on G57 to control for the stress of blood sampling. Litters were transferred to large plastic bins with wood chip bedding on the day of birth [postnatal day 0 (P0)]. Beginning at P1, offspring were weighed and monitored daily for general health. The offspring were weaned from their mothers on P17 and were separated based on gender on P22. Offspring of each gender and for all treatment groups were housed randomly in groups of up to four animals per bin.
Locomotor activity testing. On each of P10 (preweaning), P20 (postweaning), and P60 (adulthood), horizontal and vertical spontaneous locomotor activity were measured for 1 hr by placing individual offspring in a 42 × 42 cm open field apparatus (Columbus Instruments, Columbus, OH). Infrared photobeams 3 cm above the floor of the instrument measured movement in the horizontal plane, and photobeams placed at 10 cm (P10 and 20) or 12.5 cm (P60) above the floor measured movement in the vertical plane. The test was recorded by videotape for 1 hr in an isolated, quiet environment. Videotapes were reviewed, and the cumulative number of photobeam breaks in each plane was recorded at each 10 min interval.
Tissue collection. On P61, individual offspring were killed by decapitation under halothane anesthesia. The brains were rapidly excised, weighed, and dissected. Cerebral cortices were isolated from each brain, frozen in liquid N2, weighed, and stored at −70°C until analyzed. For each animal, the right cerebral cortex was allocated to the radioligand binding assay experiments, and the left cerebral cortex was used in the immunoblot analysis.
Preparation of crude cell membrane fraction for radioligand binding assays. Right cerebral cortex from each animal was thawed and homogenized in 20 ml of homogenization buffer (10 mm Tris-HCl, 300 mmsucrose, and 2 mm EDTA, pH 7.4) per gram of tissue. This homogenate was centrifuged at 1000 × gfor 10 min at 4°C. The resulting supernatant was centrifuged at 20,000 × g for 20 min at 4°C. The resulting pellet was then resuspended in 3 ml of homogenization buffer and frozen at −70°C until analyzed.
Radioligand binding assays. [3H]Muscimol binding to GABAA receptors was determined by first washing the cell membrane preparation as follows: individual aliquots were diluted with five volumes of wash buffer (50 mmTris-HCl and 2 mm EDTA, pH 7.4), mixed, and centrifuged at 13,000 × g for 10 min at 4°C. This washing procedure was repeated twice, and the final pellet was resuspended in binding assay buffer A (10 mmTris-HCl and 150 mm NaCl, pH 7.4). The protein concentration of each sample was determined by a spectrophotometric protein dye-binding assay based on the method of Bradford (1976), using bovine serum albumin as the standard. [3H]Muscimol saturation binding was determined in reaction vessels containing 100–150 μg of the washed cell membrane fraction, [3H]muscimol in concentrations ranging from 1 to 60 nm (20 Ci/mmol; New England Nuclear, Boston, MA), and binding assay buffer A to give a total volume of 500 μl. Nonspecific binding of [3H]muscimol was determined in the presence of 100 μm GABA. Samples were incubated for 60 min at 4°C, and the reaction was terminated by rapid vacuum filtration through Whatman GF/B glass fiber filters that were prewet with 5 ml of ice-cold binding assay buffer A. The filters were washed twice with 5 ml of ice-cold binding assay buffer A, and the radioactivity remaining on the filters was quantified by liquid scintillation spectrometry using a Beckman LS 6500 scintillation counter.
[3H]MK-801 binding to NMDA receptors was determined by first washing the cell membrane fraction as described above with the following changes: binding assay buffer B (20 mm HEPES and 1 mm EDTA, pH 7.4) was used, and the samples were incubated for 30 min at 37°C before the first centrifugation. [3H]MK-801 binding was determined in reaction vessels containing 200 μg of the washed cell membrane fraction, 5 nm[3H]MK-801 (21.7 Ci/mmol; New England Nuclear), 100 μm glutamate, 100 μm glycine, and binding assay buffer B to give a total volume of 500 μl. Nonspecific binding of [3H]MK-801 was determined in the presence of 100 μm MK-801. Samples were incubated for 3 hr at 37°C, the reaction was terminated by rapid vacuum filtration, and the samples were quantified as described above.
Isolation of protein for immunoblot analysis. Left cerebral cortex from each animal was thawed and homogenized in 3 ml/gm of lysis buffer [10 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, 1% (w/v) SDS, 1% (w/v) sodium deoxycholate, and 1% (v/v) Triton X-100 plus protease inhibitors (1 μg of aprotinin, 5 μg of leupeptin, and 10 μg of PMSF/ml of lysis buffer)]. The homogenate was centrifuged at 13,000 × g for 30 min at 4°C, and the supernatant was collected. The protein concentration of each supernatant was determined by the Lowry et al. (1951) assay, using bovine serum albumin as the standard. Samples were then frozen and stored at −70°C until analyzed.
Immunoblot analysis. Immunoblot analysis of the GABAA receptor α1 subunit was performed by SDS-PAGE using a Bio-Rad (Mississauga, Ontario, Canada) Mini Protean III apparatus. Individual protein samples were heated at 100°C for 5 min in loading buffer [65.8 mmTris-HCl, 10% (v/v) glycerol, 2% (w/v) SDS, 4% (v/v) 2-mercaptoethanol, and 0.025% (w/v) bromophenol blue, pH 6.8]. For each sample, 30 μg of protein in 20 μl of loading buffer was loaded onto a 10% SDS-polyacrylamide gel. Each sample was analyzed in triplicate, and each gel contained samples from three animals (one animal from each of the three treatment groups). The protein samples were separated in the presence of a running buffer [15 mm Tris base, 115 mmglycine, and 0.06% (w/v) SDS, pH 8.3] at room temperature for 60 min at 100 V.
The separated protein samples were then transferred onto a polyvinylidene difluoride carrier membrane (Amersham Pharmacia Biotech, Baie d'Urfe, Quebec, Canada). Protein transfer was performed using the Bio-Rad Mini Trans Blot system in the presence of ice-cold transfer buffer [25 mm Tris base, 192 mmglycine, and 20% (v/v) methanol, pH 8.3] for 60 min at 100 V. Nonspecific sites were blocked by incubating the membrane in blocking solution [5% (w/v) skim milk powder, Tris-buffered saline containing Tween 20 (TBS-T) (50 mm Tris-HCl, 400 mm NaCl, 0.05% (v/v) Tween 20, pH 7.6)] for 1 hr at room temperature. The membrane was washed with TBS-T and then incubated with 2 μg/ml of a rabbit polyclonal, affinity-purified antibody raised against amino acids 1–15 of the rat GABAA receptor α1 subunit (Upstate Biotechnology, Lake Placid, NY) in blocking solution overnight at 4°C. The membrane was washed with TBS-T and then incubated with a 1:4000 dilution in blocking solution of a goat anti-rabbit IgG (H+L) secondary antibody conjugated with horseradish peroxidase (Bio-Rad) for 1 hr at room temperature. The membrane was washed thoroughly with TBS-T and developed using the ECL chemiluminescence detection system and Hyperfilm ECL x-ray film (Amersham Pharmacia Biotech). The amount of total protein loaded on each gel was within the linear range of optical density for all proteins examined in this study.
To control for differences in protein loading, each membrane was stripped and reprobed for β-actin as follows: the membrane was washed with TBS-T and incubated in stripping solution (200 mmglycine, pH 2.6) for 1 hr at room temperature. The blot was washed with TBS-T and probed for β-actin as described above. A 1:4000 dilution of a mouse monoclonal anti-β-actin antibody was used (Sigma-Aldrich Canada, Oakville, Ontario, Canada), followed by a 1:4000 dilution of a goat anti-mouse IgG (H+L) secondary antibody (Bio-Rad).
The immunoblot bands were quantified by measuring their relative optical density (ROD) using Corel (Ottawa, Ontario, Canada) Photo-Paint 9. ROD measurements for the GABAA receptor subunits were normalized to the ROD of the β-actin signal within each lane, because chronic prenatal ethanol exposure did not affect β-actin content in the cerebral cortex (data not shown).
Immunoblot analysis of the GABAA receptor β2/3 subunits was performed as described above for the α1 subunit with the following alterations. For each sample, 20 μg of protein was loaded onto the SDS-polyacrylamide gel after incubation in loading buffer (without 2-mercaptoethanol) for 30 min at 37°C. A mouse monoclonal antibody raised against whole GABAA receptor β2 and β3 subunits was used at 2 μg/ml (Upstate Biotechnology), and the goat anti-mouse IgG (H+L) secondary antibody was used at a 1:4000 dilution. After detection and stripping, the membrane was probed for β-actin using a 1:2000 dilution of the mouse monoclonal anti-β-actin antibody and a 1:4000 dilution of the goat anti-mouse secondary antibody.
Data analysis. The locomotor activity, body, brain, and cerebral cortical weight data are presented as the mean ± SEM of 8–13 offspring obtained from five different litters for each treatment group. The radioligand binding and immunoblot data are presented as the mean ± SEM of eight animals from five different litters for each treatment group. TheBmax andKD for [3H]muscimol binding were obtained by nonlinear regression analysis of individual saturation binding isotherms (Prism 2.0; GraphPad Software, San Diego, CA). Statistical analysis of the locomotor activity data was performed using two-way ANOVA (Prism 2.0). All other parametric statistical analyses were performed using one-way, randomized-design ANOVA, followed by the Newman–Keuls post hoc test for a statistically significantF statistic (Prism 2.0). The data were analyzed for homogeneity of variance before conducting the ANOVA tests. Correlational analyses were performed using the Pearson correlation test (Prism 2.0). The level for a statistical difference in the data was set at p < 0.05. Unless otherwise noted, statistical difference is cited in the text when the data for the ethanol treatment group were different from the data for both the isocaloric-sucrose/pair-fed and water treatment groups. Because there was no effect of gender on any of the observed parameters in any treatment group, the data of the male and female offspring in each treatment group were combined for the purpose of statistical analysis.
RESULTS
Maternal food intake and weight gain
Maternal average daily caloric intake and weights throughout gestation are displayed in Figure 1. Pregnant guinea pigs in the isocaloric-sucrose/pair-fed treatment group consumed food that was equal to that of the ethanol treatment group because of the pair-feeding regimen. The maternal average daily caloric intake for animals in the ethanol and isocaloric-sucrose/pair-fed treatment groups was 106.5 ± 7.8 kcal/d compared with 125.8 ± 9.7 kcal/d for animals in the water treatment group (Fig.1A) (p < 0.05). There was no difference in maternal body weight at G0. However, pregnant guinea pigs in the ethanol and isocaloric-sucrose/pair-fed treatment groups gained significantly less weight during gestation than the pregnant guinea pigs in the water treatment group (Fig. 1B) (p < 0.05).
Fig. 1.
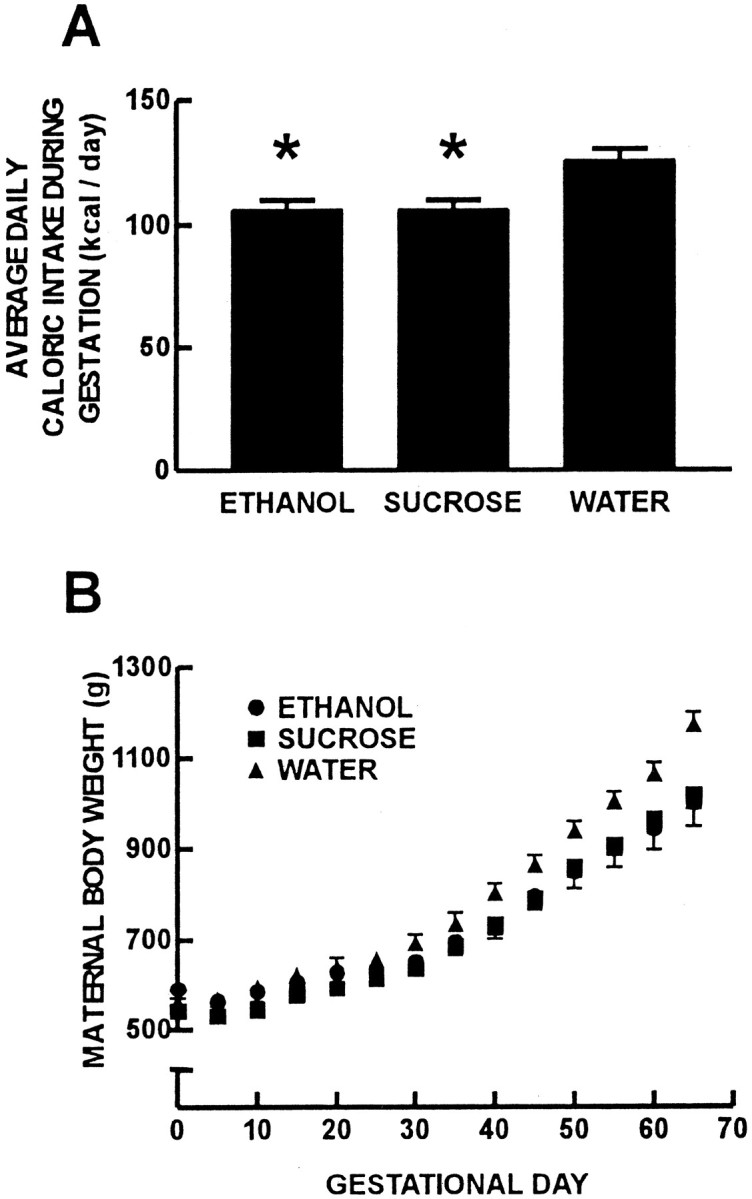
Pregnant guinea pig nutritional data for the five dams in each treatment group. Maternal average daily caloric intake during gestation (A). Pregnant guinea pigs in the ethanol treatment group consumed less food than those in the water treatment group (*p < 0.05). Pregnant animals in the isocaloric-sucrose/pair-fed treatment group were given and consumed food equal to that of the ethanol treatment group by nature of the pair-feeding regimen. B, Maternal body weight for the three treatment groups throughout gestation.Circles represent ethanol, squaresrepresent isocaloric-sucrose/pair-fed, andtriangles represent water treatment groups. Pregnant guinea pigs in the ethanol and isocaloric-sucrose/pair-fed treatment groups gained less weight than those in the water treatment group (p < 0.05).
Maternal blood ethanol concentration
The maternal blood ethanol concentration of the guinea pigs in the ethanol treatment group 1 hr after the second divided dose administrated on G57 was 328 ± 55 mg/dl (71.3 ± 12.0 mm). This maternal blood ethanol concentration value was similar to that found in previous experiments from our laboratory using the same chronic ethanol treatment regimen in the guinea pig, in which specific behavioral and neurochemical teratogenic effects were observed (Abdollah et al., 1993; Catlin et al., 1993; Kimura and Brien, 1998;Gibson et al., 2000).
Pregnancy outcome
There was no maternal lethality or spontaneous abortion in any treatment group. Chronic maternal ethanol administration had no effect on gestation length, litter size, or the ratio of male to female offspring. There were three incidents of perinatal death (death at parturition) in the ethanol treatment group and one incident in the water treatment group.
Locomotor activity
Offspring horizontal and vertical spontaneous locomotor activity data are presented in Figure 2. At each of P10, 20, and 60, chronic prenatal ethanol administration resulted in increased cumulative spontaneous locomotor activity in both the horizontal and vertical planes compared with the isocaloric-sucrose/pair-fed and water treatment groups (p < 0.05).
Fig. 2.

Offspring cumulative horizontal and vertical spontaneous locomotor activity measured in an open-field apparatus for a 60 min period. Activity was measured on each of P10 (preweaning), P20 (postweaning), and P60 (adulthood). Circles represent ethanol, squares represent isocaloric-sucrose/pair-fed, and triangles represent water treatment groups. The cumulative horizontal and vertical spontaneous locomotor activity data of the offspring in the ethanol treatment group were greater compared with the data for the isocaloric-sucrose/pair-fed and water treatment groups at each postnatal age (p < 0.05).
Body, brain, and cerebral cortical weights
Offspring body, brain, and cerebral cortical weights are presented in Table 1. Both chronic prenatal ethanol treatment and isocaloric-sucrose administration with pair feeding resulted in decreased body weight at P1 compared with the water treatment group (p < 0.05). There was no effect of chronic prenatal treatment on body weight at any other postnatal age examined. At P61, chronic prenatal ethanol treatment resulted in decreased brain and cerebral cortical weights compared with the isocaloric-sucrose/pair-fed and water treatment groups (p < 0.05).
Table 1.
Comparison of chronic prenatal administration of ethanol, isocaloric-sucrose/pair feeding, and water on offspring body, brain, and cerebral cortical weights
| Weight (gm) | Chronic treatment | ||
|---|---|---|---|
| Ethanol (n = 8) | Sucrose (n = 12) | Water (n = 13) | |
| P1 body | 93.1 ± 4.3* | 90.8 ± 4.6* | 104.6 ± 1.8 |
| P10 body | 182.5 ± 8.5 | 162.1 ± 8.5 | 176.9 ± 3.1 |
| P20 body | 288.8 ± 12.3 | 267.5 ± 11.2 | 273.5 ± 4.9 |
| P60 body | 609.4 ± 29.2 | 589.6 ± 18.5 | 563.8 ± 14.8 |
| P60 brain | 3.22 ± 0.051-160 | 3.72 ± 0.04 | 3.74 ± 0.04 |
| P60 cerebral cortex | 1.76 ± 0.031-160 | 2.02 ± 0.03 | 2.03 ± 0.03 |
p < 0.05 compared with the water treatment group.
F1-160: p < 0.05 compared with the isocaloric sucrose and water treatment groups.
[3H]Muscimol binding to GABAA receptors
Mean binding isotherm curves for [3H]muscimol binding to cerebral cortical GABAA receptors at P61 are presented in Figure 3A. Representative Scatchard transformations are presented in Figure 3B. Chronic prenatal ethanol treatment increased theBmax for [3H]muscimol binding to the high-affinity GABA binding site on GABAAreceptors compared with the isocaloric-sucrose/pair-fed and water treatment groups (Fig.4A) (p < 0.05). There was no effect of prenatal ethanol treatment on the KD (affinity) for [3H]muscimol binding to these binding sites. The [3H]muscimolKD values were 11.9 ± 1.3 nm (ethanol group), 12.1 ± 1.4 nm (isocaloric-sucrose/pair-fed group), and 10.1 ± 1.4 nm (water group).
Fig. 3.
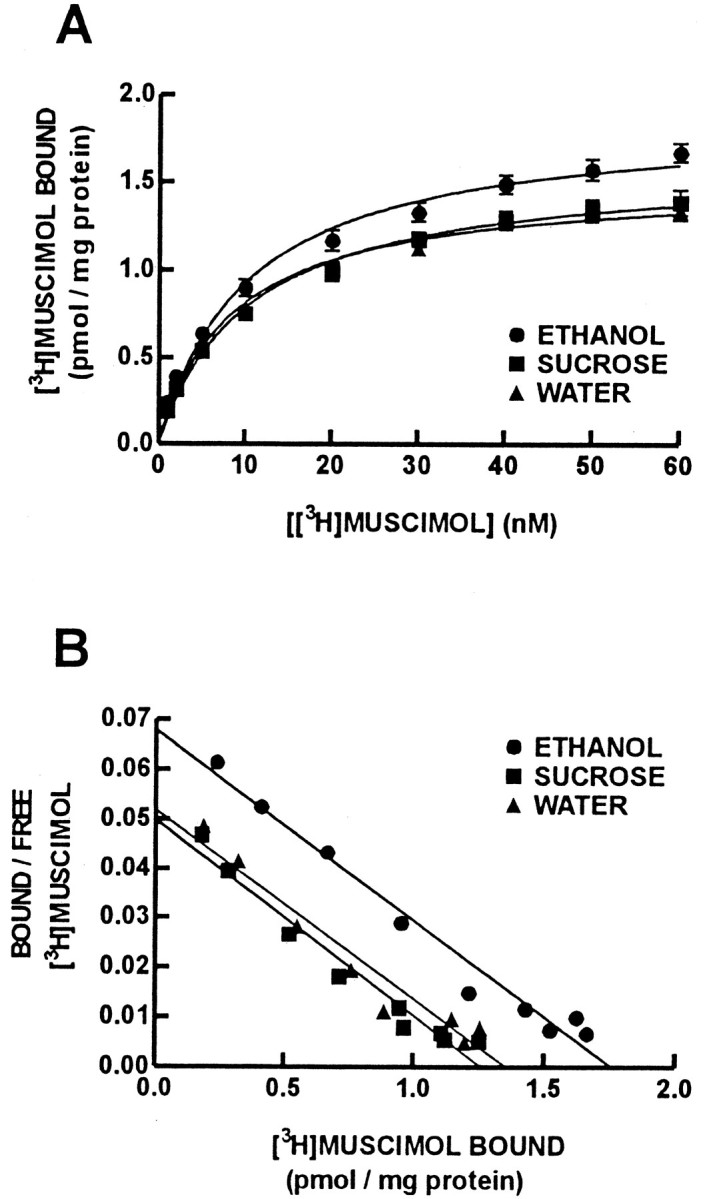
Saturation binding isotherms for [3H]muscimol binding to the high-affinity site on the GABAA receptor (A). Data are presented as the mean specific binding of [3H]muscimol ± SEM for eight offspring in each treatment group. Circles represent ethanol,squares represent isocaloric-sucrose/pair-fed, andtriangles represent water treatment groups. The number of [3H]muscimol binding sites (Bmax) was increased for offspring in the ethanol treatment group (Fig. 4) (p < 0.05), whereas the affinity of [3H]muscimol for the GABAA receptor was not affected by prenatal ethanol treatment. Representative Scatchard transformations of the [3H]muscimol saturation data also are presented (B).
Fig. 4.
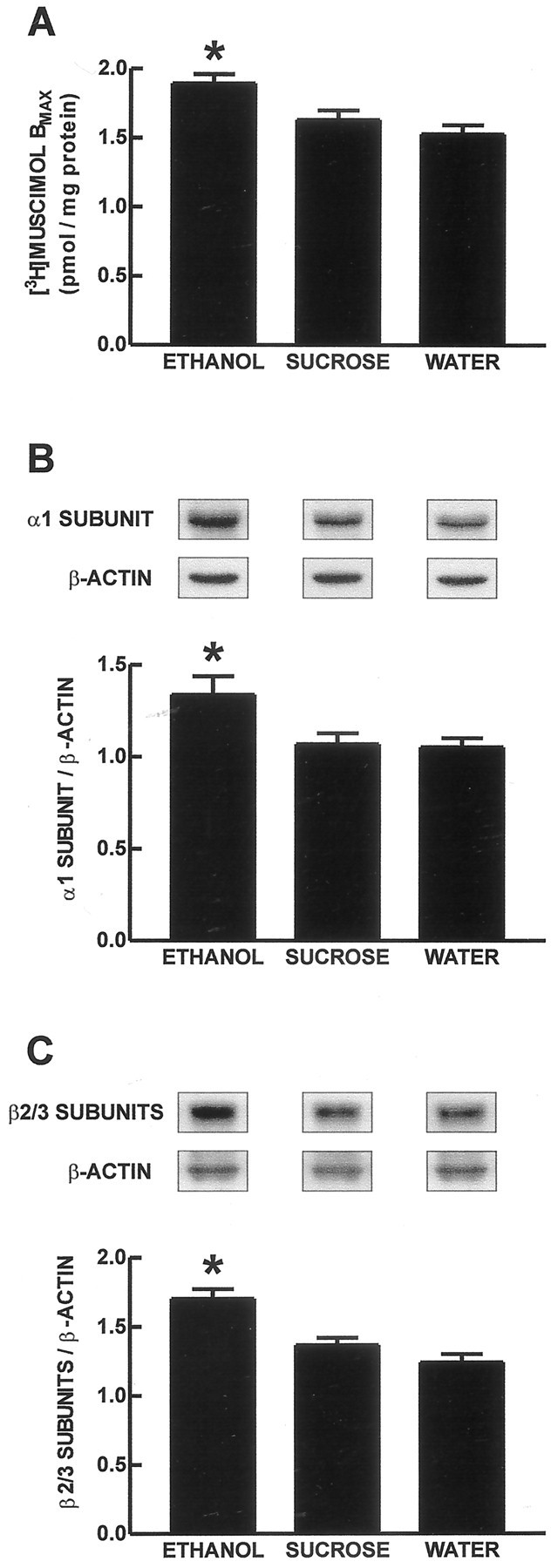
GABAA receptor [3H]muscimol binding and relative protein content for GABAA receptor α1 and β2/3 subunits.A, Bmax values for [3H]muscimol binding to the high-affinity binding site of the GABAA receptor in the adult guinea pig (P61) cerebral cortical cell membrane protein. The data are presented as the mean ± SEM of eight offspring. Representative immunoblot bands and mean ± SEM (n = 8 offspring in each treatment group) data for GABAA receptor α1 (B) and β2/3 (C) subunit proteins in adult guinea pig (P61) cerebral cortex. The data are expressed as relative amount of GABAA receptor subunit protein normalized to β-actin protein. *p < 0.05 for the increase in each of [3H]muscimolBmax (A), and the relative amounts of protein for the GABAA receptor α1 (B) and β2/3 (C) subunits in the ethanol treatment group compared with the isocaloric-sucrose/pair-fed and water treatment groups.
GABAA receptor α1 and β2/3 subunit proteins
GABAA receptor α1 subunit protein immunoblot analysis resulted in a strong signal at 51 kDa. Representative x-ray immunoblot bands and group data for the GABAA receptor α1 subunit protein are presented in Figure 4B. Chronic prenatal ethanol administration increased the relative amount of GABAA receptor α1 subunit protein normalized to β-actin (1.34 ± 0.10;n = 8) compared with the isocaloric-sucrose/pair-fed (1.07 ± 0.06; n = 8) and water (1.05 ± 0.05; n = 8) treatment groups (p< 0.05).
Immunoblot analysis of the GABAA receptor β2/3 subunit proteins resulted in a signal with a molecular weight of 55–57 kDa. Representative immunoblot bands and data are presented in Figure4C. Chronic prenatal ethanol administration increased the relative amount of these proteins (1.70 ± 0.07; n= 8) compared with the isocaloric-sucrose/pair-fed (1.36 ± 0.05;n = 8) and water (1.24 ± 0.06; n= 8) treatment groups (p < 0.05).
[3H]MK-801 binding to NMDA receptors
The amount of [3H]MK-801 bound to cerebral cortical NMDA receptors at P61 was not different among the three prenatal treatment groups. The amounts of [3H]MK-801 bound at 5 nmconcentration were 751.4 ± 38.6 fmol/mg of protein (ethanol group), 784.9 ± 26.7 fmol/mg of protein (isocaloric-sucrose/pair-fed group), and 810.1 ± 38.1 fmol/mg of protein (water group).
Correlational analyses
Correlational analyses were performed to compare cerebral cortical weight with cumulative horizontal and vertical spontaneous locomotor activity at P60 for individual offspring in which a complete set of behavioral, cerebral cortical weight, and neurochemical data were obtained (Fig. 5). There was a negative correlation between adult offspring cerebral cortical weight and the cumulative horizontal (Fig. 5A) (r = 0.629;p < 0.05) and vertical (Fig. 5B) (r = 0.489; p < 0.05) spontaneous locomotor activity. Correlational analyses were performed to compare adult guinea pig cerebral cortical weight with GABAA receptor binding and GABAA receptor subunit protein content (Fig.6). There was a negative correlation between cerebral cortical weight and both GABAAreceptor number ([3H]muscimolBmax) (Fig. 6A) (r = 0.550; p < 0.05) and GABAA receptor β2/3 subunit protein content (Fig. 6C) (r = 0.472; p < 0.05). There was no correlation between cerebral cortical weight and GABAA receptor α1 subunit protein content (Fig.6B) (r = 0.343). Correlational analysis also was performed to identify relationships between theBmax for [3H]muscimol and the relative amount of protein for the GABAA receptor α1 and β2/3 subunits (Fig. 7). There was no correlation between [3H]muscimolBmax and the relative amount of GABAA receptor α1 subunit protein (Fig.7A) (r = 0.284). However, there was a positive correlation between the Bmaxfor [3H]muscimol and the relative amount of GABAA receptor β2/3 subunit proteins (Fig.7B) (r = 0.622; p < 0.05).
Fig. 5.

Linear-linear plots for cerebral cortical weight and cumulative horizontal (A) and vertical (B) spontaneous locomotor activity data at 60 min of the 24 adult offspring of the three treatment groups.Circles represent ethanol, squaresrepresent isocaloric-sucrose/pair-fed, and trianglesrepresent water treatment groups. There was a negative correlation between cerebral cortical weight and spontaneous locomotor activity in both the horizontal (A) (r = 0.629; p < 0.05) and vertical (B) (r = 0.489;p < 0.05) planes.
Fig. 6.
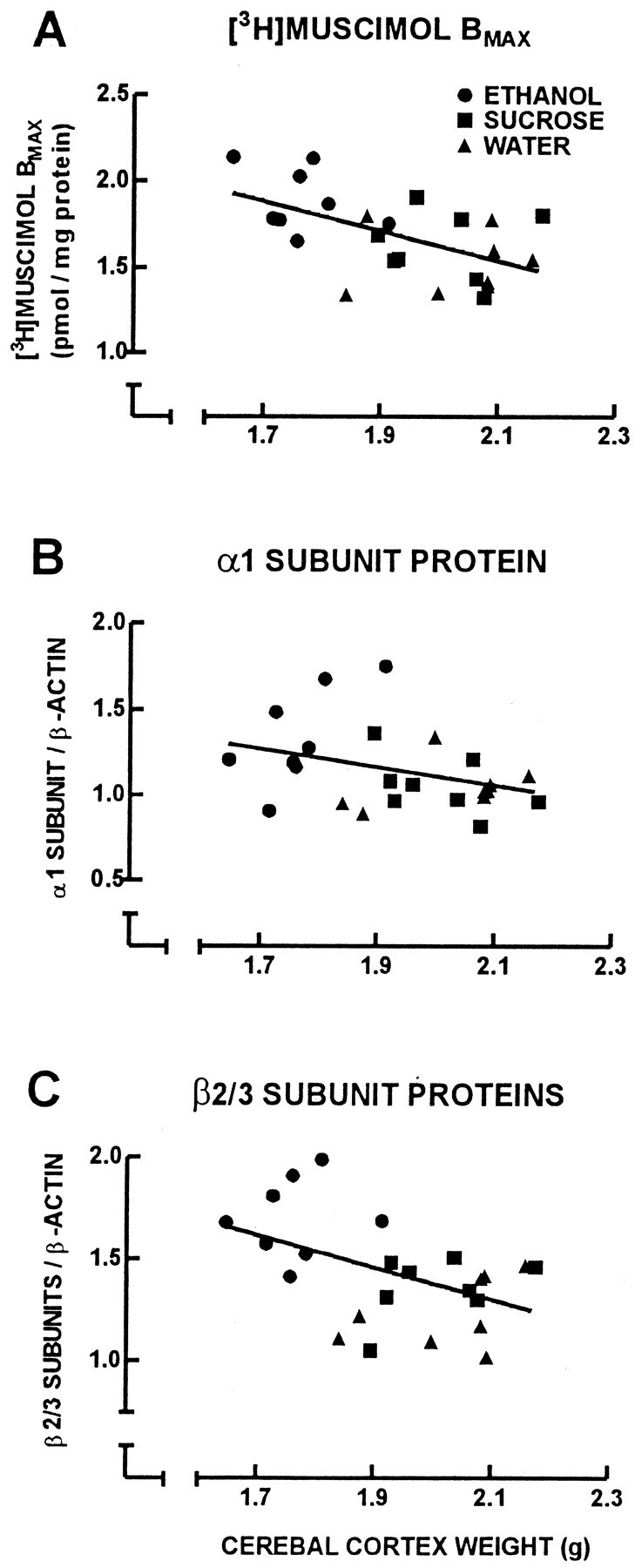
Linear-linear plots for cerebral cortical weight and [3H]muscimol Bmax(A), and relative amounts of protein for the GABAA receptor α1 (B) and β2/3 subunits (C) of the 24 adult offspring of the ethanol (circles), isocaloric-sucrose/pair-fed (squares), and water (triangles) treatment groups. There was a negative correlation between cerebral cortical weight and [3H]muscimolBmax (A) (r = 0.550; p < 0.05), and between cerebral cortical weight and the relative amount of protein for the GABAA receptor β2/3 subunits (C) (r = 0.472;p < 0.05).
Fig. 7.
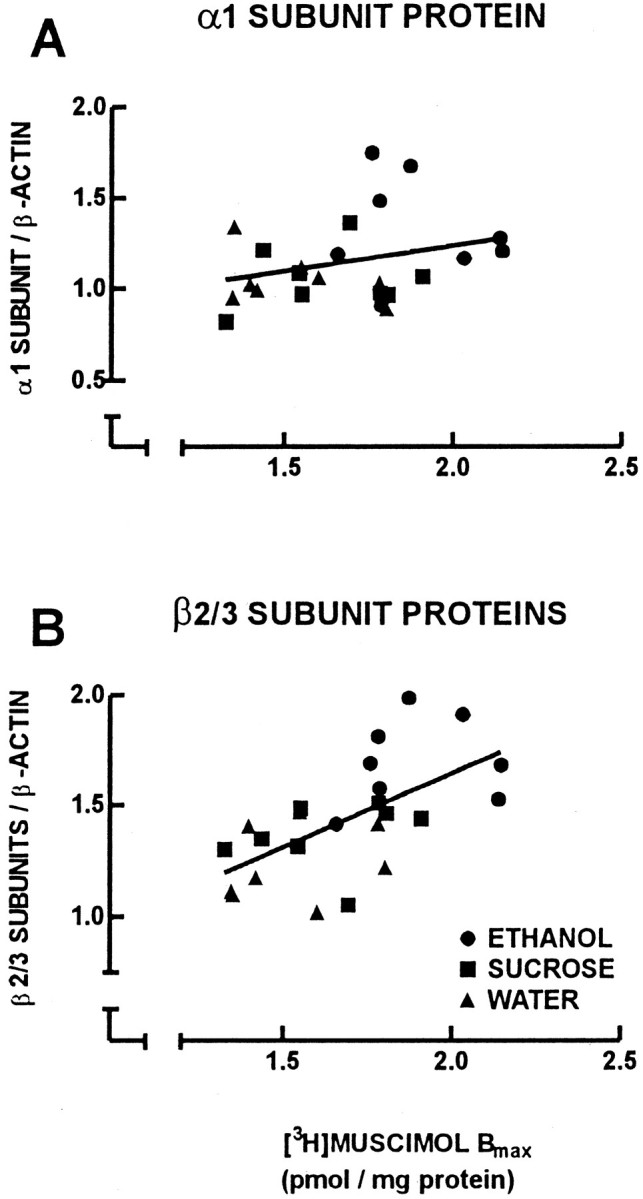
Linear-linear plots for [3H]muscimol Bmax and relative amounts of protein for the GABAA receptor α1 (A) and β2/3 (B) subunits of the 24 adult offspring of ethanol (circles), isocaloric-sucrose/pair-fed (squares), and water (triangles) treatment groups. There was a positive correlation between [3H]muscimolBmax and the relative amount of protein for the GABAA receptor β2/3 subunits (B) (r = 0.622;p < 0.05).
DISCUSSION
Chronic prenatal exposure to ethanol throughout gestation, via maternal ethanol administration, resulted in an increase in GABAA receptor number in the adult guinea pig cerebral cortex. Concurrent with this chronic prenatal ethanol-induced increase in receptor number was an increase in the relative expression of protein for cerebral cortical GABAA receptor α1 and β2/3 subunits in these same animals. This effect was found in the adult offspring 61 d after they were born, which demonstrates that prenatal exposure to ethanol can produce long-term neurochemical alterations. Indeed, young adults diagnosed with FAS during childhood have persistent behavioral deficits, including impairments in attention and memory (Kerns et al., 1997).
The decrease in maternal average daily food intake and corresponding decrease in maternal body weight gain in the ethanol and isocaloric-sucrose/pair-fed treatment groups compared with the water treatment group indicates that the isocaloric-sucrose/pair-fed treatment group was an appropriate nutritional control in this study. Moreover, offspring in the isocaloric-sucrose/pair-fed treatment group had birth weights that were similar to those in the ethanol treatment group and less than those in the water treatment group (Table 1). Although offspring body weights normalized by P10, chronic prenatal ethanol exposure resulted in persistent effects on decreasing brain and cerebral cortical weights, as seen at P61 (Table 1).
In this study, the persistent long-term effects of chronic prenatal ethanol exposure to increase locomotor activity and to decrease brain and cerebral cortical weights were of similar magnitude to results found in previous experiments from our laboratory using the same dosing regimen (Abdollah et al., 1993; Catlin et al., 1993; Butters et al., 2000). These changes are considered to be key manifestations of ethanol neurobehavioral teratogenicity. In this current study we found a strong negative correlation between cerebral cortical weight and locomotor activity and negative correlations between cerebral cortical weight and both the GABAA receptor number and GABAA receptor β2/3 subunit protein expression. These data demonstrate a relationship between brain injury and altered behavior in this animal model and suggest that the magnitude of changes in GABAA receptor expression are related to the extent of brain injury after chronic prenatal ethanol exposure.
Previous studies from our laboratory have shown that chronic prenatal ethanol exposure increases the number of benzodiazepine-binding GABAA receptors in the adult (P61) guinea pig cerebral cortex but not at P11 or P21 (Bailey et al., 1999). GABAA receptors normally contain α and β subunits but also require γ (with or without θ) subunits to confer benzodiazepine binding (Barnard et al., 1998; Bonnert et al., 1999). Using [3H]muscimol, a ligand that is selective for the high-affinity GABA binding site on the GABAA receptor, it was possible in the present study to determine the relative number of all GABAA receptors in the adult guinea pig cerebral cortex (i.e., those with and without benzodiazepine binding sites). The α1 and β2/3 subunits were chosen for examination in this study because they are the most predominant GABAAreceptor subunit subtypes in their respective subunit families in the adult cerebral cortex (Laurie et al., 1992) and would therefore be the most likely receptor subunits responsible for an increase in GABAA receptor number. This hypothesis was supported for the β2/3 subunits, for which there was a positive correlation between the number of [3H]muscimol binding sites and the relative amount of protein for the GABAA receptor β2/3 subunits of the individual animals of the ethanol, isocaloric-sucrose/pair-fed, and water treatment groups. The α1 subunit protein expression did not correlate with [3H]muscimol binding, which suggests that other α subunits also contribute to the observed increased number of [3H]muscimol binding sites. This idea is also supported by our previous study (Bailey et al., 1999), in which we reported that chronic prenatal ethanol exposure produced a decrease in the relative proportion of zolpidem-sensitive GABAA receptors (putative α1 subunit-containing GABAA receptors) in the adult guinea pig cerebral cortex.
GABAA receptor expression has been demonstrated to increase in response to chronic GABAA receptor hypofunction in vivo (Miller et al., 1989). The observed increase in GABAA receptor number in the adult guinea pig cerebral cortex produced by chronic prenatal ethanol exposure may therefore be a compensatory mechanism for a potential decrease in GABAergic neurotransmission and/or GABAA receptor function at earlier time points in development. Chronic prenatal exposure to ethanol in a rat model has been demonstrated to decrease the whole-cell patch clamp response to GABA in medial septum/diagonal band neurons in juvenile offspring but not in adult offspring (Hsiao et al., 1998). The anxiolytic response to a naturally occurring neuroactive steroid, 3α-hydroxy-5α-pregnan-20-one (allopregnanolone), which potentiates GABAA receptor function, was also decreased in neonatal rats that were exposed to ethanol during gestation (Zimmerberg et al., 1995). Interestingly, GABAA receptor function has been demonstrated to be increased in adult offspring that were prenatally exposed to ethanol. The behavioral effect of benzodiazepines to reduce anxiety was found to be increased in adult rats that were exposed to ethanol in utero (Osborn et al., 1998). Adult rats that were exposed to ethanol during the last third of gestation exhibited an increased electrophysiological response to GABA in frontal and somatosensory cortical neurons (Janiri et al., 1994). One report demonstrated that prenatal exposure to ethanol did not affect GABA-elicited Cl− flux through GABAA receptors in adult rat offspring but decreased modulation of Cl− flux by benzodiazepines and neuroactive steroids in the cerebral cortex (Allan et al., 1998). The disagreement among these studies in adult offspring may reflect differences in prenatal ethanol treatment regimens and differences in blood ethanol concentrations achieved by these different regimens, but also may suggest that there are brain regional differences in the long-term effects of prenatal exposure to ethanol.
One possible morphological explanation for a decrease in GABAergic input and subsequent upregulation of GABAAreceptors is a decrease in the number of cells that express GABAA receptors and/or use GABA as a neurotransmitter. Affected cell types in the cerebral cortex may include GABAergic interneurons, glutamatergic pyramidal cells, or glial cells (Bureau et al., 1995; Hornung and Fritschy, 1996). Chronic prenatal exposure to ethanol in the rat decreases neuronal and glial cell number in the somatosensory cortex of adult offspring (Miller and Potempa, 1990) and, more specifically, in the number of parvalbumin-positive neurons in the cingulate cortex, an index of normally functioning GABAergic neurons (Moore et al., 1998). Immunohistochemical experiments are being conducted in our laboratory to identify the cell types that may be affected by chronic prenatal ethanol exposure in the adult guinea pig somatosensory cortex. A decrease in GABAergic input also could result from a decrease in GABA synthesis and/or release from existing GABAergic interneurons. However, preliminary experiments in our laboratory in the adult guinea pig cerebral cortex indicate that protein content for glutamic acid decarboxylase, the enzyme responsible for GABA synthesis from glutamate, is not affected by chronic prenatal ethanol exposure (data not shown). The effect of chronic prenatal ethanol exposure on synaptic GABA release in the cerebral cortex has not been examined.
In this study, adult guinea pig cerebral cortical NMDA receptor number was not statistically different after chronic prenatal ethanol exposure. In the adult rat, chronic prenatal ethanol exposure decreases NMDA receptor number and produces electrophysiologic deficits in the hippocampus (Morrisett et al., 1989; Savage et al., 1991). Previous studies in our laboratory suggest that chronic prenatal ethanol exposure decreases glutamate and NMDA binding in the near-term fetal hippocampus (Abdollah and Brien, 1995) but increases MK-801 binding in the near-term fetal cerebral cortex (Chiu et al., 1999). Collectively, these data indicate that there may be brain regional and developmental age selectivity for the effects of prenatal ethanol exposure on NMDA receptors. Recently, it was proposed that ethanol acts by a dual mechanism of blockade of NMDA receptors and excessive activation of GABAA receptors to produce apoptotic neurodegeneration in the developing rat forebrain (Ikonomidou et al., 2000). The results of the present study demonstrate that there are differential long-term effects of chronic prenatal ethanol exposure on the NMDA and GABAA receptors in the adult cerebral cortex.
In conclusion, these findings in the guinea pig are novel and could have an important impact on elucidating the long-term neurobehavioral effects of chronic prenatal ethanol exposure. The GABAA receptor system is the major inhibitory neurotransmitter system in the cerebral cortex and is present in 20–50% of cerebral cortical synapses (Bloom and Iversen, 1971; Halasy and Somogyi, 1993). The results of this study suggest that there may therefore be altered inhibitory neurotransmission and GABAA receptor pharmacology within this brain region both during normal physiological activity and in response to neuroactive xenobiotics. The observed increases in GABAA receptor number and expression of GABAA receptor α1 and β2/3 subunit proteins may be causally related, at least in part, to the known consequences of chronic prenatal ethanol exposure in the human, including deficits in cognition, attention, learning, and problem solving. This neurotransmitter receptor system may therefore be a potential target for therapeutic intervention to lessen the long-term neurobehavioral effects of prenatal ethanol exposure.
Footnotes
This work was supported by the Canadian Institutes of Health Research Operating Grant MT-15150. C.D.C.B. is the recipient of a Doctoral Research Award from the Canadian Institutes of Health Research and the NeuroScience Canada Foundation.
Correspondence should be addressed to Dr. James N. Reynolds, Department of Pharmacology and Toxicology, Queen's University, Kingston, Ontario, Canada K7L 3N6. E-mail: jnr@post.queensu.ca.
REFERENCES
- 1.Abdollah S, Brien JF. Effect of chronic maternal ethanol administration on glutamate and N-methyl-d-aspartate binding sites in the hippocampus of the near-term fetal guinea pig. Alcohol. 1995;12:377–382. doi: 10.1016/0741-8329(95)00021-i. [DOI] [PubMed] [Google Scholar]
- 2.Abdollah S, Catlin MC, Brien JF. Ethanol neuro-behavioural teratogenesis in the guinea pig: behavioural dysfunction and hippocampal morphological change. Can J Physiol Pharmacol. 1993;71:776–782. doi: 10.1139/y93-116. [DOI] [PubMed] [Google Scholar]
- 3.Allan AM, Wu H, Paxton LL, Savage DD. Prenatal ethanol exposure alters the modulation of the γ-aminobutyric acidA receptor-gated chloride ion channel in adult rat offspring. J Pharmacol Exp Ther. 1998;284:250–257. [PubMed] [Google Scholar]
- 4.Bailey CDC, Brien JF, Reynolds JN. Altered GABAA-benzodiazepine receptor number and pharmacology in the adult guinea pig cerebral cortex after chronic prenatal ethanol exposure. Alcohol Clin Exp Res. 1999;23:1816–1824. [PubMed] [Google Scholar]
- 5.Barnard EA, Skolnick P, Olsen RW, Mohler H, Sieghart W, Biggio G, Braestrup C, Bateson AN, Langer SZ. International Union of Pharmacology. XV. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. Pharmacol Rev. 1998;50:291–313. [PubMed] [Google Scholar]
- 6.Bloom FE, Iversen LL. Localizing 3H-GABA in nerve terminals of rat cerebral cortex by electron microscopic autoradiography. Nature. 1971;229:628–630. doi: 10.1038/229628a0. [DOI] [PubMed] [Google Scholar]
- 7.Bonnert TP, McKernan RM, Farrar S, le Bourdelles B, Heavens RP, Smith DW, Hewson L, Rigby MR, Sirinathsinghji DJS, Brown N, Wafford KA, Whiting PJ. θ, a novel γ-aminobutyric acid type A receptor subunit. Proc Natl Acad Sci USA. 1999;96:9891–9896. doi: 10.1073/pnas.96.17.9891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 9.Bureau M, Laschet J, Bureau-Heeren M, Hennuy B, Minet A, Wins P, Grisar T. Astroglial cells express large amounts of GABAA receptor proteins in mature brain. J Neurochem. 1995;65:2006–2015. doi: 10.1046/j.1471-4159.1995.65052006.x. [DOI] [PubMed] [Google Scholar]
- 10.Butters NS, Gibson MAS, Reynolds JN, Brien JF. Effects of chronic prenatal ethanol exposure on hippocampal glutamate release in the postnatal guinea pig. Alcohol. 2000;21:1–9. doi: 10.1016/s0741-8329(99)00096-8. [DOI] [PubMed] [Google Scholar]
- 11.Catlin MC, Abdolloh S, Brien JF. Dose-dependent effects of prenatal ethanol exposure in the guinea pig. Alcohol. 1993;10:109–115. doi: 10.1016/0741-8329(93)90089-7. [DOI] [PubMed] [Google Scholar]
- 12.Chiu J, Brien JF, Wu P, Eubanks JH, Zhang L, Reynolds JN. Chronic ethanol exposure alters MK-801 binding sites in the cerebral cortex of the near-term fetal guinea pig. Alcohol. 1999;17:215–221. doi: 10.1016/s0741-8329(98)00050-0. [DOI] [PubMed] [Google Scholar]
- 13.Costa ET, Olivera DS, Meyer DA, Ferreira VM, Soto EE, Frausto S, Savage DD, Browning MD, Valenzuela CF. Fetal alcohol exposure alters neurosteroid modulation of hippocampal N-methyl-d-aspartate receptors. J Biol Chem. 2000;275:38268–38274. doi: 10.1074/jbc.M004136200. [DOI] [PubMed] [Google Scholar]
- 14.Devaud LL, Smith FD, Grayson DR, Morrow AL. Chronic ethanol consumption differentially alters the expression of γ-aminobutyric acidA receptor subunit mRNAs in rat cerebral cortex: competitive, quantitative reverse transcriptase-polymerase chain reaction analysis. Mol Pharmacol. 1995;48:861–868. [PubMed] [Google Scholar]
- 15.Devaud LL, Fritschy JM, Sieghart W, Morrow AL. Bidirectional alterations of GABAA receptor subunit peptide levels in rat cortex during chronic ethanol consumption and withdrawal. J Neurochem. 1997;69:126–130. doi: 10.1046/j.1471-4159.1997.69010126.x. [DOI] [PubMed] [Google Scholar]
- 16.Elvidge H. Production of dated pregnant guinea pigs without postpartum matings. J Inst Animal Tech. 1972;23:111–117. [Google Scholar]
- 17.Gibson MAS, Butters NS, Reynolds JN, Brien JF. Effects of chronic prenatal ethanol exposure on locomotor activity, and hippocampal weight, neurons, and nitric oxide synthase activity of the young postnatal guinea pig. Neurotoxicol Teratol. 2000;22:183–192. doi: 10.1016/s0892-0362(99)00074-4. [DOI] [PubMed] [Google Scholar]
- 18.Halasy K, Somogyi P. Distribution of GABAergic synapses and their targets in the dentate gyrus of rat: a quantitative immunoelectron microscopic analysis. J Hirnforsch. 1993;34:299–308. [PubMed] [Google Scholar]
- 19.Hornung J-P, Fritschy J-M. Developmental profile of GABAA-receptors in the Marmoset monkey: expression of distinct subtypes in pre- and postnatal brain. J Comp Neurol. 1996;367:413–430. doi: 10.1002/(SICI)1096-9861(19960408)367:3<413::AID-CNE7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 20.Hsiao SH, Mahoney JC, West JR, Frye GD. Development of GABAA receptors on medial septum/diagonal band (MD/DB) neurons after postnatal ethanol exposure. Brain Res. 1998;810:100–113. doi: 10.1016/s0006-8993(98)00891-9. [DOI] [PubMed] [Google Scholar]
- 21.Hsiao SH, West JR, Mahoney JC, Frye GD. Postnatal ethanol exposure blunts upregulation of GABAA receptor currents in Purkinje neurons. Brain Res. 1999;832:124–135. doi: 10.1016/s0006-8993(99)01480-8. [DOI] [PubMed] [Google Scholar]
- 22.Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 23.Janiri L, Gobbi G, Persico AM, Santarelli M, Minciacchi D, Tempesta E. Alterations of neocortical neuronal responses to acetylcholine and GABA in rats born to alcohol-dependent mothers. Alcohol Alcohol. 1994;29:611–619. [PubMed] [Google Scholar]
- 24.Jones KL, Smith DW. Recognition of the fetal alcohol syndrome in early infancy. Lancet. 1973;2:999–1001. doi: 10.1016/s0140-6736(73)91092-1. [DOI] [PubMed] [Google Scholar]
- 25.Kerns KA, Don A, Mateer CA, Streissguth AP. Cognitive deficits in nonretarded adults with fetal alcohol syndrome. J Learn Disabil. 1997;30:685–693. doi: 10.1177/002221949703000612. [DOI] [PubMed] [Google Scholar]
- 26.Kimura KA, Brien JF. Hippocampal nitric oxide synthase in the fetal guinea pig: effects of chronic prenatal ethanol exposure. Dev Brain Res. 1998;106:39–46. doi: 10.1016/s0165-3806(97)00184-3. [DOI] [PubMed] [Google Scholar]
- 27.Laurie DJ, Wisden W, Seeburg PH. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal developments. J Neurosci. 1992;12:4151–4172. doi: 10.1523/JNEUROSCI.12-11-04151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 29.Mattson SN, Riley EP, Delis DC, Stern C, Jones KL. Verbal learning and memory in children with fetal alcohol syndrome. Alcohol Clin Exp Res. 1996;20:810–816. doi: 10.1111/j.1530-0277.1996.tb05256.x. [DOI] [PubMed] [Google Scholar]
- 30.Mehta AK, Ticku MK. An update on GABAA receptors. Brain Res. 1999;832:164–167. doi: 10.1016/s0006-8993(99)01410-9. [DOI] [PubMed] [Google Scholar]
- 31.Mhatre MC, Ticku MK. Chronic ethanol treatment up-regulates the GABA receptor β subunit expression. Mol Brain Res. 1994;23:246–252. doi: 10.1016/0169-328x(94)90231-3. [DOI] [PubMed] [Google Scholar]
- 32.Mhatre MC, Pena G, Sieghart W, Ticku MK. Antibodies specific for GABAA receptor alpha subunits reveal that chronic alcohol treatment down-regulates alpha-subunit expression in rat brain regions. J Neurochem. 1993;61:1620–1625. doi: 10.1111/j.1471-4159.1993.tb09795.x. [DOI] [PubMed] [Google Scholar]
- 33.Miller LG, Greenblatt DJ, Roy RB, Gaver A, Lopez F, Shader RI. Chronic benzodiazepine administration. III. Upregulation of gamma-aminobutyric acidA receptor binding and function associated with chronic benzodiazepine antagonist administration. J Pharmacol Exp Ther. 1989;248:1096–1101. [PubMed] [Google Scholar]
- 34.Miller MW, Potempa G. Numbers of neurons and glia in mature rat somatosensory cortex: effects of prenatal exposure to ethanol. J Comp Neurol. 1990;293:92–102. doi: 10.1002/cne.902930108. [DOI] [PubMed] [Google Scholar]
- 35.Moore DB, Quintero MA, Ruygrok AC, Walker DW, Heaton MB. Prenatal ethanol exposure reduces parvalbumin-immunoreactive GABAergic neuronal number in the adult rat cingulate cortex. Neurosci Lett. 1998;249:25–28. doi: 10.1016/s0304-3940(98)00378-4. [DOI] [PubMed] [Google Scholar]
- 36.Morrisett RA, Martin D, Wilson WA, Savage DD, Swartzwelder HS. Prenatal exposure to ethanol decreases the sensitivity of the adult rat hippocampus to N-methyl-d-aspartate. Alcohol. 1989;6:415–420. doi: 10.1016/0741-8329(89)90013-x. [DOI] [PubMed] [Google Scholar]
- 37.Osborn JA, Yu C, Gabriel K, Weinberg J. Fetal ethanol effects on benzodiazepine sensitivity measured by behavior on the elevated plus-maze. Pharmacol Biochem Behav. 1998;60:625–633. doi: 10.1016/s0091-3057(98)00039-2. [DOI] [PubMed] [Google Scholar]
- 38.Rabow LE, Russek SJ, Farb DH. From ion currents to genomic analysis: recent advances in GABAA receptor research. Synapse. 1995;21:189–274. doi: 10.1002/syn.890210302. [DOI] [PubMed] [Google Scholar]
- 39.Savage DD, Montano CY, Otero MA, Paxton LL. Prenatal ethanol exposure decreases hippocampal NMDA-sensitive [3H]-glutamate binding site density in 45-day-old rats. Alcohol. 1991;8:193–201. doi: 10.1016/0741-8329(91)90806-8. [DOI] [PubMed] [Google Scholar]
- 40.Steenaart NAE, Clarke DW, Brien JF. Gas-liquid chromatographic analysis of ethanol and acetaldehyde in blood with minimal artifactual acetaldehyde formation. J Pharmacol Meth. 1985;14:199–212. doi: 10.1016/0160-5402(85)90033-6. [DOI] [PubMed] [Google Scholar]
- 41.Steinhausen HC, Spohr HL. Long-term outcome of children with fetal alcohol syndrome: psychopathology, behavior, and intelligence. Alcohol Clin Exp Res. 1998;22:334–338. doi: 10.1111/j.1530-0277.1998.tb03657.x. [DOI] [PubMed] [Google Scholar]
- 42.Streissguth AP, Aase JM, Clarren SK, Randels SP, LaDue RA, Smith DF. Fetal alcohol syndrome in adolescents and adults. JAMA. 1991;265:1961–1967. [PubMed] [Google Scholar]
- 43.Zimmerberg B, Drucker PC, Weider JM. Differential behavioral effects of the neuroactive steroid allopregnanolone on neonatal rats prenatally exposed to alcohol. Pharmacol Biochem Behav. 1995;51:463–468. doi: 10.1016/0091-3057(95)00008-k. [DOI] [PubMed] [Google Scholar]